

Abstract
Esophageal cancer (EC) is a common cancer and is histopathologically classified into esophageal squamous cell carcinoma and esophageal adenocarcinoma. EC is a worldwide public health issue because of late diagnosis and lack of effective therapy. In contrast to standard tumor biopsies, liquid biopsies are emerging as a tool which is minimally invasive that can complement or even substitute more classical approaches. Specifically, cell‐free DNA (cfDNA) has shown promise in cancer‐related clinical applications. Indeed, cfDNA has been shown to be an effective circulating biomarker for non‐invasive cancer diagnosis and monitoring of cancer patients. Although the clinical application of cfDNA has been reported on other cancers, few studies have evaluated its use in EC. Here, we review this relevant literature and discuss limitations and advantages of its application in the diagnosis and monitoring of EC.

Abbreviations
- EC
Esophageal cancer
- ESCC
esophageal squamous cell carcinoma
- EAC
esophageal adenocarcinoma
- cfDNA
circulating cell‐free DNA
- ctDNA
circulating tumor DNA
- TNM
tumor, node and metastasis
- CRT
chemoradiotherapy
- NSCLC
non‐small cell lung cancer
- PRISMA
preferred reporting items for systematic reviews and meta‐analyses
- MeSH
medical subject headings
- VAFs
variant allele frequencies
- PCR
polymerase chain reaction
- NGS
next‐generation sequencing
- ddPCR
digital droplet polymerase chain reaction
- WGS
whole genome sequencing
- SALP
single strand adaptor library preparation
- TP53
tumor protein 53
- FAT3
fat atypical cadherin 3
- MLL3
mixed lineage leukemia 3
- AJUBA
ajuba LIM protein
- TGPS
targeted gene panel sequencing
- WES
whole exome sequencing
- 5hmC
5‐hydroxylmethylcytosine
- 5mC
5‐methylcytosine
- SiMSen‐seq
simple, multiplexed, PCR‐based barcoding of DNA for sensitive mutation detection using sequencing
- MB
molecular barcodes
- WBC
white blood cells
- CT
computed tomography
- PET‐CT
Positron emission tomography‐computed tomography
- AUC
area under curve
- CpG
cytosine adjacent to guanine
- LINE‐1
long interspersed nuclear element
- GEA
gastric and esophageal adenocarcinomas
- ARID1A
AT‐rich interaction domain 1A
- ERBB4
erb‐B2 receptor tyrosine kinase 4
- NR
not reported
1. BACKGROUND
Esophageal cancer (EC) is a common and highly aggressive malignancy that causes over 400,000 deaths annually. EC is classified into two major histopathological subtypes: esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC). Although both subtypes lead to poor outcomes (5‐year survival rate of about 15%), they have different cell of origin, epidemiology, and tumor molecular biology [1]. ESCC is the predominant subtype that derives from the squamous epithelial cells in the esophagus. In contrast, EAC originates from glandular cells present in the gastro‐esophageal junction and has been linked to gastric acid reflux at the lower esophagus [2]. Their epidemiological characteristics are also different. Around 79% of global ESCC cases are found in Southeast and Central Asia, and it is also present in Southeast Africa and South America [3]. In contrast, EAC is a major subtype in North and West Europe, North America and Oceania, accounting for about 46% of global cases [4]. ESCC incidence is declining in most parts of the world, whereas EAC incidence has risen sharply in developed countries over the past four decades [5].
The precise mechanisms underlying the pathogenesis of EC are still unclear, although both environmental and genetic factors are suspected to be involved. Tobacco and alcohol consumption are major environmental risk factors, and environmental and dietary factors have been used to explain geographical differences in mode, incidence, and sex ratios [6, 7]. EC management depends on patient and tumor characteristics, especially the tumor‐node‐metastasis (TNM) stage. At early stages, tumors may be suitable for endoscopic resection, whereas locally advanced EC is treated with surgical resection, chemotherapy, chemoradiotherapy (CRT), or a combination of them. Patients with unresectable EC are treated with systemic chemotherapy [8].
In 1948, Mandel and Metais [9] discovered the presence of circulating cell‐free DNA (cfDNA) in human blood samples. Thirty years later, it was realized that cfDNA in blood samples from cancer patient was more abundant than in those from healthy individuals [10]. The idea that tumor‐derived cfDNA, known as circulating tumor DNA (ctDNA), is present in the circulation came from the observation that cfDNA derived from cancer patients harbored tumor‐specific genetic alterations [11, 12]. In patients with bladder, colorectal, and non‐small cell lung cancers (NSCLC), both cfDNA and ctDNA was detected in stool and several types of fluids, such as recent urine [13], saliva [14], cerebrospinal [15] and pleural fluids [16]. The exact origin and molecular releasing mechanism of cfDNA is still poorly understood. In both normal and malignant cells, cfDNA is thought to derive from apoptosis and necrosis [16] or linked to secretion of exosomes [17, 18, 19].
It is possible that the mechanism for DNA condensation and release into blood, and perhaps cell origin itself, determines cfDNA fragment size [20]. Indeed, the source and mechanism of production of cfDNA could mark it with specific signatures, potentially providing valuable information about cell type, gene expression, or response to therapy [21, 22, 23].
Typically, cfDNA is observed as double‐stranded fragments approximately 150‐200 base pairs (bp) long, as expected for nucleosome‐associated DNA [24]. Crucially, the fraction of cfDNA which is released from primary tumors or metastases, referred to as ctDNA, harbors genetic alterations that can be potentially used as diagnostic, prognostic, and predictive biomarkers [11, 12]. Thus, cfDNA analysis may be used to assess the genetic profile of a tumor from a simple blood draw, without the need for an invasive biopsy. However, although the feasibility of using cfDNA to detect resistance mutations in treated cancer patients has been reported [25], whether cfDNA‐based and tissue biopsy‐derived mutational profiles are equivalent remains debatable [26].
Although studies on cfDNA analysis in the context of EC are still scarce, the present review summarizes both feasibility and potential use of a molecular biomarker in EC clinical practice.
2. LITERATURE ACQUISITION
This review was conducted following the guidelines in the Preferred Reporting Items for Systematic Reviews and Meta‐analyses (PRISMA). Pubmed, Medline, and Embase databases were searched until March 2020, and EC medical subject heading (MeSH) terms were combined with terms cfDNA or ctDNA (Figure 1). All titles and abstracts were screened by two of the authors (Z.Y. and X.W.) to determine eligibility, and divergences were resolved by discussion until consensus was reached. After removing duplicates, 59 titles and abstracts were screened, and 20 full‐text papers were reviewed describing the clinical utility of cfDNA of typical length 150‐200 bp in EC patients. Non‐English publications were excluded. Z.Y. and X.W. reviewed the papers independently, and 9 papers that met all criteria for inclusion were selected, with extracted study characteristics shown in Tables 1 and 2. From the objectives and results, studies were classified as diagnostic, monitoring, or prognostic. Studies were classified as diagnostic when cfDNA was used to detect or diagnose EC versus healthy controls, as monitoring when cfDNA was used to monitor disease status or response to therapy, and as prognostic when the association of cfDNA dynamics with residual disease after curative resection or outcomes after systemic therapy was studied. Endnote (version 9.0, Thomson Reuters, New York City, NY, USA) was used to select and screen the literature.
FIGURE 1.
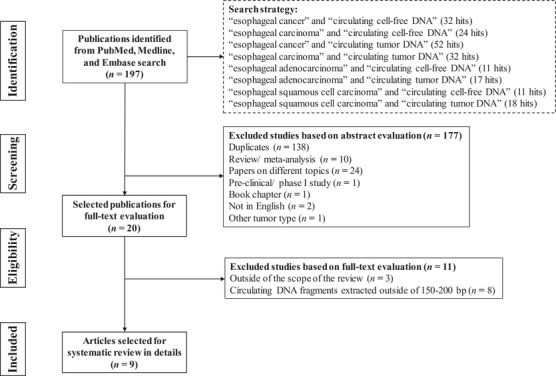
PRISMA flow diagram of the selection of studies on the clinical applications of cfDNA in EC. PRISMA = Preferred Reporting Items for Systematic Reviews and Meta‐analyses, cfDNA = circulating cell‐free DNA, EC = esophageal cancer
TABLE 1.
Baseline characteristics of the included studies on the clinical applications of cfDNA in EC
| Included study | Objective | Subtype of EC | Number of patients | Treatment | Timing of sampling | Marker/gene(s) of interest | VAFs | ctDNA positivity | Conclusion(s) |
|---|---|---|---|---|---|---|---|---|---|
| Pasternack et al. [42] | Prognosis | ESCC, EAC and BE | 27 (7 ESCC, 19 EAC, 1 BE) | Resection, neoadjuvant therapy, and definitive CRT | Pre‐treatment | Panel of 12 cancer‐related genes | NGS: 1.0%‐2.4%; ddPCR: <1.5% mostly | 3/27 (11.1%) | Compared to NGS, ddPCR has a higher sensitivity to detect cfDNA mutations in EC. Detection of somatical alterations in cfDNA during primary staging is indicative for post‐resection tumor recurrence. |
| Wu et al. [47] | Diagnosis | EC | 16 | Resection | Pre‐ and post‐resection | 23 epigenetically and 28 genetically altered EC‐specific genes | NR | NR | The adapted SALP‐seq is successfully applied to the analysis of cfDNA in EC and may support diagnosis and clinical cancer studies in the future. |
| Tian et al. [55] | Diagnosis | ESCC, EAC, small cell carcinoma, and neuroendocrine carcinoma | 150 (137 ESCC, 9 EAC, 3 small cell carcinoma, 1 neuroendocrine carcinoma) | Resection and CRT | Pre‐treatment | 5hmC‐based biomarkers | NR | NR | 5hmC‐based biomarkers in cfDNA may help early diagnose EC. The prediction performance achieved a sensitivity of 93.75% and specificity of 85.71% (AUC = 0.972). |
| Ueda et al. [56] | Diagnosis and prognosis | ESCC | 13 | Resection and neoadjuvant therapy | Pre‐ and throughout treatment | Panel of 53 cancer‐related genes | 0.12%‐7.2% | 11/13 (83.3%) | NGS using a multigene panel is an effective method for detecting somatic mutations in plasma cfDNA. The use of cfDNA analysis in clinical assessments of the tumor burden may help predict tumor recurrence in ESCC. |
| Hagi et al. [58] | Diagnosis | ESCC | 5 | Resection and neoadjuvant therapy | Pre‐ and post‐treatment | TP53 | 0‐1.97% | Pre‐treatment: 2/5 (67%); post‐treatment: 3/5 (14%) | Molecular barcode sequencing enabled comprehensive and highly sensitive detection of ctDNA in ESCC patients. |
| Egyud et al. [60] | Monitoring and prognosis | EAC | 38 | Resection, neoadjuvant therapy, and palliative therapy | Pre‐resection, pre‐treatment, and throughout treatment | TP53, ARID1A, ERBB4 | 0.05%‐5.30% | 18/38 (47%) | ctDNA correlates with disease burden and can be a dynamic biomarker to monitor treatment response and disease recurrence in patients with EAC. |
| Luo et al. [61] | Diagnosis and monitoring | ESCC | 11 | Resection | Pre‐ and post‐resection | Panel of 94 cancer‐related genes | NR | NR | Combining ctDNA with other ESCC‐specific biomarkers would achieve a more accurate diagnosis. The VAFs of some mutations were lower than or equaled 0 in post‐ resection plasma. |
| Meng et al. [62] | Monitoring | ESCC | 17 | Resection | Pre‐ and post‐resection | Panel of 483 cancer‐related genes | 0.24%‐4.91% | Pre‐resection: 8/12 (67%); post‐ resection: 2/14 (14%) | cfDNA could potentially be used to monitor disease load, even in early‐stage patients. |
| Azad et al. [63] | Prognosis | ESCC and EAC | 45 (10 ESCC, 35 EAC) | Resection and CRT | Pre‐ and post‐CRT | Panel of 607 cancer‐related genes | 0‐0.91% | 27/45 (60%) | Detection of ctDNA from patients who underwent CRT for EC was associated with tumor progression, metastasis, and disease‐specific survival. |
cfDNA = circulating cell‐free DNA, ctDNA = circulating tumor DNA, EC = esophageal cancer, ESCC = esophageal squamous cell carcinoma, EAC = esophageal adenocarcinoma, BE = Barrett's esophagus, TP53 = tumor protein 53, ARID1A = AT‐rich interaction domain 1A, ERBB4 = erb‐B2 receptor tyrosine kinase 4, CRT = chemoradiotherapy, 5hmC = 5‐hydroxymethylcytosine, VAFs = variant allele frequencies, NGS = next generation sequencing, NR = not reported, ddPCR = digital droplet polymerase chain reaction, SALP‐seq = single strand adaptor library preparation sequencing, AUC = area under curve.
TABLE 2.
Plasma cfDNA isolation and ctDNA sequencing methods used in the included studies on the clinical applications of cfDNA in EC
| Included study | Spin protocol | Amount of plasma | Isolation kit | cfDNA amount | Sequencing methods |
|---|---|---|---|---|---|
| Pasternack et al. [42] | 3000 rpm 10 min, 16,000 ×g 10 min | 4 mL | QIAsymphony PAXgene blood ccfDNA kit (Qiagen) | 12 ng (mean) | ddPCR and MiSeq |
| Wu et al. [47] | 1600 ×g 15 min, 16,000 ×g 10 min | 200 μL | Plasma circulating DNA kit (TIANGEN) | NR | SALP‐seq |
| Tian et al. [55] | 1350 ×g 12 min, 13,500 ×g 12 min | NR | QiaAmp circulating nucleic acid kit (Qiagen) | NR | Nano‐hmC‐Seal |
| Ueda et al. [56] | 2500 ×g 10 min, 16,000 ×g 10 min | NR | QiaAmp circulating nucleic acid kit (Qiagen) | NR | Targeted sequencing |
| Hagi et al. [58] | 1600 ×g 10 min, 16,000 ×g 10 min | 1.2 mL | QiaAmp circulating nucleic acid kit (Qiagen) | 14.76‐50.76 ng | MiSeq and molecular barcode sequencing using Ion Torrent Proton sequencer |
| Egyud et al. [60] | 1600 ×g 10 min, 3600 ×g 10 min | NR | QiaAmp circulating nucleic acid kit (Qiagen) | NR | ddPCR and SiMSen‐seq |
| Luo et al. [61] | 1600 ×g 20 min | 0.5 or 1 mL | QiaAmp circulating nucleic acid kit (Qiagen) | NR | Whole‐exome sequencing and targeted sequencing (TruSight Cancer panel) |
| Meng et al. [62] | 900 ×g 10 min, 16,000 ×g 10 min | 1‐3 mL | QiaAmp circulating nucleic acid kit (Qiagen) | 4.86‐38.6 ng | Targeted sequencing |
| Azad et al. [63] | 1800 ×g 10 min | NR | QiaAmp circulating nucleic acid kit (Qiagen) | NR | CAPP‐seq |
cfDNA = circulating cell‐free DNA, ctDNA = circulating tumor DNA, NR = not reported, SALP‐seq = single‐strand adaptor library preparation sequencing, ddPCR = digital droplet polymerase chain reaction, SiMSen‐seq = simple multiplexed PCR‐based barcoding of DNA for sensitive mutation detection using sequencing, CAPP‐seq = cancer personalized profiling by deep sequencing. Nano‐hmC‐Seal is a highly sensitive and selective chemical labeling and capture approach for genome‐wide profiling of 5‐hydroxylmethylcytosine using DNA isolated from ∼1000 cells.
3. cfDNA DETECTION AND ANALYSIS
3.1. Sample preparation and cfDNA isolation
Because of its low concentration and short half‐life, isolation of cfDNA is usually performed using the QIAamp circulating nucleic acid kit from QIAGEN (Dusseldorf, North Rhine‐Westphalia, Germany). Two crucial issues are (i) the stability of the cfDNA and (ii) the possibility of germline DNA contamination resulting from blood cell lysis during isolation. Thus, when cfDNA is isolated from blood collected in standard collection tubes (e.g., K2EDTA vacutainer), plasma must be centrifuged twice and separated within 4 h. The need of rapid processing generates both logistic challenges and pre‐analytic variability since differences in processing time can cause variations in cfDNA concentration and purity [27, 28]. However, specialized collection tubes which contain fixatives (e.g., Streck cell‐free DNA BCT tubes) that stabilize both cfDNA and blood cells for up to 14 days at room temperature for convenient shipping or storage are available [29]. To assess cfDNA quality and quantity, the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) is commonly used. This is combined with the Qubit DNA Assay (Thermo Fisher Scientific, Waltham, MA, USA), which has a DNA‐specific dye that can quantify double‐stranded cfDNA even at low concentrations. The typical yield of cfDNA is 1‐100 ng/mL of plasma, and the fragment size distribution is centered around 166 bp [30].
In EC patients, the contribution of ctDNA to cfDNA is typically small and shows high inter‐patient variability. Therefore, ultrasensitive approaches are needed to detect mutations, copy number variations (CNV), and other alterations present in ctDNA at very low variant allele frequencies (VAFs). Such methods are mainly based on polymerase chain reaction (PCR) and next‐generation sequencing (NGS).
3.2. Techniques based on PCR assays
Quantitative PCR (qPCR) [31], BEAMing [32], and droplet digital PCR (ddPCR) [33] can detect individual point mutations in cfDNA and identify and quantify alterations at VAFs of 0.01% or less [32, 33]. In early applications, qPCR demonstrated that cfDNA levels in EC patients were significantly higher than those in healthy individuals [34, 35]. However, its high variability and the fact that increased cfDNA levels are also present in other non‐cancer‐related pathological conditions [36, 37, 38, 39, 40, 41] have prevented its use as a biomarker.
ddPCR detects mutations in cfDNA with high sensitivity, but it is only suitable when the mutations are already known from previous tumor tissue‐based analyses [42]. Also, the location of somatic mutations in EC‐related driver genes span large regions of the genome [43], which are difficult to track by PCR‐based methods. If mutations are not known or when large regions have to be analyzed, NGS‐based methods are preferable (see below).
3.3. Next‐generation sequencing (NGS)
The overwhelming majority of cancers harbor genetic and epigenetic alterations in both coding and non‐coding regions. These alterations can influence normal gene expression and contribute to cancer occurrence and development [44]. Genetic changes in cfDNA can potentially be used to detect cancers or track their dynamics during treatment in real time [45, 46]. High‐throughput NGS is highly sensitive and specific for cfDNA sequencing, and can use samples obtained from liquid biopsy. In particular, deep whole‐genome sequencing (WGS) can identify signature mutations and detect chromatin openness state, an important epigenetic marker, whereas targeted gene panel sequencing (TGPS) can detect known cancer‐specific mutations. Also, cfDNA bisulfite sequencing can characterize methylation status, another important cancer epigenetic marker, in specific regions of DNA [47]. However, deep WGS of cfDNA is expensive, and most studies have used NGS panels containing only a set of common cancer‐related genes.
Since cfDNA has a small and defined size (see above), no further DNA fragmentation is required before double‐stranded sequencing library construction. The key steps to generate these libraries include DNA‐end repair, A‐addition, adapter ligation, reaction cleanup, removal of the adapters and adapter dimers, and library amplification. The typical sequencing library concentration is 50‐200 nmol/L when obtained from 1‐10 ng of cfDNA [48]. The number of library amplification PCR cycles can be changed depending on the yield required. However, although NGS has been used to test cfDNA with high sensitivity and specificity [30], the low concentration of cfDNA in blood is a challenge for the construction of sequencing libraries with high quality and complexity, especially in EC. For example, mutations due to clonal hematopoiesis, e.g., in tumor protein 53 (TP53), found in matched white blood cells (WBCs) must be excluded [49]. This requires simultaneous sequencing of DNA extracted from WBCs [50] to correctly attribute the cfDNA mutations to tumors.
4. THE APPLICATION OF cfDNA TO DIAGNOSIS
4.1. EC
5‐hydroxymethylcytosine (5hmC) is an intermediate of active DNA demethylation and a novel cancer epigenetic marker [51, 52]. Modes of this intermediate in cfDNA can identify cancer‐associated signatures in multiple malignancies [53, 54]. Tian et al. [55] mapped the cfDNA 5hmC profiles from a cohort of 150 newly diagnosed EC patients (137 ESCC, 9 EAC, 3 small cell carcinoma, and 1 neuroendocrine carcinoma) and 177 healthy individuals. Using a novel approach, nano‐hmC‐Seal, EC‐associated 5hmC signature was identified. It was used to diagnose EC with a sensitivity of 93.75% and a specificity of 85.71% (area under curve [AUC] = 0.972), suggesting its potential use as a biomarker for minimally invasive EC diagnosis.
Recently, a single‐strand adaptor library preparation (SALP) method was used to construct an NGS library of cfDNA in EC patients [47]. Genetic and epigenetic differences between 16 EC patients and 4 healthy individuals were detected, and 23 epigenetically and 28 genetically altered EC‐associated genes were identified. Overall, the study suggested that adapted SALP‐seq can potentially be used in minimally invasive diagnosis of EC and other cancers.
4.2. ESCC
In ESCC patients, a multi‐gene panel was used to detect somatic mutations in plasma cfDNA [56]. The cfDNA genetic profiles matched the status of the cancer and showed the diagnostic utility of mutations in 4 genes (TP53, fat atypical cadherin 3 [FAT3], mixed lineage leukemia 3 [MLL3], and ajuba LIM protein [AJUBA]). In both cfDNA and tumor tissues, the use of these mutations achieved 78.9% sensitivity, 100% specificity, and 92.3% diagnostic accuracy [56].
Identification of true mutations is difficult when using integrated analysis via NGS because of the low ctDNA concentrations in ESCC patients. When NGS is used in combination with molecular barcodes (MB), mutations can be detected with a high sensitivity in a relatively wide range of genes [57]. NGS without MB produced background errors of 3.22% as the maximum frequency, which was reduced to 0.08% after inclusion of MB [58], suggesting that this combined approach can be used to detect cfDNA mutations in ESCC patients.
5. THE APPLICATION OF cfDNA TO MONITORING THERAPEUTIC RESPONSE
5.1. EAC
Mutations in ctDNA of 38 EAC patients were detected using ddPCR and SiMSen‐seq (simple, multiplexed, PCR‐based barcoding of DNA for sensitive mutation detection) [59]. ctDNA could be detected at all stages of the disease, and both detection rate and ctDNA quantity were increased at later stages. This study showed that ctDNA quantification could be used in some patients under treatment to determine therapeutic response and to detect recurrence after definitive treatment. Levels of ctDNA are dynamic and can be analyzed before imaging to monitor therapeutic response and recurrence [60].
5.2. ESCC
A deep sequencing comparison of pre‐ and postoperative plasma ctDNA in 11 ESCC patients showed that some ctDNA mutations were reduced in frequency or even disappeared postoperatively [61]. VAFs of some mutations were extremely lower or even equaled 0 in postoperative plasma. However, deep targeted sequencing showed altered driver genes that were also found in other cancers, suggesting that the application of ctDNA mutation detection to the diagnosis of ESCC lacks specificity.
In another study, targeted sequencing of pre‐ and postoperative cfDNA of matched tumor tissues and WBCs was performed using a panel of 483 cancer‐related genes in 17 ESCC patients [62]. A subset of cancer‐specific mutations could be detected in preoperative cfDNA from most patients at stages II and III. The majority of those mutations were not detected in blood samples collected as early as 3‐4 h postoperatively. The association of somatic mutations in cfDNA with tumor load suggested cfDNA as a potential biomarker for monitoring tumor burden, even in ESCC patients at early stages.
6. THE APPLICATION OF cfDNA TO PROGNOSIS
6.1. EC
Mutations in genomic DNA from tissue biopsies and in cfDNA were analyzed using a 12‐gene panel with NGS and ddPCR [42]. In 21 EC patients with identified mutations (7 ESCC, 19 EAC, and 1 Barret's esophagus), cfDNA mutations were observed with a higher rate in ddPCR (8/21, 38%) than in NGS (3/21, 14%). During the follow‐up period, 67% of patients with somatic cfDNA alterations during primary staging suffered from an early recurrence and had a shorter progression‐free survival as compared with the patients who had cfDNA without somatic alterations. In contrast, tumor recurrence was only observed in 10% of patients with no detected mutations, suggesting that these cfDNA alterations in patients with locally advanced EC can predict recurrence postoperatively. [42].
6.2. EAC
Unfortunately, relapse including local or distant recurrence happens often even if EC patients undergo potentially curative esophagectomy. Recurrence can be identified not only from dysphagia symptoms or weight loss but also from computed tomography (CT) or positron emission tomography‐computed tomography (PET‐CT). Levels of ctDNA could be detected in some EAC patients before recurrence was identified by imaging [60], suggesting ctDNA as a valid biomarker. The possibility that ctDNA analysis can predict recurrence in patients with localized EC before imaging was examined by Azad et al. [63]. The ctDNA of EC patients who underwent chemoradiotherapy (CRT) was analyzed, and ctDNA and metabolic imaging were combined to further improve risk stratification. In 35 of those patients, the detection of ctDNA was associated with tumor progression, metastasis, and short survival. ctDNA evidence was on average 2.8 months before imaging evidence of tumor progression. In 10 EAC patients who underwent CRT without resection, the combination of ctDNA and metabolic imaging could predict progression in all patients. Also, ctDNA detection after CRT was more predictive of distant metastasis than local progression.
6.3. ESCC
Azad et al. [63] reported similar results in 10 ESCC patients who underwent either CRT followed by esophagectomy (n = 8) or definitive CRT (n = 2). In the latter, the combination of ctDNA and metabolic imaging after treatment predicted progression in all patients. Combined with the EAC findings, these results suggested that ctDNA can be used to identify EC patients at risk of tumor progression after therapy [63].
Ueda et al. [56] detected somatic mutations in cfDNA from both primary tumors and recurrent lesions. VAFs of concordant mutations in serial plasma samples were useful not only for assessing tumor status but also for predicting ESCC recurrence. In particular, concordant mutations in cfDNA with high frequency appeared 6 months prior to the detection of recurrences by imaging tests in 2 ESCC patients.
7. DISCUSSION AND PERSPECTIVES
Analysis of somatic mutations in tumor tissue has been recently implemented in clinical oncology. However, tissue biopsy is limited because it is difficult to obtain and is affected by sampling bias due to temporal and spatial tumor heterogeneity [64]. Thus, alternative approaches, such as liquid biopsy, are currently assessed for applicability in clinical practice. Liquid biopsy is minimally invasive, safe, and may overcome the difficulties derived from intratumoral heterogeneity [65]. Also, combined with the potential of longitudinal evaluations during treatment, cfDNA may predict therapeutic response and modes of resistance earlier than imaging studies [60, 63]. The isolation of cfDNA from cancer patients has led to the concept of “ctDNA”, which harbors genetic and epigenetic alterations associated with cancer that can be used for diagnostic and monitoring purposes. Thus clinicians no longer depend exclusively on invasive tissue biopsies, but can analyze cfDNA routinely to follow the reaction of a cancer to a given therapy and monitor the development of resistance. This is especially useful in some tumors where biopsies are difficult or unsafe. Also, compared to a needle biopsy taken from a single lesion, cfDNA analysis can not only better describe the intratumoral heterogeneity harbored by various subclonal populations in a tumor but also enable detection or monitoring in individuals with no clinically evident disease [65, 66].
cfDNA is thought to be released into the blood circulation from apoptotic or necrotic cells, and its size distribution suggests the source is apoptotic caspase‐dependent cleavage [24]. For this reason, we excluded from this review studies which used larger circulating DNA fragments; e.g., DNA fragments from 200 bp to 50 kb, obtained using the QIAamp DNA Blood Kits (Qiagen).
Current methods to improve ctDNA detection mostly focus on genomic alterations using cfDNA with double‐stranded fragments of approximately 150‐200 bp. However, differences in size could be used to increase detection sensitivity of ctDNA and improve genomic analysis of cancer [67].
Multiple mechanisms of release or sample processing methods may lead to distinct fragment sizes [20] which can affect PCR amplification efficiency during library preparation. Smaller fragments are generally amplified more efficiently, resulting in an over‐representation of short fragments.
Theoretically, ctDNA detection sensitivity is mainly determined by (1) amount of input cfDNA, (2) sequencing depth, (3) number of mutations tracked, and (4) assay background (i.e., sequencing or variant calling errors) [68]. Sensitivity can be increased by increasing sequencing panel size or designing personalized ctDNA assays which track more mutations, as previously reported for other cancer types [68, 69]. Notably, ctDNA could be used to monitor underlying tumor load and to accurately predict recurrence [70]. Furthermore, ctDNA analysis can be used to monitor response to anticancer therapies more frequently and safely in clinical practice, which would reduce morbidity compared to invasive tissue biopsies [45]. Unfortunately, not many cfDNA or ctDNA studies have been undertaken in EC patients so far compared to other cancer types, but it is nevertheless a promising field in biomarker research.
Recently, efforts have been directed to develop cancer non‐invasive screening at early stages, but this is difficult [46]. Early EC detection by liquid biopsy remains elusive, and endoscopy is still the gold standard for EC diagnosis [71]. However, the esophageal mucosa can be friable because of ulceration or necrosis, and a minimum of 6 tissue biopsies are recommended for histological confirmation [72]. Endoscopy is often used, but it is invasive and expensive and requires skilled operators. Less invasive approaches would enable screening for a larger part of the population. However, quantification of cfDNA in EC patients is not sufficiently specific [36, 37, 38, 39, 40, 41] despite having been observed to be higher than in healthy individuals [34, 35].
As described above, Ueda et al. [56] used high‐throughput sequencing technologies to detect frequently mutated genes in ESCC, identifying genetic profiles of cfDNA which can accurately reflect tumor status of EC patients and providing the basis for the development of an ESCC diagnostic tool. By labeling each DNA fragment with a barcode sequence, reads from the same DNA fragment can be grouped, eliminating background errors [57]. The combination of NGS with MB enables highly sensitive detection of ctDNA in a relatively wide range of genes, which may be useful for the diagnosis of ESCC [58].
Epigenetic alterations are early events during carcinogenesis [73], and in cancer patients these are reflected in cfDNA [74]. Wu et al. [47] used a new method to construct an NGS library of cfDNA and identified 23 epigenetically and 28 genetically altered EC‐specific genes in EC clinical samples and EC‐related molecular markers reported on chromatin status. Chromatin openness state of EC patients differed from that in healthy individuals, although these results were not verified in cancerous tissues of the same patients [47].
Methylation of cytosine adjacent to guanine (CpG sites) is important for cell type‐specific gene regulation and is a hallmark of cell identity [75]. Cancer cells exhibit two types of DNA methylation alteration: site‐specific hypermethylation at the promoter of oncosuppressor genes and global DNA hypomethylation [76, 77]. Interestingly, DNA methylation patterns detected in cfDNA are highly concordant with those found in matched primary tumor tissues [78, 79]. Certain cfDNA methylation markers have been associated with EC and other types of cancer, which can be used for its detection and localization [80, 81, 82, 83]. Tian et al. [55] mapped the 5hmC profiles in cfDNA from EC patients, demonstrating that 5hmCs in cfDNA are promising biomarkers for EC diagnosis.
In Barrett's esophagus, squamous epithelium of the esophagus is replaced by metaplastic columnar epithelium, and this is the only well‐defined precursor of EAC [8]. Recently, an analysis of methylation patterns of long interspersed nuclear element (LINE‐1) sequences, a good surrogate indicator of global DNA methylation, showed that hypomethylation of these sequences was present in cfDNA in 19 EAC patients [84]. Also, longitudinal studies on 2 BE patients suggested an association between methylation status of LINE‐1 sequences in cfDNA and progression to EAC [77]. Although further studies are needed, these results suggest a potential method for early EAC detection.
Efforts have been focused to develop cfDNA for early diagnosis of BE and EAC, with potential applicability to primary care. Genome‐wide cfDNA methylation profiles were highly consistent with DNA methylation profiles detected in corresponding tumor tissues. Moreover, differential cfDNA methylation profiles could distinguish EAC and BE from controls, as well as EAC from BE [85]. These results suggest that differential cfDNA methylation profiling may be useful for noninvasive screening of EAC and EAC premalignant lesions.
Successful monitoring of therapeutic response provides physicians with a rapid evaluation of the therapeutic effects and possible modification of therapeutic regimens [60]. For repeat or serial testing during one or more lines of therapy, cfDNA analysis is advantageous over invasive tumor biopsies. The latter is increasingly utilized as it can also reveal significant mutations that guide therapeutic regimens in these patients [25]. Recently, data from one study with longitudinal EAC samples indicated that ctDNA has the potential to be a dynamic biomarker to monitor treatment response in patients with EAC, and VAFs of some mutations were lowered or eliminated in ESCC patients’ postoperative plasma [60]. Similarly, somatic mutations could be detected in preoperative cfDNA in patients with ESCC at stage IIA‐IIIB and were at a lower frequency in postoperative cfDNA in another study [62]. These results demonstrated that ctDNA is a valuable biomarker for tracking tumor status and evaluating therapeutic effect. In addition, cfDNA can capture genetic heterogeneity associated with therapeutic resistance. The use of cfDNA was suggested as a tool to identify therapeutic targets that are not detected by standard tissue biopsy in gastric and esophageal adenocarcinomas (GEA) with metastatic lesions [86], representing more accurately GEA disseminated disease.
Thus, studies suggest that cfDNA analysis may enable detection of genetic alterations specific to molecular subtypes of cancer which are associated with mechanism of resistance and may be therapeutic targets. This is important for future treatments developed specifically for subtypes of EC identified and tracked from a routine blood test. Integration of real‐time cfDNA analysis into standard clinical management may become a valuable tool for capturing tumor heterogeneity and monitoring therapeutic response.
One application of cfDNA analysis may be to detect tumor in patients without clinical evidence, e.g., as a predictive tool in the early detection of recurrence after surgery or adjuvant therapy [65]. Primary treatment of cure for EC is surgical resection. In high‐risk patients with advanced EC, adjuvant CRT can reduce the risk of recurrence after surgery [1, 2, 8]. However, it is currently not possible to determine which patients harbor minimal residual disease postoperatively or which patients have been cured. Therefore, decisions about adjuvant CRT are mainly based on clinical risk stratification, which may not be accurate.
Detection of ctDNA can be used to determine minimal residual disease after CRT and is strongly predictive for progression and disease‐specific survival, and a combined ctDNA‐imaging mode may enable effective risk stratification of EC patients treated with CRT alone [63]. Similarly, multiple studies have shown that detection of tumor‐specific mutations in cfDNA after surgery can predict prognosis in EC. For example, in ESCC patients, an increased frequency of concordant somatic mutations in cfDNA occurred 6 months earlier than recurrences identified by imaging studies [56], and detection of somatic mutations in the cfDNA of EC patients at the time of primary staging may be indicative of a increased risk of postoperative tumor recurrence [42]. In summary, analysis of cfDNA has the potential to become a useful tool in the postoperative management of EC patients, especially in prognosis prediction. Prospective studies are needed to evaluate the utility of residual ctDNA detection after surgery to guide adjuvant CRT for EC.
In the last decade, cfDNA has become a hot topic in oncology, and it is difficult to keep pace with the number of papers that are published. However, our basic knowledge on cfDNA or ctDNA is still far from complete. The mechanisms leading to the release of cell‐free DNA into circulation are not known, and technical and methodological issues have to be solved. So far, there is no consensus on a “gold standard” for the isolation of cfDNA. Additionally, computational approaches should be improved to perform genetic and epigenetic analysis deconvoluting cancer‐specific signals from the mixture of cancer and normal signals present in cfDNA.
Finally, the use of healthy individuals as controls in many studies is questionable, since biomarkers may not be able to differentiate cancer patients and patients with premalignant lesions, or patients with benign disease affecting target organs. Therefore, the choice of an appropriate control population for the discovery of new biomarkers is very important.
8. CONCLUSIONS
Advances in the field of cfDNA can produce new molecular biomarkers for use in early diagnosis, monitoring, and accurate prognosis prediction of EC. Mutational analysis of EC‐related genes has shown that ctDNA is present in cfDNA. The genetic profile of ctDNA includes both point mutations and somatic copy number alterations in EC. Although more studies are needed, cfDNA analysis has a potential to be used as a non‐invasive technology for EC.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
FUNDING
Not applicable.
AUTHORS' CONTRIBUTIONS
ZY, YL and JH conceived this study. ZY, XW and XG drafted the manuscript. YL, JM, FT, QX, SG, JH revised the manuscript. All authors read and approved the final manuscript.
Yuan Z, Wang X, Geng X, et al. Liquid biopsy for esophageal cancer: Is detection of circulating cell‐free DNA as a biomarker feasible?. Cancer Commun. 2021;41:3–15. 10.1002/cac2.12118
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Rustgi AK, El‐Serag HB. Esophageal carcinoma. N Engl J Med. 2014:371(26):2499‐2509. [DOI] [PubMed] [Google Scholar]
- 2. Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013:381(9864):400‐12. [DOI] [PubMed] [Google Scholar]
- 3. Arnold M, Soerjomataram I, Ferlay J, Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut. 2015:64(3):381‐87. [DOI] [PubMed] [Google Scholar]
- 4. Edgren G, Adami HO, Weiderpass E, Nyren O. A global assessment of the oesophageal adenocarcinoma epidemic. Gut. 2013:62(10):1406‐14. [DOI] [PubMed] [Google Scholar]
- 5. Clocchiatti A, Cora E, Zhang Y, Dotto GP. Sexual dimorphism in cancer. Nat Rev Cancer. 2016:16(5):330‐39. [DOI] [PubMed] [Google Scholar]
- 6. Dorak MT, Karpuzoglu E. Gender differences in cancer susceptibility: an inadequately addressed issue. Front Genet. 2012:3:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pohl H, Wrobel K, Bojarski C, Voderholzer W, Sonnenberg A, Rosch T, et al. Risk factors in the development of esophageal adenocarcinoma. Am J Gastroenterol. 2013:108(2):200‐7. [DOI] [PubMed] [Google Scholar]
- 8. Smyth EC, Lagergren J, Fitzgerald RC, Lordick F, Shah MA, Lagergren P, et al. Oesophageal cancer. Nat Rev Dis Primers. 2017:3:17048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mandel P, Metais P. [Nuclear acids in human blood plasma.] C R Seances Soc Biol Fil. 1948:142(3‐4):241‐43. [in French] [PubMed] [Google Scholar]
- 10. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977:37(3):646‐50. [PubMed] [Google Scholar]
- 11. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008:14(9):985‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med. 2014:6(224):224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Husain H, Melnikova VO, Kosco K, Woodward B, More S, Pingle SC, et al. Monitoring Daily Dynamics of Early Tumor Response to Targeted Therapy by Detecting Circulating Tumor DNA in Urine. Clin Cancer Res. 2017:23(16):4716‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y, Springer S, Mulvey CL, Silliman N, Schaefer J, Sausen M, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci Transl Med. 2015:7(293):293ra104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Mattos‐Arruda L, Mayor R, Ng CKY, Weigelt B, Martinez‐Ricarte F, Torrejon D, et al. Cerebrospinal fluid‐derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun. 2015:6:8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001:61(4):1659‐65. [PubMed] [Google Scholar]
- 17. Gahan PB, Anker P, Stroun M. Metabolic DNA as the origin of spontaneously released DNA? Ann N Y Acad Sci. 2008:1137:7‐17. [DOI] [PubMed] [Google Scholar]
- 18. Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, et al. Identification of double‐stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014:289(7):3869‐3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa‐Silva B, et al. Double‐stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014:24(6):766‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016:35(3):347‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, et al. Inferring expressed genes by whole‐genome sequencing of plasma DNA. Nat Genet. 2016:48(10):1273‐78. [DOI] [PubMed] [Google Scholar]
- 22. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell‐free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues‐Of‐Origin. Cell. 2016:164(1‐2):57‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bronkhorst AJ, Wentzel JF, Aucamp J, van Dyk E, du Plessis L, Pretorius PJ. Characterization of the cell‐free DNA released by cultured cancer cells. Biochim Biophys Acta. 2016:1863(1):157‐65. [DOI] [PubMed] [Google Scholar]
- 24. Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Analysis of the size distributions of fetal and maternal cell‐free DNA by paired‐end sequencing. Clin Chem. 2010:56(8):1279‐86. [DOI] [PubMed] [Google Scholar]
- 25. Volckmar AL, Sultmann H, Riediger A, Fioretos T, Schirmacher P, Endris V, et al. A field guide for cancer diagnostics using cell‐free DNA: From principles to practice and clinical applications. Genes Chromosomes Cancer. 2018:57(3):123‐39. [DOI] [PubMed] [Google Scholar]
- 26. Chen XX, Zhong Q, Liu Y, Yan SM, Chen ZH, Jin SZ, et al. Genomic comparison of esophageal squamous cell carcinoma and its precursor lesions by multi‐region whole‐exome sequencing. Nat Commun. 2017:8(1):524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. El Messaoudi S, Rolet F, Mouliere F, Thierry AR. Circulating cell free DNA: Preanalytical considerations. Clin Chim Acta. 2013:424:222‐30. [DOI] [PubMed] [Google Scholar]
- 28. Swinkels DW, Wiegerinck E, Steegers EA, de Kok JB. Effects of blood‐processing protocols on cell‐free DNA quantification in plasma. Clin Chem. 2003:49(3):525‐26. [DOI] [PubMed] [Google Scholar]
- 29. Alidousty C, Brandes D, Heydt C, Wagener S, Wittersheim M, Schafer SC, et al. Comparison of Blood Collection Tubes from Three Different Manufacturers for the Collection of Cell‐Free DNA for Liquid Biopsy Mutation Testing. J Mol Diagn. 2017:19(5):801‐4. [DOI] [PubMed] [Google Scholar]
- 30. Volik S, Alcaide M, Morin RD, Collins C. Cell‐free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol Cancer Res. 2016:14(10):898‐908. [DOI] [PubMed] [Google Scholar]
- 31. Page K, Hava N, Ward B, Brown J, Guttery DS, Ruangpratheep C, et al. Detection of HER2 amplification in circulating free DNA in patients with breast cancer. Br J Cancer. 2011:104(8):1342‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single‐molecule PCR on microparticles in water‐in‐oil emulsions. Nat Methods. 2006:3(7):551‐59. [DOI] [PubMed] [Google Scholar]
- 33. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011:83(22):8604‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tomita H, Ichikawa D, Ikoma D, Sai S, Tani N, Ikoma H, et al. Quantification of circulating plasma DNA fragments as tumor markers in patients with esophageal cancer. Anticancer Res. 2007:27(4C):2737‐41. [PubMed] [Google Scholar]
- 35. Lan YT, Chen MH, Fang WL, Hsieh CC, Lin CH, Jhang FY, et al. Clinical relevance of cell‐free DNA in gastrointestinal tract malignancy. Oncotarget. 2017:8(2):3009‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fatouros IG, Destouni A, Margonis K, Jamurtas AZ, Vrettou C, Kouretas D, et al. Cell‐free plasma DNA as a novel marker of aseptic inflammation severity related to exercise overtraining. Clin Chem. 2006:52(9):1820‐24. [DOI] [PubMed] [Google Scholar]
- 37. Lo YM, Rainer TH, Chan LY, Hjelm NM, Cocks RA. Plasma DNA as a prognostic marker in trauma patients. Clin Chem. 2000:46(3):319‐23. [PubMed] [Google Scholar]
- 38. Fournie GJ, Martres F, Pourrat JP, Alary C, Rumeau M. Plasma DNA as cell death marker in elderly patients. Gerontology. 1993:39(4):215‐21. [DOI] [PubMed] [Google Scholar]
- 39. Atamaniuk J, Vidotto C, Tschan H, Bachl N, Stuhlmeier KM, Muller MM. Increased concentrations of cell‐free plasma DNA after exhaustive exercise. Clin Chem. 2004:50(9):1668‐70. [DOI] [PubMed] [Google Scholar]
- 40. Jung K, Stephan C, Lewandowski M, Klotzek S, Jung M, Kristiansen G, et al. Increased cell‐free DNA in plasma of patients with metastatic spread in prostate cancer. Cancer Lett. 2004:205(2):173‐80. [DOI] [PubMed] [Google Scholar]
- 41. Laktionov PP, Tamkovich SN, Rykova EY, Bryzgunova OE, Starikov AV, Kuznetsova NP, et al. Extracellular circulating nucleic acids in human plasma in health and disease. Nucleosides Nucleotides Nucleic Acids. 2004:23(6‐7):879‐83. [DOI] [PubMed] [Google Scholar]
- 42. Pasternack H, Fassunke J, Plum PS, Chon SH, Hescheler DA, Gassa A, et al. Somatic alterations in circulating cell‐free DNA of oesophageal carcinoma patients during primary staging are indicative for post‐surgical tumour recurrence. Sci Rep. 2018:8(1):14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Talukdar FR, di Pietro M, Secrier M, Moehler M, Goepfert K, Lima SSC, et al. Molecular landscape of esophageal cancer: implications for early detection and personalized therapy. Ann N Y Acad Sci. 2018:1434(1):342‐59. [DOI] [PubMed] [Google Scholar]
- 44. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016:17(10):630‐41. [DOI] [PubMed] [Google Scholar]
- 45. Rothwell DG, Ayub M, Cook N, Thistlethwaite F, Carter L, Dean E, et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med. 2019:25(5):738‐43. [DOI] [PubMed] [Google Scholar]
- 46. Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, et al. Direct detection of early‐stage cancers using circulating tumor DNA. Sci Transl Med. 2017:9(403). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu J, Dai W, Wu L, Li W, Xia X, Wang J. Decoding genetic and epigenetic information embedded in cell free DNA with adapted SALP‐seq. Int J Cancer. 2019:145(9):2395‐2406. [DOI] [PubMed] [Google Scholar]
- 48. Fang N, Loffert D, Akinci‐Tolun R, Heitz K, Wolf A. Construction of a Sequencing Library from Circulating Cell‐Free DNA. Curr Protoc Mol Biol. 2016:114:7 25 21‐27 25 13. [DOI] [PubMed] [Google Scholar]
- 49. Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High‐intensity sequencing reveals the sources of plasma circulating cell‐free DNA variants. Nat Med. 2019:25(12):1928‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bellosillo B, Montagut C. High‐accuracy liquid biopsies. Nat Med. 2019:25(12):1820‐21. [DOI] [PubMed] [Google Scholar]
- 51. Chen K, Zhang J, Guo Z, Ma Q, Xu Z, Zhou Y, et al. Loss of 5‐hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res. 2016:26(1):103‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vasanthakumar A, Godley LA. 5‐hydroxymethylcytosine in cancer: significance in diagnosis and therapy. Cancer Genet. 2015:208(5):167‐77. [DOI] [PubMed] [Google Scholar]
- 53. Song CX, Yin S, Ma L, Wheeler A, Chen Y, Zhang Y, et al. 5‐Hydroxymethylcytosine signatures in cell‐free DNA provide information about tumor types and stages. Cell Res. 2017:27(10):1231‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li W, Zhang X, Lu X, You L, Song Y, Luo Z, et al. 5‐Hydroxymethylcytosine signatures in circulating cell‐free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017:27(10):1243‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tian X, Sun B, Chen C, Gao C, Zhang J, Lu X, et al. Circulating tumor DNA 5‐hydroxymethylcytosine as a novel diagnostic biomarker for esophageal cancer. Cell Res. 2018:28(5):597‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ueda M, Iguchi T, Masuda T, Nakahara Y, Hirata H, Uchi R, et al. Somatic mutations in plasma cell‐free DNA are diagnostic markers for esophageal squamous cell carcinoma recurrence. Oncotarget. 2016:7(38):62280‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011:108(23):9530‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hagi T, Kurokawa Y, Takahashi T, Saito T, Yamashita K, Tanaka K, et al: Molecular Barcode Sequencing for Highly Sensitive Detection of Circulating Tumor DNA in Patients with Esophageal Squamous Cell Carcinoma. Oncology. 2020:98(4):222‐29. [DOI] [PubMed] [Google Scholar]
- 59. Stahlberg A, Krzyzanowski PM, Egyud M, Filges S, Stein L, Godfrey TE. Simple multiplexed PCR‐based barcoding of DNA for ultrasensitive mutation detection by next‐generation sequencing. Nat Protoc. 2017:12(4):664‐82. [DOI] [PubMed] [Google Scholar]
- 60. Egyud M, Tejani M, Pennathur A, Luketich J, Sridhar P, Yamada E, et al. Detection of Circulating Tumor DNA in Plasma: A Potential Biomarker for Esophageal Adenocarcinoma. Ann Thorac Surg. 2019:108(2):343‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Luo H, Li H, Hu Z, Wu H, Liu C, Li Y, et al. Noninvasive diagnosis and monitoring of mutations by deep sequencing of circulating tumor DNA in esophageal squamous cell carcinoma. Biochem Biophys Res Commun. 2016:471(4):596‐602. [DOI] [PubMed] [Google Scholar]
- 62. Meng P, Wei J, Geng Y, Chen S, Terpstra MM, Huang Q, et al. Targeted sequencing of circulating cell‐free DNA in stage II‐III resectable oesophageal squamous cell carcinoma patients. BMC Cancer. 2019:19(1):818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Azad TD, Chaudhuri AA, Fang P, Qiao Y, Esfahani MS, Chabon JJ, et al. Circulating Tumor DNA Analysis for Detection of Minimal Residual Disease After Chemoradiotherapy for Localized Esophageal Cancer. Gastroenterology. 2020:158(3):494‐505 e496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hao JJ, Lin DC, Dinh HQ, Mayakonda A, Jiang YY, Chang C, et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat Genet. 2016:48(12):1500‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Corcoran RB, Chabner BA. Application of Cell‐free DNA Analysis to Cancer Treatment. N Engl J Med. 2018:379(18):1754‐65. [DOI] [PubMed] [Google Scholar]
- 66. Elshimali YI, Khaddour H, Sarkissyan M, Wu Y, Vadgama JV. The clinical utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int J Mol Sci. 2013:14(9):18925‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018:10(466). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016:34(5):547‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Abbosh C, Birkbak NJ, Wilson GA, Jamal‐Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early‐stage lung cancer evolution. Nature 2017:545(7655):446‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016:8(346):346ra392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Morita FH, Bernardo WM, Ide E, Rocha RS, Aquino JC, Minata MK, et al. Narrow band imaging versus lugol chromoendoscopy to diagnose squamous cell carcinoma of the esophagus: a systematic review and meta‐analysis. BMC Cancer. 2017:17(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Graham DY, Schwartz JT, Cain GD, Gyorkey F. Prospective evaluation of biopsy number in the diagnosis of esophageal and gastric carcinoma. Gastroenterology. 1982:82(2):228‐31. [PubMed] [Google Scholar]
- 73. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000:100(1):57‐70. [DOI] [PubMed] [Google Scholar]
- 74. Liu L, Toung JM, Jassowicz AF, Vijayaraghavan R, Kang H, Zhang R, et al. Targeted methylation sequencing of plasma cell‐free DNA for cancer detection and classification. Ann Oncol. 2018:29(6):1445‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018:392(10149):777‐86. [DOI] [PubMed] [Google Scholar]
- 76. Van Tongelen A, Loriot A, De Smet C. Oncogenic roles of DNA hypomethylation through the activation of cancer‐germline genes. Cancer Lett. 2017:396:130‐37. [DOI] [PubMed] [Google Scholar]
- 77. Boldrin E, Curtarello M, Dallan M, Alfieri R, Realdon S, Fassan M, et al. Detection of LINE‐1 hypomethylation in cfDNA of Esophageal Adenocarcinoma Patients. Int J Mol Sci. 2020:21(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Board RE, Knight L, Greystoke A, Blackhall FH, Hughes A, Dive C, Ranson M: DNA methylation in circulating tumour DNA as a biomarker for cancer. Biomark Insights. 2008:2:307‐19. [PMC free article] [PubMed] [Google Scholar]
- 79. Gormally E, Caboux E, Vineis P, Hainaut P. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: practical aspects and biological significance. Mutat Res. 2007:635(2‐3):105‐17. [DOI] [PubMed] [Google Scholar]
- 80. Liggett TE, Melnikov AA, Marks JR, Levenson VV. Methylation patterns in cell‐free plasma DNA reflect removal of the primary tumor and drug treatment of breast cancer patients. Int J Cancer. 2011:128(2):492‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lee BB, Lee EJ, Jung EH, Chun HK, Chang DK, Song SY, et al. Aberrant methylation of APC, MGMT, RASSF2A, and Wif‐1 genes in plasma as a biomarker for early detection of colorectal cancer. Clin Cancer Res. 2009:15(19):6185‐91. [DOI] [PubMed] [Google Scholar]
- 82. Melnikov AA, Scholtens D, Talamonti MS, Bentrem DJ, Levenson VV. Methylation profile of circulating plasma DNA in patients with pancreatic cancer. J Surg Oncol. 2009:99(2):119‐22. [DOI] [PubMed] [Google Scholar]
- 83. Liggett TE, Melnikov A, Yi Q, Replogle C, Hu W, Rotmensch J, et al. Distinctive DNA methylation patterns of cell‐free plasma DNA in women with malignant ovarian tumors. Gynecol Oncol. 2011:120(1):113‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009:10(10):691‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhai R, Zhao Y, Su L, Cassidy L, Liu G, Christiani DC. Genome‐wide DNA methylation profiling of cell‐free serum DNA in esophageal adenocarcinoma and Barrett esophagus. Neoplasia. 2012:14(1):29‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pectasides E, Stachler MD, Derks S, Liu Y, Maron S, Islam M, et al. Genomic Heterogeneity as a Barrier to Precision Medicine in Gastroesophageal Adenocarcinoma. Cancer Discov 2018:8(1):37‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.