

Abstract
Background
The interaction between activating receptor NKp30 and its major tumor ligand B7‐H6 is important for NK cell‐mediated tumor rejection. However, the regulation of B7‐H6 by tumor therapeutics remains largely unknown. In this study, we investigated the regulation of B7‐H6 by all‐trans retinoic acid (atRA), a terminal differentiation inducer of tumor cells that is extensively used for clinical leukemia therapy.
Methods
We investigated the role of NKp30:B7‐H6 axis in NK cell‐mediated tumor lysis against leukemia cells and the influence of atRA treatment on the cytotoxicity of NK cells using NK cell lines (NK92 and NKG) and leukemia cell lines (U‐937 and THP‐1). We evaluated the effect of atRA treatment on the expression of B7‐H6 using real‐time PCR, flow cytometry and western blotting. We used CRISPR/Cas9 to knockdown B7‐H6 expression and siRNA to knockdown c‐Myc in U‐937 cells to evaluate the role of B7‐H6 and c‐Myc in atRA‐induced tumor resistance against NK cells.
Results
NK cell‐mediated U‐937 cell lysis was mainly dependent on NKp30/B7‐H6 interaction. Blockade of B7‐H6 by monoclonal antibody significantly impaired NK cytotoxicity. atRA treatment induced U‐937 resistance to NK cell cytotoxicity by reducing B7‐H6 expression, and showed no effect on NK cytotoxicity against B7‐H6 knockdown U‐937 cells. Epigenetic modifications, such as DNA methylation and histone deacetylase (HDAC), were not responsible for atRA‐mediated B7‐H6 down‐regulation as inhibitors of these pathways could not restore B7‐H6 mRNA expression. On the other hand, atRA treatment reduced c‐Myc expression, which in turn inhibited the transcription of B7‐H6 on leukemia cells.
Conclusion
atRA treatment promotes tumor cell resistance against NK cell‐mediated lysis by down‐regulating B7‐H6 expression via the c‐Myc signaling pathway, suggesting that more attention needs to be paid to the immunological adverse effects in the clinical use of atRA treatment.
Keywords: retinoic acid, natural killer cell, leukemia, B7‐H6, c‐Myc
Abbreviations
- AML
acute myeloid leukemia
- APL
acute promyelocytic leukemia
- ATCC
American Type Culture Collection
- atRA
all‐trans retinoic acid
- E:T
effector/target
- FBS
fetal bovine serum
- 5‐FU
fluorouracil
- HDAC
histone deacetylase
- IFN‐γ
interferon‐γ
- mAb
monoclonal antibody
- NCR
natural cytotoxicity receptor
- NK
natural killer
- NKG2D
natural killer group 2 member D
- RAR
retinoic acid receptor
- RARE
RA response element
- RXR
retinoic X receptor
- SAHA
suberoylanilide hydroxamic acid
- shRNA
small hairpin RNA
- siRNA
small interfering RNA
- TNF‐α
tumor necrosis factor‐α
- TSA
trichostatin A
- VPA
valproic acid
1. INTRODUCTION
Natural killer (NK) cells play important roles in fighting against tumor cells. They recognize tumor cells by activating receptors such as NKp30 and natural killer group 2 member D (NKG2D) [1, 2, 3, 4, 5, 6, 7, 8, 9]. NKp30 recognizes tumor ligand B7‐H6, which expresses exclusively on tumor cells [10, 11, 12]. The NKp30/B7‐H6 interaction leads to NK cell activation and tumor cell lysis [10, 13]. A study by Matta et al. [12] demonstrated that induced expression of B7‐H6 on the surface of monocytes triggers the activation of cells with NKp30 reporter and this activation could be blocked by anti‐B7‐H6 blocking monoclonal antibodies (mAbs). B7‐H6 has its advantages as a checkpoint target compared with NKp30 since it is undetectable on normal human tissues but selectively expressed on stressed cells [14], including tumor cell lines [10, 15‐18] and many kinds of primary cancer cells, such as the cancer cells of leukemia [10], neuroblastoma [17], hepatocellular carcinoma [19], melanoma [20], lung cancer [21], ovarian carcinoma [22], and breast cancer [23]. Therefore, B7‐H6 is considered a promising target for tumor therapy and more attention has been paid to the regulation of its expression.
The expression of B7‐H6 in tumor cells is promoted by stress inducers [16], such as cisplatin, fluorouracil (5‐FU), irradiation, non‐lethal heat shock, as well as inflammatory cytokines tumor necrosis factor‐α (TNF‐α), while histone deacetylase (HDAC) inhibitors down‐regulate B7‐H6 expression [15]. B7‐H6 expression on tumor cells is closely related to their susceptibility to NK cell lysis. The small hairpin RNA (shRNA) treatment of B7‐H6 effectively dampens the sensitization of tumor cells to NK‐mediated lysis [16]. It has been recently reported that oncogenic transcriptional factor c‐Myc drives B7‐H6 transcription in tumor cells [18]. Additionally, inhibition or knockdown of c‐Myc in tumor cells impairs NKp30‐mediated degranulation of NK cells [18, 24]. However, the regulation of B7‐H6 by tumor therapeutics remains largely unknown.
All‐trans retinoic acid (atRA)‐mediated differentiation therapy is the front‐line therapy for acute promyelocytic leukemia (APL). It leads to complete remission rates >90% and long‐term remission rates above 80% in combination with chemotherapy [25, 26, 27, 28]. Due to numerous achievements in APL treatment, atRA is tested in non‐APL acute myeloid leukemia (AML) and many solid tumors. However, clinical trials using atRA alone or in combination with chemotherapy have somehow shown disappointing results [28].
In this study, we investigated the regulation of B7‐H6 expression by atRA treatment in leukemia cells and its influence on tumor resistence to NK cell lysis. We found that NKp30:B7‐H6 axis plays an important role in NK cell‐mediated U‐937 cell lysis, and atRA treatment impairs the cytotoxicity of NK cells against U‐937 cells by reducing B7‐H6 expression through the regulation of c‐Myc. Our study revealed potential immunological adverse effects in the clinical use of atRA treatment.
2. MATERIALS AND METHODS
2.1. Cell lines
NK cell line NKG was established and maintained in our labratory as previously described [29] and cultured in α‐MEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 15% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA), 15% horse serum (Thermo Fisher Scientific, Waltham, MA, USA) and 100 U/mL rhIL‐2 (Kingsley, Yixing, Jiangsu, China). NK cell line NK92 was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in α‐MEM supplemented with 12.5% FBS, 12.5% horse serum, 0.2 mmol/L inositol (Sigma Aldrich, St. Louis, MO, USA), 0.1 mmol/L 2‐mercaptoethanol (Sigma Aldrich, St. Louis, MO, USA), 0.02 mmol/L folic acid (Sigma Aldrich, St. Louis, MO, USA) and 100 U/mL rhIL‐2. The leukemia cell lines U‐937 and THP‐1 were obtained from ATCC and cultured in RPMI‐1640 medium (Hyclone, South Logan, UT, USA) containing 10% FBS. The erythroleukemia cell line K562 were obtained from National Collection of Authenticated Cell Cultures (Shanghai, China) and cultured in IMDM medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS. The human embryonic kidney cell line HEK293T were obtained from National Collection of Authenticated Cell Cultures (Shanghai, China) and cultured in DMEM medium (Hyclone, South Logan, UT, USA) containing 10% FBS. All cell lines used in this study were maintained at 37°C in 5% CO2.
2.2. Antibody generation and labeling
Mouse anti‐human B7‐H6 antibody (anti‐B7‐H6#1, anti‐B7‐H6#2 and anti‐B7‐H6#3) were generated and verified in our laboratory. Briefly, BALB/c mice were immunized with B7‐H6‐hIgG1‐Fc fusion protein and spleen cells were fused with Sp/20 cells by Absea‐antibody (Beijing, China). Hybridoma culture supernants were selected for the binding with HL60‐B7‐H6 cells. Antibodies derived from hybridoma cells were tested for their ability to detect B7‐H6 as well as to inhibit NK cell‐mediated cytotoxicity. Anti‐B7‐H6#1 and anti‐B7‐H6#2 were able to inhibit NK cell‐mediated tumor lysis in 4 h cytotoxicity assay, and anti‐B7‐H6#3 was able to detect B7‐H6 in western blotting assay. Anti‐B7‐H6#1 antibody was labeled with Andy Fluor 647 NHS Ester (ABP Biosciences, Rockville, MD, USA) according to Protein Labeling Protocol. 100 μg antibody in 90 μL PBS was added to 1 vial of Andy Fluor 647 NHS Ester and incubated for 30 min at room temperature in a dark space. 10 μL Quench buffer was added to stop labeling and residue flour was removed by centrifuge at 10,000 g, 4℃, using an ultra‐filtration vial. The labeled antibody was stored at 1 mg/mL in PBS containing 0.5 mg/mL BSA and 0.02% NaN3 (Sigma Aldrich, St. Louis, MO, USA) at 4℃.
2.3. Flow cytometry
2 × 105 cells in 90 μL were incubated with 10 μL mouse serum (Future, Guangzhou, Guangdong, China) at 4℃ for 30 min. 0.5 μL Alexa 647‐conjugated anti‐B7‐H6 antibody was added and incubated at 4℃ for 30 min avoiding light. The cells were then washed with PBS. Flow cytometry was performed using a FACS LSRII (BD, Franklin Lakes, NJ, USA) and analyzed using the Flowjo software (BD, Franklin Lakes, NJ, USA).
2.4. siRNA and transfection
Small interfering RNA (siRNA) was used for transient knockdown of c‐Myc. c‐Myc siRNA (sc‐29226) and control siRNA (sc‐37007) were purchased from the Santa Cruz Biotechnology (Santa Cruz, CA, USA) and used at a final concentration of 100 nmol/L. The cells were transfected using Amaxa® Cell Line Nucleofector® Kit C (VCA‐1004, Lonza, Basel, Switzerland) according to the manufacturer's instructions. Transfection efficiency was determined by real‐time PCR, flow cytometry and western blot.
2.5. Construction of sgRNA expression vector
The coding sequence of Ncr3lg1 (coding gene of B7‐H6) was cloned into BsmBI‐pre‐digested pLentiCRISPRv2 plasmid (VT8107, YouBio, Changsha, Hunan, China). The target sequence of sgRNA was 20 bp and two complementary oligonucleotides (top, 5’‐CACCGTACCCATAGACGTGATGTTG‐3’ and bottom, 5’‐AAACCAACATCACGTCTATGGGTAC‐3’) were annealed to generate dsDNA which had 4 bp overhangs on both ends. pLentiCRISPRv2 plasmid was digested by BsmBI (R0739L, NEB, Ipswich, MA, USA) and dsDNA was ligased to the vector by T4 DNA ligase (EL0011, Thermo Fisher Scientific, Waltham, MA, USA).
2.6. Lentiviral package and transduction of cells
Lentiviral supernatants were generated by co‐transfecting HEK293T cells with pLentiCRISPRv2, psPAX2 (VT1444, YouBio, Changsha, Hunan, China) and pMD2G (VT1443, YouBio, Changsha, Hunan, China) plasmids using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA). Culture supernatants collected 48 h after transfection were added directly to U‐937 cells plated in 6‐well culture plates, and Polybrene (Sigma Aldrich, St. Louis, MO, USA) was added at a final concentration of 5 μg/mL for 24 h. Transduced cells were selected by the addition of 5 μg/mL puromycin (Invitrogen, Carlsbad, CA, USA). U‐937 cells transduced with B7‐H6 sgRNA were named U‐937‐δB7H6. Transfection efficiency was determined by flow cytometry.
2.7. Real‐time PCR assay
Total RNA was isolated using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. RNA was reverse transcribed using M‐MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA) and the resulting cDNA was used for real‐time PCR. SYBR Premix Ex TaqII (Takara, Kusatsu, Shiga, Japan) was used for real‐time PCR. Real‐time PCR was performed using LightCycler 96 (Roche, Basel, Switzerland), the cycling parameters were 95°C for 60s, followed by 40 cycles of 95°C for 5s and 60°C for 30s. Relative quatification of mRNA level was analyzed by supporting software (Roche, Basel, Switzerland). The following primers were used: B7‐H6 forward primer, 5’‐TTTTCCATTCATTGGTGGCCTA‐3’; B7‐H6 reverse primer, 5’‐TGCCCGAGTGCAAAAGAATATG‐3’; c‐Myc forward primer, 5’‐TCAAGAGGCGAACACCAAC‐3’; c‐Myc reverse primer, 5’‐GGCCTTTTCATTGTTTTCCA‐3’; β‐actin forward primer, 5’‐CTGGAACGGTGAAGGTGACA‐3’; and β‐actin reverse primer, 5’‐AAGGGACTTCCTGTAACAATGCA‐3’.
2.8. Western blotting
Cells were washed twice with ice‐cold PBS and then lysed in RIPA Lysis Buffer (Beyotime, Shanghai, China) supplemented with 0.25 mg/mL PMSF (Sigma Aldrich, St. Louis, MO, USA) and 1× protease inhibitor (Roche, Basel, Switzerland). An equal volume of 2 sample loading buffer (62.5 mmol/L Tris‐HCl, 25% glycerol, 2% SDS, 0.02% bromphenol blue, 5% β‐mercaptoethanol, pH 6.8) was added, and cleared extracts were frozen at ‐80°C. Protein concentration was determined using the BCA method. Equal amounts of protein (100 μg) were denatured in 95°C water bath for 5 min, electrophoresed with 4‐20% SurePAGE (GenScript, Nanjing, Jiangsu, China) in 1× MOPS (Sangon, Shanghai, China), and blotted to PVDF membrane (Millipore, ISEQ00010, USA). Anti‐B7‐H6#3 (1:2000) and c‐Myc (1:1000; AF6513, Beyotime, Shanghai, China) antibodies were used as primary antibodies respectively. For chemiluminescence, secondary antibodies HRP‐goat‐anti‐mouse IgG (1:5000; Boster Bio, Pleasanton, CA, USA) and HRP‐goat‐anti‐rabbit IgG (1:5000; Boster Bio, Pleasanton, CA, USA) were applied respectively, and ECL substrate (Bio‐Rad, Hercules, CA, USA) was used for detection.
2.9. Cytotoxicity assay
1 × 106 target cells in 1 mL serum‐free DMEM medium (Hyclone, South Logan, UT, USA) were incubated with CFSE (Thermo Fisher Scientific, Waltham, MA, USA) at a final concentration of 5 μmol/L for 30 min at 37℃. Staining was terminated by adding ice‐cold FBS. Labeled cells were then washed 3 times with complete DMEM medium. Target cells were diluted with complete RPMI‐1640 medium and added to 96‐well round plate at 1 × 104/well. NK92 and NKG cells were used as effector cells and co‐cultured with target cells at different effector:target (E:T) ratios for 4 h. 1 μL 7‐AAD (BD pharmingen, San Diego, CA, USA) was added to each well and cytotoxicity was determined using flow cytometry.
2.10. Statistical analysis
Statistical analysis and figure creation were performed using GraphPad Prism 6.0 (La Jolla, CA, USA) and SPSS Statistics 21 (IBM, Armonk, NY, USA). Comparisons between two groups were analyzed using the unpaired t‐test. Comparisons between three or more groups were analyzed using either the one‐way or two‐way ANOVA test. Results are expressed as means ± SEM. Values of P <0.05 were considered statistically significant.
3. RESULTS
3.1. NK cell‐mediated tumor lysis depends on NKp30/B7‐H6 interaction
NK cell‐mediated tumor rejection depends on activating receptors such as NKp30 and NKG2D [1, 2, 3, 4, 5, 6, 7, 8, 9]. To define the critical receptor‐ligand interaction between NK cells and tumor cells, we tested the effect of B7‐H6 mAb using cytotoxicity assay. As shown in Figure 1A and B, blockade of B7‐H6 significantly impaired the lysis of U‐937 cells mediated by NK92 or NKG cells (P <0.0001). Furthermore, blockade of B7‐H6 significantly impaired the lysis of THP‐1 cells mediated by NK92 cells (P <0.01; Supplementary Figure S1). Taking together, these data suggest that NKp30/B7‐H6 interaction played an important role in NK cell‐mediated tumor lysis.
FIGURE 1.
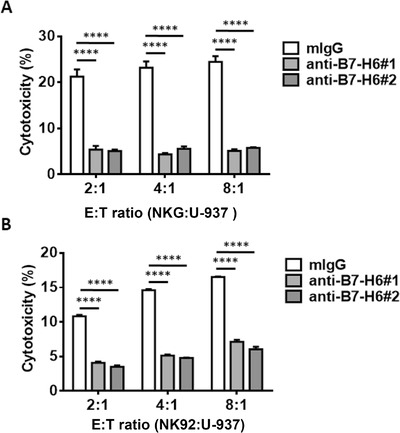
Blockade of B7‐H6 significantly impairs the cytotoxicity of NK cells against U‐937 cells. Lysis of U‐937 cells by NKG (A) or NK92 cells (B) was tested in a 4‐hour cytotoxicity assay. 5 μg/mL mouse anti‐human B7‐H6 antibodies (anti‐B7‐H6#1 and anti‐B7‐H6#2) or control antibodies (mIgG) was added to the culture medium. Data were analyzed for significance by the one‐way ANOVA test. Results are representative of at least three independent experiments with a minimum of two technical replicates and expressed as the mean±SEM. E:T, effector/target; ****, P <0.0001.
3.2. atRA treatment promotes tumor cell resistance against NK cell cytotoxicity
To define the influence of atRA treatment on NK cell cytotoxicity against tumor cells, we treated U‐937 and THP‐1 cells with 10 μmol/L atRA for 24 h and tested their sensitivity to NK cells in a 4‐hour cytotoxicity assay. Lysis of U‐937 cells mediated by NKG cells was significantly impaired by atRA treatment at E:T ratios of 8:1 and 16:1 (P <0.001; Figure 2A), while lysis of U‐937 cells mediated by NK92 cells was significantly impaired by atRA treatment at all the E:T ratios tested (all P <0.05; Figure 2B). Furthermore, atRA treatment significantly impaired the lysis of THP‐1 cells mediated by NK92 cells at E:T ratios of 2:1 and 8:1 (P < 0.05; Supplementary Figure S2). Taking together, these data suggest that atRA treatment promoted tumor resistance against NK cell cytotoxicity.
FIGURE 2.
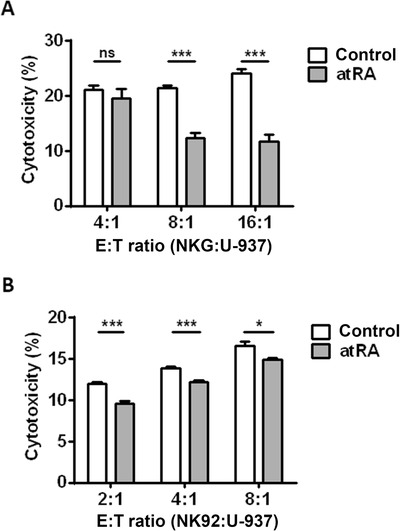
atRA induces U‐937 cell resistance against NK cell cytotoxicity. U‐937 cells were treated with 10 μmol/L atRA for 24 h or left untreated (Control) and subjected to NKG (A) or NK92 (B)‐mediated lysis in a 4‐hour cytotoxicity assay. Data were analyzed for significance by the unpaired t‐test. Results are representative of at least three independent experiments with a minimum of two technical replicates and expressed as the mean±SEM. E:T, effector/target; *, P <0.05; ***, P <0.001; ns, not significant.
3.3. atRA treatment down‐regulates B7‐H6 expression in U‐937 and THP‐1 cells
To explore the influence of atRA treatment on B7‐H6 expression, we treated U‐937 and THP‐1 cells with 10 μmol/L atRA for 24 h respectively, and analyzed B7‐H6 cell‐surface expression by flow cytometry. atRA treatment significantly down‐regulated B7‐H6 cell‐surface expression in both U‐937 and THP‐1 cells (P <0.0001; Figure 3A and B). To explore whether atRA treatment down‐regulates B7‐H6 at the transcriptional level, tumor cells were treated with 10 μmol/L atRA and subjected to analysis by real‐time PCR. 24 h and 48 h of atRA treatment both resulted in the reduced B7‐H6 mRNA level in U‐937 cells (P <0.0001; Figure 4A). We further treated U‐937 cells with various concentrations (0.039, 0.156, 0.625, 2.5, and 10 μmol/L) of atRA for 24 h and found that atRA down‐regulated B7‐H6 mRNA level in a dose‐dependent manner (Figure 4B). The influence of atRA treatment on THP‐1 cells was also investigated, 2.5 or 10 μmol/L atRA treatment significantly reduced B7‐H6 mRNA level in THP‐1 cells (Figure 4C). However, atRA treatment showed no negative effect on B7‐H6 mRNA level in human immortalized myelogenous leukemia cell line K562 (Supplementary Figure 3A). Taking together, these data suggest that atRA treatment reduced B7‐H6 expression in both U‐937 and THP‐1 cells.
FIGURE 3.
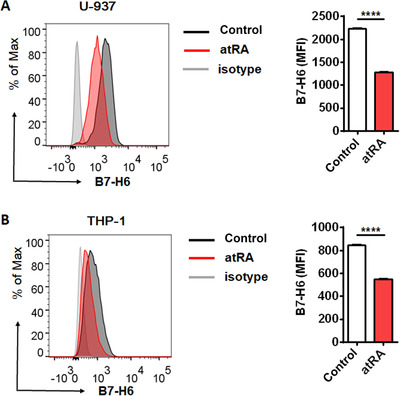
atRA down‐regulates B7‐H6 cell‐surface expression on U‐937 and THP‐1 cells. U‐937 (A) and THP‐1 (B) cells were treated with 10 μmol/L atRA or left untreated (Control) for 24 h, followed by the analysis of cell surface B7‐H6 expression through flow cytometry. Data were analyzed for significance by the unpaired t test. Results are representative of at least three independent experiments with a minimum of two technical replicates and are expressed as the mean±SEM. MFI, mean fluorescence intensity; ****, P <0.0001.
FIGURE 4.
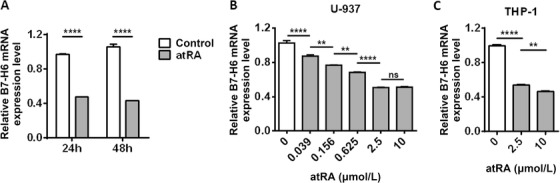
atRA down‐regulates B7‐H6 mRNA expression level in U‐937 and THP‐1 cells. A. U‐937 cells were treated with 10 μmol/L atRA or left untreated (Control) for 24 h or 48h. B7‐H6 mRNA level was analyzed by real‐time PCR. B‐C. U‐937 (B) and THP‐1 cells (C) were treated with atRA at various concentrations for 24 h and B7‐H6 mRNA level was analyzed by real‐time PCR. Data were analyzed for significance by the unpaired t test or the one‐way ANOVA test. Results are representative of at least three independent experiments with a minimum of two technical replicates and are expressed as the mean±SEM. **, P <0.01; ****, P <0.0001; ns, not significant.
3.4. atRA treatment promotes tumor cell resistance to NK cell‐mediated cytotoxicity by reducing B7‐H6 expression
To explore and verify the underlying mechanism, we generated B7‐H6 knockdown U‐937 cells (U‐937‐δB7H6) by transducing U‐937 cells with Crispr/Cas9 plasmid expressing B7‐H6 sgRNA (Figure 5A). U‐937 and U‐937‐δB7H6 cells were then treated with 10 μmol/L atRA for 24 h and subjected to NK92 mediated lysis in a 4‐hour cytotoxicity assay. The cytotoxicity of NK92 cells against U‐937‐δB7H6 cells was lower than that against U‐937 cells (Figure 5B and C). Furthermore, the use of atRA treatment impaired NK92 cytotoxicity against U‐937 cells, however, it showed no effect on the lysis of U‐937‐δB7H6 cells (Figure 5B and C), suggesting that atRA treatment promoted tumor cell resistance to NK cell‐mediated cytotoxicity by regulating B7‐H6 expression.
FIGURE 5.
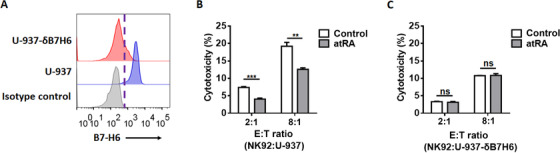
atRA shows no effect on the cytotoxicity of NK cells against B7‐H6 knockdown U‐937 cells. A. B7‐H6 was knockdown in U‐937 cells (U‐937‐δB7H6) by transducing the cells with Crispr/Cas9 plasmid expressing B7‐H6 sgRNA. B7‐H6 cell‐surface expression of U‐937 and U‐937‐δB7H6 cells was verified through flow cytometry. B‐C. U‐937 (B) and U‐937‐δB7H6 (C) cells were treated with 10 μmol/L atRA for 24 h or left untreated (Control) and subjected to NK92 mediated lysis in a 4‐hour cytotoxicity assay. Data were analyzed for significance by the unpaired t test. Results are representative of at least three independent experiments with a minimum of two technical replicates and are expressed as the mean±SEM. **, P <0.01; ***, P <0.001; ns, not significant.
3.5. atRA‐mediated B7‐H6 down‐regulation does not depend on DNA methylation or HDAC
atRA has been reported to regulate gene expression through epigenetic modifications such as DNA methylation and HDAC [28, 30‐32]. To investigate whether atRA‐mediates B7‐H6 down‐regulation relies on epigenetic modifications, we treated U‐937 cells with DNA methylation inhibitors and HDAC inhibitor, and then analyzed B7‐H6 mRNA by real‐time PCR. DNA methylation inhibitors, including Azacitidine, Zebularine, and RG‐108, inhibit DNA methylation through different mechanisms and therefore were all tested. None of these inhibitors restored B7‐H6 mRNA level which was suppressed by atRA treatment (Figure 6A‐C), suggesting that down‐regulation of B7‐H6 mRNA by atRA treatment was not dependent on DNA methylation.
FIGURE 6.
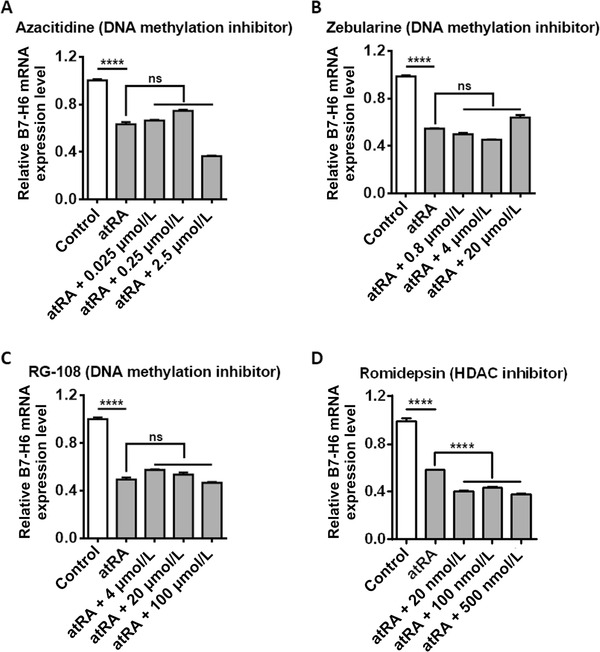
atRA‐mediated B7‐H6 down‐regulation is neither dependent on DNA methylation nor HDAC. U‐937 cells were treated with 10 μmol/L atRA, 10 μmol/L atRA in combination with various concentrations of Azacitidine (A), Zebularine (B), RG‐108 (C), or Romidepsin (D) for 24 h. B7‐H6 mRNA expression was analyzed by real‐time PCR. U‐937 cells treated with 10 μmol/L atRA or left untreated (Control) were used as control. Data were analyzed for significance by the two‐way ANOVA test. Results are representative of at least three independent experiments with a minimum of two technical replicates and are expressed as the mean±SEM. ****, P <0.0001; ns, not significant.
To study whether HDAC is involved, U‐937 cells were treated with HDAC inhibitor Romidepsin. Romidepsin alone reduced B7‐H6 mRNA level (data not shown), consistent with the report that HDAC inhibitors (suberoylanilide hydroxamic acid, trichostatin A, and valproic acid) down‐regulate B7‐H6 transcription in HeLa cells [15]. The combination of atRA and Romidepsin further reduced B7‐H6 mRNA level compared with atRA treatment alone (Figure 6D). Together, these data suggest that the down‐regulation of B7‐H6 by atRA treatment was neither dependent on DNA methylation nor HDAC.
3.6. atRA inhibits B7‐H6 transcription by down‐regulating c‐Myc
It was recently reported that the oncogenic transcription factor c‐Myc drives B7‐H6 transcription in tumor cells [24, 33]. We further investigated whether the transcriptional factor c‐Myc was involved in B7‐H6 transcriptional regulation in leukemia cell lines. We transfected U‐937 cells with c‐Myc siRNA and then analyzed B7‐H6 mRNA expression. As shown in Figure 7A and B, c‐Myc knockdown significantly reduced B7‐H6 mRNA and protein levels (P <0.0001). To study whether atRA down‐regulates c‐Myc expression, we treated U‐937 cells with 10 μmol/L atRA for 24 h and 48 h respectively, and then analyzed c‐Myc mRNA level by real‐time PCR. Both 24 h and 48 h of atRA treatment significantly reduced c‐Myc level in U‐937 cells (P <0.0001; Figure 7C). However, atRA treatment showed no effect on the c‐Myc mRNA level in K562 cells (Supplementary Figure S3B). Furthermore, atRA treatment significantly reduced c‐Myc protein expression in U‐937 cells (Figure 7D). THP‐1 cells were treated with either 2.5 μmol/L or 10 μmol/L atRA for 24 h and c‐Myc mRNA expression were evaluated by real‐time PCR. Both 2.5 μmol/L and 10 μmol/L atRA treatment significantly reduced c‐Myc mRNA in THP‐1 cells (Figure 7E). Taking together, these data suggest that atRA treatment down‐regulated B7‐H6 transcription possibly through inhibiting the c‐Myc signaling pathway.
FIGURE 7.
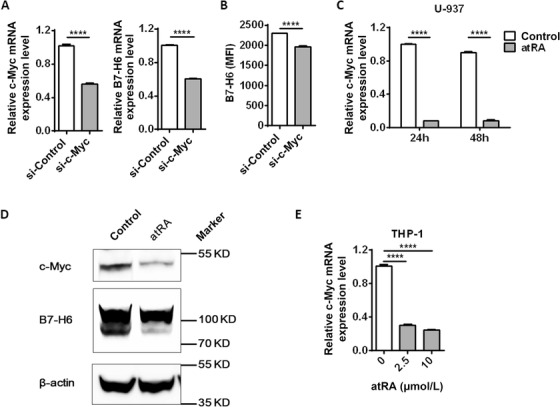
atRA inhibits B7‐H6 transcription by down‐regulating c‐Myc. A‐B. U‐937 cells were transfected with c‐Myc siRNA (si‐c‐Myc) or control siRNA (si‐Control) and harvested for 24 h. c‐Myc mRNA and B7‐H6 mRNA expression were analyzed by real‐time PCR (A). B7‐H6 cell‐surface expression was analyzed by flow cytometry (B). C. U‐937 cells were treated with 10 μmol/L atRA or left untreated (Control) for 24 h or 48 h, and c‐Myc mRNA expression was analyzed by real‐time PCR. D. U‐937 cells were treated with atRA or left untreated (Control) for 24 h, and then c‐Myc protein expression was analyzed by western blotting. E. THP‐1 cells were treated with atRA at various concentrations for 24 h and c‐Myc mRNA was analyzed by real‐time PCR. Data were analyzed for significance by the unpaired t test or the one‐way ANOVA test. Results are representative of at least three independent experiments with a minimum of two technical replicates and are expressed as the mean±SEM. ****, P <0.0001.
4. DISCUSSION
The cytotoxicity of NK cells depends on the balance between activating and inhibitory signals. The natural cytotoxicity receptors (NCRs) and NKG2D are the major activating receptors involved in tumor cell surveillance [14]. The interaction between NKp30 and tumor cell‐derived B7‐H6 results in the production of cytotoxic mediators, such as perforins, granzymes, TNF‐α, and interferon‐γ (IFN‐γ) [34]. Our study showed that NK cell‐mediated lysis of tumor cells (U‐937 and THP‐1) was mainly dependent on NKp30/B7‐H6 interaction and atRA treatment induced U‐937 and THP‐1 resistance against NK cell cytotoxicity by reducing the expression of NKp30 ligand B7‐H6. It has been reported that transcriptional factor c‐Myc is responsible for driving B7‐H6 expression in tumor cells, our study showed that atRA treatment down‐regulated B7‐H6 transcription by reducing c‐Myc expression.
The epigenetic modifications by atRA have been deeply investigated. The receptor of atRA, retinoic acid receptor (RAR), forms a heterodimer with retinoic X receptor (RXR) and binds to the RA response element (RARE) of the promoter region [35]. In the absence of ligand binding, RAR/RXR forms a complex with co‐repressors including epigenetic modifiers that mediate DNA methylation or HDACs and suppress gene expression [28, 32, 31]. However, our data indicate that atRA‐mediated B7‐H6 down‐regulation was neither dependent on DNA methylation nor DHAC.
c‐Myc reduction is one of the hallmarks of atRA‐induced terminal differentiation [36]. However, c‐Myc is not down‐regulated in atRA‐resistant tumors upon atRA treatment. It has been reported that atRA alone does not lead to c‐Myc reduction in solid tumor cells, such as MCF‐7, while the combination of atRA and type I IFN leads to c‐Myc down‐regulation [33, 37]. As c‐Myc drives the B7‐H6 transcription in tumor cells and c‐Myc mRNA level is not affected by atRA treatment in K562 cells, atRA resistance explains the unaffected B7‐H6 expression in tumor cell line K562.
In conclusion, we found that lysis of U‐937 cells by NK92 or NKG cells was largely dependent on NKp30/B7‐H6 interaction, blockade of B7‐H6 by mAb significantly impaired NK cytotoxicity. atRA treatment impaired U‐937 lysis by NK92 or NKG cells and reduced B7‐H6 expression in U‐937 and THP‐1 cells on both protein and mRNA levels. Furthermore, atRA treatment showed no effect on NK cytotoxicity against B7‐H6 knockdown U‐937 cells. Epigenetic modifications such as DNA methylation and HDAC were not responsible for atRA‐mediated down‐regulation of B7‐H6 expression, as inhibitors of these pathways could not restore B7‐H6 expression. On the other hand, atRA treatment reduced c‐Myc expression, the transcriptional factor driving B7‐H6 expression in tumor cells, and therefore inhibited the transcription of B7‐H6 in leukemia cells. Our study suggests that atRA impaired NK cell‐mediated tumor rejection through down‐regulating B7‐H6 expression via the c‐Myc signaling pathway.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This work was supported by the National Key R&D Program of China (2019YFA0508502), CAMS Innovation Fund for Medical Sciences (2019‐I2M‐5‐073), and the National Natural Science Foundation of China (31700754, 81788101, and 81821001).
AUTHORS' CONTRIBUTIONS
GC and RS designed the study. GC and YC performed the experiments and analyzed the data. GC and HS wrote and revised the manuscript. ZT, HW, RS, and XZ supervised the study. RS and HS critically reviewed the manuscript. All authors read and approved the final manuscript.
Supporting information
Supporting Figure S1
Supporting Figure S2
Supporting Figure S3
ACKNOWLEDGMENTS
Not applicable.
Cao G, Cheng Y, Zheng X, et al. All‐trans retinoic acid induces leukemia resistance to NK cell cytotoxicity by down‐regulating B7‐H6 expression via c‐Myc signaling. Cancer Commun. 2021;41:51–61. 10.1002/cac2.12121
Contributor Information
Rui Sun, Email: sunr@ustc.edu.cn.
Haoyu Sun, Email: haoyusun@ustc.edu.cn.
REFERENCES
- 1. Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1(1):41‐9. [DOI] [PubMed] [Google Scholar]
- 2. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503‐10. doi:http://www.nature.com/ni/journal/v9/n5/supinfo/ni1582_S1.html. [DOI] [PubMed] [Google Scholar]
- 3. Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? The Journal of Clinical Investigation. 2008;118(6):1991‐2001. 10.1172/JCI35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL et al. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science. 2011;331(6013):44‐9. 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell‐based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10(3):230‐52. 10.1038/cmi.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MORETTA L, Pietra G, Montaldo E, Vacca P, Pende D, Falco M et al. Human NK cells: From surface receptors to the therapy of leukemias and solid tumors. Frontiers in Immunology. 2014;5 10.3389/fimmu.2014.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Habif G, Crinier A, Andre P, Vivier E, Narni‐Mancinelli E. Targeting natural killer cells in solid tumors. Cellular & molecular immunology. 2019;16(5):415‐22. 10.1038/s41423-019-0224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kucan Brlic P, Lenac Rovis T, Cinamon G, Tsukerman P, Mandelboim O, Jonjic S. Targeting PVR (CD155) and its receptors in anti‐tumor therapy. Cellular & molecular immunology. 2019;16(1):40‐52. 10.1038/s41423-018-0168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prajapati K, Perez C, Rojas LBP, Burke B, Guevara‐Patino JA. Functions of NKG2D in CD8(+) T cells: an opportunity for immunotherapy. Cellular & molecular immunology. 2018;15(5):470‐9. 10.1038/cmi.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B et al. The B7 family member B7‐H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. The Journal of Experimental Medicine. 2009;206(7):1495‐503. 10.1084/jem.20090681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cellular & molecular immunology. 2019;16(5):430‐41. 10.1038/s41423-019-0206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matta J, Baratin M, Chiche L, Forel JM, Cognet C, Thomas G et al. Induction of B7‐H6, a ligand for the natural killer cell‐activating receptor NKp30, in inflammatory conditions. Blood. 2013;122(3):394‐404. 10.1182/blood-2013-01-481705. [DOI] [PubMed] [Google Scholar]
- 13. Kellner C, Maurer T, Hallack D, Repp R, van de Winkel JGJ, Parren PWHI et al. Mimicking an Induced Self Phenotype by Coating Lymphomas with the NKp30 Ligand B7‐H6 Promotes NK Cell Cytotoxicity. The Journal of Immunology. 2012;189(10):5037‐46. 10.4049/jimmunol.1201321. [DOI] [PubMed] [Google Scholar]
- 14. Kaifu T, Escaliere B, Gastinel LN, Vivier E, Baratin M. B7‐H6/NKp30 interaction: a mechanism of alerting NK cells against tumors. Cellular and Molecular Life Sciences. 2011;68(21):3531‐9. 10.1007/s00018-011-0802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fiegler N, Textor S, Arnold A, Rölle A, Oehme I, Breuhahn K et al. Downregulation of the activating NKp30 ligand B7‐H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood. 2013;122(5):684‐93. [DOI] [PubMed] [Google Scholar]
- 16. Cao G, Wang J, Zheng X, Wei H, Tian Z, Sun R. Tumor Therapeutics Work as Stress Inducers to Enhance Tumor Sensitivity to Natural Killer (NK) Cell Cytolysis by Up‐regulating NKp30 Ligand B7‐H6. Journal of Biological Chemistry. 2015;290(50):29964‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Semeraro M, Rusakiewicz S, Minard‐Colin V, Delahaye NF, Enot D, Vély F et al. Clinical impact of the NKp30/B7‐H6 axis in high‐risk neuroblastoma patients. Science translational medicine. 2015;7(283):283ra55‐ra55. 10.1126/scitranslmed.aaa2327. [DOI] [PubMed] [Google Scholar]
- 18. Textor S, Bossler F, Henrich K‐O, Gartlgruber M, Pollmann J, Fiegler N et al. The proto‐oncogene Myc drives expression of the NK cell‐activating NKp30 ligand B7‐H6 in tumor cells. OncoImmunology. 2016;5(7):e1116674 10.1080/2162402X.2015.1116674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen L, Feng J, Xu B, Zhou Y, Zheng X, Wu C et al. Expression of B7‐H6 expression in human hepatocellular carcinoma and its clinical significance. Cancer cell international. 2018;18:126 10.1186/s12935-018-0627-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schlecker E, Fiegler N, Arnold A, Altevogt P, Rose‐John S, Moldenhauer G et al. Metalloprotease‐Mediated Tumor Cell Shedding of B7‐H6, the Ligand of the Natural Killer Cell–Activating Receptor NKp30. Cancer Research. 2014;74(13):3429‐40. 10.1158/0008-5472.can-13-3017. [DOI] [PubMed] [Google Scholar]
- 21. Zhang X, Zhang G, Qin Y, Bai R, Huang J. B7‐H6 expression in non‐small cell lung cancers. International Journal of Clinical and Experimental Pathology. 2014;7(10):6936‐42. [PMC free article] [PubMed] [Google Scholar]
- 22. Pesce S, Tabellini G, Cantoni C, Patrizi O, Coltrini D, Rampinelli F et al. B7‐H6‐mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. OncoImmunology. 2015;4(4):e1001224 10.1080/2162402X.2014.1001224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun J, Tao H, Li X, Wang L, Yang J, Wu P et al. Clinical significance of novel costimulatory molecule B7‐H6 in human breast cancer. Oncology letters. 2017;14(2):2405‐9. 10.3892/ol.2017.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen HQ, Guo YD, Sun J, Dong J, Bao QH, Zhang XG et al. Preferential Expression of B7‐H6 in Glioma Stem‐Like Cells Enhances Tumor Cell Proliferation via the c‐Myc/RNMT Axis. Journal of Immunology Research. 2020;2020. doi:Artn 232867510.1155/2020/2328675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L et al. Use of all‐trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72(2):567‐72. [PubMed] [Google Scholar]
- 26. Testa U, Lo‐Coco F. Prognostic factors in acute promyelocytic leukemia: strategies to define high‐risk patients. Annals of hematology. 2016;95(5):673‐80. 10.1007/s00277-016-2622-1. [DOI] [PubMed] [Google Scholar]
- 27. Kayser S, Schlenk RF, Platzbecker U. Management of patients with acute promyelocytic leukemia. Leukemia. 2018;32(6):1277‐94. 10.1038/s41375-018-0139-4. [DOI] [PubMed] [Google Scholar]
- 28. Ni X, Hu G, Cai X. The success and the challenge of all‐trans retinoic acid in the treatment of cancer. Critical reviews in food science and nutrition. 2018:1‐10. 10.1080/10408398.2018.1509201. [DOI] [PubMed] [Google Scholar]
- 29. Cheng M, Ma J, Chen YY, Zhang JH, Zhao WD, Zhang J et al. Establishment, Characterization, and Successful Adaptive Therapy Against Human Tumors of NKG Cell, a New Human NK Cell Line. Cell Transplant. 2011;20(11‐12):1731‐46. 10.3727/096368911x580536. [DOI] [PubMed] [Google Scholar]
- 30. Erkelens MN, Mebius RE. Retinoic Acid and Immune Homeostasis: A Balancing Act. Trends in Immunology. 2017. [DOI] [PubMed] [Google Scholar]
- 31. Di Masi A, Leboffe L, De Marinis E, Pagano F, Cicconi L, Rochette‐Egly C et al. Retinoic acid receptors: from molecular mechanisms to cancer therapy. Molecular aspects of medicine. 2015;41:1‐115. [DOI] [PubMed] [Google Scholar]
- 32. Tang X‐H, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annual Review of Pathology: Mechanisms of Disease. 2011;6:345‐64. [DOI] [PubMed] [Google Scholar]
- 33. Shang YF, Baumrucker CR, Green MH. c‐Myc is a major mediator of the synergistic growth inhibitory effects of retinoic acid and interferon in breast cancer cells. Journal of Biological Chemistry. 1998;273(46):30608‐13. 10.1074/jbc.273.46.30608. [DOI] [PubMed] [Google Scholar]
- 34. Pinheiro PF, Justino GC, Marques MM. NKp30 ‐ A prospective target for new cancer immunotherapy strategies. Br J Pharmacol. 2020;177(20):4563‐80. 10.1111/bph.15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Minucci S, Leid M, Toyama R, Saint‐Jeannet JP, Peterson VJ, Horn V et al. Retinoid X receptor (RXR) within the RXR‐retinoic acid receptor heterodimer binds its ligand and enhances retinoid‐dependent gene expression. Mol Cell Biol. 1997;17(2):644‐55. 10.1128/mcb.17.2.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dimberg A, Bahram F, Karlberg I, Larsson LG, Nilsson K, Oberg F. Retinoic acid‐induced cell cycle arrest of human myeloid cell lines is associated with sequential down‐regulation of c‐Myc and cyclin E and posttranscriptional up‐regulation of p27(Kip1). Blood. 2002;99(6):2199‐206. 10.1182/blood.V99.6.2199. [DOI] [PubMed] [Google Scholar]
- 37. van Gils N, Verhagen H, Smit L. Reprogramming acute myeloid leukemia into sensitivity for retinoic‐acid‐driven differentiation. Experimental hematology. 2017;52:12‐23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figure S1
Supporting Figure S2
Supporting Figure S3
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.