

Supplemental Digital Content is available in the text.
Keywords: esters, heart failure, ketone bodies, myocardial infarction, myocardium
Background:
Accumulating evidence suggests that the failing heart reprograms fuel metabolism toward increased utilization of ketone bodies and that increasing cardiac ketone delivery ameliorates cardiac dysfunction. As an initial step toward development of ketone therapies, we investigated the effect of chronic oral ketone ester (KE) supplementation as a prevention or treatment strategy in rodent heart failure models.
Methods:
Two independent rodent heart failure models were used for the studies: transverse aortic constriction/myocardial infarction (MI) in mice and post-MI remodeling in rats. Seventy-five mice underwent a prevention treatment strategy with a KE comprised of hexanoyl-hexyl-3-hydroxybutyrate KE (KE-1) diet, and 77 rats were treated in either a prevention or treatment regimen using a commercially available β-hydroxybutyrate-(R)-1,3-butanediol monoester (DeltaG; KE-2) diet.
Results:
The KE-1 diet in mice elevated β-hydroxybutyrate levels during nocturnal feeding, whereas the KE-2 diet in rats induced ketonemia throughout a 24-hour period. The KE-1 diet preventive strategy attenuated development of left ventricular dysfunction and remodeling post-transverse aortic constriction/MI (left ventricular ejection fraction±SD, 36±8 in vehicle versus 45±11 in KE-1; P=0.016). The KE-2 diet therapeutic approach also attenuated left ventricular dysfunction and remodeling post-MI (left ventricular ejection fraction, 41±11 in MI-vehicle versus 61±7 in MI-KE-2; P<0.001). In addition, ventricular weight, cardiomyocyte cross-sectional area, and the expression of ANP (atrial natriuretic peptide) were significantly attenuated in the KE-2–treated MI group. However, treatment with KE-2 did not influence cardiac fibrosis post-MI. The myocardial expression of the ketone transporter and 2 ketolytic enzymes was significantly increased in rats fed KE-2 diet along with normalization of myocardial ATP levels to sham values.
Conclusions:
Chronic oral supplementation with KE was effective in both prevention and treatment of heart failure in 2 preclinical animal models. In addition, our results indicate that treatment with KE reprogrammed the expression of genes involved in ketone body utilization and normalized myocardial ATP production following MI, consistent with provision of an auxiliary fuel. These findings provide rationale for the assessment of KEs as a treatment for patients with heart failure.
What Is New?
Ketone ester (KE)–enriched diets that induce sustained ketonemia reduce pathological cardiac remodeling and enhance ventricular function in multiple heart failure models across both mice and rats using both prevention and treatment strategies.
In rats with heart failure, KE–enriched diets induce the expression of genes involved in the myocardial uptake and oxidation of ketones.
KE–enriched diets restore myocardial ATP production to normal values, consistent with the provision of an auxiliary fuel.
What are the Clinical Implications?
These results provide a rationale for the development of ketone therapies for heart failure in humans.
Our findings suggest that the increase in myocardial ketone oxidation observed in patients with heart failure is an adaptive response to overcome bioenergetic insufficiency.
Growing evidence indicates that derangements in cardiac fuel metabolism and bioenergetics contribute to the development of heart failure (HF).1–7 Studies conducted in animal models and in humans have revealed reprogramming of myocardial fuel utilization in the hypertrophied and failing heart with a shift away from the chief fuel metabolic pathway, fatty acid oxidation, to increased reliance on glycolysis.1–11 Recently, we and others have found that the failing rodent and human heart increases ketone body utilization.12–14 In addition, we have found that a ketogenic diet, which increases circulating levels of the ketone body β-hydroxybutyrate (βHB), retarded the development of HF in mice, and infusion of βHB markedly enhanced cardiac function and reduced elevated ventricular filling pressures in a canine tachypacing model of progressive HF.15 Recently, acute infusion of βHB to humans with HF resulted in impressive hemodynamic improvement.16 These collective results suggest that increasing delivery of ketone bodies may prevent the development of HF.
Given the growing evidence that ketones are beneficial in HF, efforts to develop chronic therapeutic strategies to increase myocardial ketone body delivery are warranted. A ketogenic diet, while increasing circulating levels of ketone bodies, introduces significant nutrient imbalances including a marked enrichment in long-chain fatty acids together with reduced carbohydrate and protein composition. Direct infusion of sodium βHB, while potentially useful for acute HF, is too invasive for chronic use.17 βHB or acetoacetate are also available as oral salt formulations but have limited bioavailability, an adverse taste, and can result in substantial sodium loading. Ketone esters (KEs) offer a more practical approach for chronic oral therapies. The first commercially available oral KE is βHB-butanediol.18–21 The βHB-butanediol ester is thought to be cleaved in the gut by esterases to yield βHB and 1,3-butanediol—the latter of which is further metabolized to βHB in the liver by alcohol and aldehyde dehydrogenases. Studies conducted in rodents and in humans have shown that this ester, when given orally admixed in liquid or solid diets, results in a substantial transient increase in circulating βHB.19,22–24 Some evidence, including studies in humans, suggests that oral administration of βHB-butanediol ester increases cognitive function and exercise performance, but their effects in HF have not been established.19–21,23,25
The goal of this study was to determine the impact of chronic oral administration of KE in rodent models of HF. We sought to assess the efficacy of a KE diet for both prevention and treatment of HF, given that the effects of chronic ketone body therapy as a treatment intervention have not been established. We report that diets enriched in 2 separate KEs were effective in partially preventing and treating HF in a mouse model of combined ventricular pressure overload/ischemia and in rats following MI. Evidence is also presented to support the conclusion that the benefit of KE treatment in HF is due, at least in part, to improving cardiac energetics. These results provide rationale for the development of ketone therapies for HF in humans.
Methods
The data generated and analyzed during the current study are available from the corresponding author on reasonable request. The description of the materials and methods is presented below and in the Data Supplement.
Ethical Statement
Mouse experiments were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. The rat experimental protocol was approved by the Animal Ethical Committee of the University of Groningen (permit number: 16487-02-002). The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85–23, revised 1996). We followed Animal Research: Reporting In Vivo Experiments guidelines when reporting this study.
KE Administration
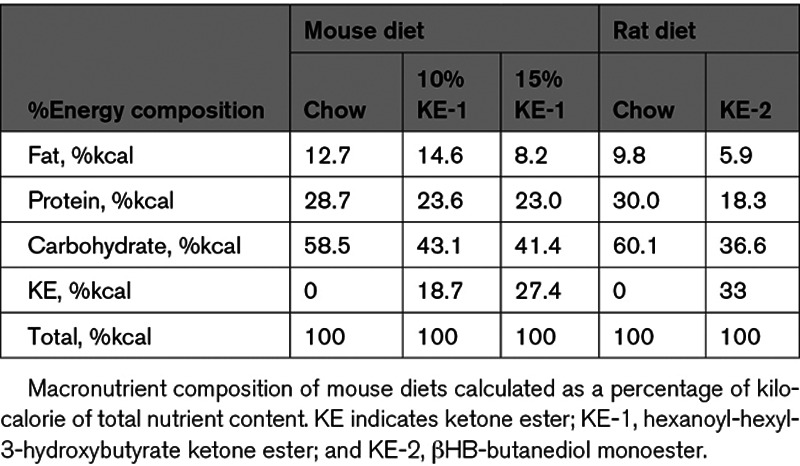
A hexanoyl-hexyl-3-hydroxybutyrate KE (KE-1; AK Scientific, Union City, CA; Figure 1A) diet was custom-synthesized for the mouse experiments and mixed with LabDiet 5010 (St. Louis, MO) by adding either 10% w/w or 15% w/w KE-1. More details can be found in the Data Supplement. For rat experiments, H.V.M.N. βHB-butanediol monoester (DeltaG; KE-2) was purchased from H.V.M.N. (San Francisco, CA) and mixed with standard rat chow (Ssniff, Germany) and sugar-free jelly crystals (Hartley’s, United Kingdom) to stimulate food intake as described elsewhere.26 The calorie composition of the diets is listed in Table 1.
Figure 1.
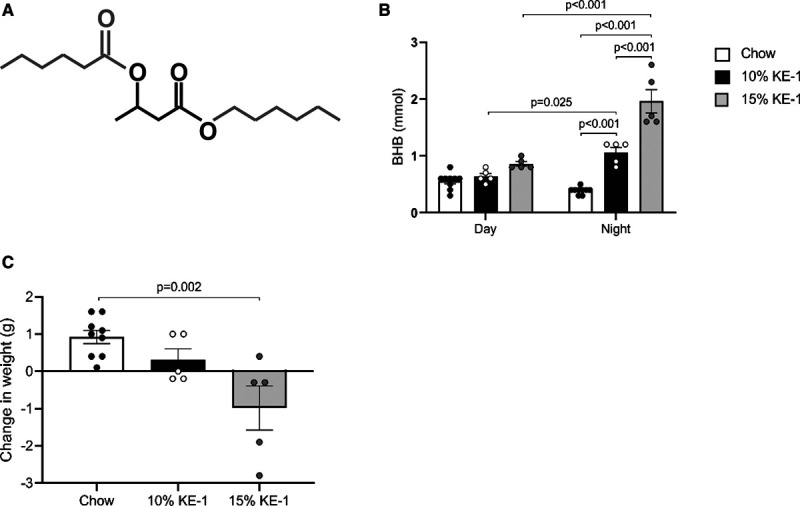
Ketone ester diet increases nocturnal circulating β-hydroxybutyrate (βHB or BHB) in mice. A, Structure of the hexanoyl-hexyl-3-hydroxybutyrate ketone ester (KE-1) depicting a central βHB flanked by two 6-carbon moieties. B, Levels of blood βHB measured after 1 wk of feeding normal chow, 10% w/w KE-1 diet, or 15% w/w KE-1 diet (n=5–9). βHB measurements were taken during the day (09:00–13:00) and at night (00:00–02:00). Two-way ANOVA with Tukey multiple comparison test. C, Body weight normalized to starting weight measured after 1 wk of feeding normal chow, 10% w/w KE-1 diet, or 15% w/w KE-1 diet. Data are presented as mean±SEM. Comparisons between groups were performed using 1-way ANOVA with Tukey multiple comparison test.
Table 1.
Composition of Mouse and Rat Diets
Animal Studies
Seventy-five male C57BL/6NJ mice (Jackson Laboratories, Bar Harbor, ME) aged 7 weeks were randomized to chow or KE-1 diet starting 1 week before surgery. HF was achieved via transverse aortic constriction (TAC) combined with an apical myocardial infarction (MI) as described previously.15,27 Sham surgery consisted of anesthesia, intubation, and intercostal thoracotomy as per the TAC/MI procedure without performing aortic constriction or coronary ligation. The experimental protocol is listed in Figure 2A.
Figure 2.
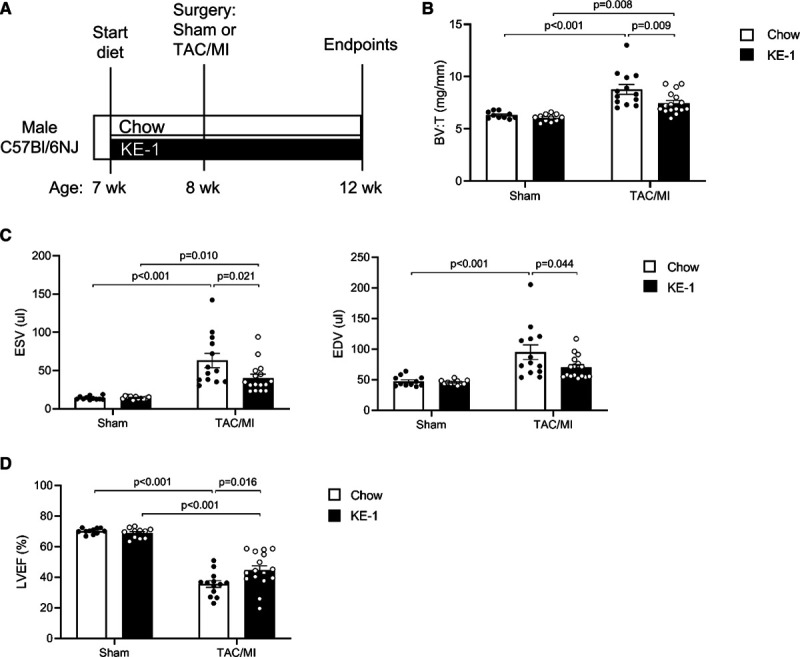
Preventive treatment with ketone ester diet attenuates pathological cardiac remodeling and left ventricular dysfunction in a mouse model of heart failure. A, Schematic showing the experimental timeline. Male C57Bl/6NJ mice aged 7 wk were randomized to diet of either standard chow or 10% hexanoyl-hexyl-3-hydroxybutyrate ketone ester (KE-1) diet. One week later, mice received either sham surgery or transverse aortic constriction (TAC)/myocardial infarction (MI) surgery. B, Ratio of biventricular weight (BV) to tibia length (T). C, Left ventricular (LV) end-systolic volume (ESV) and end-diastolic volume (EDV) 4 wk after TAC/MI surgery as determined by echocardiography. D, LV ejection fraction (LVEF) 4 wk post-TAC/MI surgery (n=11–16). Data are presented as mean±SEM. Comparisons between groups were performed using 2-way ANOVA with Tukey multiple comparison test.
Seventy-seven male Sprague-Dawley rats (Envigo, the Netherlands) were randomized to KE-2 treatment starting before surgery (early) or 2 weeks after surgery (late). HF or sham surgery was performed under isoflurane (2.5%) inhalation anesthesia as described previously.28,29 After left-sided thoracotomy, HF was induced by permanent ligating of the proximal portion of the left coronary. Sham-operated rats underwent the same procedure but without the actual ligation. The experimental protocol is illustrated in Figure 3A.
Figure 3.
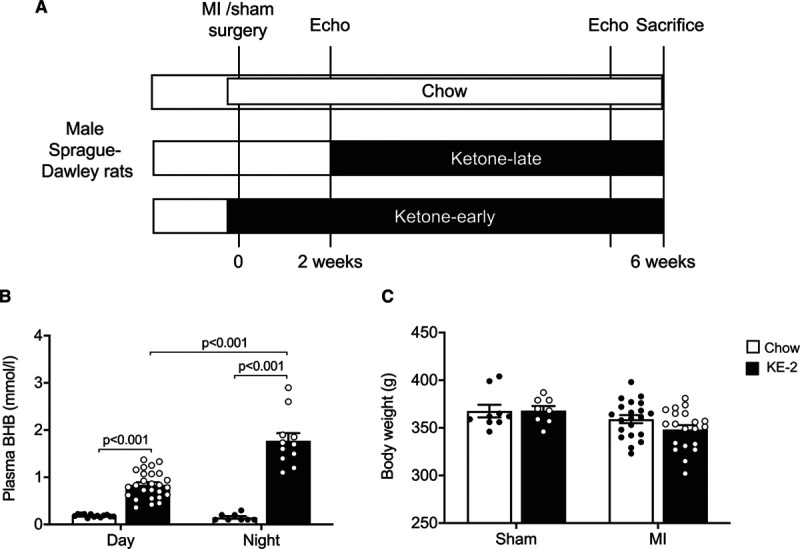
Ketone ester supplementation induces sustained ketonemia in rats. A, Experimental design of early and late β-hydroxybutyrate (βHB or BHB)–butanediol monoester (KE-2) diet supplementation regimens in rats with heart failure (HF) after myocardial infarction (MI). B, Plasma βHB concentrations measured after 10 d of KE-2 supplementation during the day (09:00–10:30) and at night (22:00–22:30; n=8–26). C, Body weight at the end of the study (n=8–20). Data are presented as mean±SEM. Comparisons between groups were performed using 2-way ANOVA with Tukey multiple comparison test. Echo indicates echocardiography.
Cardiac End Points
Echocardiography, invasive hemodynamics, and cardiac gravimetric and histological measurements were performed as described elsewhere.15,27–30
Statistical Analysis
Data are presented as mean±SD unless otherwise indicated. Two-way ANOVA followed by Tukey post hoc test was used to compare the effects of 2 variables (surgical intervention and treatment). For other normally distributed variables, 1-way ANOVA followed by Tukey post hoc test was used. When skew distributed, a nonparametric Kruskal-Wallis test followed by a Dunn test with correction for multiple comparisons was used. Wilcoxon signed-rank test was used to evaluate left ventricular (LV) ejection fraction (LVEF) post-MI versus before termination. Differences were considered significant at P<0.05. GraphPad Prism, version 8 (San Diego, CA), was used to perform all statistical analyses.
Results
Preventive Intervention With a KE Diet Attenuates Pathological Cardiac Remodeling and Dysfunction in a Mouse Model of HF
After 1 week of feeding, circulating βHB levels were significantly higher in 10% KE-1–fed mice compared with chow-fed control mice (mean, 1.1±0.2 versus 0.4±0.1 mmol/L; Figure 1B). The circulating βHB concentration was further increased in mice fed the 15% KE-1 diet (2.0±0.5 mmol/L; Figure 1B). Notably, both KE-1 diets only induced ketonemia during the main feeding period (nocturnal hours). Compared with chow-fed controls, mice fed the 15% KE-1 diet had a modest but significant weight loss during the 1-week feeding period (Figure 1C). To limit confounding effects from differences in body weight between treatment groups, the 10% KE-1 diet was selected for further studies.
We next sought to determine whether the oral KE supplementation would impact pathological cardiac remodeling en route to HF in a well-established murine model of TAC/MI. Male wild-type C57BL/6NJ mice were fed normal chow or a 10% KE-1 diet starting 1 week before TAC/MI surgery and throughout the duration of the 4-week postsurgical period (Figure 2A). The KE-1 diet induced consistent ketonemia during the dark cycle throughout the duration of the experimental protocol (Figure IA in the Data Supplement). Compared with chow-fed controls, mice fed KE-1 diet gained less weight although this weight difference stabilized during the 5-week treatment period (Figure IB in the Data Supplement). There were no differences in survival between groups fed chow or the KE-1 diet (Figure IC in the Data Supplement). Following TAC/MI, cardiac hypertrophy was significantly attenuated in mice fed KE-1 diet compared with chow-fed mice (Figure 2B). In addition, the KE-1 diet–treated group exhibited a significant improvement in pathological cardiac remodeling and left ventricular function as evidenced by reduced LV end-systolic and end-diastolic volumes (Figure 2C) and improved LVEF, respectively (Figure 2D). The KE-1 diet did not result in significant changes in cardiac function in sham-operated mice.
Development of a KE Supplementation Protocol to Induce Sustained Ketonemia in Rats
We next sought to develop a KE supplementation regimen that maintained ketonemia in rodents over a 24-hour period. For these studies, adult male Sprague-Dawley rats were used. The rats received either control chow or a KE-2 diet that was supplemented with the βHB-butanediol monoester as 30% of calories (Table 1).
A schematic representation of the treatment strategy with KE-2 in rats is depicted in Figure 3A. A detailed analysis of the ketonemic response was performed for 10 days. Rats consumed an average of 3.4±1 g of KE-2 per day, and the daily caloric intake was comparable among the groups (Table II in the Data Supplement). During the nocturnal feeding hours, the circulating βHB concentrations increased from 0.2±0.03 mmol/L in rats fed control chow to 1.8±0.2 mmol/L in rats fed the KE-2 diet (Figure 3B). Circulating ketone levels remained elevated during the day (Figure 3B). Importantly, we did not observe a significant effect on body weight after KE-2 supplementation (Figure 3C). Liver, kidney, and spleen weight were not affected by ketone treatment, and we did not observe any significant difference in plasma creatinine or electrolyte levels (all P>0.05; Table II in the Data Supplement). As expected, treatment with KE reduced plasma insulin levels without significant changes in glucagon (Table II in the Data Supplement).
Treatment With a KE Diet Attenuates Pathological Cardiac Remodeling and Dysfunction in a Rat Model of HF
Our results demonstrating that the KE-1 diet partially prevented cardiac dysfunction and remodeling following TAC/MI in mice led us to assess the potential of KE supplementation to serve as a treatment of HF. The 24-hour ketosis achieved with the KE-2 diet provided further rationale for this experiment. The treatment study was conducted in a well-established post-MI HF model in rats using the experimental design shown in Figure 3A. We used 2 treatment strategies: (1) early administration of the KE-2 diet administered 2 days before the MI surgery (KE-2-early; Figure 3A) and (2) administration of the KE-2 diet at 2 weeks post-surgery to assess the effect of KE-2 as a true treatment for post-MI remodeling (KE-2-late; Figure 3A). Early treatment with KE-2 did not result in significant changes in myocardial infarct size induced by a permanent coronary artery ligation (Figure 4A and 4B). Consistent with the results of the preventive KE-1 strategy in the mouse model, LV function and remodeling indices were improved in the KE-2-early group compared with Chow-fed controls (Figure 4B through 4E).
Figure 4.
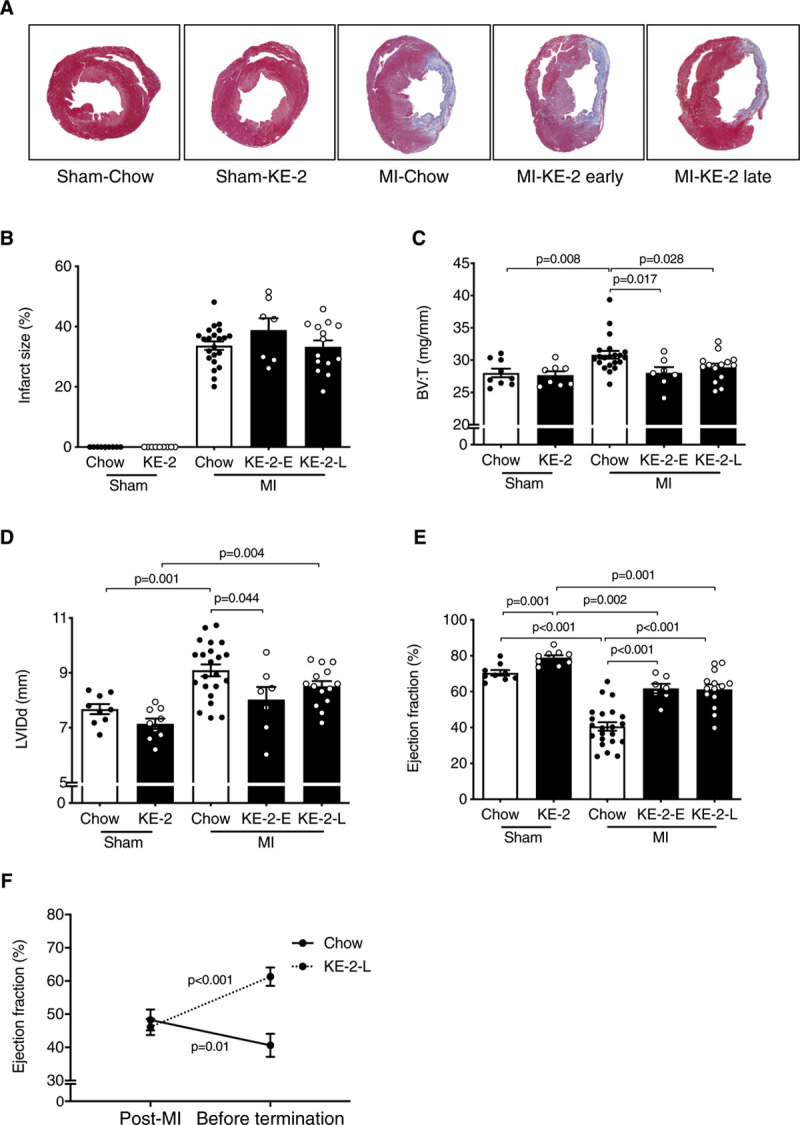
Ketone ester treatment improves pathological cardiac remodeling and left ventricular (LV) dysfunction in a rat model of heart failure. A, Representative LV sections stained with Masson trichrome. B, Quantification of infarct size from Masson trichrome stained section (n=7–22). C, Ratio of biventricular weight (BV) to tibia length (T; n=7–21). D, LV internal dimensions in diastole (LVIDd) as determined by echocardiography (n=7–22). E, LV ejection fraction (LVEF; n=7–22). Comparisons between groups were performed using 1-way ANOVA with Tukey multiple comparison test. F, Longitudinal change in LVEF between 2 wk post-myocardial infarction (MI) and termination in the βHB-butanediol monoester (KE-2)-late (KE-2-L) compared with the chow control group, tested using Wilcoxon signed-rank test. Data are presented as mean±SEM. KE-2-E indicates KE-2-early.
The impact of a treatment strategy in the context of stable cardiac dysfunction was next assessed using the KE-2-late protocol. The average MI size was 34±1% and was comparable between MI-chow and MI-KE-2 diet groups (Figure 4A and 4B). As expected, MI resulted in ventricular hypertrophy, dilatation of the LV, along with significant diminution in LVEF (Figure 4C through 4E). Treatment with KE-2 improved cardiac function and reduced cardiac remodeling (Figure 4C through 4E). Analysis of longitudinal changes in LVEF during active treatment with the KE-2-late protocol revealed modest further worsening of cardiac function in the MI-chow group, whereas improvement of cardiac function was observed with the KE-2 diet (Figure 4F). Notably, the KE-2 also slightly improved LVEF in the sham group (Figure 4E).
Hemodynamic studies were also conducted to assess the remodeling response in the KE-2 study. As expected, the infarcted rats were hemodynamically compromised, as reflected by a decrease in contractility and relaxation (dP/dtmax−min) and substantial reduction in systolic blood pressure (Table 2). Treatment with KE-2 diet reduced heart rate and improved contractility as indicated by a significantly enhanced LV dP/dtmax (Table 2). Systolic and diastolic blood pressures were similar among the groups (Table 2).
Table 2.
Hemodynamic Parameters for Sham-Operated and Post-MI Rats
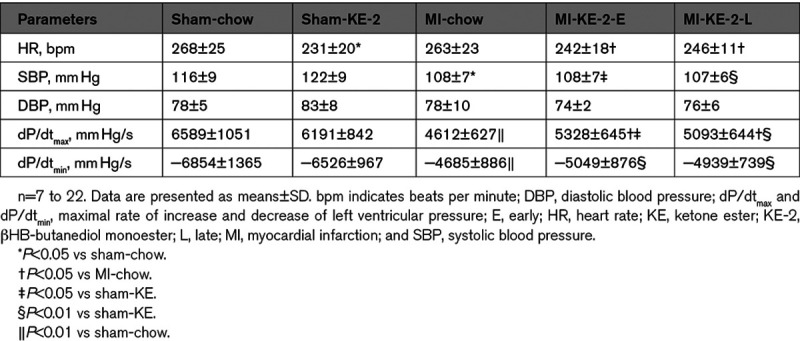
Treatment With KE Reduces Cardiac Hypertrophy Independent of the Fibrotic Response
We next evaluated the impact of KE-2 treatment on cellular and molecular markers of pathological cardiac remodeling. The ventricular weight was increased in the MI-chow control group compared with sham-chow group at 6 weeks post-MI (Figure 4C). Accordingly, this difference was also reflected in mean cardiomyocyte cross-sectional area (Figure 5A and 5B). The KE-2 diet treatment resulted in significant reductions in both ventricular weight and a 22% reduction in cardiomyocyte cross-sectional area (Figures 4C, 5A, and 5B). MI-chow rats exhibited an increase in atrial weight/tibia length ratio, which was normalized to sham values by KE-2 (Table II in the Data Supplement). In contrast to the observed amelioration of ventricular hypertrophy with KE-2 diet, interstitial fibrosis in the LV myocardial regions remote from the infarct was increased to a similar degree in both chow and KE-2–treated groups (Figure 5A and 5C). Gene markers of the hypertrophic and fibrotic response paralleled the histological responses to KE-2 diet post-MI (Figure 5D and 5E; Figure IIA in the Data Supplement). Consistent with the ventricular hypertrophy findings, LV expression of Nppa, atrial natriuretic peptide (ANP), was induced to a lesser extent in the KE-2-early group compared with controls (Figure 5D). In contrast, induction of mRNAs encoding Col1a1 (collagen type I alpha 1) and Timp1 (tissue inhibitor of metalloproteinase 1), known markers of the fibrotic response, was comparable between all MI groups (Figure 5E; Figure IIA in the Data Supplement).
Figure 5.
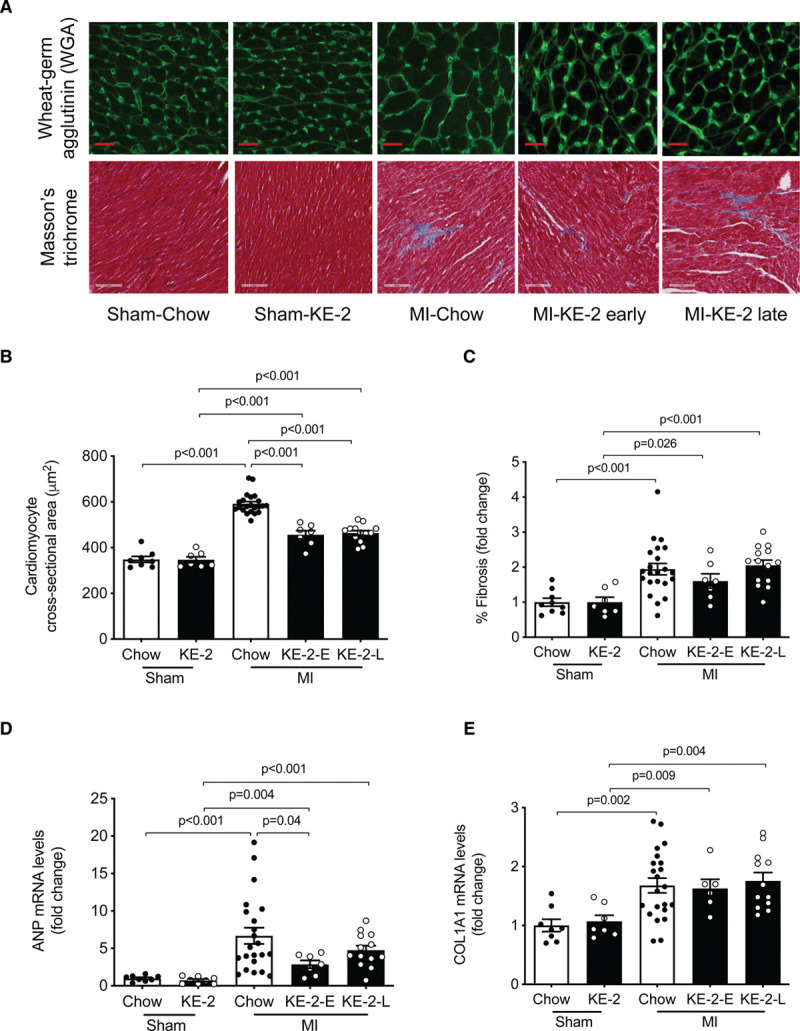
Ketone ester treatment attenuates cardiac hypertrophy independent of fibrotic response. A, Representative left ventricular (LV) sections stained with wheat germ agglutinin (WGA) and Masson trichrome to assess cardiomyocyte hypertrophy and fibrosis, respectively. B, Quantification of cardiomyocyte cross-sectional area from WGA-stained section (n=7–22). C, Quantification of fibrosis in noninfarcted LV from Masson trichrome–stained section (n=7–22). The graph denotes fold change over sham-chow control group. D and E, Measurement of mRNA levels to assess molecular markers for remodeling and fibrosis, respectively, normalized to 36B4 (n=7–22). The graph denotes fold change over sham-chow group. Data are presented as mean±SEM. Comparisons between groups were performed using 1-way ANOVA with Tukey multiple comparison test. ANP indicates atrial natriuretic peptide; COL1A1, collagen type I alpha 1; KE-2-E, βHB-butanediol monoester-early; KE-2-L, βHB-butanediol monoester-late; and MI, myocardial infarction.
Evidence for Enhanced Cardiac Energetics After Treatment With KE
Mitochondrial DNA integrity was not affected by KE-2 treatment, and the total mitochondrial content was unaltered (Figure IIB and IIC in the Data Supplement). Expression of the gene encoding MCT1 (monocarboxylate transporter 1), Slc16a1, the main transporter for ketone bodies to enter cardiomyocytes, was increased in the MI-chow compared with the sham-chow group (Figure 6A). Treatment with KE-2 resulted in a significant further increase in Slc16a1 expression in the MI-KE-2 group but did not result in significant changes in the sham-KE-2 group (Figure 6A). In addition, the expression of the gene encoding the enzyme catalyzing the first step in ketone oxidation, Bdh1, was modestly but significantly increased in the MI-chow group, and treatment with KE-2 resulted in a further increase in both sham and MI groups (Figure 6B). Furthermore, protein expression of the enzyme catalyzing the second step of ketone oxidation, succinyl CoA:3-oxo acid CoA transferase, was significantly increased in both sham and MI groups treated with KE-2 (Figure 6C). To determine the effect of KE supplementation on myocyte energetics, myocardial ATP levels were measured. Consistent with increased utilization of βHB as a fuel, the reductions in myocardial ATP levels observed in the MI-chow group were normalized to sham values in the MI-KE-2 group (Figure 6D).
Figure 6.
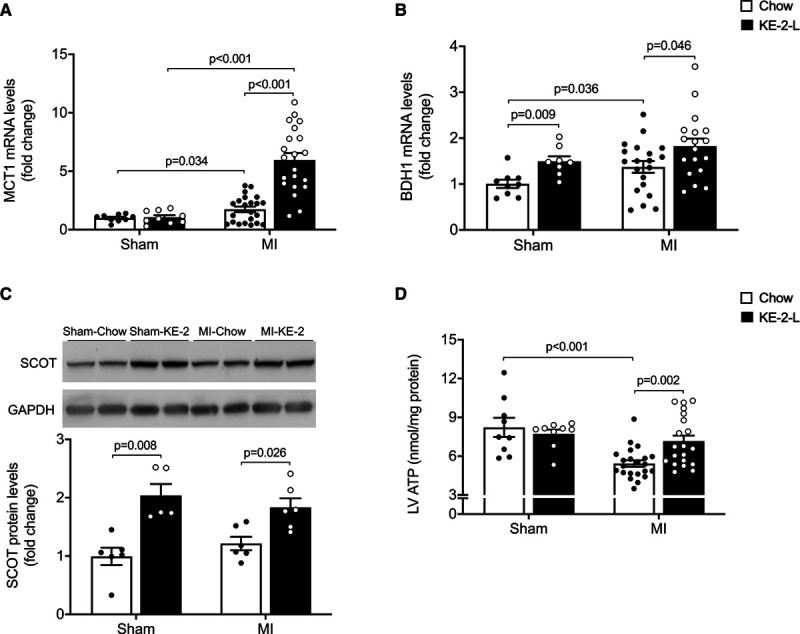
Ketone ester treatment enhances cardiac energy. Levels of mRNA encoding MCT1 (monocarboxylate transporter 1; n=9–21; A) and BDH1 (3-hydroxybutyrate dehydrogenase 1; n=9–21; B) in left ventricular (LV) free wall remote from the infarction (as determined by quantitative polymerase chain reaction) shown as arbitrary units normalized to the value of sham-chow control (+1.0). C, Top, Representative immunoblot analyses of succinyl CoA:3-oxo acid CoA transferase (SCOT) and GAPDH from protein extracts derived from the noninfarcted LV free wall. Bottom, Average SCOT protein expression normalized to GADPH and presented as fold change over the sham-chow group (n=5–6). D, ATP levels in the LV tissue (n=9–21). Data are presented as mean±SEM. Comparisons between groups were performed using Kruskal-Wallis test with Dunn multiple comparisons test. MI indicates myocardial infarction.
Discussion
This study was designed to determine whether chronic administration of a KE-supplemented diet could reduce the development and the severity of HF in preclinical small animal models. To achieve this goal, we first sought to formulate a KE-supplemented diet that was able to induce persistent ketonemia in rodents. The second major goal was to determine whether KE could not only impact the development of HF but also serve as a treatment initiated after the onset of HF. Our results demonstrated that (1) KE-enriched diets can be formulated to induce sustained ketonemia, including 24-hour exposure in rodents; (2) chronic KE supplementation was efficacious in both preventive and treatment strategies for HF in rodent models; (3) diets formulated with 2 different KE preparations, in 2 independent HF models across both mice and rats, reduced pathological cardiac remodeling and enhanced ventricular function; (4) KE supplementation reduced pathological ventricular hypertrophy but did not impact fibrosis during post-MI remodeling; and (5) chronic treatment with a βHB-butanediol monoester (KE-2 diet) led to induction of genes involved in the myocardial uptake and oxidation of βHB and recovered myocardial ATP levels post-MI.
To effectively test the impact of chronic oral ketone delivery on HF, it was necessary to ensure that sufficient and sustained ketonemia was induced. The use of KE-1, hexanoyl-hexyl-(R)-3-hydroxybutyrate, in the mouse diet resulted in a significant elevation in circulating βHB but only during periods of feeding. KE-1 was used because it generates one molecule of βHB by esterase cleavage and drives hepatic ketogenesis by oxidation of the hexanoate and hexanol moieties liberated from the initial cleavage. Whereas the KE-1 diet resulted in a significant short-term increase in blood βHB, attempts at increasing dose to attain more sustained elevations into the nonfeeding period were limited by weight loss despite a similar caloric content/weight compared with the standard chow diet. It is likely that the weight loss relates, at least in part, to the known anorexigenic effects of ketone bodies.19,31 We modified the KE administration approach by testing a different KE-2 diet, which contained βHB-butanediol monoester, and assessed whether the effects of ketone supplementation were also apparent in a different rodent species. The βHB-butanediol monoester has been used previously by others in animal models and has been shown to be safe and well tolerated in humans.32,33 To maintain food intake, the KE-2 diet was supplemented with a sugar-free jelly crystal to enhance taste. This approach resulted in a similar caloric intake for both chow and KE-2–treated groups. Accordingly, the KE-2 diet did not cause significant weight loss and resulted in sustained therapeutic ketonemia over a 24-hour period. Our results do not allow us to determine whether the differences in duration of ketonemia and weight loss between the KE-1 and KE-2 diets are related to species or specific KE formulations. Future studies aimed at addressing these questions, especially in humans, will be necessary for further development of this therapeutic approach. Notably, despite achieving ketonemia limited only to the nocturnal feeding period, the KE-1 diet was effective in the mouse HF model as a preventive treatment.
A major question related to this KE study is how do ketone bodies improve HF? Studies of aging and neurodegenerative disease in animal models suggest that ketones may be cytoprotective by several mechanisms including antioxidant and anti-inflammatory actions or through epigenetic effects related to histone deacetylase inhibition.34–36 We did not observe significant changes in mitochondrial damage as a readout for oxidative stress in rats treated with KE-2. Furthermore, we did not observe differences in myocardial infarct size induced by the early treatment, although it should be noted that our permanent ligation model is not the most suitable to study early infarct formation. In addition, acute administration of βHB may reduce systemic vascular resistance in HF as suggested by large animal15 and human studies.16,37 Ketone bodies can also be used by the heart as a fuel. It is well known that the failing heart reprograms to a lower capacity for oxidizing fatty acids—the chief fuel for the normal heart.8 Several lines of evidence, including the cardiomyopathy phenotype of human genetic defects in mitochondrial β-oxidation,38 suggest that reduced capacity for myocardial fatty acid utilization contributes to a vicious cycle of energy starvation and pathological remodeling. Accordingly, ketones could supply an alternate mitochondrial energy substrate to the failing heart. In support of this mechanism, infusion of βHB in a canine tachypacing HF model resulted in a marked increase in ventricular function and reduced glucose utilization suggestive of substrate competition.15 In addition, administration of βHB to isolated mitochondria in the context of limited substrate availability improved the redox state, mitochondrial membrane potential, and proton motive force for ATP synthesis.15 The results described herein provide additional support for ketone bodies serving as an alternate fuel for the failing heart. First, cardiac expression of the main βHB transporter, MCT1, and the ketolytic enzymes Bdh1 and SCOT were increased in the post-MI rat heart and further increased by the KE-2 diet. Ketolytic enzymes have also been shown to be induced in HF in mice and humans.13,14 Second, KE-2 diet normalized reduced ATP levels in the post-MI rat heart. These results are supportive of the conclusion that KE supplementation provides an ancillary fuel to the failing heart. However, other cardioprotective or extracardiac hemodynamic mechanisms may also contribute. Interestingly, we also found that KE supplementation reduced cardiac hypertrophy, both in mice and rats. In the rat post-MI model, this effect was shown to be uncoupled from the fibrotic response. It is tempting to speculate that the increased mitochondrial oxidation of βHB reduces glucose utilization, thus impacting the hypertrophic growth response. Indeed, pathological cardiac hypertrophic response is diminished in recent studies in which high rates of fatty acid oxidation are maintained.39,40 It is possible that increasing ketone-generated acetyl-CoA input into the TCA cycle would have a similar effect.
Translational Implications
The therapeutic paradigm for the treatment of HF has recently shifted from inhibition of potentially deleterious neurohormonal activation toward stimulation of endogenous protective responses. Indeed, substantial improvements in outcome have been achieved by increasing natriuretic peptides with neprilysin inhibition41 and more recently by activating guanyl cyclase.42 Promoting an adaptive metabolic switch toward enhanced myocardial ketone oxidation would nicely fit into this paradigm. Indirect evidence that increasing circulating ketone concentrations may benefit HF patients comes from the mild but persistent ketonemia induced by SGLT2 (sodium-glucose cotransporter 2) inhibitors. SGLT2 inhibitors have been shown to improve cardiac function in small and large animal models of HF, accompanied by increased cardiac ketone utilization and partial normalization of myocardial ATP.28,43 The SGLT2 inhibitor dapagliflozin was also recently found to reduce HF hospitalizations and prevent cardiovascular mortality in patients with HF and reduced LVEF,44 both with or without diabetes at baseline.45 While it is tempting to speculate that these benefits are mediated by ketones, many other potential mechanisms of action may exist. Further mechanistic studies are required to determine the true contribution of ketone oxidation to the salutary effects of SGLT2 inhibition.46
The only direct clinical evidence that exogenous ketones could benefit HF patients comes from a small Danish study in patients with HF and reduced LVEF who received sodium-βHB for up to 3 hours, during which invasive cardiac pressure measurements and echocardiography were performed.16 Acute βHB infusion improved biventricular function and resulted in a dose-dependent increase in cardiac output, without major hemodynamic side effects. In this study, we sought to assess the chronic effects of βHB on pathological cardiac remodeling. While intravenous βHB infusion warrants further investigation as a treatment for selected patients with acute HF, the KE βHB-butanediol or related esters are excellent candidates for initial clinical trials in chronic HF. βHB-butanediol is commercially available, and a single oral dose of βHB-butanediol was shown to be sufficient to improve exercise performance in athletes and in patients with long-chain acyl-CoA dehydrogenase deficiency.25,32 Furthermore, chronic βHB-butanediol treatment is safe and well tolerated by healthy volunteers and efficacy trials in various patient populations are currently enrolling patients. Several additional KE preparations are currently under development, including esterification of medium-chain fatty acids to βHB to boost hepatic ketogenesis.
Study Limitations
Next to limitations inherent to the use of animal models to study human diseases, our study has several specific limitations. First, our results did not allow us to assess effects on myocardial ischemic damage given that we used models of permanent coronary artery ligation. It has been reported, however, that ketones exert cardioprotective properties in the setting of ischemia/reperfusion injury.47 Second, while the increase in cardiac ATP levels and the improvements in cardiac function induced by ketone supplementation strongly suggest that the heart is utilizing KE as a fuel, we do not provide direct proof that ketone oxidation is increased nor did we quantify the contribution of ketone oxidation to the increase in myocardial ATP levels. This would require mass spectrometry–based quantitative proteomics and metabolomics analyses or studies in mice incapable of oxidizing BHB.13,15 In our opinion, this is beyond the scope of the current investigation, and such effects have been demonstrated with BHB infusion in a canine pacing model of HF.22
Conclusions
We show that chronic oral supplementation with KEs prevents and treats HF in 2 preclinical small animal models. Our results also indicate that chronic treatment with KEs reprograms the expression of genes involved in ketone body uptake and oxidation and restores myocardial ATP production following MI consistent with provision of an auxiliary fuel. Taken together, these findings suggest that treatment with KEs could be of benefit for patients with HF.
Acknowledgments
We acknowledge Dr Cher-Rin Chong (Basil Hetzel Institute for Translational Research, The Queen Elizabeth Hospital, Australia, and Adelaide Medical School, University of Adelaide, Adelaide, Australia) for her support and expert advice throughout this study and thank Dr Michael G. Dickinson (University Medical Center Groningen) for validating the echo study. We thank Silke U. Oberdorf-Maass, Marloes Schouten, Martin Dokter, and Janny Takens (University Medical Center Groningen) for expert technical assistance. We acknowledge the Mouse Cardiovascular Phenotyping Core in the Cardiovascular Institute at the Perelman School of Medicine for the mouse surgeries and echocardiography.
Sources of Funding
Dr Matsuura is supported by the Penn Cardiovascular Medicine Training Program (NIH T32 HL007843). Dr de Boer is supported by the Netherlands Heart Foundation (CVON DOSIS, grant 2014-40; CVON SHE-PREDICTS-HF, grant 2017-21; and CVON RED-CVD, grant 2017-11) and the Innovational Research Incentives Scheme program of the Netherlands Organization for Scientific Research (NWO VIDI, grant 917.13.350). Dr Westenbrink is supported by the Netherlands Organisation for Scientific Research (NWO VENI, grant 016.176.147) and the Netherlands Heart Foundation Senior Clinical Scientist Grant (2019T064). Dr Kelly is supported by NIH R01 HL151345 and HL128349.
Disclosures
The University Medical Center Groningen, which employs Dr Yurista, Dr Silljé, K.T. Nijholt, Dr van Veldhuisen, Dr de Boer, and Dr Westenbrink, has received research grants and fees from Abbott, AstraZeneca, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche. Dr de Boer received personal fees from Abbott, AstraZeneca, and Novartis. Drs Newman and Verdin are cofounders with equity interest in BHB Therapeutics, Inc. Dr Verdin is an advisor to KetoFuel, Inc. Dr Newman serves on the scientific advisory board of Virta Health, Inc, and Roche.
Supplemental Materials
Methods
Figures I and II
Tables I and II
Supplementary Material
Nonstandard Abbreviations and Acronyms
- βHB
- β-hydroxybutyrate
- ANP
- atrial natriuretic peptide
- COL1A1
- collagen type I alpha 1
- HF
- heart failure
- KE
- ketone ester
- KE-1
- hexanoyl-hexyl-3-hydroxybutyrate ketone ester
- KE-2
- βHB-butanediol monoester
- LV
- left ventricle
- LVEF
- left ventricular ejection fraction
- MCT1
- monocarboxylate transporter 1
- MI
- myocardial infarction
- SGLT2
- sodium-glucose cotransporter 2
- TAC
- transverse aortic constriction
- TIMP1
- tissue inhibitor of metalloproteinase 1
Drs Yurista and Matsuura contributed equally to this work.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCHEARTFAILURE.120.007684.
For Sources of Funding and Disclosures, see page 123.
Contributor Information
Salva R. Yurista, Email: s.r.yurista@umcg.nl.
Timothy R. Matsuura, Email: Timothy.Matsuura@pennmedicine.upenn.edu.
Herman H.W. Silljé, Email: h.h.w.sillje@umcg.nl.
Kirsten T. Nijholt, Email: k.t.nijholt@umcg.nl.
Kendra S. McDaid, Email: Kendra.mcdaid@pennmedicine.upenn.edu.
Swapnil V. Shewale, Email: vsw@pennmedicine.upenn.edu.
Teresa C. Leone, Email: terleone@pennmedicine.upenn.edu.
John C. Newman, Email: jnewman@buckinstitute.org.
Eric Verdin, Email: EVerdin@buckinstitute.org.
Dirk J. van Veldhuisen, Email: d.j.van.veldhuisen@umcg.nl.
Rudolf A. de Boer, Email: r.a.de.boer@umcg.nl.
Daniel P. Kelly, Email: dankelly@pennmedicine.upenn.edu.
References
- 1.Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol. 1970;218:153–159. doi: 10.1152/ajplegacy.1970.218.1.153 [DOI] [PubMed] [Google Scholar]
- 2.Taegtmeyer H, Overturf ML. Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension. 1988;11:416–426. doi: 10.1161/01.hyp.11.5.416 [DOI] [PubMed] [Google Scholar]
- 3.Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;2672 pt 2H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742 [DOI] [PubMed] [Google Scholar]
- 4.Christe ME, Rodgers RL. Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J Mol Cell Cardiol. 1994;26:1371–1375. doi: 10.1006/jmcc.1994.1155 [DOI] [PubMed] [Google Scholar]
- 5.Paolisso G, Gambardella A, Galzerano D, D’Amore A, Rubino P, Verza M, Teasuro P, Varricchio M, D’Onofrio F. Total-body and myocardial substrate oxidation in congestive heart failure. Metabolism. 1994;43:174–179. doi: 10.1016/0026-0495(94)90241-0 [DOI] [PubMed] [Google Scholar]
- 6.Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. Energy metabolism in the hypertrophied heart. Heart Fail Rev. 2002;7:161–173. doi: 10.1023/a:1015380609464 [DOI] [PubMed] [Google Scholar]
- 7.Chandler MP, Kerner J, Huang H, Vazquez E, Reszko A, Martini WZ, Hoppel CL, Imai M, Rastogi S, Sabbah HN, et al. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am J Physiol Heart Circ Physiol. 2004;287:H1538–H1543. doi: 10.1152/ajpheart.00281.2004 [DOI] [PubMed] [Google Scholar]
- 8.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. doi: 10.1161/01.cir.94.11.2837 [DOI] [PubMed] [Google Scholar]
- 9.Wallhaus TR, Taylor M, DeGrado TR, Russell DC, Stanko P, Nickles RJ, Stone CK. Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation. 2001;103:2441–2446. doi: 10.1161/01.cir.103.20.2441 [DOI] [PubMed] [Google Scholar]
- 10.Dávila-Román VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:271–277. doi: 10.1016/s0735-1097(02)01967-8 [DOI] [PubMed] [Google Scholar]
- 11.de las Fuentes L, Herrero P, Peterson LR, Kelly DP, Gropler RJ, Dávila-Román VG. Myocardial fatty acid metabolism: independent predictor of left ventricular mass in hypertensive heart disease. Hypertension. 2003;41:83–87. doi: 10.1161/01.hyp.0000047668.48494.39 [DOI] [PubMed] [Google Scholar]
- 12.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart. Circ Hear Fail. 2014;7:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Krüger M, Hoppel CL, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bedi KC, Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133:706–716. doi: 10.1161/CIRCULATIONAHA.115.017545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, Recchia FA, Kelly DP. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight. 2019;4:e124079 doi: 10.1172/jci.insight.124079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen R, Møller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, Harms HJ, Frøkiær J, Eiskjaer H, Jespersen NR, et al. Cardiovascular effects of treatment with the ketone body 3-hydroxybutyrate in chronic heart failure patients. Circulation. 2019;139:2129–2141. doi: 10.1161/CIRCULATIONAHA.118.036459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdul Kadir A, Clarke K, Evans RD. Cardiac ketone body metabolism. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165739 doi: 10.1016/j.bbadis.2020.165739 [DOI] [PubMed] [Google Scholar]
- 18.Veech RL, Mehlman MA. Liver metabolite content, redox and phosphorylation states in rats fed diets containing 1,3-butanediol and ethanol. In: Energy Metabolism and the Regulation of Metabolic Processes in Mitochondria. 1972. Elsevier; 171–183. [Google Scholar]
- 19.Kashiwaya Y, Pawlosky R, Markis W, King MT, Bergman C, Srivastava S, Murray A, Clarke K, Veech RL. A ketone ester diet increases brain malonyl-CoA and uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. J Biol Chem. 2010;285:25950–25956. doi: 10.1074/jbc.M110.138198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shivva V, Cox PJ, Clarke K, Veech RL, Tucker IG, Duffull SB. The population pharmacokinetics of D-β-hydroxybutyrate following administration of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. AAPS J. 2016;18:678–688. doi: 10.1208/s12248-016-9879-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stubbs BJ, Cox PJ, Evans RD, Santer P, Miller JJ, Faull OK, Magor-Elliott S, Hiyama S, Stirling M, Clarke K. On the metabolism of exogenous ketones in humans. Front Physiol. 2017;8:848 doi: 10.3389/fphys.2017.00848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kashiwaya Y, Bergman C, Lee JH, Wan R, King MT, Mughal MR, Okun E, Clarke K, Mattson MP, Veech RL. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2013;34:1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murray AJ, Knight NS, Cole MA, Cochlin LE, Carter E, Tchabanenko K, Pichulik T, Gulston MK, Atherton HJ, Schroeder MA, et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. 2016;30:4021–4032. doi: 10.1096/fj.201600773R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pawlosky RJ, Kemper MF, Kashiwaya Y, King MT, Mattson MP, Veech RL. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J Neurochem. 2017;141:195–207. doi: 10.1111/jnc.13958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B, West J, McLure SW, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2016;24:256–268. doi: 10.1016/j.cmet.2016.07.010 [DOI] [PubMed] [Google Scholar]
- 26.Clarke K, Tchabanenko K, Pawlosky R, Carter E, Knight NS, Murray AJ, Cochlin LE, King MT, Wong AW, Roberts A, et al. Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul Toxicol Pharmacol. 2012;63:196–208. doi: 10.1016/j.yrtph.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinheimer CJ, Lai L, Kelly DP, Kovacs A. Novel mouse model of left ventricular pressure overload and infarction causing predictable ventricular remodelling and progression to heart failure. Clin Exp Pharmacol Physiol. 2015;42:33–40. doi: 10.1111/1440-1681.12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yurista SR, Silljé HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, van Goor H, van Veldhuisen DJ, de Boer RA, Westenbrink BD. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. 2019;21:862–873. doi: 10.1002/ejhf.1473 [DOI] [PubMed] [Google Scholar]
- 29.Yurista SR, Silljé HHW, Nijholt KT, Dokter MM, van Veldhuisen DJ, de Boer RA, Westenbrink BD. Factor Xa inhibition with apixaban does not influence cardiac remodelling in rats with heart failure after myocardial infarction. Cardiovasc Drugs Ther. 2020. doi: 10.1007/s10557-020-06999-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yurista SR, Silljé HHW, van Goor H, Hillebrands JL, Heerspink HJL, de Menezes Montenegro L, Oberdorf-Maass SU, de Boer RA, Westenbrink BD. Effects of sodium-glucose co-transporter 2 inhibition with empaglifozin on renal structure and function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Cardiovasc Drugs Ther. 2020;34:311–321. doi: 10.1007/s10557-020-06954-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis RAH, Deemer SE, Bergeron JM, Little JT, Warren JL, Fisher G, Smith DL, Jr, Fontaine KR, Dickinson SL, Allison DB, et al. Dietary R, S-1,3-butanediol diacetoacetate reduces body weight and adiposity in obese mice fed a high-fat diet. FASEB J. 2019;33:2409–2421. doi: 10.1096/fj.201800821RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bleeker JC, Visser G, Clarke K, Ferdinandusse S, de Haan FH, Houtkooper RH, IJlst L, Kok IL, Langeveld M, van der Pol WL, et al. Nutritional ketosis improves exercise metabolism in patients with very long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2020;43:787–799. doi: 10.1002/jimd.12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soto-Mota A, Vansant H, Evans RD, Clarke K. Safety and tolerability of sustained exogenous ketosis using ketone monoester drinks for 28 days in healthy adults. Regul Toxicol Pharmacol. 2019;109:104506 doi: 10.1016/j.yrtph.2019.104506 [DOI] [PubMed] [Google Scholar]
- 34.Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–269. doi: 10.1038/nm.3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, Qi S, Li J, Colak G, Chen Y, et al. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol Cell. 2016;62:194–206. doi: 10.1016/j.molcel.2016.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gormsen LC, Svart M, Thomsen HH, Søndergaard E, Vendelbo MH, Christensen N, Tolbod LP, Harms HJ, Nielsen R, Wiggers H, et al. Ketone body infusion with 3-hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: a positron emission tomography study. J Am Heart Assoc. 2017;6:e005066 doi: 10.1161/JAHA.116.005066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Epstein FH, Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330:913–919. doi: 10.1056/NEJM199403313301308 [DOI] [PubMed] [Google Scholar]
- 39.Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, Hsu YA, Kolwicz SC, Jr, Raftery D, Tian R. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res. 2020;126:182–196. doi: 10.1161/CIRCRESAHA.119.315483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kolwicz SC, Airhart S, Tian R. Ketones step to the plate. Circulation. 2016;133:689–691. doi: 10.1161/CIRCULATIONAHA.116.021230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, Martinez F, Packer M, Pfeffer MA, Pieske B, et al. ; PARAGON-HF Investigators and Committees. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. 2019;381:1609–1620. doi: 10.1056/NEJMoa1908655 [DOI] [PubMed] [Google Scholar]
- 42.Armstrong PW, Pieske B, Anstrom KJ, Ezekowitz J, Hernandez AF, Butler J, Lam CSP, Ponikowski P, Voors AA, Jia G, et al. ; VICTORIA Study Group. Vericiguat in patients with heart failure and reduced ejection fraction. N Engl J Med. 2020;382:1883–1893. doi: 10.1056/NEJMoa1915928 [DOI] [PubMed] [Google Scholar]
- 43.Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, Flores E, Garcia-Ropero A, Sanz J, Hajjar RJ, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. 2019;73:1931–1944. doi: 10.1016/j.jacc.2019.01.056 [DOI] [PubMed] [Google Scholar]
- 44.Petrie MC, Verma S, Docherty KF, Inzucchi SE, Anand I, Belohlávek J, Böhm M, Chiang CE, Chopra VK, de Boer RA, et al. Effect of dapagliflozin on worsening heart failure and cardiovascular death in patients with heart failure with and without diabetes. JAMA. 2020;323:1353–1368. doi: 10.1001/jama.2020.1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, et al. ; DAPA-HF Trial Committees and Investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008. doi: 10.1056/NEJMoa1911303 [DOI] [PubMed] [Google Scholar]
- 46.Zelniker TA, Braunwald E. Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J Am Coll Cardiol. 2020;75:422–434. doi: 10.1016/j.jacc.2019.11.031 [DOI] [PubMed] [Google Scholar]
- 47.Yu Y, Yu Y, Zhang Y, Zhang Z, An W, Zhao X. Treatment with D-β-hydroxybutyrate protects heart from ischemia/reperfusion injury in mice. Eur J Pharmacol. 2018;829:121–128. doi: 10.1016/j.ejphar.2018.04.019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.