

Summary
Aberrant redox signaling underlies the pathophysiology of many chronic metabolic diseases, including type 2 diabetes (T2D). Methodologies aimed at rebalancing systemic redox homeostasis have had limited success. A noninvasive, sustained approach would enable the long-term control of redox signaling for the treatment of T2D. We report that static magnetic and electric fields (sBE) noninvasively modulate the systemic GSH-to-GSSG redox couple to promote a healthier systemic redox environment that is reducing. Strikingly, when applied to mouse models of T2D, sBE rapidly ameliorates insulin resistance and glucose intolerance in as few as three days with no observed adverse effects. Scavenging paramagnetic byproducts of oxygen metabolism with SOD2 in hepatic mitochondria fully abolishes these insulin sensitizing effects, demonstrating that mitochondrial superoxide mediates induction of these therapeutic changes. Our findings introduce a remarkable redox-modulating phenomenon that exploits endogenous electromagneto-receptive mechanisms for the noninvasive treatment of T2D, and potentially other redox-related diseases.
Keywords: redox, type 2 diabetes, insulin resistance, electromagnetic fields, ROS, superoxide, glutathione, radical pair mechanism, mitochondria, liver
Introduction
Life on Earth evolved in the presence of a static magnetic (B) field and a vertically oriented electrostatic (E) field (Doglioni et al., 2016; Honkonen et al., 2018; Thébault et al., 2015). Yet, the interaction of both fields with living systems is one of the least understood in biology. Many lifeforms across all major phyla sense static electromagnetic fields (EMFs) for cellular migration (Lin et al., 2017; Lin et al., 2018), geospatial mapping and long-distance navigation (Mouritsen, 2018; Wiltschko and Wiltschko, 1972). Although poorly understood, the directional responses induced by EMFs are purportedly mediated by modulating of endogenous redox reactions, specifically through quantum spin-state interactions with paramagnetic radicals (Harkins and Grissom, 1994; Hiscock et al., 2016; Maeda et al., 2008; Mouritsen, 2018; Schulten et al., 1978). However, the physiological function of electromagnetic sensing beyond spatial mapping remains unknown.
Glucose metabolism also evolved in the presence of EMFs and is increasingly understood to be regulated by redox homeostasis; that is, the balance of pro-oxidants and antioxidants (Jones and Sies, 2015; Oberley, 1988; Watson, 2014). Aberrant redox homeostasis contributes to the development of insulin resistance and hyperglycemia (Anderson et al., 2009; Henriksen et al., 2011; Houstis et al., 2006). Patients with type 2 diabetes (T2D) have elevated levels of pro-oxidants, such as superoxide (O2•−) and hydrogen peroxide (H2O2), and lower levels of antioxidants, primarily glutathione (Goldstein et al., 2005; Henriksen et al., 2011; Monnier et al., 2006; Samiec et al., 1998; Sampson et al., 2002; Sekhar et al., 2011). This redox imbalance leads to an oxidized systemic environment that modifies cysteine residues, disrupting protein function and insulin action (Finkel, 2011; van der Reest et al., 2018). Rebalancing the systemic redox environment by the continuous administration of antioxidants enhances insulin sensitivity in patients with T2D, demonstrating that redox homeostasis regulates glucose metabolism and insulin effectiveness (Barbagallo et al., 1999; De Mattia et al., 1998; Paolisso et al., 1992). Unfortunately, redox-based interventions for the management of T2D have not been translated to clinical settings because they require frequent or continuous administration due to short-half-lives (e.g. GSH <10 mins) and are associated with side effects which negatively impacts adherence (Buglak et al., 2018; Elbatreek et al., 2019; Sabaté, 2003). New methods are needed to modulate systemic redox homeostasis to address an underlying pathomechanism of T2D.
Based on the involvement of redox systems in facilitating both the biological effects of EMFs and in regulating glucose metabolism, we reasoned that EMFs may modulate glucose metabolism via redox systems. However, the evidence supporting a physiological effect of EMFs in glucose regulation is scant. Attempts to investigate the potential effects of EMFs on glucose metabolism have yielded conflicting findings with some studies demonstrating that EMFs raise fasting blood glucose and others suggesting that EMFs have no effect (Elferchichi et al., 2011; Gerardi et al., 2008; Harakawa et al., 2005; Lai et al., 2016; Li et al., 2016; Shi et al., 2015; Szemerszky et al., 2010). These studies utilized different field parameters, lacked sensitive measures of glycemia and insulin sensitivity and did not utilize insulin resistant animal models. Thus, the effect of EMFs on glucose regulation has not been systematically evaluated in vivo.
Here we set out to investigate the biological effects of static magnetic and electrostatic fields. In particular, we investigated the effect of a combined application of magnetostatic and electrostatic fields on glycemia using three mouse models of T2D: the obese-diabetic mouse model of the pleiotropic human disorder Bardet-Biedl Syndrome (BBS) (Carter et al., 2012; Starks et al., 2015), the leptin-receptor deficient (db/db) model, and the 60% high fat diet-fed dietary (hereafter referred to as HFD) model. We report that the combined application of static magnetic and electric fields (sBE) ameliorates hyperglycemia and insulin resistance in these models via redox-dependent mechanisms.
Results
Exposure to sBE Improves Glycemia
Adult normoglycemic mice [normal chow diet-fed (hereafter referred to NCD) and wildtype (WT)], as well as two obese mouse models of type 2 diabetes, the HFD and db/db models, were continuously exposed to sBE; B field, 3 mT, and vertically-oriented E field, 7 kV/m) for 30 days (Figure 1A). These field strengths were chosen to be approximately 100x the strength of the Earth’s B and vertical E fields in order to minimize noise due to natural fluctuations (Honkonen et al., 2018; Thébault et al., 2015). Control animals were housed in similar but inert devices to control for potential environmental variations (i.e. temperature, humidity, lighting, sound, tactile, scent etc.). All mice were housed in non-metallic cages to prevent electromagnetic interference and mice were able to roam freely while inside their cages. Glycemia was evaluated by measurement of fasting blood glucose (FBG) and performance of glucose tolerance tests (GTTs). To our surprise, exposure to sBE significantly reduced FBG by 45% in the BBS model compared to control (Figure 1B) and by 33% in both the HFD and db/db mice while reversing glucose intolerance in the latter two models (Figure 1C and 1D). No significant changes in FBG or glucose tolerance were observed in non-diabetic mice exposed to sBE (Figure 1C and 1D).
Figure 1. sBE exposure improves glucose tolerance.
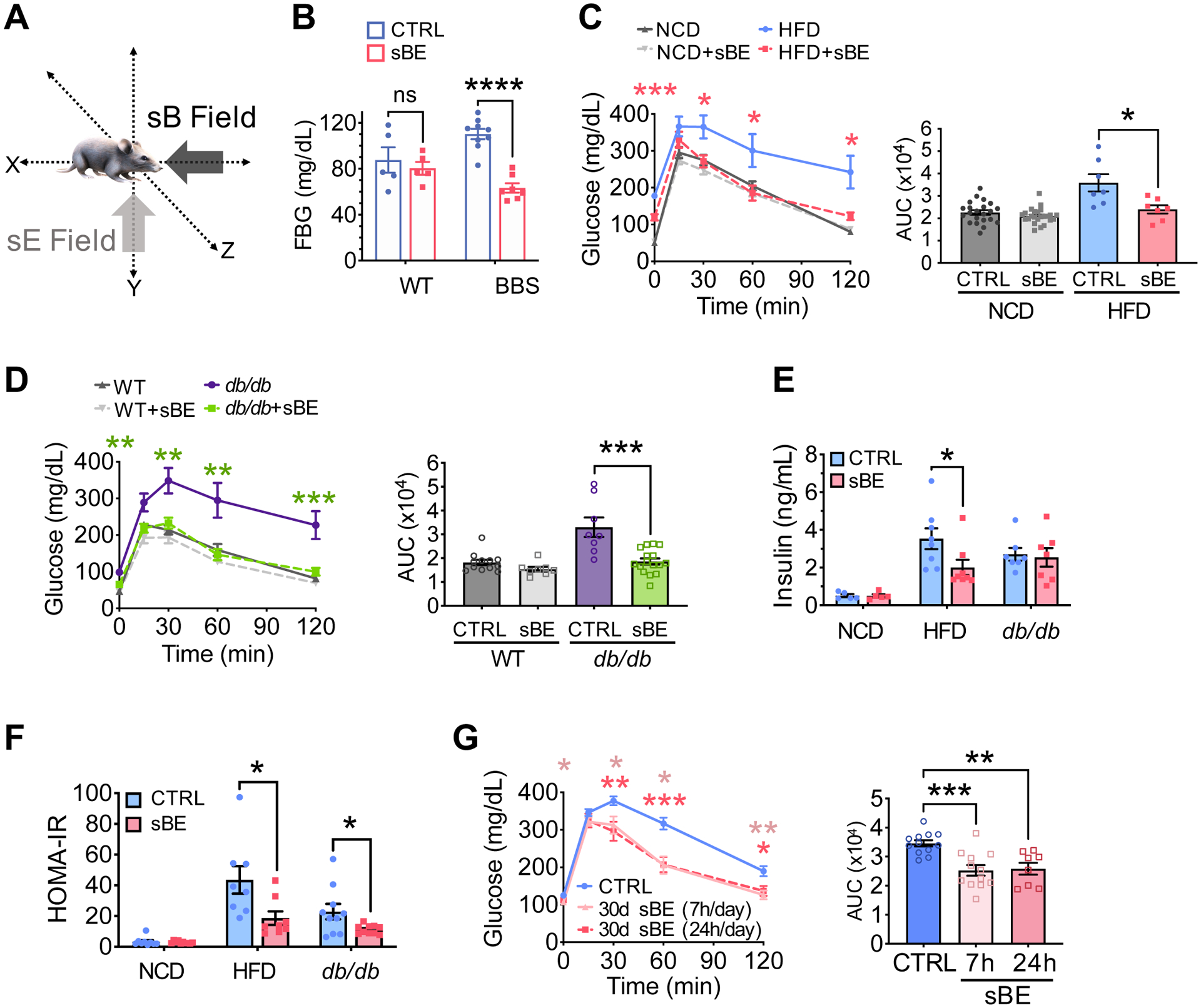
(A) Illustration of static magnetic (sB) and vertically oriented electrostatic fields (sE), in combination termed sBE.
(B) Fasting blood glucose (FBG) levels after a 16 h fast in Bardet-Biedl Syndrome (BBS) mice and their wildtype (WT) littermates after 30 days of sBE exposure (WT mice, n = 5/group; BBS mice, n ≥ 7/group).
(C) Glucose tolerance test (GTT) and FBG shown as timepoint 0 min (left) and area under the curve (AUC) (right) for normal chow diet (NCD) and high fat diet (HFD) mice exposed to sBE for 30 days (NCD mice, n ≥ 22/group; HFD mice, n = 7/group).
(D) GTT and FBG shown as timepoint 0 min (left) and area under the curve (AUC) (right) for WT and leptin-receptor deficient mice (db/db) exposed to sBE for 30 days (n ≥ 8 mice/group).
(E) Plasma insulin levels after a 16 h fast in NCD, HFD, and db/db mice exposed to sBE for 30 days (n ≥ 8 mice/group).
(F) Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) of NCD, HFD, and db/db mice exposed to sBE for 30 days (n ≥ 8 mice/group).
(G) GTT and FBG (left) and area under the curve (AUC) (right) for HFD mice exposed to 30 days of sBE for 7 h per day or 24 h per day (n ≥ 8 mice/group).
Data presented as mean ± SEM. Data panels (A–F) analyzed by two-tailed, unpaired t-test. Data panel (G) analyzed by one-way ANOVA with Tukey’s correction for multiple comparisons. ns = not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To determine whether both fields were necessary to achieve these effects, HFD mice were exposed to magnetostatic (sB), electrostatic (sE) or combined sBE fields for 30 days at which point GTTs were performed. Interestingly, sB fields significantly worsened glycemia and glucose tolerance, whereas sE fields had no significant effects relative to untreated mice (Figure S1A). These findings are consistent with prior studies that showed that exposure to magnetostatic fields worsened glycemia (Elferchichi et al., 2011). We found that only combined sBE fields significantly improved glucose tolerance (Figure S1A). Therefore, we employed sBE fields for all experiments henceforth.
To determine whether long-term exposure was effective, HFD mice were exposed to sBE for 22 weeks. Treated mice showed a 40% reduction in FBG compared to untreated mice, suggesting that the anti-hyperglycemic effects of sBE are durable over 5 months of continuous sBE exposure (Figure S1B and S1C). This finding indicates that long-term sBE exposure does not lead to metabolic compensations that attenuate the anti-hyperglycemic effects. After 22 weeks, sBE exposure was ceased and resulted in the rebounding of FBG within 7 days of sBE withdrawal (Figure S1B and S1C). A withdrawal effect was also investigated in db/db mice in a pair-wise manner in which GTTs were performed at three timepoints: prior to sBE treatment, at 16 days of treatment and after 4 days of treatment cessation (Figure S1D). These experiments show that 16 days of sBE exposure improved glucose tolerance by 36% relative to pre-treatment glucose tolerance (Figure S1E). Treatment was then ceased, and glucose tolerance rebounded to pre-treatment levels within 4 days (Figure S1E). Together, these findings indicate that the glycemic effects induced by sBE are durable and rebound within 7 days of treatment cessation.
Next, we investigated whether sBE exposure exerts glycemic effects by raising insulin levels. HFD and db/db mice were continuously exposed to sBE for 30 days and plasma insulin levels were assessed following a 16 h fast. Fasting plasma insulin levels were not elevated in any animal exposed to sBE and were 43% lower in sBE-exposed HFD mice (Figure 1E). Lower fasting insulin levels, as observed in treated HFD mice, are consistent with improved insulin sensitivity. Insulin levels were not lower in treated db/db mice likely reflecting model-specific differences in the response to sBE due to changes in leptin signaling, differences in the severity of insulin resistance, and more severe islet dysfunction (Ishida et al., 2004). Nevertheless, these findings suggest that sBE exposure acts by enhancing insulin sensitivity rather than acting as an insulin secretagogue. To estimate the effect of sBE exposure on insulin sensitivity, homeostatic model assessment of insulin resistance (HOMA-IR) was calculated and revealed that sBE ameliorated insulin resistance in both the HFD and db/db insulin resistant mouse models when compared to untreated mice (Figure 1F). Thus, the anti-hyperglycemic effects of sBE treatment are likely due to the enhancement of insulin action.
These remarkable changes prompted us to evaluate a more amenable treatment regimen. Current medications for T2D require patients to self-administer medications throughout the day and thus have significant adherence challenges that impact treatment success (Sabaté, 2003). Due to its potential to be automated, we reasoned that sBE exposure could serve as a passive modality to manage T2D during sleep if treatment time could be reduced. Therefore, HFD mice were exposed to sBE for 7 h/day for 30 days, during the time that mice typically sleep (7 am – 2 pm). Notably, mice exposed for 7 h/day showed similar improvements in glucose tolerance compared to continuously-exposed mice, indicating that continuous exposure is not needed to achieve therapeutic effects (Figure 1G).
sBE Exposure Enhances Insulin Sensitivity and Ameliorates Insulin Resistance
To definitively determine whether sBE exposure modulates insulin sensitivity, we performed euglycemic-hyperinsulinemic clamps (Figure S2A). NCD- and HFD mice were exposed to sBE for up to 30 days and then clamped following a 6 h fast. During the clamped period, 30-day sBE exposed mice required striking increases in glucose infusion rates (GIRs) of 17% in NCD and 104% in HFD mice in order to maintain euglycemia in response to steady state insulin (Figure 2A, 2B, S2B and S2C). These improvements in insulin sensitivity occurred in as few as 3 days of continuous exposure and were as effective when exposed for 7 h/day (Figure 2B). Consistent with enhanced insulin sensitivity, sBE exposure markedly enhanced insulin-stimulated glucose disposal by 14% in NCD and by 62% in HFD mice after as early as 3 days of exposure (Figure 2C and 2D). In addition, sBE exposure did not enhance insulin suppression of glucose production in NCD mice and only showed an effect in HFD mice after 30 days of exposure, indicating that the primary driver of the elevated GIR following sBE exposure was insulin-stimulated glucose disposal (Figure 2E and 2F). Interestingly, sBE did not significantly enhance insulin-stimulated glucose uptake in any tissue measured in NCD or HFD mice (Figure 3A and 3B). However, exposure to sBE significantly enhanced glucose incorporation into glycogen in liver but not in skeletal muscle in both NCD and HFD mice (Figure 3C and 3D). Only the combined sBE fields elicited a significant elevation in hepatic glycogen in HFD mice (Figure 3E). Elevations in hepatic glycogen were also found in db/db mice following sBE exposure and in primary human hepatocytes, suggesting a liver-specific effect that extends beyond rodents (Figure 3F and 3G). These data demonstrate that sBE exposure markedly enhances insulin sensitivity and implicate the liver as a major target.
Figure 2. sBE exposure enhances insulin sensitivity.
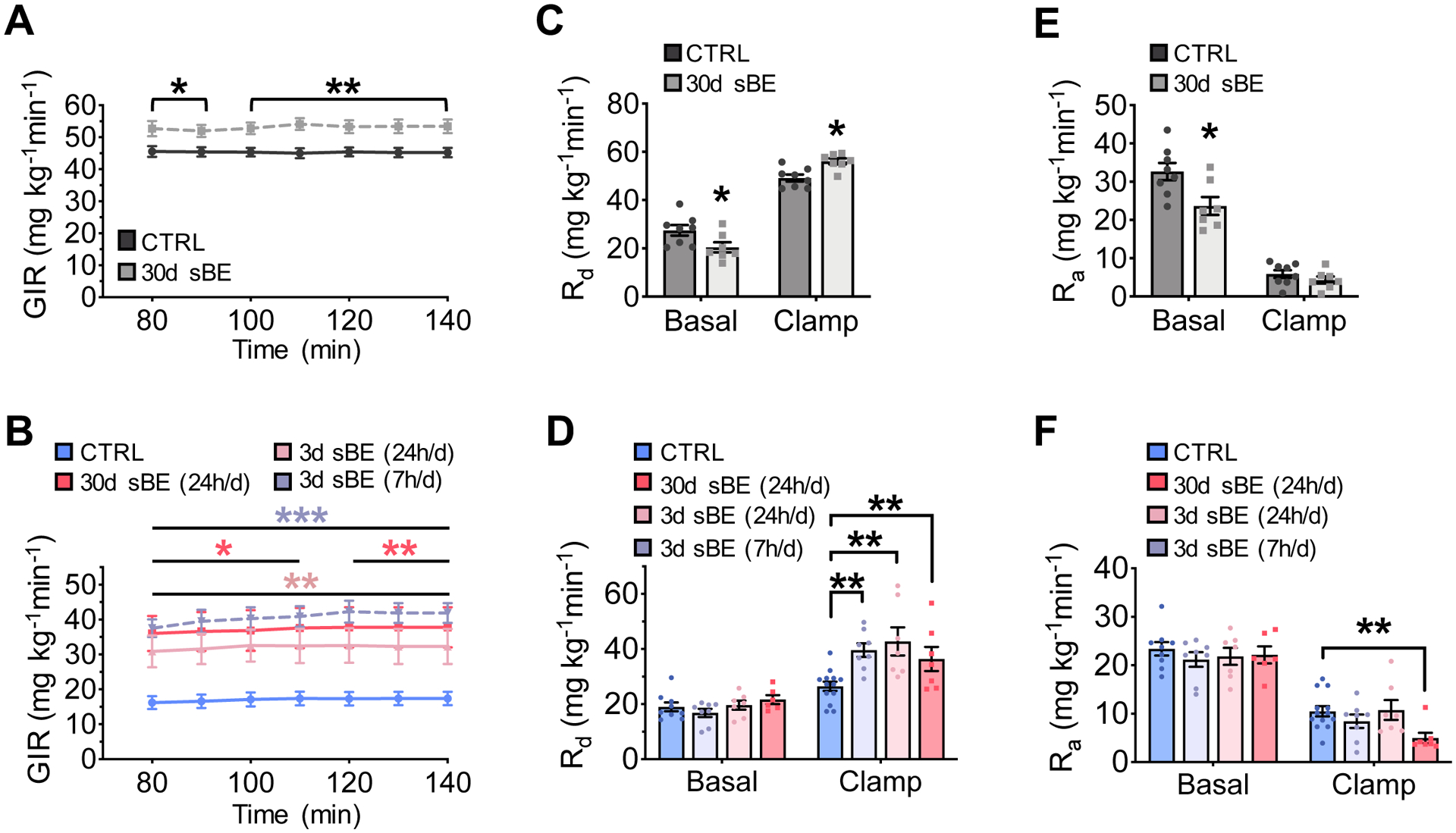
(A–F) Euglycemic-hyperinsulinemic clamps were performed on NCD and HFD mice exposed to sBE following a 6 h fast.
(A) Glucose infusion rate (GIR) in NCD mice exposed to 30 days of continuous sBE (n ≥ 7 mice/group).
(B) GIR in HFD mice exposed to sBE continuously (24 h/day) for 30 days, continuously (24 h/day) for 3 days, or for 7 h/day for a total of 3 days (n ≥ 7mice/group).
(C) Rate of glucose disposal (Rd) in NCD mice (n ≥ 7mice/group).
(D) Rd in HFD mice (n ≥ 7mice/group).
(E) Rate of glucose appearance (Ra) in NCD mice (n ≥ 7mice/group).
(F) Ra in HFD mice (n ≥ 7mice/group).
Data presented as mean ± SEM. NCD mice data analyzed by two-tailed, unpaired Student’s t-test. HFD mice data analyzed by one-way ANOVA with Dunnett’s correction for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3. sBE exposure enhances glucose incorporation into liver glycogen.
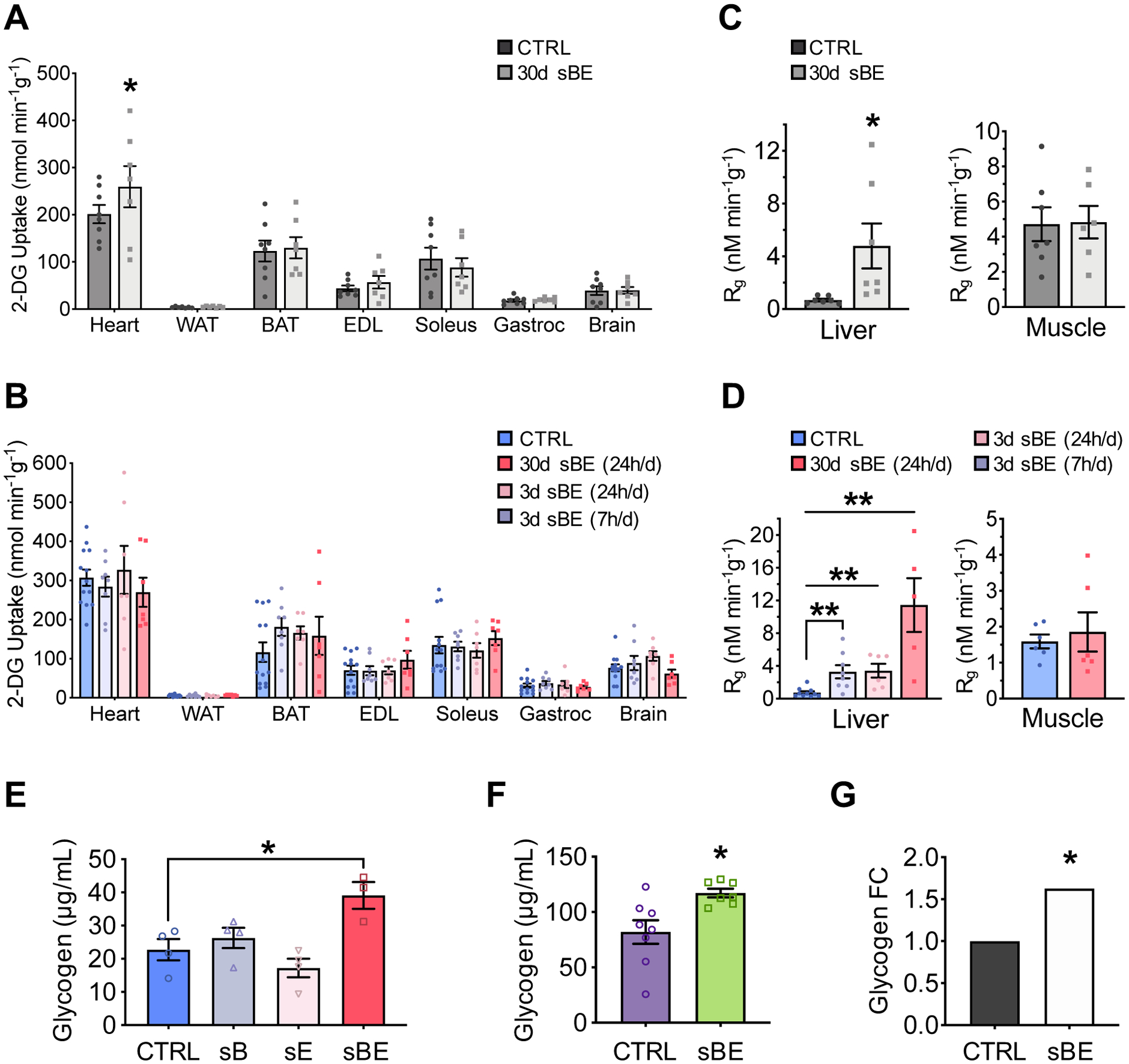
(A–D) Euglycemic-hyperinsulinemic clamped conditions.
(A,B) 14C-2-deoxyglucose uptake into tissues in: (A) NCD mice (n ≥ 7/group) and (B) HFD mice (n ≥ 7/group).
(C,D) 14C-2-deoxyglucose incorporation into liver and muscle glycogen in (C) NCD mice (n ≥ 7/group) and (D) HFD mice (n ≥ 6/group).
(E) Total liver glycogen levels in HFD mice treated with static magnetic fields (sB), static electric fields (sE), or both static magnetic and electric fields (sBE) for 25 days (n ≥ 7/group).
(F) Total liver glycogen levels in db/db mice treated with sBE for 30 days (n ≥ 7/group).
(G) Glycogen produced by primary human hepatocytes after 6 hours of sBE exposure presented as fold change (n = 5 biological replicates across 5 different days).
Data presented as mean ± SEM. Data panels (A), (C), and (F) analyzed by two-tailed, unpaired Student’s t-test. Data panels (B–E) analyzed by one-way ANOVA with Dunnett’s correction for multiple comparisons. Data panel (G) analyzed by mixed effects modeling. *P < 0.05, **P < 0.01.
sBE Exposure Does Not Enhance Phosphorylation of Insulin Signaling Intermediates
Insulin signaling was assessed in 3-day sBE exposed NCD- and HFD mice following a 15-minute intravenous stimulation with insulin or vehicle (PBS). Insulin stimulation enhanced insulin signaling as measured by protein phosphorylation in 3 key tissues that regulate glucose homeostasis: liver, white adipose tissue (WAT) and skeletal muscle. Despite enhancing insulin sensitivity, sBE exposure did not enhance phosphorylation of AKT and GSK3β in liver, white adipose tissue or skeletal muscle relative to untreated mice, suggesting the existence of an insulin sensitizing mechanism that does not involve an increase in the phosphorylation of insulin signaling intermediates (Figure S2D–S2L).
sBE Exposure Does Not Result in Adverse Effects
We investigated whether the striking improvements in insulin sensitivity driven by exposure to sBE were the result of adverse effects. Mice were continuously exposed to sBE for at least 30 days and then evaluated. No abnormal histopathological findings were observed, blood pressure was not changed, and echocardiography revealed normal cardiac function in mice exposed to sBE (Figure S3A and Table S1). Glucose excretion was not higher in sBE-exposed animals, ruling out elevated glucosuria (Figure S3B–S3D). NCD and db/db mice showed no changes in body weight, food intake or energy expenditure (Figure S3E–S3G). However, HFD mice displayed lower body weight, higher energy expenditure, food intake and a lower respiratory quotient, suggesting model-specific differences (Figure S3G). Based on the observed effects of sBE exposure on liver glucose metabolism, the ultrastructure of hepatic mitochondria was evaluated and revealed normal mitochondrial morphology (Figure S3H and S3I). Thus, exposure to sBE is well-tolerated and the effects on insulin action are unlikely to be secondary to adverse effects.
sBE Exposure Lowers Oxidative Distress and Activates NRF2
Given that changes in insulin signaling could not explain the insulin sensitizing effects of sBE, we turned our mechanistic focus to other pathways that could regulate insulin sensitivity. Redox imbalance and oxidative distress have been suggested to contribute to the pathogenesis of insulin resistance (Anderson et al., 2009; Henriksen et al., 2011; Houstis et al., 2006; Madiraju et al., 2018; Rindler et al., 2013). To determine whether sBE exerts biological effects through redox-dependent mechanisms, we measured circulating F2-isoprostanes (F2-IsoPs), a product of ROS-driven oxidation of arachidonic acid that is used as a clinical marker of lipid peroxidation and systemic oxidative distress (Milne et al., 2007). Metabolic health and insulin sensitivity negatively correlate with circulating levels of F2-IsoPs (Monnier et al., 2006; Sampson et al., 2002; Sandu et al., 2005). F2-IsoPs were measured in NCD- and HFD mice exposed to sBE for 3 days followed by a 16 h fast and 4 h re-feed to assess the effect of sBE on the whole-body oxidative burden in the fasted and fed states. HFD mice showed higher levels of F2-IsoPs indicating greater systemic oxidative distress in the diabetic state, consistent with prior studies (Sampson et al., 2002; Sandu et al., 2005; Sutton et al., 2018). Relative to untreated controls, sBE exposure reduced circulating F2-IsoPs by 19% and 20% in the fasting state of NCD- and HFD mice respectively and reduced circulating F2-IsoPs by 37% in the refed state of HFD mice (Figure 4A). To determine whether these striking changes were related to an adaptive redox response, we probed for expression of nuclear factor erythroid 2-related factor (NRF2), a master regulator of cellular redox homeostasis (Jones and Sies, 2015). Upon activation of this transcription factor, NRF2 translocates to the nucleus where it regulates expression of antioxidant response elements to initiate a redox response (Yun and Finkel, 2014). Three-day exposure of HFD mice to sBE significantly elevated the levels of nuclear NRF2 in the liver (Figure 4B). These findings indicate that exposure to sBE activates an adaptive redox response to lower oxidative distress in diabetic animals.
Figure 4. sBE exposure alters the systemic redox state to enhance the insulin response.
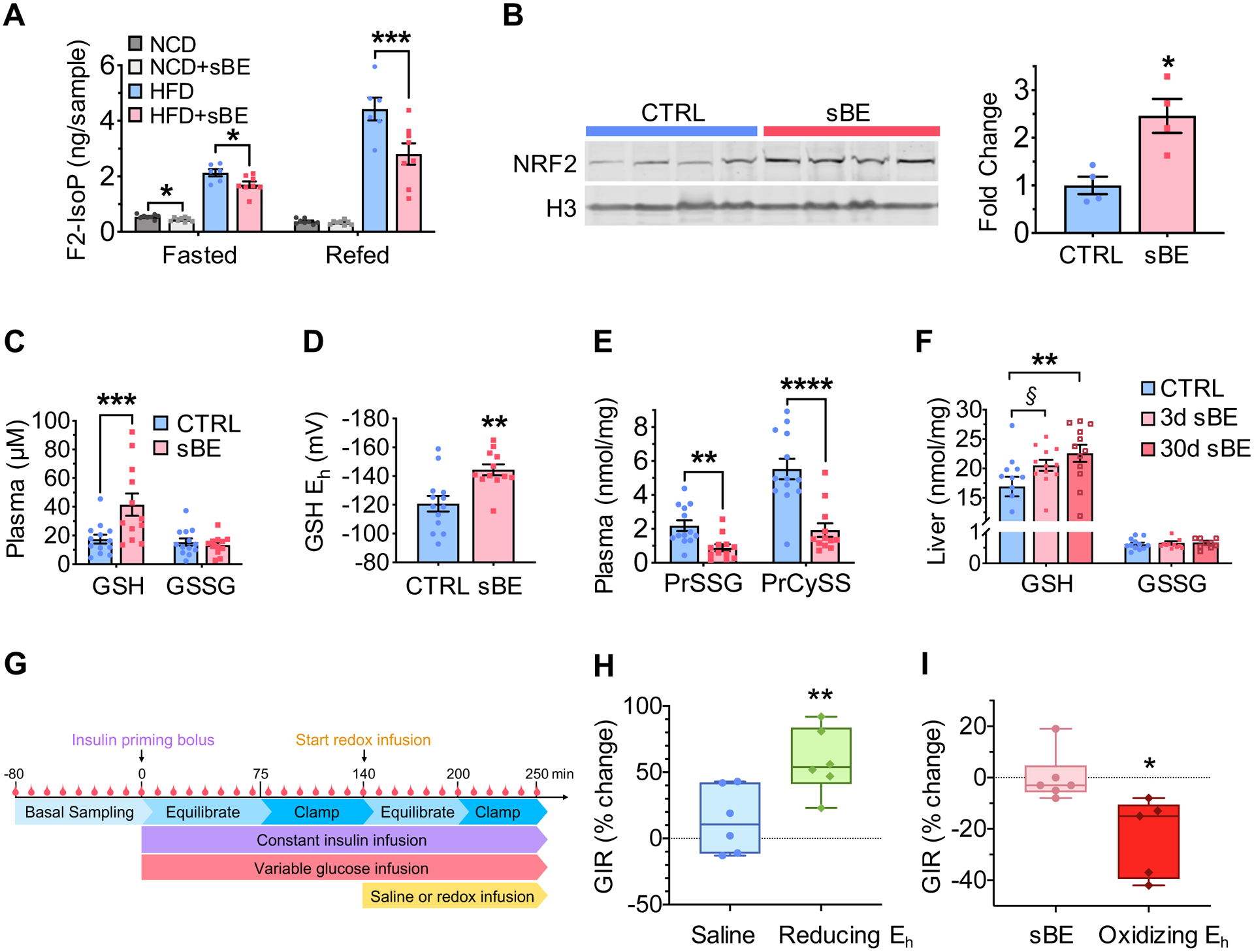
(A) Plasma F2-isoprostanes in 3 day sBE-exposed NCD- and HFD mice fasted for 16 h and refed for 4 h (n = 8 mice/group).
(B) Western blot image (left) and quantification (right) of 3-day sBE-exposed HFD mouse liver nuclear fractions NRF2 normalized to histone 3 (H3) (n = 4 mice/group).
(C–E) Measurements of analytes in plasma collected from control HFD mice and 3 day sBE-exposed HFD mice.
(C) Plasma concentration of glutathione (GSH) and glutathione disulfide (GSSG) (n ≥ 12 mice/group).
(D) Half-cell reduction potential of GSH in plasma (n ≥ 12 mice/group).
(E) Protein glutathionylation (PrSSG) and protein cysteinylation (PrCySS) in plasma (n ≥ 12 mice/group).
(F) Livers were collected from 3 day and 30 day sBE-exposed HFD mice and assessed for GSH and GSSG presented as a concentration (nmol per mg of protein) (n ≥ 10 mice/group). §P = 0.07. **P < 0.01
(G) Schematic of euglycemic-hyperinsulinemic clamp procedure with the infusion of a reducing or oxidizing redox solution.
(H–I) Euglycemic-hyperinsulinemic clamp paired studies performed on HFD mice.
(H) Percent change in glucose infusion rate (GIR) of untreated HFD mice basally clamped at 150 mg/dL plasma glucose and then re-clamped at this concentration during an infusion of saline or a reducing solution of GSH/GSSG (reducing Eh) (Saline infusion, n = 6 mice; Reducing Eh infusion, n = 6 mice).
(I) Percent change in GIR from 3 day sBE-exposed HFD mice basally clamped at 150 mg/dL plasma glucose and then re-clamped at this concentration during an infusion of saline or an oxidizing solution of GSH/GSSG (oxidizing Eh) (sBE-exposed with saline infusion, n ≥ 5 mice; sBE-exposed with oxidizing Eh infusion, n ≥ 5 mice).
Data presented as mean ± SEM. Data panels (A) and (F) analyzed by with Sidak’s multiple comparison t-tests. Data panels (B–E) analyzed by two-tailed, unpaired Student’s t-test. Data panels (H) and (I) analyzed by two-tailed, paired Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
sBE Exposure Induces a Systemic Redox Response that is Insulin Sensitizing
The systemic redox environment is regulated by rapidly responsive antioxidant systems which act as reducing agents to neutralize oxidants (Jones and Sies, 2015). The glutathione-to-glutathione disulfide (GSH/GSSG) and cysteine-to-cystine (Cys/CySS) are thiol redox couples that represent two of the major antioxidant systems in mammals (Jones and Sies, 2015; Schafer and Buettner, 2001). Both the GSH/GSSG and Cys/CySS systems are present in virtually every cell and are highly compartmentalized, including extracellular compartments, such as plasma (Hansen et al., 2006; Jones and Go, 2010). Moreover, thiol-mediated cysteine modifications constitute an alternative signaling mechanism that may modulate protein function independently of protein phosphorylation (Dalle-Donne et al., 2009; Jones and Sies, 2015). We reasoned that these redox systems represent a likely candidate to explain the therapeutic effects of sBE exposure on systemic oxidative distress. Therefore, we investigated whether exposure to sBE induces adaptive changes in these critical redox couples. Three days of sBE exposure elevated circulating GSH by 2.4-fold, resulting in a −25 mV more reducing redox environment compared to unexposed diabetic mice, as calculated by the Nernst equation (Schafer and Buettner, 2001) (Figure 4C and 4D). Interestingly, the more ubiquitous redox couple, Cys/CySS, showed no change, suggesting a specific systemic effect on the GSH/GSSG redox couple (Figure S4A and S4B). In addition, both circulating protein S-glutathionylation (PrSSG) and S-cysteinylation (PrCySS) were 60% and 65% lower, respectively, in HFD mice treated with sBE for 3 days compared with untreated mice, consistent with a systemic shift toward a more reducing circulating redox environment (Figure 4E). sBE exposure trended toward higher GSH levels in the liver at 3 days of exposure (P = 0.07) and significantly elevated GSH levels in the liver at 30 days of exposure (Figure 4F). The change in S-glutathionylation and S-cysteinylation of hepatic proteins was not statistically significant following sBE exposure (Figure S4C). These data indicate that exposure to sBE alters the hepatic redox environment and suggest that the liver is a source of the sBE-driven changes in the systemic redox environment. To investigate whether enzymatic shifts could explain these redox changes, we assessed the activity of liver GSSG reductase, thioredoxin reductase and glucose-6-phosphate dehydrogenase and found no change in the activity any of these redox-regulating enzymes (Figure S4D–S4F). These data further support the concept that sBE may alter redox homeostasis independently of changes in the activity of enzymes that regulate glutathione redox metabolism.
The striking elevation in circulating GSH prompted us to investigate whether the observed changes in the plasma GSH/GSSG redox potential could directly account for the insulin sensitizing effects of sBE. A more reducing GSH/GSSG redox potential was achieved in vivo by infusion of redox adjusted solutions of GSH/GSSG into HFD mice for approximately one hour under euglycemic-hyperinsulinemic clamped conditions (Figure 4G). Remarkably, one-hour infusion of a more reducing redox environment similar to that achieved by sBE, mimicked the insulin sensitizing effects of sBE exposure (Figure 4H). These findings are consistent with human studies showing that infusion of GSH into patients with T2D is insulin sensitizing (De Mattia et al., 1998; Paolisso et al., 1992). Conversely, reversal of the sBE-induced changes in circulating redox homeostasis by infusion of a solution of GSH/GSSG adjusted to achieve a more oxidized redox environment attenuated the effects of sBE on insulin action (Figure 4I). These data reveal that the systemic redox environment exerts rapid control of the insulin response and indicate that exposure to sBE exerts insulin sensitizing effects by promoting a more reducing systemic redox environment.
sBE Exposure Alters ROS Homeostasis
Thiol antioxidants are activated in response to the presence of excess oxidants, including reactive oxygen species (ROS). ROS have long been proposed to play a causal role in the pathogenesis of insulin resistance by poorly understood mechanisms (Anderson et al., 2009; Henriksen et al., 2011; Houstis et al., 2006; Jones and Sies, 2015). Given that many ROS possess free electrons, e.g. O2•−, these species have been proposed to mediate the biological responses to weak electromagnetic fields (Maeda et al., 2008; Mouritsen, 2018; Solov’yov and Schulten, 2009). Due to their paramagnetic nature and involvement in the pathophysiology of T2D, we investigated whether sBE exposure alters O2•− homeostasis. When exposed to sBE at 3.0 mT but not 0.5 mT, the oxidation of dihydroethidium (DHE) and MitoSOX in Hepa1–6 liver cells was significantly lower than untreated cells, suggesting lower steady-state levels of both cytosolic and mitochondrial O2•− (Figures 5A and S5A). Moreover, sBE enhanced oxidation of Amplex Red, an indicator of steady-state levels of H2O2 at 3.0 mT but not 0.5 mT compared to untreated cells (Figure 5A and S5A). These findings are consistent with the ratio of production being altered by disproportionation of O2•− to H2O2 (Buettner et al., 2006). We then assessed the effects of sBE on markers of O2•− in vivo using HFD mice. In vivo and ex vivo imaging revealed that sBE exposure lowered the oxidation of DHE in liver but not in kidney or heart at 3 days of treatment, consistent with in vitro data (Figures 5B, 5C and S5B). No change in the activity or expression of superoxide dismutases (SOD1 and SOD2) or catalase was found in sBE exposed livers (Figure 5D–5F). These data indicate that sBE induces non-enzymatic shifts in liver O2•− metabolism.
Figure 5. sBE exposure alters ROS homeostasis without altering ROS enzyme function.
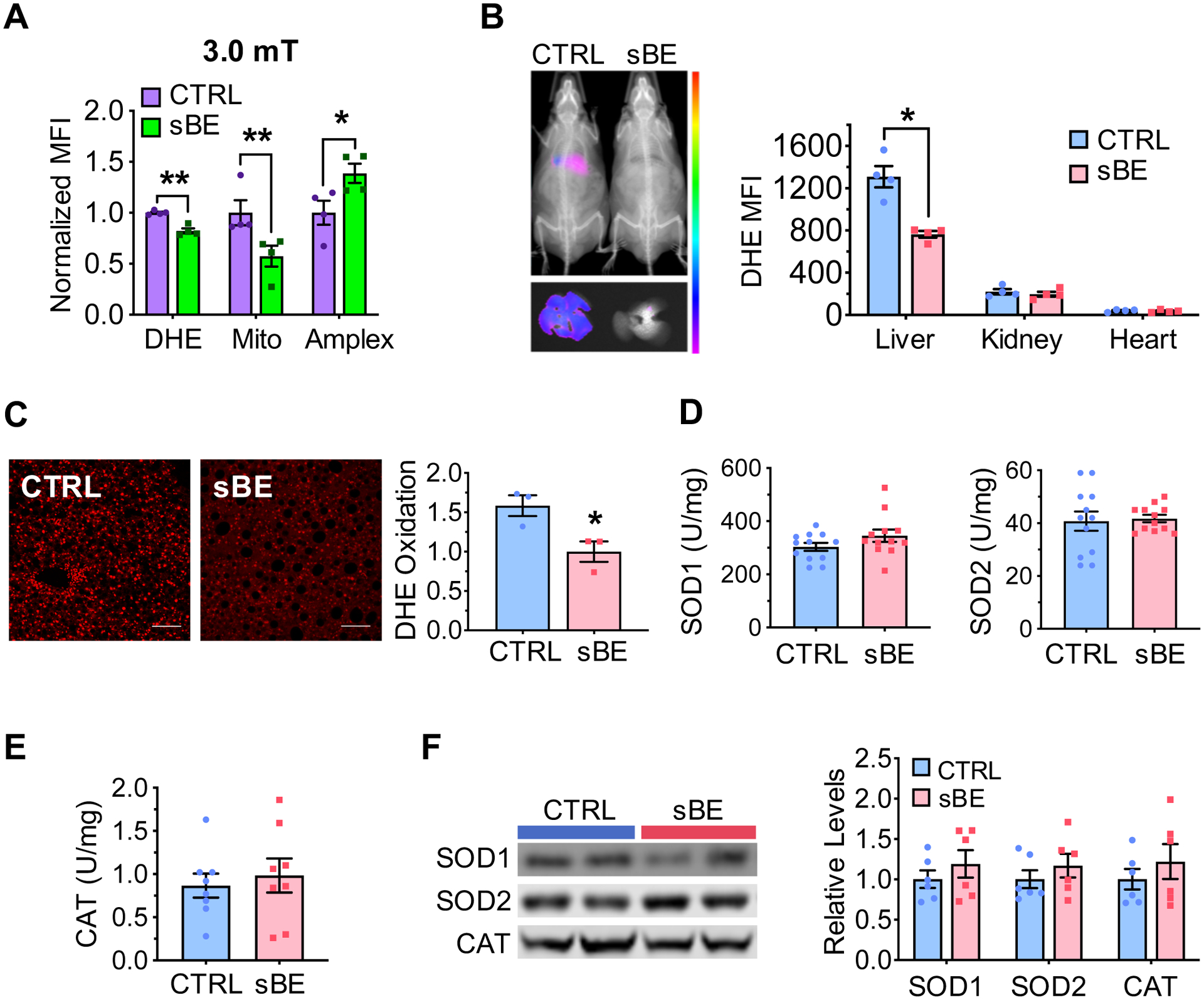
(A) Normalized mean fluorescent intensity (MFI) of dihydroethidium (DHE), MitoSox (Mito), and Amplex Red (Amplex) in Hepa1–6 cells exposed to 3.0 mT sBE (n = 4 biological replicates/group).
(B) Representative images (left) of DHE oxidation in 3 day sBE-exposed HFD mice in vivo, whole mouse (top) and liver (bottom) and MFI quantification (right) in liver, kidneys, and heart (n = 4 mice/group).
(C) Representative images (left) of liver sections (scale, 100 μm) collected from HFD mice exposed to 3 days of sBE and stained with DHE and quantification (right) of the mean fluorescent intensity as a surrogate of DHE oxidation (n = 300 nuclei/mouse, n = 3 mice/group).
(D–F) HFD mice were exposed to sBE for 3 days and assessed for ROS related liver enzyme function.
(D) Liver superoxide dismutase 1 (SOD1) (left) and SOD2 activity (right) (n = 12 mice/group).
(E) Liver catalase activity (n = 8 mice/group).
(F) Western blot images (left) and quantifications (right) for protein expression of SOD1, SOD2, and catalase (CAT) in liver (n = 6/group).
Data presented as mean ± SEM. Data panel (A) analyzed with Sidak’s multiple comparison t-tests. Data panel (B–F) analyzed by two-tailed, unpaired Student’s t-test. *P < 0.05, **P < 0.01.
Mitochondrial ROS in the Liver Mediates the Insulin Sensitizing Effects of sBE
We hypothesized that O2•− is a key magneto-receptive radical that serves as a sensor to mediate the metabolic effects of sBE due to its unpaired electron (Maeda et al., 2008; Solov’yov and Schulten, 2009; Usselman et al., 2016). To test the involvement of O2•−, HFD mice were treated with two different pharmacologic superoxide dismutase (SOD) mimetics, GC4403 (specific) and TEMPOL (non-specific) to scavenge O2•− throughout the 3-day sBE exposure period. GC4403 is a manganese(II) pentaazamacrocyclic compound that freely crosses cell membranes and is highly specific for superoxide as the manganese center of GC4403 cycles between Mn(II) and Mn(III) to convert O2•− to H2O2 (Coleman et al., 2013; Mapuskar et al., 2017; Weekley et al., 2017). TEMPOL, or 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl, is a membrane permeable antioxidant that is a metal-independent SOD mimetic and has been shown in several studies to scavenge O2•− (Aksu et al., 2015; Kaewpila et al., 2008; Marciniak et al., 2016). We found that treatment with either superoxide scavenger fully attenuated the insulin sensitizing effects of sBE (Figures 6A, 6B and S6A–S6C).
Figure 6. Hepatic superoxide mediates the insulin sensitizing effects of sBE exposure.
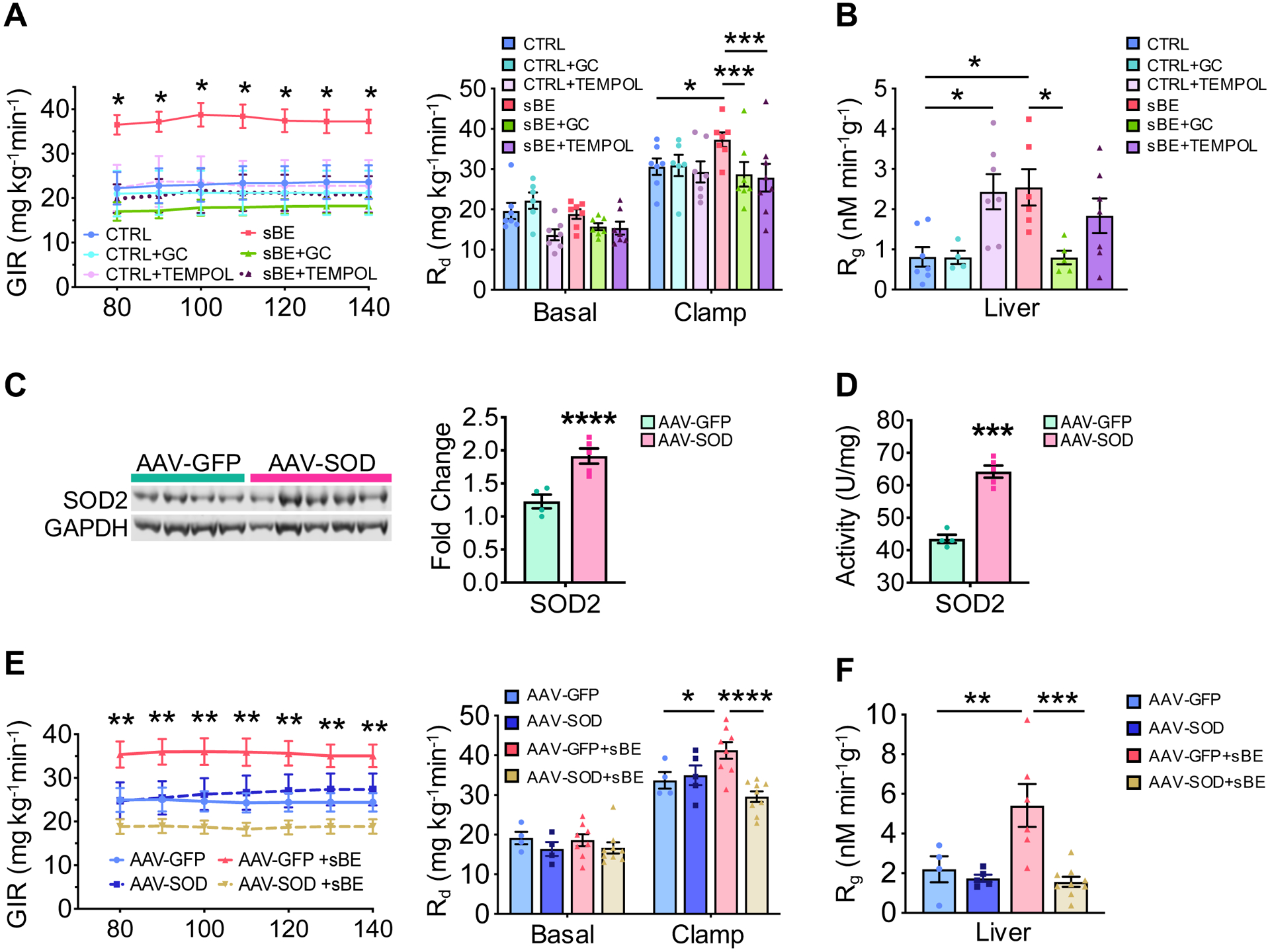
(A–B) Euglycemic-hyperinsulinemic clamps were performed after a 6 h fast on HFD mice exposed concurrently to sBE for 3 days and a pharmacological superoxide dismutase mimetic, GC or TEMPOL.
(A) GIRs (left) and glucose rate of disappearance (Rd) (right) (n ≥ 6 mice/group).
(B) 14C-2-deoxyglucose incorporation into liver glycogen (n ≥ 6 mice/group).
(C) Liver protein expression of SOD2 in HFD mice overexpressing liver-specific superoxide dismutase 2 (AAV-SOD) or control green fluorescent protein (AAV-GFP) (AAV-GFP, n = 4 mice; AAV-SOD, n = 5 mice).
(D) Liver activity of SOD2 in AAV-GFP and AAV-SOD mice (AAV-GFP, n = 4 mice; AAV-SOD, n = 5 mice).
(E–F) Euglycemic-hyperinsulinemic clamps were performed after a 6 h fast on AAV-SOD or AAV-GFP HFD mice after 3 days of sBE exposure.
(E) GIRs (left) and Rd (right) (AAV-GFP, n = 4 mice; AAV-SOD, n = 5 mice; AAV-GFP+sBE, n = 8 mice; AAV-SOD+sBE, n = 9 mice).
(F) 14C-2-deoxyglucose incorporation into liver glycogen (AAV-GFP, n = 4 mice; AAV-SOD, n = 5 mice; AAV-GFP+sBE, n = 8 mice; AAV-SOD+sBE, n = 9 mice).
Data presented as mean ± SEM. Data panels (A–B) and (E–F) analyzed by two-way ANOVA with Sidak’s multiple comparisons. Data panels (C–D) analyzed by two-tailed, unpaired Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Based on our data implicating the liver and mitochondria as important targets of sBE exposure, we investigated whether hepatic mitochondrial superoxide was required for the insulin-sensitizing effects of sBE. To demonstrate target specificity to mitochondrial O2•−, human SOD2 was overexpressed in HFD mice using the liver tropic adeno-associated virus vector (AAV), AAV2/8 serotype, with the liver specific chimeric promoter alpha-1-antitrypsin fused with an albumin enhancer (Figure 6C and 6D) (Kramer et al., 2003). SOD2 is highly specific to mitochondrial O2•− scavenging as this enzyme only has SOD activity once the mitochondrial leader sequence is cleaved, metalated, and folded in the inner mitochondrial membrane (Fujii et al., 1998; Wispe et al., 1989). Following viral transduction, HFD mice were exposed to sBE for 3 days, followed by euglycemic-hyperinsulinemic clamps to assess insulin sensitivity. Overexpression of SOD2 in liver completely abolished the insulin sensitizing effects of sBE, including the full attenuation of effects on GIR, glucose disposal, and glucose incorporation into liver glycogen (Figures 6E, 6F and S6D–S6F). These findings demonstrate that mitochondrial superoxide in the liver is a key mediator of the insulin sensitizing effects of sBE.
Discussion
Life evolved on Earth over billions of years in the presence of weak magnetostatic and electrostatic fields (Doglioni et al., 2016; Honkonen et al., 2018; Thébault et al., 2015). However, the biological effects of these fields are not well understood. In this study, we demonstrate that combined magnetostatic and electrostatic fields exert remarkable insulin sensitizing effects via redox dependent mechanisms. These striking effects are rapid, occurring by 3 days of treatment for 7 hours of exposure per day without adverse effects. We also found that exposure to sBE exerts rapid and robust effects on glycogen synthesis, enhancing hepatic glycogenesis in vivo in mice as well as in primary human hepatocytes. These findings are consistent with an enhanced insulin response and suggest that the effects of sBE may extend beyond rodents.
We found that sBE exerts insulin sensitizing effects, at least in part by modulating the systemic GSH/GSSG redox environment. Three days of exposure to sBE resulted in striking increases in plasma GSH levels, resulting in a considerably more reducing systemic redox environment. Moreover, systemic infusion of redox-adjusted solutions under clamped conditions rapidly altered insulin sensitivity. These findings are consistent with prior studies that show that the extracellular redox environment regulates insulin sensitivity and glycogen synthesis (Barbagallo et al., 1999; De Mattia et al., 1998; Monnier et al., 2006; Nocito et al., 2015; Paolisso et al., 1992; Sampson et al., 2002). It has been purported that redox systems regulate metabolism via redox-dependent protein post-translational modifications (PTMs) on cysteine residues, primarily, S-glutathionylation and S-cysteinylation. These modifications modulate protein functional changes for the rapid regulation of metabolic enzyme activity and energy metabolism (Dalle-Donne et al., 2009; Jones and Sies, 2015). Indeed, our findings reveal that sBE exposure decreases S-glutathionylation and S-cysteinylation on circulating proteins within 3 days of exposure. These findings are in line with 1) a healthy, more reducing systemic environment in which these redox PTMs are removed and 2) a signaling mechanism by which sBE rapidly regulates protein function and insulin sensitivity via non-phosphorylation PTMs (Dalle-Donne et al., 2009; Jones and Sies, 2015). The short time frame of the modified insulin response following redox adjusted infusions coupled with our observations that sBE alters redox PTMs point toward the existence of a rapidly responsive, redox-dependent switch that regulates insulin sensitivity and can be modulated by sBE. Thus, rapid, non-genetic mechanisms, such as redox-dependent post-translational modifications, are likely involved in regulating the function of proteins involved in the regulation of insulin sensitivity. As the major affected redox couple is GSH/GSSG (as opposed to Cys/CySS), GSH-sensitive targets are likely involved.
Thiol redox adaptations often occur to maintain redox homeostasis in response to oxidative events, including perturbations in ROS equilibrium (Jones and Sies, 2015). Altering ROS homeostasis, particularly O2•− products, has been found to serve as a key initiating signal for the activation of a hormetic response originating in mitochondria, termed mito-hormesis, that enhances the antioxidant capacity to provide protection against the deleterious consequences of oxidative distress (Cox et al., 2018; Ristow, 2014; Spitz et al., 1987; Yun and Finkel, 2014). Consistent with the activation of mito-hormesis, we found that sBE exposure alters levels of mitochondrial ROS in liver, increases activation of hepatic NRF2 (as indicated by increased levels of nuclear NRF2), enhances the GSH/GSSG antioxidant capacity (as evidenced by changes in the plasma redox potential) and results in the amelioration of oxidative distress (as evidenced by the reduction of circulating isoprostanes). Activation of mito-hormesis and induction of an antioxidant response, particularly in the GSH redox system, have been shown to have beneficial effects on metabolism and insulin sensitivity (Barbagallo et al., 1999; Cox et al., 2018; De Haes et al., 2014; De Mattia et al., 1998; Paolisso et al., 1992). However, the mechanistic link between ROS remains incompletely understood.
ROS vary in terms of their reactivity and molecular targets; thus, modestly altering ROS stoichiometries may result in profoundly different biological effects (Brandes Ralf et al., 2018; Jones and Sies, 2015). In line with this and prior work on EMFs, we demonstrated that sBE alters ROS homeostasis in a manner consistent with the altered metabolism of O2•− disproportionation (Buettner et al., 2006; Güler et al., 2006; Lin et al., 2018; Usselman et al., 2016). Due to the diversity of interactions and the high degree of reactivity, the biological effects of a given set of ROS signals are challenging to decode. It is equally challenging to identify the specific type of ROS that is responsible for mediating a biological response to exogenous stimuli (Brandes Ralf et al., 2018). It is for these reasons that no studies to date have been able to definitively demonstrate the involvement of a particular type of ROS in mediating the effects of externally applied electromagnetic fields.
Based on prior studies, we hypothesized that O2•− acts as a paramagnetic sensor to initiate or mediate the metabolic effects of sBE. This concept was tested by scavenging O2•− systemically using pharmacological molecules and specifically in the liver via genetic approaches during sBE exposure. Removal of the suspected mediating signal (i.e. O2•−) with mitochondrial SOD2, fully attenuated the insulin-sensitizing effects of sBE, supporting the conclusion that mitochondrial O2•− in the liver is a crucial signal that mediates the effects of sBE. These findings extend the previous understanding of the effects of EMFs on ROS by demonstrating that a metabolic phenotype (i.e. insulin responsiveness) depends on the homeostasis of a paramagnetic radical (i.e. O2•−). To our knowledge, this is the first study to fully attenuate and modulate any biological effect induced by EMFs, and this was done so by scavenging mitochondrial O2•− and by modulating the systemic redox environment. Therefore, we propose that exposure to sBE alters the mileu of mitochondrial ROS in liver which serves as a biological signal to trigger an adaptive systemic redox response that is insulin sensitizing. Further work is needed to elucidate the exact mechanisms at play.
While we have determined that O2•− is a key mediator of these effects, the physical mechanisms underlying the interaction between sBE and paramagnetic ROS are not well understood and remain a major challenge to investigate due to current technological limitations. There are two common hypotheses underlying this interaction. The first relies on the existence of a magnetoreceptor that physically responds to EMFs; for example, opening or closing of a channel. However, weak fields such as those used in our study are likely not strong enough to physically alter the gating properties of channels (Meister, 2016). The second and more plausible hypothesis is the radical-pair mechanism (RPM), which has been proposed to underlie magnetoreception in migrating animals. This hypothesis states that weak EMFs alter the spin states of radical intermediates, thereby changing their reaction products and downstream effects (Harkins and Grissom, 1994; Schulten et al., 1978). Consistent with RPM, sBE exposure changes the reaction products and may alter the rate of formation of O2•−, shifting the redox environment to enhance insulin sensitivity. Thus, we propose that sBE exerts physiological effects, at least in part, by spin-state interaction with O2•− or its rate of formation via RPM. Although we found that O2•− is a key player, the metabolic effects of sBE may involve other reactive species including, reactive nitrogen species and radicals (e.g. semiquinone flavin, FADH•) (Elferchichi et al., 2011). In addition to identifying the involvement of O2•−, we identify hepatic mitochondria as a target of sBE. This is likely because the liver is a metabolic nexus and a large source of O2•−, due to the presence of detoxifying enzymes and respiratory reactions that produce O2•− as a reaction byproduct. Further work is needed to fully understand these mechanisms. Nevertheless, our findings point toward the existence of evolutionarily encoded electromagneto-receptive mechanisms in mammals that regulate the insulin response.
Despite numerous options available, many patients with T2D fail treatment, leaving them with a significantly higher risk of cardiovascular disease and premature death (Gregg et al., 2014; Sabaté, 2003). There is a pressing need to develop simple therapeutic strategies that 1) target the underlying causes of T2D, including redox imbalance and 2) do not negatively impact adherence. To address these issues, attempts have been made to develop simple methods to adjust glycemia. However, recent methods do not target the underlying redox imbalance and require the use of exogenous transgenes, ferromagnetic nanoparticles or the implantation of electrosensitive cells, thus limiting their clinical utility (Krawczyk et al., 2020; Stanley et al., 2012; Stanley et al., 2016; Stanley et al., 2015). Our studies show that sBE therapy, which can be fully automated, represents an entirely noninvasive means to ameliorate insulin resistance through an interaction with endogenous paramagnetic molecules (e.g. O2•−) and through the adaptive restoration of healthy redox homeostatic mechanisms (e.g. GSH/GSSG). Thus, the findings presented here identify a novel physiological effect of magnetostatic and electrostatic fields that may be exploited for the automated long-term, noninvasive management of T2D and potentially other redox-related conditions.
Limitations of Study
There are limitations to our study. Notably, we demonstrate that static magnetic and electric fields exert robust insulin sensitizing effects using mouse models of insulin resistance. Studies in large mammals and humans are needed to test safety and efficacy of this noninvasive modality. Further, a dose-response of each component of sBE and its timing may need to be optimized. Also, while we demonstrate that mitochondrial superoxide in the liver mediates the therapeutic effects of sBE, it is possible that other forms of ROS play an important role. Future studies should seek to identify additional key players in this effect. Additionally, it is difficult to define a mechanism underlying the interactions between sBE and ROS due to technological limitations of measuring spin-state in vivo. Future studies will be required to define the biological effects of modulating the spin-state of intracellular molecules. Finally, while we identify the existence of a redox switch that mediates the insulin sensitizing effects of exposure to sBE, the exact proteins that mediate these effects remain undefined. Future studies are needed to identify proteins that constitute the insulin sensitizing redox switch that is activated by exposure to sBE.
STAR Methods
RESOURCE AVAILABILITY
Lead Contacts
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contacts, Calvin S. Carter (calvin-carter@uiowa.edu) and Val C. Sheffield (val-sheffield@uiowa.edu).
Materials Availability
Mouse lines used in this study are commercially available at the Jackson Laboratory. The BBS mice previously generated by the Sheffield Laboratory have been deposited at Jackson Laboratory and are also commercially available.
Data and Code Availability
No new code was created in this paper.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
C57BL/6J (#000664), HFD-fed (#380050) and db/db (#000697) mice were obtained from Jackson Labs and housed at The University of Iowa Animal Facility under controlled light conditions (12 h light/12 h dark) and fed ad libitum. Animals were habituated to the animal facility for at least one week upon arrival before beginning experiments. Bbs1M390R/M390R mice were previously generated by the Sheffield Laboratory from in-house matings under the same environmental conditions. Male C57BL/6J and HFD mice were used. Male and female db/db mice were used. Experiments were performed using mice that were in good health condition between approximately 16–20 weeks of age. HFD mice were placed on the HFD diet beginning at 6 weeks of age and experiments were performed when mice were on the diet for approximately 10–14 weeks. All groups were randomly assigned based on age and body weight. All experiments followed the guidelines of the Institutional Animal Care and Use Committee.
Cell Culture
HEPA1–6 (sex: female, ATCC CRL-1830, RRID:CVCL_0327) cells were grown in DMEM with 10% fetal bovine serum (Gibco) at 37 °C and 5% CO2. Cells were grown in a 12-well format and seeded ~300,000/well for studies.
Primary human hepatocytes were derived from male donors (Axol Biosciences). These cells were thawed at 37 °C in specific thawing and growing medium purchased from Axol Bioscience. For experiments, hepatocytes were seeded in 6-well dishes at ~1 million cells/well. Hepatocytes were grown to full confluency before starting studies.
METHOD DETAILS
sBE Exposure
A static magnetic field was provided by a power supply (Sorensen) and applied to a multi-turn solenoid coil to produce a field strength of 0.5 – 5.0 mT, equating to approximately 10x – 100x of the magnetic field of the Earth. The magnetic B field was calculated by the following equation: where μ0 is the permeability of free space, N is the number of turns, I is the current in the coil, len is the length of the solenoid and ϕ is the angle from a normal to the axis of the solenoid and the winding of the solenoid. A vertical electrostatic field was provided by a modified power supply (Analog Technologies) to produce a field strength of 5.0 – 10.0 kV/m, equating to approximately 100x of the vertical electric field of the Earth. Samples were placed within the solenoid. For mouse experiments, mice were housed in non-magnetic cages (Innovive). sBE was applied whole-body to mice housed in cages. Mice could freely roam cages as orientation of mice relative to the B-field did not interfere with exposure. All controls were placed in inert devices controlled for environmental conditions (i.e. temperature, humidity, lighting).
Euglycemic-hyperinsulinemic clamps
A catheter was placed into the jugular vein of mice. Mice were allowed to fully recover from surgery for 6-days prior to the clamp procedure. Clamps were performed in 6 h fasted, unrestrained, conscious mice. Whole-body glucose flux was traced by infusion of D-[3-3H]-glucose. After an 80-min basal sampling period, insulin administration was initiated with a 40 mU bolus followed by 4 mU/(kg min) (for mice on HFD) or 25 mU bolus followed by 2.5 mU/(kg min) (for NCD) continuous infusion. At 65 min prior to clamp conclusion, [1-14C]-2-deoxy-D-glucose was infused in a single bolus in 1 min. 14C-2-deoxy-D-glucose-6-phosphate tracer enrichment was used to measure glucose uptake into specific tissues. Briefly, at the beginning of clamp period (i.e. once the steady state was achieved) a bolus of 14C-2-deoxy-D-glucose-6-phosphate (2DGP) was administered and at the end of the clamp whole anatomically defined tissues of interest were harvested (heart, gonadal white fat, brown fat, EDL, soleus and gastrocnemius muscles, brain) and immediately frozen and stored at −80 °C until use. For the tracer enrichment analyses, care was taken to ensure that same regions of larger tissues (such as heart, white and brown fat, brain gastrocnemius muscle) were included when not whole samples were used. Samples were subjected to mechanical homogenization and lysis using 0.6 M perchloric acid, neutralized using 10 M KOH, then 2DGP was precipitated using the Ba(OH)2-ZnSO4 method (Ayala et al., 2006). The tissue-specific glucose clearance was quantitated by dividing tissue 2DGP radioactivity by the integrated area of the plasma radioactivity and then normalized to a rate of glucose uptake by multiplying by the average plasma glucose concentration during the decay period (Rg).
Glucose disappearance (Rd) and appearance (Ra) were determined by employing a 3H labeled glucose tracer. The 3H radioactivity levels in plasma and infusate were determined by liquid scintillation counting. Plasma glucose concentrations were measured using a glucose analyser (Analox). These values were used for the calculation of Rd and Ra as described (Ayala et al., 2011; Ayala et al., 2006; Ayala et al., 2010; McGuinness et al., 2019). Rd and Ra rates were calculated using Steele’s equations (Steele et al., 1956). Plasma insulin was measured by chemiluminescence ELISA.
14C-2-Deoxy-Glucose (2DG) incorporation into glycogen and glycogen assay
2DG incorporation into glycogen was measured as previously described (Hans-Georg Joost, 2012). Briefly, 0.2 – 0.4g liver or 0.5 – 1.0 g muscle tissues were boiled in 3 mL of 30% KOH for 20 min. 2 mL of 100% EtOH was added to the tissue lysate (1 mL) and samples were then incubated in overnight at 4 °C. If less tissue was available, the volume of KOH used was adjusted. Samples were centrifuged at 18,000 g for 30 min and precipitate was placed in boiling water for 10 min to evaporate remaining EtOH and then resuspended in 1 mL of water at room temperature. Two mL of 100% EtOH were added, and then incubated on ice for 2 h followed by a 30 min centrifugation, boiling off EtOH and placed in 1 mL of water. This process was repeated once more followed by the addition of 500 uL of water to re-suspend the precipitant and radioactivity in a 500 μL sample was determined using liquid scintillation. Obtained dpm values were then normalized to weight of tissue used (dpm/mg, then the Rg value for glucose incorporation into glycogen was calculated in the same manner as described above for glucose incorporation into tissues.
Primary human hepatocytes (Axol Biosciences) were thawed and cultured per manufacturer’s protocol. Briefly, hepatocytes were thawed at 37 °C and seeded at approximately 1 million cells/6-well. Hepatocytes were grown to full confluency before starting sBE exposure. Hepatocytes were washed twice with PBS (Gibco), then serum and glucose starved for 2 h. After starvation, hepatocytes were treated with glycogen synthesis medium containing DMEM low glucose supplemented with 10 nM insulin, 10 nM dexamethasone, and 2 mM L-glutamine for 6 hours with or without sBE treatment. Hepatocytes were harvested and assessed for glycogen production using a kit (Cayman No. 700480) per manufacturer’s instructions.
Glucose tolerance tests
Blood glucose was measured following a 6 h fast using a glucometer (Freestyle Lite) via tail vein. Mice were then intraperitoneally injected with 2 g/kg glucose solution for GTT. Blood glucose was measured at 15 – 120 min post-injection.
Plasma and serum analysis
Mouse blood was collected via tail vein and left to sit at room temperature for 30 min for serum collection or combined with heparin or EDTA (0.5 mM final) on ice for plasma extraction. Blood then was spun at 400 g for 20 min 4 °C to separate serum or plasma. Plasma insulin (Crystal Chem) levels were assessed following a 16 h fast via kits per manufacturer’s instructions.
Homeostatic Model Assessment of Insulin Resistance (HOMA-IR)
HOMA-IR index was calculated as follows: fasting insulin (mU/L) × fasting glucose (nmol/L)/22.5.
F2-Isoprostane measurements
F2-Isoprostanes were measured in the Vanderbilt University Medical Center Eicosanoid Core Laboratory using gas chromatography-mass spectrometry as previously described (Milne et al., 2007).
In vitro ROS measurements
HEPA1–6 (ATCC CRL-1830), were cultured in DMEM with 10% fetal bovine serum (FBS; Gibco) at 37 °C and 5% CO2. Cells were seeded at approximately 300k cells/12 well. Steady-state levels of cellular H2O2 production were measured using the horseradish peroxidase-linked Amplex Red fluorometric assay (Cayman) per manufacturer’s instructions. Steady-state levels of intracellular levels of O2•− were measured using dihydroethidium and MitoSOX (Thermo Fisher) and fluorescence was recorded using a microplate reader (Tecan).
In vivo ROS measurements
ROS was measured in vivo using the Carestream MS FX-Pro preclinical imaging system (Carestream Health; Rochester, NY). DHE (5 mg/kg) was administered i.p. to mice two times separated by 12 h. Fluorescent images were captured 5 h post second DHE administration on four isoflurane anesthetized mice. Capture settings were: acquisition time: 30 s, ex: 520 nm, em: 600 nm filter and overlayed with an x-ray image. Mice were euthanized and liver, kidney, and heart tissue were collected. Tissues were immediately imaged using the same settings as for live imaging. Images were quantified by measuring the mean fluorescent intensity of each organ (Fiji, ImageJ) corrected for size and normalized to background fluorescence.
Tissue DHE staining
Liver tissue from untreated HFD mice and 3-day sBE-treated HFD mice were frozen in Tissue-Teck O.C.T. compound. OCT-frozen liver tissues were cut into 8 μM sections. One control sample and one sBE exposed sample were cut and placed on the same slide. Tissue sections were stained with 10 μM DHE in PBS + 5 mM pyruvate for 10 min prior to analysis by confocal microscopy (Olympus FLUOVIEW FV1000). For a positive control, tissue sample were co-treated with 10 μM antimycin A during DHE staining. Three images were taken per sample and each image was quantified by measuring the mean fluorescent intensity of 300 cell nuclei (Fiji, ImageJ), and normalized to background fluorescence.
SOD mimetics
GC4403 (Galera Therapeutics) and TEMPOL (Sigma) were intraperitoneally administered to mice at 10 mg/kg and 150 mg/kg, respectively. Each drug was administered 15 min prior to the start of sBE exposure, then once per day for the remainder of the three-day sBE exposure.
Histology
Tissues were fixed in 4% paraformaldehyde for at least 24 h prior to paraffin embedding and sectioning (7 μm). Sections were stained with hematoxylin and eosin.
Transmission Electron Microscopy
Mouse tissues were harvested in Karnovsky’s Fixative (2.5% glutaraldehyde and 2% paraformaldehyde) overnight then underwent fixation in sodium cacodylate buffer and light osmication (0.5% osmium tetroxide). Tissues were then dehydrated through an ethanol series, embedded in resin and polymerized for 48 h. Ultrathin sections were cut and examined under a JEOL 1230 transmission electron microscope in the Central Microscopy Core Facility (University of Iowa). Measurements of mitochondrial area and mitochondrial number were made with the Multi Measure ROI tool of ImageJ.
Echocardiography and Blood Pressure Measurements
Echocardiography on C57BL/6J mice was performed as previously described (Hill et al., 2000; Weiss et al., 2006). Briefly, mice were sedated by 0.15 mg subcutaneous injection of midazolam. To improve the acoustic interface, the anterior chest of the mouse was shaved and warmed gel was applied. Images were acquired at ~200 s−1 using a 15 MHz linear-array transducer, coupled to a Sonos 5500R ultrasonograph. Parasternal long- and short-axis 2D images were obtained for analysis. Heart rate was assessed by pulse-wave Doppler interrogation of mitral inflow. Blood pressure was measured via tail cuff using a BP-2000 system (Visitech Systems) in control or 30-day sBE exposed HFD mice following a five-day acclimatization period and two-day rest period. Reported values represent blood pressure measurements averaged over the course of a five-day experimental period.
Metabolic chambers
Mice were acclimatized for 24 h and then monitored in environmentally controlled Promethion or Comprehensive Laboratory Animal Monitoring System (CLAMS) metabolic chambers. Each chamber is fitted with indirect calorimetry as well as monitors of food consumption, locomotor activity, and gas exchange. Animals were monitored continuously for 72 h.
Plasmids and viruses
The human SOD2 cDNA was amplified from a human cDNA liver library (Clontech 639307) and cloned into pFBAAV-mcs-BgHpA provided by The University of Iowa Viral Vector Core. Next a chimeric liver specific promoter was created by fusing an albumin enhancer with a minimal alpha1 antitrypsin promoter (Kramer et al., 2003). The liver-tropic recombinant adeno-associated virus, AAV2/8 was packaged with GFP or SOD2 by The University of Iowa Viral Vector Core. The final titers were 5.1 × 1013 vg/mL and 4.2 × 1013 vg/mL for GFP and SOD2 viruses, respectively.
Insulin stimulation and tissue collection
Insulin signaling was assessed in 10-day sBE exposed NCD and HFD mice following a 15 min intravenous stimulation with insulin or vehicle (PBS). Briefly, mice were sedated and IV injected with insulin (1 mU/g weight) or a similar volume of vehicle. After 15 min of circulation, mice were euthanized and liver, white adipose tissue, and gastrocnemius skeletal muscle were collected and flash frozen in liquid nitrogen. Tissues were stored at −80 °C until protein isolation.
Protein extraction and Western blotting
Mouse liver, white adipose and gastrocnemius skeletal muscle was lysed in homogenization buffer. Protein concentrations were determined using the Bradford assay (Thermo Fisher) before separation by SDS-PAGE. Proteins were transferred to nitrocellulose membranes (Licor) and blocked for 10 min in blocking buffer (Licor) diluted in TBST. Membranes were then incubated with Revert stain (Licor) per manufacturer’s instructions to quantitate total protein levels. Membranes were then incubated overnight with primary antibodies followed by a 60 min incubation at room temperature with fluorescently conjugated secondary antibodies (Licor). The following primary antibodies from Cell Signaling Technologies were used at a 1:1000 dilution: phosphorylated AKT (S473), phosphorylated GSK3β (S9) and GSK3β. The following primary antibodies from ProteinTech were used at a 1:1000 dilution: AKT, SOD1, SOD2 and catalase. Fluorescent intensity was quantified using Image Studio (Licor).
Nuclear protein isolation and Western blotting
Liver nuclear protein fractions were isolated from 3 day sBE treated HFD mice using a modified version of the Verma, et al. protocol (Verma et al., 2013). Briefly, ~100 mg of liver were homogenized in 500 μL of hypotonic buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2% NP40, 1 mM EDTA, 5% sucrose, 0.5 mM DTT, 1x cOmplete protease inhibitor from Roche, and 1x PhosSTOP phosphatase inhibitor from Sigma-Aldrich). Liver homogenates were laid gently over a cushion buffer (10 mM Tris, pH 7.5, 15 mM NaCl, 60 mM KCl, 1 mM EDTA, 10% sucrose) prior to centrifugation at 5000 rpm at 4 °C for 10 min. The supernatant containing the cytosolic fraction was removed. The nuclear pellet was washed with 1 mL of hypotonic buffer and centrifuged at 5000 rpm at 4 °C for 5 min to remove cytoplasmic contaminants. The nuclear pellet was resuspended in 200 μL of a buffer containing 50 mM Tris, pH 8.0, 1 mM EDTA, 15% glycerol, 500 mM NaCl, 1x protease inhibitor, and 1x phosphatase inhibitor. The nuclear solution was sonicated briefly while kept on an ice slurry and then incubated at 4 °C for 30 min with rotation. For every 100 μL of nuclear solution, 25 U of benzonase (Millipore/Novagen) was added and incubated at 4 °C for 60 min to remove genomic DNA contaminants. Genomic DNA contaminants were pelleted by centrifugation at 7800 rpm at 4 °C for 10 min. Supernatant was collected and protein concentration was determined by Bradford protein assay. The following primary antibodies were used: NRF2 (1:500, Proteintech) and H3 (1:2000, Abcam). Fluorescent intensity was quantified using Image Studio (Licor).
Redox Measurements
Preparation of samples for HPLC measurement and HPLC methodologies were performed as outlined previously (Jones et al., 2000). In brief, cysteine, cystine, GSH and GSSG were assayed by HPLC as S-carboxymethyl and N-dansyl derivatives using gamma-Glu-Glu as an internal standard. Concentrations were determined via quantification of the internal standard and for redox measurements, they were used in the Nernst equation (Eo – RT/nF ln [reduced]2/[oxidized]) to yield a half-cell reduction potential (Eh) for the couple, where Eo is the standard half-cell reduction potential for the redox couple, R is the gas constant, T is the absolute temperature, n is 2 for the number of electrons transferred, and F is Faraday’s constant. pH was estimated as 7.4, thus the Eo = −264 mV for GSSG/2GSH and −250 mV for CySS/2Cys (Schafer and Buettner, 2001). Redox states in tissues samples were measured by collecting samples in 5% perchloric acid containing 10 μM gamma-Glu-Glu and homogenized. Samples were centrifuged to collect precipitated proteins and the supernatant was derivatized for HPLC. Proteins concentrations were measured via BCA assay and were used to estimate cellular volume for GSH, GSSG, Cys and CySS quantification as described elsewhere (Kirlin et al., 1999). For quantification of redox sensitive, post-translational modifications of proteins, S-glutathionylation and S-cysteinylation were measured from plasma/tissue proteins collected during PCA precipitation. In brief, samples were centrifuged and the obtained proteins pellets were then washed with ice-cold perchloric acid. Samples were then resuspended in 0.50 mL of 0.1 M NaOH after which 0.25 mL were transferred to a new tube containing 0.25 mL 0.1 M phosphate buffer (pH 6.0) with 5 mM dithiothreitol. Samples were incubated for 30 min at room temperature. To reprecipitate the proteins, 0.50 mL of 10% perchloric acid/boric acid (with 10 μM gamma-Glu-Glu) was added. After centrifugation, supernatants were derivatized for HPLC analysis. Data are presented as nmol GSH/mg protein and nmol Cys/mg protein.
GSH/GSSG Redox Infusions
Infusates of GSSG + GSH having a specific half-cell reduction potential for the GSH/GSSG couple were prepared in PBS containing 0.1% BSA (vehicle). Concentrations of GSSG and GSH were varied to achieve a more reducing GSH/GSSG redox potential of approximately −170 mV or a more oxidizing GSH/GSSG redox potential of approximately −80 mV. These solutions were infused into the jugular vein at the rate of 3 μL/min into HFD mice that were subjected to euglycemic hyperinsulinemic clamp with vehicle infusion only. GSH/GSSG infusions were started following successful clamping and were re-clamped after approximately 60 – 70 min. Glucose infusion rates for both first step and second step of the clamp were used to evaluate the effects of each GSH/GSSG half-cell reduction redox potential on insulin sensitivity.
Activity Assays
For all enzymatic activity assays, liver tissue was homogenized in 50 mM potassium phosphate buffer, pH 7.8, containing 1.34 mM DETAPAC. Protein concentrations were determined by Lowry assay.
SOD activity was measured by the Spitz and Oberley method using whole homogenates prepared from tissue as previously described (Spitz and Oberley, 1989). Catalase activity was assessed by UV spectroscopy as previously described (Aebi, 1984). Briefly, 500 μL of a 30 mM H2O2 stock was added to samples and the loss of absorbance at 240 nm at 25°C was monitored on a Beckman DU-600 UV-Vis spectrophotometer (Beckman-Coulter). Activities were calculated by fitting data to the first order kinetics. Glutathione reductase activity was assessed by measuring the disappearance of NADPH as previously described (Ray and Prescott, 1975). Briefly, whole liver lysate was added to a solution containing 70 mM potassium phosphate buffer, pH 7.6, with 2.4 mM EDTA, 0.1% BSA, 94 μM NADPH, and 1 mm GSSG and the loss of absorbance at 340 nM at 25 °C was assessed using UV-Vis spectroscopy. Thioredoxin reductase activity was measured by using a kit (Sigma CS0170) per manufacturer’s instructions. Glucose-6-phosphate dehydrogenase activity was measured using the Glock and McLean method (Glock and Mc, 1953). Briefly, whole liver homogenate was added to a solution containing 75 mM Tris, pH 8.0, with 0.75 mM MgCl2, 1.5 mM NADP+, and 1.2 mM glucose 6-phosphate (G6P). Activity was determined by the rate of NADPH formation at 340 nm. To ensure the specificity of glucose-6-phosphate dehydrogenase activity measurement, homogenates were additionally run in the presence of 1.2 mM 6-phosphogluconic acid, with and without G6P.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Information
Mice were randomly assigned to groups and were age and body-weight matched. No power analysis was performed to determine sample size. The sample size in each study was based on experience with previous glycemic studies in mouse models. Experimenters were blinded to the treatment groups for the following studies: euglycemic hyperinsulinemic clamps, glycogen analysis, DHE analysis, TEM analysis, histopathological analysis, echochardiography, blood pressure measurements, protein assays, redox measurements and isoprostane measurements. Data were tested for normality and variance before subsequent analyses by several statistical tests. Data with normal distribution and equal variance were analyzed using two-tailed, unpaired Student’s t-tests. Multiple t-tests were corrected for multiple comparisons by the Holm-Sidak method. Paired data were analyzed by paired t-tests. Data with more than two groups were analyzed by one-way or two-way ANOVA with Dunnett’s correction or Tukey’s post-hoc analysis. Above statistical tests were performed using Prism 8 version 8.3.1. Repeated measures were analyzed using a mixed effects model using the R package Ime4 (version 1.1.21) to allow for biological replicates and P-value computed using ImerTest (version 3.1.0.). P-values are indicated and data are shown as mean ± SEM unless otherwise noted.
Supplementary Material
Acknowledgements
We would like to thank the Janice and Herbert Wilson Family Foundation, the Chris and Charles Chessman Foundation and the Roy J. Carver Charitable Trust for their generous support. This work was also supported by the American Diabetes Association (119PMF030), the Francois Abboud Cardiovascular Center and The University of Iowa Research Foundation (CSC); Medical Scientist Training Program NIH 5T32GM007337 (SCH); NIH R01EY11298, NIH R01EY017168 and NIH P30EY025580 (VCS). We would like to thank Dr. Dennis Riley at Galera Therapeutics, Inc. for providing the SOD mimetic (GC4403). DWD and DRS are supported by The Holden Comprehensive Cancer Center at The University of Iowa and its National Cancer Institute Award P30CA086862. GRB and DRS are supported by NCI P01CA217797, P42ES013661 (GRB), R01CA169046 (GRB), and R01CA182804 (DRS). SAW is supported by NIH1S10OD026835-01. KCH is supported by F30CA213817. EBT is supported by NIH R01 DK104998. AJR is supported by American Diabetes Association postdoctoral fellowship 1-18-PDF-060. GLM is supported by the Vanderbilt University Medical Center Diabetes Research and Training Center (NIDDK Grant DK-20593). AWN is supported by NIH R01DK115791 and the Fraternal Order of Eagles Diabetes Research Center, KR is supported by NIH P01 HL084207 and VA Merit grant BX004249 and EDA, an established investigator of the AHA is supported by the Fraternal Order of Eagles Diabetes Research Center, the Teresa Benoit Diabetes research Fund and NIH R01 HL127764 and HL112413. We thank Drs. David Kooyman, Geoffrey Duyk, Charles Burant, Francois Abboud, Bryan Allen, Nina Nuangchamnong, Ryan Sheldon, Brian O’Neill, Janelle Garrison, Kathy Zimmerman, Michael McCormick, Jamie Soto, Valerie Buffard and Ariana Mitra for their feedback. We thank Dr. Anthony Calvin Carter for his guidance and support and dedicate this work to him.
Footnotes
Declaration of Interests
C.S.C., S.C.H., V.C.S., C.C.S. and M.J.M. have patents pending related to this work. C.S.C., S.C.H. and W.A.C. are founders of Geminii, Inc. D.R.S. has a Sponsored Research Agreement with Galera Therapeutics.
Data availability
Data that support the findings of this study are available from the corresponding authors (C.S.C., V.C.S.) upon reasonable request.
References
- Aebi H (1984). Catalase in vitro. Methods Enzymol 105, 121–126. [DOI] [PubMed] [Google Scholar]
- Aksu U, Ergin B, Bezemer R, Kandil A, Milstein DMJ, Demirci-Tansel C, and Ince C (2015). Scavenging reactive oxygen species using tempol in the acute phase of renal ischemia/reperfusion and its effects on kidney oxygenation and nitric oxide levels. Intensive Care Med Exp 3, 57–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd, Kang L, Rabinovitch PS, Szeto HH, et al. (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, Malabanan C, James FD, Ansari T, Fueger PT, McGuinness OP, and Wasserman DH (2011). Hyperinsulinemic-euglycemic clamps in conscious, unrestrained mice. Journal of visualized experiments : JoVE, 3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, McGuinness OP, and Wasserman DH (2006). Considerations in the Design of Hyperinsulinemic-Euglycemic Clamps in the Conscious Mouse. Diabetes 55, 390. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, and McGuinness OP (2010). Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech 3, 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo M, Dominguez Ligia J, Tagliamonte Maria R, Resnick Lawrence M, and Paolisso G (1999). Effects of Vitamin E and Glutathione on Glucose Metabolism. Hypertension 34, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Brandes Ralf P, Rezende F, and Schröder K (2018). Redox Regulation Beyond ROS. Circulation Research 123, 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner GR, Ng CF, Wang M, Rodgers VG, and Schafer FQ (2006). A new paradigm: manganese superoxide dismutase influences the production of H2O2 in cells and thereby their biological state. Free radical biology & medicine 41, 1338–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buglak NE, Batrakova EV, Mota R, and Bahnson ESM (2018). Insights on Localized and Systemic Delivery of Redox-Based Therapeutics. Oxidative medicine and cellular longevity 2018, 2468457–2468457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS, Vogel TW, Zhang Q, Seo S, Swiderski RE, Moninger TO, Cassell MD, Thedens DR, Keppler-Noreuil KM, Nopoulos P, et al. (2012). Abnormal development of NG2+PDGFR-[alpha]+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nat Med 18, 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MC, Olivier AK, Jacobus JA, Mapuskar KA, Mao G, Martin SM, Riley DP, Gius D, and Spitz DR (2013). Superoxide Mediates Acute Liver Injury in Irradiated Mice Lacking Sirtuin 3. Antioxidants & Redox Signaling 20, 1423–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CS, McKay SE, Holmbeck MA, Christian BE, Scortea AC, Tsay AJ, Newman LE, and Shadel GS (2018). Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell metabolism 28, 776–786.e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo G, Giustarini D, and Milzani A (2009). Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends in Biochemical Sciences 34, 85–96. [DOI] [PubMed] [Google Scholar]
- De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, Braeckman BP, Schoofs L, and Temmerman L (2014). Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proceedings of the National Academy of Sciences 111, E2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mattia G, Bravi MC, Laurenti O, Cassone-Faldetta M, Armiento A, Ferri C, and Balsano F (1998). Influence of reduced glutathione infusion on glucose metabolism in patients with non—insulin-dependent diabetes mellitus. Metabolism: clinical and experimental 47, 993–997. [DOI] [PubMed] [Google Scholar]
- Doglioni C, Pignatti J, and Coleman M (2016). Why did life develop on the surface of the Earth in the Cambrian? Geoscience Frontiers 7, 865–873. [Google Scholar]
- Elbatreek MH, Pachado MP, Cuadrado A, Jandeleit-Dahm K, and Schmidt HHHW (2019). Reactive Oxygen Comes of Age: Mechanism-Based Therapy of Diabetic End-Organ Damage. Trends in Endocrinology & Metabolism 30, 312–327. [DOI] [PubMed] [Google Scholar]
- Elferchichi M, Mercier J, Bourret A, Gross R, Lajoix A-D, Belguith H, Abdelmelek H, Sakly M, and Lambert K (2011). Is static magnetic field exposure a new model of metabolic alteration? Comparison with Zucker rats. International Journal of Radiation Biology 87, 483–490. [DOI] [PubMed] [Google Scholar]
- Finkel T (2011). Signal transduction by reactive oxygen species. The Journal of cell biology 194, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii J, Ikeda Y, Watanabe T, Kawasaki Y, Suzuki K, Fujii C, Takahashi M, and Taniguchi N (1998). A Defect in the Mitochondrial Import of Mutant Mn-Superoxide Dismutase Produced in Sf21 Cells1. The Journal of Biochemistry 124, 340–346. [DOI] [PubMed] [Google Scholar]
- Gerardi G, De Ninno A, Prosdocimi M, Ferrari V, Barbaro F, Mazzariol S, Bernardini D, and Talpo G (2008). Effects of electromagnetic fields of low frequency and low intensity on rat metabolism. Biomagnetic Research and Technology 6, 3–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glock GE, and Mc LP (1953). Further studies on the properties and assay of glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase of rat liver. The Biochemical journal 55, 400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BJ, Mahadev K, and Wu X (2005). Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes 54, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg EW, Li Y, Wang J, Rios Burrows N, Ali MK, Rolka D, Williams DE, and Geiss L (2014). Changes in Diabetes-Related Complications in the United States, 1990–2010. New England Journal of Medicine 370, 1514–1523. [DOI] [PubMed] [Google Scholar]
- Güler G, Seyhan N, and Aricioğlu A (2006). Effects of static and 50 Hz alternating electric fields on superoxide dismutase activity and TBARS levels in guinea pigs. Gen Physiol Biophys 25, 177–193. [PubMed] [Google Scholar]
- Hans-Georg Joost HA-H, Annette Schurmann (2012). Animal Models in Diabetes Research. (Humana Press; ). [Google Scholar]
- Hansen JM, Go Y-M, and Jones DP (2006). Nuclear And Mitochondrial Compartmentation Of Oxidative Stress And Redox Signaling. Annual review of pharmacology and toxicology 46, 215–234. [DOI] [PubMed] [Google Scholar]
- Harakawa S, Inoue N, Hori T, Tochio K, Kariya T, Takahashi K, Doge F, Martin DE, Saito A, Suzuki H, et al. (2005). Effects of exposure to a 50 Hz electric field on plasma levels of lactate, glucose, free Fatty acids, triglycerides and creatine phosphokinase activity in hind-limb ischemic rats. The Journal of veterinary medical science 67, 969–974. [DOI] [PubMed] [Google Scholar]
- Harkins TT, and Grissom CB (1994). Magnetic field effects on B12 ethanolamine ammonia lyase: evidence for a radical mechanism. Science (New York, N.Y.) 263, 958–960. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ, Diamond-Stanic MK, and Marchionne EM (2011). Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free radical biology & medicine 51, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, and Weiss RM (2000). Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation 101, 2863–2869. [DOI] [PubMed] [Google Scholar]
- Hiscock HG, Worster S, Kattnig DR, Steers C, Jin Y, Manolopoulos DE, Mouritsen H, and Hore PJ (2016). The quantum needle of the avian magnetic compass. Proceedings of the National Academy of Sciences 113, 4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkonen I, Kuvshinov A, Rastätter L, and Pulkkinen A (2018). Predicting Global Ground Geoelectric Field With Coupled Geospace and Three-Dimensional Geomagnetic Induction Models. Space Weather 16, 1028–1041. [Google Scholar]
- Houstis N, Rosen ED, and Lander ES (2006). Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948. [DOI] [PubMed] [Google Scholar]
- Ishida H, Takizawa M, Ozawa S, Nakamichi Y, Yamaguchi S, Katsuta H, Tanaka T, Maruyama M, Katahira H, Yoshimoto K, et al. (2004). Pioglitazone improves insulin secretory capacity and prevents the loss of beta-cell mass in obese diabetic db/db mice: Possible protection of beta cells from oxidative stress. Metabolism: clinical and experimental 53, 488–494. [DOI] [PubMed] [Google Scholar]
- Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, and Sternberg P (2000). Redox state of glutathione in human plasma. Free Radic Biol Med 28, 625–635. [DOI] [PubMed] [Google Scholar]
- Jones DP, and Go YM (2010). Redox compartmentalization and cellular stress. Diabetes, obesity & metabolism 12 Suppl 2, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP, and Sies H (2015). The Redox Code. Antioxidants & redox signaling 23, 734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaewpila S, Venkataraman S, Buettner GR, and Oberley LW (2008). Manganese Superoxide Dismutase Modulates Hypoxia-Inducible Factor-1α Induction via Superoxide. Cancer research 68, 2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, and Jones DP (1999). Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic Biol Med 27, 1208–1218. [DOI] [PubMed] [Google Scholar]
- Kramer MG, Barajas M, Razquin N, Berraondo P, Rodrigo M, Wu C, Qian C, Fortes P, and Prieto J (2003). In vitro and in vivo comparative study of chimeric liver-specific promoters. Mol Ther 7, 375–385. [DOI] [PubMed] [Google Scholar]
- Krawczyk K, Xue S, Buchmann P, Charpin-El-Hamri G, Saxena P, Hussherr M-D, Shao J, Ye H, Xie M, and Fussenegger M (2020). Electrogenetic cellular insulin release for real-time glycemic control in type 1 diabetic mice. Science (New York, N.Y.) 368, 993. [DOI] [PubMed] [Google Scholar]
- Lai J, Zhang Y, Zhang J, Liu X, Ruan G, Chaugai S, Tang J, Wang H, Chen C, and Wang DW (2016). Effects of 100-μT extremely low frequency electromagnetic fields exposure on hematograms and blood chemistry in rats. Journal of Radiation Research 57, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Lei T, Xie K, Wu X, Tang C, Jiang M, Liu J, Luo E, and Shen G (2016). Effects of extremely low frequency pulsed magnetic fields on diabetic nephropathy in streptozotocin-treated rats. BioMedical Engineering OnLine 15, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B. j., Tsao S. h., Chen A, Hu S-K, Chao L, and Chao P.-h.G. (2017). Lipid rafts sense and direct electric field-induced migration. Proceedings of the National Academy of Sciences, 201702526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Dong L, Xu Y, and Di G (2018). Studies on effects of static electric field exposure on liver in mice. Scientific reports 8, 15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Qiu Y, Perry RJ, Rahimi Y, Zhang X-M, Zhang D, Camporez J-PG, Cline GW, Butrico GM, Kemp BE, et al. (2018). Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nature Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Henbest KB, Cintolesi F, Kuprov I, Rodgers CT, Liddell PA, Gust D, Timmel CR, and Hore PJ (2008). Chemical compass model of avian magnetoreception. Nature 453, 387–390. [DOI] [PubMed] [Google Scholar]
- Mapuskar KA, Flippo KH, Schoenfeld JD, Riley DP, Strack S, Hejleh TA, Furqan M, Monga V, Domann FE, Buatti JM, et al. (2017). Mitochondrial Superoxide Increases Age-Associated Susceptibility of Human Dermal Fibroblasts to Radiation and Chemotherapy. Cancer research 77, 5054–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak A, Walczyna B, Rajtar G, Marciniak S, Wojtak A, and Lasiecka K (2016). Tempol, a Membrane-Permeable Radical Scavenger, Exhibits Anti-Inflammatory and Cardioprotective Effects in the Cerulein-Induced Pancreatitis Rat Model. Oxidative medicine and cellular longevity 2016, 4139851–4139851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness OP, Ayala JE, and Wasserman DH (2019). Glucose Clamping The Conscious Mouse: A Laboratory Course. V. MPC-NIH, ed. [Google Scholar]
- Meister M (2016). Physical limits to magnetogenetics. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GL, Sanchez SC, Musiek ES, and Morrow JD (2007). Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nature protocols 2, 221–226. [DOI] [PubMed] [Google Scholar]
- Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol J-P, and Colette C (2006). Activation of Oxidative Stress by Acute Glucose Fluctuations Compared With Sustained Chronic Hyperglycemia in Patients With Type 2 Diabetes. JAMA : the journal of the American Medical Association 295, 1681–1687. [DOI] [PubMed] [Google Scholar]
- Mouritsen H (2018). Long-distance navigation and magnetoreception in migratory animals. Nature 558, 50–59. [DOI] [PubMed] [Google Scholar]
- Nocito L, Kleckner AS, Yoo EJ, Jones Iv AR, Liesa M, and Corkey BE (2015). The extracellular redox state modulates mitochondrial function, gluconeogenesis, and glycogen synthesis in murine hepatocytes. PloS one 10, e0122818–e0122818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberley LW (1988). Free radicals and diabetes. Free radical biology & medicine 5, 113–124. [DOI] [PubMed] [Google Scholar]
- Paolisso G, Di Maro G, Pizza G, D’Amore A, Sgambato S, Tesauro P, Varricchio M, and D’Onofrio F (1992). Plasma GSH/GSSG affects glucose homeostasis in healthy subjects and non-insulin-dependent diabetics. American Journal of Physiology-Endocrinology and Metabolism 263, E435–E440. [DOI] [PubMed] [Google Scholar]
- Ray LE, and Prescott JM (1975). Isolation and some characteristics of glutathione reductase from rabbit erythrocytes (38548). Proc Soc Exp Biol Med 148, 402–409. [DOI] [PubMed] [Google Scholar]
- Rindler PM, Crewe CL, Fernandes J, Kinter M, and Szweda LI (2013). Redox regulation of insulin sensitivity due to enhanced fatty acid utilization in the mitochondria. American Journal of Physiology-Heart and Circulatory Physiology 305, H634–H643. [DOI] [PubMed] [Google Scholar]
- Ristow M (2014). Unraveling the Truth About Antioxidants: Mitohormesis explains ROS-induced health benefits. Nature Medicine 20, 709. [DOI] [PubMed] [Google Scholar]
- Sabaté E (2003). Adherence to long-term therapies: evidence for action. (World Health Organization; ). [Google Scholar]
- Samiec PS, Drews-Botsch C, Flagg EW, Kurtz JC, Sternberg P, Reed RL, and Jones DP (1998). Glutathione in Human Plasma: Decline in Association with Aging, Age-Related Macular Degeneration, and Diabetes. Free Radical Biology and Medicine 24, 699–704. [DOI] [PubMed] [Google Scholar]
- Sampson MJ, Gopaul N, Davies IR, Hughes DA, and Carrier MJ (2002). Plasma F2 Isoprostanes. Diabetes Care 25, 537. [DOI] [PubMed] [Google Scholar]
- Sandu O, Song K, Cai W, Zheng F, Uribarri J, and Vlassara H (2005). Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes 54, 2314–2319. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, and Buettner GR (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biology and Medicine 30, 1191–1212. [DOI] [PubMed] [Google Scholar]
- Schulten K, Swenberg Charles E, and Weller A (1978). A Biomagnetic Sensory Mechanism Based on Magnetic Field Modulated Coherent Electron Spin Motion In Zeitschrift für Physikalische Chemie, p. 1. [Google Scholar]
- Sekhar RV, McKay SV, Patel SG, Guthikonda AP, Reddy VT, Balasubramanyam A, and Jahoor F (2011). Glutathione Synthesis Is Diminished in Patients With Uncontrolled Diabetes and Restored by Dietary Supplementation With Cysteine and Glycine. Diabetes Care 34, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Yu H, Sun Y, Yang C, Lian H, and Cai P (2015). The Energy Metabolism in Caenorhabditis elegans under The Extremely Low-Frequency Electromagnetic Field Exposure. Scientific reports 5, 8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solov’yov IA, and Schulten K (2009). Magnetoreception through Cryptochrome May Involve Superoxide. Biophysical Journal 96, 4804–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz DR, Dewey WC, and Li GC (1987). Hydrogen peroxide or heat shock induces resistance to hydrogen peroxide in Chinese hamster fibroblasts. Journal of Cellular Physiology 131, 364–373. [DOI] [PubMed] [Google Scholar]
- Spitz DR, and Oberley LW (1989). An assay for superoxide dismutase activity in mammalian tissue homogenates. Analytical Biochemistry 179, 8–18. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Gagner JE, Damanpour S, Yoshida M, Dordick JS, and Friedman JM (2012). Radio-Wave Heating of Iron Oxide Nanoparticles Can Regulate Plasma Glucose in Mice. Science (New York, N.Y.) 336, 604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SA, Kelly L, Latcha KN, Schmidt SF, Yu X, Nectow AR, Sauer J, Dyke JP, Dordick JS, and Friedman JM (2016). Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature 531, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SA, Sauer J, Kane RS, Dordick JS, and Friedman JM (2015). Remote regulation of glucose homeostasis in mice using genetically encoded nanoparticles. Nature Medicine 21, 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starks RD, Beyer AM, Guo DF, Boland L, Zhang Q, Sheffield VC, and Rahmouni K (2015). Regulation of Insulin Receptor Trafficking by Bardet Biedl Syndrome Proteins. PLoS genetics 11, e1005311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele R, Wall JS, Bodo R.C.d., and Altszuler N (1956). Measurement of Size and Turnover Rate of Body Glucose Pool by the Isotope Dilution Method. American Journal of Physiology-Legacy Content 187, 15–24. [DOI] [PubMed] [Google Scholar]
- Sutton EF, Beyl R, Early KS, Cefalu WT, Ravussin E, and Peterson CM (2018). Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell metabolism 27, 1212–1221.e1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szemerszky R, Zelena D, Barna I, and Bárdos G (2010). Stress-related endocrinological and psychopathological effects of short- and long-term 50 Hz electromagnetic field exposure in rats. Brain Research Bulletin 81, 92–99. [DOI] [PubMed] [Google Scholar]
- Thébault E, Finlay CC, Beggan CD, Alken P, Aubert J, Barrois O, Bertrand F, Bondar T, Boness A, Brocco L, et al. (2015). International Geomagnetic Reference Field: the 12th generation. Earth, Planets and Space 67, 79. [Google Scholar]
- Usselman RJ, Chavarriaga C, Castello PR, Procopio M, Ritz T, Dratz EA, Singel DJ, and Martino CF (2016). The Quantum Biology of Reactive Oxygen Species Partitioning Impacts Cellular Bioenergetics. Scientific reports 6, 38543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Reest J, Lilla S, Zheng L, Zanivan S, and Gottlieb E (2018). Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nature communications 9, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SK, Deshmukh V, Liu P, Nutter CA, Espejo R, Hung M-L, Wang G-S, Yeo GW, and Kuyumcu-Martinez MN (2013). Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. The Journal of biological chemistry 288, 35372–35386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD (2014). Type 2 diabetes as a redox disease. The Lancet 383, 841–843. [DOI] [PubMed] [Google Scholar]
- Weekley CM, Kenkel I, Lippert R, Wei S, Lieb D, Cranwell T, Wedding JL, Zillmann AS, Rohr R, Filipovic MR, et al. (2017). Cellular Fates of Manganese(II) Pentaazamacrocyclic Superoxide Dismutase (SOD) Mimetics: Fluorescently Labeled MnSOD Mimetics, X-ray Absorption Spectroscopy, and X-ray Fluorescence Microscopy Studies. Inorganic Chemistry 56, 6076–6093. [DOI] [PubMed] [Google Scholar]
- Weiss RM, Ohashi M, Miller JD, Young SG, and Heistad DD (2006). Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation 114, 2065–2069. [DOI] [PubMed] [Google Scholar]
- Wiltschko W, and Wiltschko R (1972). Magnetic Compass of European Robins. Science (New York, N.Y.) 176, 62. [DOI] [PubMed] [Google Scholar]
- Wispe JR, Clark JC, Burhans MS, Kropp KE, Korfhagen TR, and Whitsett JA (1989). Synthesis and processing of the precursor for human mangano-superoxide dismutase. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 994, 30–36. [DOI] [PubMed] [Google Scholar]
- Yun J, and Finkel T (2014). Mitohormesis. Cell metabolism 19, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new code was created in this paper.