

Abstract
Genetic engineering of induced pluripotent stem cells (iPSCs) holds great promise for gene and cell therapy as well as drug discovery. However, there are potential concerns regarding the safety and control of gene expression using conventional vectors such as viruses and plasmids. Although human artificial chromosome (HAC) vectors have several advantages as a gene delivery vector, including stable episomal maintenance and the ability to carry large gene inserts, the full potential of HAC transfer into iPSCs still needs to be explored. Here, we provide evidence of a HAC transfer into human iPSCs by microcell-mediated chromosome transfer via measles virus envelope proteins for various applications, including gene and cell therapy, establishment of versatile human iPSCs capable of gene loading and differentiation into T cells, and disease modeling for aneuploidy syndrome. Thus, engineering of human iPSCs via desired HAC vectors is expected to be widely applied in biomedical research.
Keywords: human artificial chromosome, MV-MMCT, DMD, gene and cell therapy, T-iPSC, aneuploidy syndrome
Graphical Abstract
Engineering of human iPSCs has great potential for cell therapy and drug discovery. Kazuki and colleagues demonstrate engineering of human iPSCs via human artificial chromosome vectors with a large cargo capacity for biomedical research, such as gene and cell therapies and generation of isogenic models for aneuploid syndromes.
Introduction
Human artificial chromosomes (HACs) are non-integrating gene delivery vectors that have advantages such as no size limitation of the insert DNA, control of the loading gene copy number, and independent maintenance in host cells.1 Physiological gene expression can be achieved by introducing not only the gene-coding region but also potential physiological regulatory elements into HACs. Introduction of the desired gene copy number may also achieve the ideal gene expression level. Independent maintenance avoids disruption of the host genome and positional effects such as silencing of the transferred gene. There are two basic strategies to construct HACs: manipulation of a natural human chromosome to generate size-reduced mini-chromosomes (top-down), and assembly of new chromosomes de novo from their constituent DNA elements (bottom-up).2,3 Using the top-down approach, we constructed HACs devoid of known expressed genes by truncating natural human chromosomes at a site adjacent to the centromeric region.4 These HAC vectors contain acceptor sequences for site-specific insertion of desired gene(s), which facilitates the introduction of exogenous DNA fragments.
We used HACs to demonstrate various applications, including gene correction of Duchenne muscular dystrophy (DMD) in DMD patient-derived muscle progenitors and induced pluripotent stem cells (iPSCs), and establishment of various model animals such as humanized CYP3A mice and human antibody-producing mice or calves.5, 6, 7, 8, 9, 10 Furthermore, recent advances in gene loading systems applicable to HACs have greatly expanded the versatility of these chromosome vector systems.11, 12, 13
iPSCs provide various benefits for regenerative medicine, disease modeling, and drug screening, as they can be generated from the individual’s own tissues and are capable of infinite propagation and differentiation into various cell types.14 Future applications of iPSCs should largely rely on gene transfer technologies to exogenously introduce reporter constructs and therapeutic genes into iPSCs. Although retroviral and lentiviral vectors have been widely employed for this purpose, transgene insertion into random genomic sites can result in silencing of the transgene itself and disruption in the expression of endogenous genes neighboring the insertion site. Recently, genomic safe harbors (GSHs) have attracted attention because they are genomic sites where transgenes can be expressed predictably without disrupting the expression or regulation of adjacent genes.15 However, currently, no fully validated GSHs exist in the human genome.16 In such a context, the use of iPSCs harboring HACs may be a promising approach to provide a “genomic safe island,” a site with a huge cargo capacity for exogenous genetic material, which is independent of host chromosomes.
Microcell-mediated chromosome transfer (MMCT), a technique to transfer a specific chromosome from donor cells to target cells, has been a fundamental technology in chromosome engineering research.17 Although MMCT can generate HAC-containing hybrid cells from a wide range of cell lines and primary cells, the very low efficiency of MMCT in human iPSCs has prohibited the establishment of human iPSC lines containing HACs. Therefore, as an alternative approach, we introduced a HAC into human fibroblasts first and then generated human iPSCs from the fibroblasts containing the HAC.5 Nevertheless, it is desirable that HAC vectors can be directly introduced into human iPSCs that are well scrutinized or validated for biomedical approaches.
Recently, two groups have reported successful transfer of HACs constructed by the bottom-up approach into human iPSCs.18, 19, 20, 21 However, the specific applications and advantages of the transfer of a HAC into human iPSCs have not been demonstrated, possibly because of instability or induced gene silencing in their bottom-up HACs. Thus, introduction of stable chromosome vectors free of gene silencing into human iPSCs is highly desirable.
We previously developed an MMCT method (MV-MMCT) employing measles virus (MV) envelope proteins instead of phytohemagglutinin P (PHA-P) and polyethylene glycol (PEG) (PEG-MMCT).22 Micronuclei presenting hemagglutinin (MV-H) protein and fusion (MV-F) protein showed an increase in fusion efficiency compared with conventional PEG-MMCT (Figure 1A). Our recent study also demonstrated that chimeric H protein consisting of the anti-transferrin receptor (TfR) scFv and C terminus of the H protein achieve efficient fusion to human fibroblasts.23
Figure 1.

Combination of MV-MMCT and iPSC technologies for biomedical applications
(A) Process of conventional MMCT (PEG-MMCT) is shown at the top. The process of MV-MMCT is shown at the bottom. (B) Biomedical applications of the combination of MV-MMCT and iPSC technologies. The HAC-mediated gene and cell therapy model via MV-MMCT is shown at the top. Establishment of a versatile iPSC line carrying the basal-HAC capable of gene(s) loading is shown in the middle. Generation of disease model cells for aneuploidy syndrome by MV-MMCT is shown at the bottom.
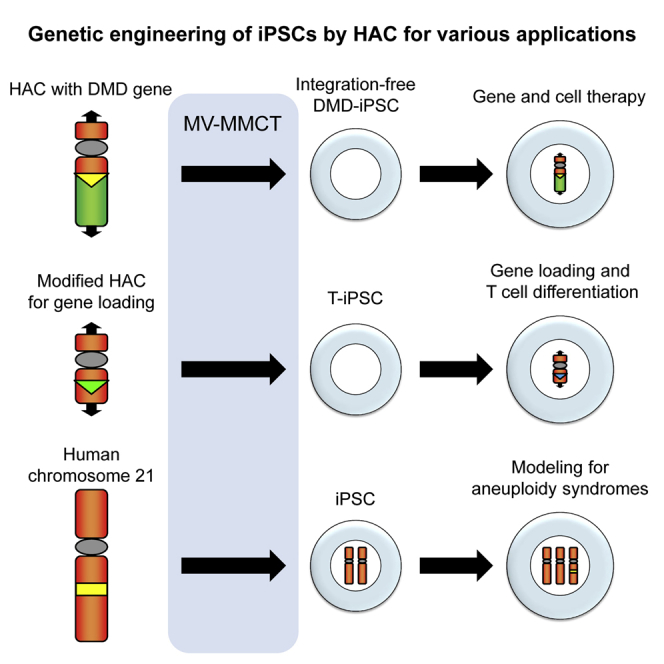
In the present study, we employed MV-MMCT to enable direct transfer of HAC vectors into human iPSC lines. To investigate the versatility of HAC vectors for genetic modification of iPSCs, we demonstrated three main applications (Figure 1B): (1) transfer of the HAC carrying 2.4 Mb of the dystrophin locus (DYS-HAC) into a human iPSC line derived from a patient with DMD for integration-free genetic correction, (2) transfer of the engineered HAC into a human iPSC line for stable expression of a desired gene on the HAC during differentiation, and (3) transfer of human chromosome 21 into an iPSC line to generate an isogenic disease model cell line for Down syndrome.
Results
Transfer of DYS-HAC into DMD-iPSCs for gene and cell therapy
First, we performed genetic correction by introducing an upgraded version of our previously constructed DYS-HAC devoid of potentially immunogenic products (i.e., DYS-HAC210), which contains the 2.4 Mb entire human dystrophin gene locus, into a novel DMD-derived iPSC line as a model for genomic-integration-free gene and cell therapy. Integration-free DMD-iPSCs were generated from fibroblasts with DMD/DYSTROPHIN gene deletion in exons 4–43 (DMD-fibroblasts) by infection with a Sendai virus (SeV) vector carrying four reprogramming factors (Yamanaka factors, i.e., OCT3/4, KLF4, c-MYC, and SOX2). Infected DMD-fibroblasts gave rise to iPSC-like colonies that were isolated and expanded to cell lines. Two clones with a normal karyotype (46, XY), deletion of DMD exons 4–43, and without residual SeV were identified. DMD-iPS#1 was used for the following experiments (Figure S1).
Expression vectors for MV-H and MV-F were introduced into donor Chinese hamster ovary (CHO) cells carrying the DYS-HAC2, and purified micronuclei presenting both MV-H and MV-F proteins were fused with DMD-iPSCs via MV-derived fusion machinery. We employed two modified chimeric MV-H proteins targeting different surface proteins, CD9-scFv and CD71-scFv, in addition to the wild-type (WT)-H. After 3–4 weeks of selection, 10, 10, and 12 drug-resistant iPSCs were obtained via WT-H, CD9-scFv, and CD71-scFv, respectively. Although the expression level of each target cell surface marker protein (CD46, CD9, and CD71) differed in DMD-iPSCs, the efficiency of chromosome transfer was not changed significantly by the three types of MV-H protein (Figure S2). Twenty-seven out of the 32 drug-resistant iPSCs were grown and expanded. Genomic PCR analysis to check the presence of the DYS-HAC2 in the iPSCs confirmed that 14 out of 27 clones were positive (Table S1). Multiplex genomic PCR analysis also showed the presence of the DYS-HAC2 (Figure 2A). Quinacrine and Hoechst (QH) staining showed a normal karyotype of the host genome containing the additional DYS-HAC2, 47, XY, +DYS-HAC2 (Figure 2B). Fluorescence in situ hybridization (FISH) analysis to confirm successful transfer of the DYS-HAC2 revealed that a single copy of the DYS-HAC2 was independently maintained in DMD-iPSCs without integration into the host genome (Figure 2C). Expression of endogenous pluripotency marker genes was detected in DMD-iPSCs carrying the DYS-HAC2 by RT-PCR (Figure 2D). To evaluate whether the DMD-iPSCs (DYS-HAC2) could differentiate into all three embryonic germ layers, a teratoma formation assay was performed. Histological analyses showed presence of all three embryonic germ layers (endoderm, mesoderm, and ectoderm), indicating that the DMD-iPSCs (DYS-HAC2) were pluripotent (Figure 2E). Taken together, the chromosome transfer of the DYS-HAC2 into DMD-iPSCs was performed successfully by the MV-MMCT method, and the corrected iPSCs maintained their pluripotent status.
Figure 2.

Transfer of the DYS-HAC2 into DMD-derived human iPSCs for gene and cell therapy
(A) Multiplex genomic PCR results of DMD iPSCs carrying the DYS-HAC2. (B) Representative karyotype analysis of DMD-iPSCs with the DYS-HAC2. (C) FISH image of metaphase of DMD iPSCs with the DYS-HAC2. Yellow arrow indicates the DYS-HAC2 and the inset shows enlarged image. Red arrows indicate the endogenous centromeres of human chromosomes 13 and 21. (D) Gene expression analyses by RT-PCR of pluripotency marker genes REX1, NANOG, OCT4, and SOX2. (E) Representative images of the teratoma formation assay of DMD-iPSCs carrying the DYS-HAC2.
Finally, we tested whether the DMD-iPSCs (DYS-HAC2) could undergo directed differentiation into a specific cell type with relevance for skeletal muscle cell therapy. To this aim, we generated human iPSC-derived inducible skeletal myogenic cells (also known as HIDEMs) using an established protocol.24 These cells show a phenotypic profile similar to muscle pericyte-derived mesoangioblasts and were the first patient-specific iPSC derivative to be genetically corrected and transplanted in a pre-clinical animal model of muscular dystrophy.9 Flow cytometry (FCM) analysis showed that HIDEMs generated from DMD-iPSCs (DYS-HAC2) had the expected pattern of cell surface markers (e.g., positive for CD13, CD44, and CD146 and negative for CD45, CD56, and SSEA4; Figure S3), which was comparable to multiple reported reference HIDEM lines.9,24 Overall, these data indicate that genomic-integration-free genetic correction of DMD iPSCs with HACs does not interfere with the potency and differentiation capacity of iPSCs, enabling also the generation of specific derivatives with muscle cell therapy relevance.
Transfer of a HAC with a gene loading site into human iPSCs for biomedical applications
To establish an iPSC line harboring a HAC on which genes of interest could be introduced efficiently (Figure 3A), we constructed a modified 21HAC24 and transferred it into a human iPSC line (Figure S4). Because iPSCs derived from T cells (T-iPSCs) show a highly efficient differentiation capacity into T cell lineages and are a potential cell source for adoptive T cell therapy,25,26 one T-iPSC line, TkT3v1-7, was used for HAC transfer as an iPSC resource.25,27 The modified 21HAC2 was transferred from CHO cells to T-iPSCs via the MV-MMCT method. Nine clones were obtained, and all of the clones expressed EGFP (Figure 3B, left column). Then, to eliminate the neomycin resistance gene from the modified 21HAC2, recombination was performed by flippase (Flp) expression (Figure S3). We designated the further modified 21HAC2 without the neomycin-resistant gene as basal-HAC.
Figure 3.

Transfer of a HAC into human iPSCs for biomedical application
(A) Outline of the chromosome transfer of the modified 21HAC2 from CHO cells to T-iPSCs and manipulations of the basal-HAC in T-iPSCs. (B) Fluorescence microscopy images of basal-HAC (left) and KO1-HAC (right) T-iPSCs on MEFs. Phase, phase contrast; GFP, enhanced green fluorescent protein; KO, Kusabira Orange fluorescent protein. (C) FISH analysis of T-iPSCs containing KO1-HAC. Blue, DAPI; red, alpha satellite probe for chromosomes 13 and 21 and HAC (p11-4); green, plasmid containing KO1 (ΦC31 attB-NeoR-pUbc-KO1). (D) Flow cytometry data of KO1-HAC T-iPSCs. Expression of fluorescent proteins in all live cells and pluripotency markers SSEA4 and TRA1-60 in GFP+KO+ cells are shown. (E) Images of an iPSC-Sac containing hematopoietic progenitor cells on 10T1/2 feeder cells. (F) Flow cytometry data of trypsinized sac cells derived from KO1-HAC T-iPSCs. Expression of fluorescent proteins in all live cells (left) and hematopoietic markers CD34 and CD43 in GFP+KO+ cells (right) are shown. (G) Expression of CD3 on T cells differentiated from KO-inserted basal-HAC T-iPSCs.
Next, we evaluated whether circular plasmid DNA with Kusabira Orange 1 (KO1) could be inserted into the basal-HAC in T-iPSCs by a simple transfection method. The KO1 expression cassette, pCART2.2-Ubc-hKO1 (ΦC31 attB-NeoR-pUbc-KO1), and PhiC31 integrase vector (pEF1-phiC31-T) were introduced into T-iPSCs with the basal-HAC via electroporation to induce PhiC31 integrase-mediated attB/attP recombination. Seven G418-resistant clones were obtained and expanded. PCR analyses revealed that the expected recombination occurred in all seven iPSC clones (Table S2). All T-iPSCs stably expressed KO (Figure 3B, right column). The basal-HAC inserted with KO1 in T-iPSCs was designated as KO1-HAC. FISH analyses showed that KO1 existed on the basal-HAC, and the KO1-HAC was independently maintained without integration into the host genome in T-iPSCs (Figure 3C).
FCM analysis showed that 98.1% of the T-iPSCs coexpressed KO and GFP (Figure 3D). Expression of pluripotency markers SSEA4 and TRA1-60 was found in T-iPSCs with KO1-HAC (Figure 3D). These data suggested that the basal-HAC was stable in iPSCs and the undifferentiated state was maintained after several steps of manipulation of the basal-HAC. To confirm the differentiation capacity into the hematopoietic lineage, T-iPSCs with KO1-HAC were cocultured with 10T1/2 feeder cells. The T-iPSCs formed a sac that contained hematopoietic progenitors, and signals of GFP and KO were colocalized (Figure 3E). The CD34+CD43+ cell population that contained hematopoietic progenitors28 existed among GFP+KO+ cells (Figure 3F). Cells from the sac were differentiated further into T cells. After 47 days of culture, the CD3 T cell marker was expressed in GFP+KO+ cells (Figure 3G). These data suggest that the basal-HAC was stably acting as a gene expression vector during differentiation from pluripotent stem cells to T cells.
Transfer of human chromosome 21 to human iPSCs for disease modeling
Transfer of human chromosome 21 into human iPSCs derived from a normal human fibroblast line (HFL1) was performed via MV-MMCT to generate disease model cells for Down syndrome. CHO cells carrying human chromosome 21 tagged with neo resistance gene were used as a donor for MV-MMCT. Karyotype analysis by QH staining in the obtained drug-resistant clone revealed a normal karyotype with an additional single copy of human chromosome 21 (Figure 4A). The clone was designated as trisomy 21-iPSCs (Ts21-iPSCs). Multicolor FISH confirmed that the karyotype was 47, XY, +21 in the Ts21-iPSCs (Figure 4B). Thus, we successfully generated isogenic Ts21-iPSCs as disease model cells for Down syndrome.
Figure 4.

Transfer of hChr.21 into normal human iPSCs for the trisomy model
(A) QH staining of WT- and Ts21-iPSC lines. (B) mFISH analyses of WT- and Ts21-iPSC lines. (C) Immunostaining of differentiated cells derived from WT- and Ts21-iPSC lines by neural induction.
Given the developmental and cognitive aspects of Down syndrome, there is interest in studying possible neuronal phenotypes. Therefore, to compare neuronal differentiation abilities, we differentiated WT-iPSCs and Ts21-iPSCs into neural precursor cells and neurons using serum-free floating culture of embryoid body-like aggregates with quick reaggregation (SFEBq).29 Small molecule inhibitors (XAV939: Wnt signaling inhibitor; LDN-193189: ALK2/3/6 inhibitor; SB431542: ALK5 inhibitor) were added to the medium for SFEBq to induce a forebrain fate.30 Both SFEBq-induced neurospheres differentiated into βIII tubulin-positive neurons under the adherent condition, which were positive for PAX6 (dorsal marker) (Figure 4C). Our results showed no apparent difference in the onset of neuronal differentiation between WT-iPSCs and Ts21-iPSCs, which is in agreement with other Ts21-iPSC lines derived from Down syndrome patients.31 Further studies are needed to elucidate whether maturation of neurons and neuronal functions are affected in the presence of an extra copy of human chromosome 21.
Discussion
We successfully transferred HACs directly into human iPSCs via MV-MMCT, thereby demonstrating various applications that may be challenging to achieve using conventional gene delivery vector systems. Thus, in combination with rapidly advancing genome editing technologies useful to modify specific sites of chromosome vectors, our HACs will enable us to genetically engineer iPSCs with increasing precision for various biomedical applications. Although other groups have recently reported the establishment of human iPSC lines retaining bottom-up HACs freely segregated from host chromosomes,19,21 it is difficult to determine whether bottom-up HACs have key advantages compared with conventional vectors based on only the limited information regarding the stability and expression of transgenes loaded on them.
Gene and iPSC therapy for DMD is a good example that demonstrates the advantages of a HAC, because of the extremely large size of the responsible DYSTROPHIN gene (2.4 Mb).32 The DYS-HAC containing the entire dystrophin locus, including potential gene control elements, reproduces the physiological gene expression in tissue- and developmental stage-specific manners.33,34 In previous work, we provided pre-clinical evidence of safety and efficacy of DYS-HAC-mediated gene and cell therapy in dystrophic mice using tissue-derived murine progenitor cells;35 we then recently translated this approach to human muscle biopsy-derived progenitor cells.10 However, tissue-derived progenitors are difficult to source and expand from DMD muscle biopsies, and to this aim we generated DMD-iPSCs via lentiviruses and retroviruses for HAC-mediated gene and cell therapy,5 which were also differentiated to expandable and transplantable inducible skeletal myogenic cells.9 Because these were generated with integrating viral vectors, the iPSCs include the risk of oncogene activation. In this study, we succeeded in generating fully integration-free iPSCs with genetic correction by combining an integration-free SeV virus vector to induce iPSCs and an integration-free HAC vector for genetic restoration.
Some steps of chromosome transfer, especially purification of micronuclei, are still laborious, which prevents this methodology becoming a widespread research tool. Therefore, as an example, we established a human iPSC line with the HAC, which enabled loading of a desired gene into the HAC in human iPSCs through only simple transfection without laborious MMCT. The marker gene was efficiently loaded on the HAC in human iPSCs, and the HAC functioned stably as a gene expression vector during differentiation to T cells, indicating the potential of the HAC for various applications using iPSC technology. The stability of marker gene expression during differentiation may rely in part on chicken-derived HS4 insulators flanked by the gene expression unit. In combination with gene editing technologies, GSHs such as the AAVS1 locus have been used to introduce an exogenous gene expression unit and optimize gene expression in human iPSCs.15 However, there has been no fully validated GSH that satisfies the functionality of the transgene and the safety by altering the host genome.16 Because HACs are maintained independently from host chromosomes and non-essential in human iPSCs, they can be considered as a “genomic safe island” in which a transgene(s) with no size limitation can be inserted.
An additional example presented in this study was production of human iPSC lines to model the aneuploidy of Down syndrome by transferring a human chromosome 21 into human iPSCs. Because we had already developed mouse A9 and CHO microcell hybrid libraries containing a single human chromosome tagged with a drug resistance marker,36,37 the human trisomy iPSC line panel with the same genetic background could be generated using the hybrid libraries to uncover the complex mechanism of various symptoms of aneuploidy syndromes in the future. Additionally, transfer of a specific chromosome into iPSCs derived from adult cells harboring accumulated specific genetic mutations and subsequent differentiation of the resultant cells into defined mature cell types may represent a relevant tool to investigate the synergistic effect of specific trisomies with certain genetic mutations on tumorigenesis.38,39
Recently, an exogenous human X chromosome was successfully transferred into human iPSCs reprogrammed from Lesch-Nyhan disease patients with a mutation in the X-linked HPRT gene, opening a possible avenue for genetic correction of several X-linked pathologies via chromosome replacement.40 Chromosome replacement to correct iPSCs with complex genetic rearrangements may be useful as a future gene and cell therapy. However, because other unrelated regions on the chromosome will be replaced, non-self-derived genes on the chromosome may be immunogenic or carcinogenic to a patient.
Taken together, iPSCs engineered to carry a HAC with a gene of interest or desired human chromosome will be useful for cell therapy, drug discovery, and basic research.
Materials and methods
Generation of iPSCs
Fibroblasts from a DMD patient with deletion of exons 4–43 in the DYSTROPHIN gene were obtained from the Coriell Institute (Camden, NJ, USA). Induction of iPSCs from DMD-fibroblasts was performed using a SeV vector system (DNAVEC) as described previously.41 Briefly, four SeV vectors containing OCT3/4, KLF4, c-MYC, and SOX2 were used to infect DMD-fibroblasts. Six days after transfection, DMD-fibroblasts were replated at 5 × 104 cells per 100-cm dish on SNL (SNL, STO-Neo-LIF; STO, Sandos inbred mice (SIM) embryo-derived 6-thioguanine- and ouabain-resistant) feeder cells. The next day, the medium was replaced with primate embryonic stem cell (ESC) medium (ReproCell, Tokyo, Japan) supplemented with 4 ng/mL bFGF. The SNL cell line for the feeder layer was obtained from the SANGER Institute (Cambridge, UK). Thirty days after transduction, human ESC-like colonies were picked up and cultured on SNL feeder cells in 24-well plates.
Immunofluorescence staining
Immunofluorescence staining was performed using a primary anti-SeV polyclonal antibody (Medical and Biological Laboratories) after fixation with 4% paraformaldehyde in PBS. A secondary anti-rabbit IgG antibody conjugated with Alexa Fluor 594 (Molecular Probes) was applied, followed by analysis under a fluorescence microscope (ECLIPSE Ti-U, Nikon).
To analyze neural differentiation, cells were fixed in 4% paraformaldehyde and washed with PBS-glycine. The fixed samples were blocked with 1% normal goat serum and 1 mg/mL BSA and then incubated with primary antibodies: anti-βIII tubulin (mouse monoclonal/1:1,000, COVANCE) and anti-PAX6 (rabbit polyclonal/1:300, ATLAS Antibodies). Secondary antibodies were goat anti-mouse Alexa Fluor Plus 488 and goat anti-rabbit Alexa Fluor 594 (Thermo Fisher Scientific). Fluorescence microscopy was performed using an LSM 780 confocal microscope (ZEISS). Images were acquired using a ×20 objective lens.
Cell culture
Each CHO cell line carrying DYS-HAC2, modified 21HAC2, or a human chromosome 21 fragment was cultured in Ham’s F-12 medium supplemented with 10% fetal bovine serum (FBS) and 800 μg/mL G418 (Promega).4,10,42 DYS-HAC2 contained a human centromere, 5ʹ-HPRT-lox71 site, CMV-neo flanked by flippase recognition target (FRT) sites, entire human dystrophin locus, and telomeres.10 21HAC2 contained a human centromere, PGK-puro, CAG-EGFP flanked by HS4 insulators, CMV-bsd, 5ʹ-HPRT-loxP site, PGK-hygro, MC1-TK, β-actin-hisD, and telomeres.4 Human iPSCs, including DMD-iPSCs generated in this study, HFL-1-fibroblast-derived iPSCs generated previously,5 and TkT3v1-725,27 were used in each experiment. Human iPSCs for MV-MMCT were grown on a dish coated with 0.5 μg/mL iMatrix-511 (Fuji Film) in Stemfit medium (Ajinomoto). The experiments to use TkT3v1-7 iPSCs in this research were approved by ethical board of Kyoto University hospital (G590) and Tottori University.
MV-MMCT
CHO cells in a 10-cm dish were cotransfected with 12 μg MV-H (WT), chimeric MV-H of anti-CD9 or anti-CD71ScFv expression vectors, and 12 μg of MV-F expression vector using Lipofectamine 2000 (Invitrogen), following the manufacturer’s protocol. After 24 h of transfection, the cells were passaged into three T-25 flasks. Purification of micronuclei was performed as described previously.22 Purified microcells were overlaid on recipient cells in a 10-cm dish and incubated for 24 h. Then, the cells were replated into three 10-cm dishes and cultured for 3–4 weeks in the presence of 50–90 μg/mL G418.
Genomic PCR
Multiplex PCR analyses to detect deletion of the exons within human dystrophin were performed in accordance with the manufacturer’s protocols (Maxim Biotech, San Francisco, CA, USA). PCR analyses to detect DYS-HAC2 using NeoF/DloxP3L primers were performed as described previously.10 PCR primer sequences to detect modified 21HAC2 and basal-HAC were as follows. TRANS L1/TRANS R1, 5′-TGG AGG CCA TAA ACA AGA C-3′ and 5′-CCC CTT GAC CCA GAA ATT CCA-3′; EF1a FW/HS4 RV, 5′-CAC TGA GTG GGT GGA GAC TGA AGT TAG G-3′ and 5′-CTT TCA GCC TAA AGC TTT TTC CCC GTA T-3′. PCR primers to confirm the gene loading of pCART2.2-Ubc-hKO1 were as follows: EF1a FW/NeoR Rv, described above, and 5′-AGC GGC GAT ACC GTA AAG CA-3′.
Cytogenetic analyses
Microcell hybrids on slides were stained with quinacrine mustard and Hoechst 33258 to enumerate chromosomes. Images were captured under an AxioImagerZ2 fluorescence microscope (Carl Zeiss, Jena, Germany) and analyzed with Ikaros software (MetaSystems, Altlussheim, Germany). mFISH analyses were performed in accordance with the manufacturer instructions (MetaSystems). Human mFISH probes were purchased from MetaSystems. Metaphase images were captured digitally with a CoolCubeI CCD camera and ISIS mFISH software (MetaSystems). FISH analyses were performed on fixed metaphases of microcell hybrids using digoxigenin-labeled (Roche, Basel, Switzerland) alphoid DNA probe p11-443 and biotin-labeled BAC DNA (RP11-954B16, located in the dystrophin genomic region) to detect DYS-HAC, and digoxigenin-labeled p11-4 and a biotin-labeled plasmid containing KO1 (ΦC31 attB-NeoR-pUbc-KO1) to confirm gene loading on the HAC.44 Chromosomal DNA was counterstained with 4’,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Images were captured under the AxioImagerZ2 fluorescence microscope.
RT-PCR
Total RNA from cultured cells was purified with Trizol reagent (Invitrogen) and treated with a Turbo DNA-free kit (Ambion) to remove genomic DNA. First-strand cDNA synthesis was undertaken using an oligo-(dT)20 primer and ReverTraAce (Toyobo, Osaka, Japan). MEFs and HFL-1 cells (fibroblasts) were used as negative controls. NAT1 was used as an internal control. PCR was performed with cDNA using ExTaq (Takara Bio, Shiga, Japan) and AmpliTaq Gold (Perkin Elmer, Waltham, MA, USA). Amplifications were performed with an annealing temperature of 58°C for 30–35 cycles, and then amplified fragments were resolved by electrophoresis on a 2% agarose gel, followed by staining with ethidium bromide. RT-PCR to detect endogenous ESC markers was performed using previously described primers.5,27
Teratoma formation assay
To produce teratomas, DMD-iPSCs carrying the DYS-HAC were injected subcutaneously into testes of severe combined immunodeficiency (SCID) mice (Charles River, Yokohama, Japan). After 8 weeks, resected teratomas were fixed in 20% formalin, processed for paraffin sectioning, and then stained with hematoxylin and eosin. All animal experiments were approved by the Institutional Animal Care and Use Committee of Tottori University.
Generation of HIDEMs
Human iPSCs were differentiated into HIDEMs as previously reported.9,24 iPSCs were pretreated with 10 μM Y-27632 (Wako) for 1 h and collected as single cells by incubation for 30–120 min in dissociation medium, PBS(−) containing 0.5 mM EDTA (Dojin), 0.1 mM β-mercaptoethanol (Sigma Aldrich), and 3% FBS. The iPSCs (6 × 105) were seeded in Minimum essential medium (MEM) alpha containing nucleosides (Thermo Fisher Scientific), 10% FBS, 1% penicillin/streptomycin (Thermo Fisher Scientific), and 0.2% β-mercaptoethanol on a well of a BioCoat Collagen I 6-well plate (Corning) pre-coated with 1% Matrigel Growth Factor-educed Basement Membrane Matrix, Lactate dehydrogenase elevating virus (LDEV)-free (Corning) and incubated for 1 week at 37°C with 5% CO2 and 5% O2. The medium was changed every 2 days. The cells were dissociated by gentle scraping and then passed through a 40-μm cell strainer (Corning). Cells (2.5 × 105) on a well of the 1% Matrigel-coated 6-well plate in MEM alpha were incubated for 1 week. On the third week, the differentiated cells were trypsinized and collected, and 9.6 × 105 cells were reseeded on a well of the coated 6-well plate in human mesoangioblast medium, Iscove’s modified Dulbecco’s medium (Sigma-Aldrich) containing 10% FBS, 2 mM glutamine, 0.1 mM β-mercaptoethanol, 1% nonessential amino acids (Gibco), human basic fibroblast growth factor (5 ng/mL) (FUJIFILM Wako), 1% penicillin/streptomycin, 0.5 μM oleic and linoleic acids (Sigma Aldrich), 1.5 μM Fe2+ (Iron[II] chloride tetrahydrate) (Sigma Aldrich), 0.12 μM Fe+++ (Iron[III] nitrate nonahydrate), and 1% insulin/transferrin/selenium (Gibco). The cells were incubated for 1 week and the medium was changed every 2 days. When the HIDEMs reached confluency, they were expanded on a Matrigel-coated well-plate at 80% confluence in human mesoangioblast medium for characterization.
Plasmid construction
For pCART1.2 construction to generate the modified HAC, an EF1a promoter-FRT-NeoR-human growth hormone (hGH) polyA-FRT unit was synthesized and cloned into the pMK-RQ vector (pCART1.1) (Thermo Fisher Scientific). The unit was cloned into NotI/AvrII sites of the pB4ins2 vector45 to be flanked by HS4 sequences. Then, loxP-3′HPRT fragment was inserted into KpnI/AscI sites of the vector (pCART1.2). For pCART2.1 construction, a ΦC31 attB sequence was synthesized and cloned into the pMA-RQ vector (Thermo Fisher Scientific). The NeoR-hGH polyA unit was cloned into XhoI/HindIII sites of the vector to generate the pCART2.1 vector. For pCART2.2 construction, the ΦC31 attB-NeoR-hGH polyA unit of the pCART2.1 vector was inserted into KpnI/AscI sites of the pB4ins2 vector. For pCART2.2-Ubc-hKO1 (ΦC31 attB-NeoR-pUbc-KO1) construction, DNA fragments of a Ubc promoter and bovine growth hormone (BGH) polyA were synthesized and sequentially cloned into SmaI and HindIII sites, respectively, of the phKO1-S1 cloning vector (MBL) to generate the pUbc-hKO1-BGHpA vector. Next, a Ubc promoter-hKO1-BGHpA unit amplified from the pUbc-hKO1-BGHpA vector was inserted into the MluI site of the pCART2.2 vector to generate the pCART2.2-Ubc-hKO1 vector. For pEF1-phiC31-T construction, an EF1a promoter fragment was amplified from the pBApo-EF1α Puro vector (TAKARA BIO) and cloned into the KpnI/BamHI sites of the pUbc-hKO1-BGHpA vector to generate the pEF1a-hKO1-BGHpA vector. Next, a ΦC31 integrase fragment amplified from the phiC31 integrase pVAX1v2 vector11 was inserted into the BamHI/NheI sites of the pEF1a-hKO1-BGHpA vector to generate the pEF1-phiC31-T vector.
Modification of 21HAC2
pCART1.2 was inserted into 21HAC2 by Cre-loxP recombination. Then, 1 × 106 CHO cells containing 21HAC2 were prepared, and 1.8 μg pCAR-T1.2 and 0.6 μg pBS185 expressing Cre recombinase were transfected with Lipofectamine 2000, following the manufacturer’s instructions. The HPRT (hypoxanthine phosphoribosyltransferase) gene was reconstituted in CHO (modified 21HAC2) cells correctly inserted with pCART1.2 and the cells resisted HAT (hypoxanthine-aminopterin-thymidine) medium. The modified 21HAC2 was transferred from the CHO cells to T-iPSCs by MV-MMCT. PCR and FISH analyses were performed to confirm successful modification in each intermediate product.
Electroporation of iPSCs
An NEPA21 electroporator was used to introduce plasmids into iPSCs. Cells (2 × 106) were prepared in 90 μL Opti-MEM (Gibco) in a 2-mm cuvette (Nepa Gene). Poring pulse conditions were 175 V, 2.5 ms pulse length, 50 ms pulse interval, two pulses, 10% attenuation rate, and (+) polarity. Transfer pulses were 20 V, 50 ms pulse length, 50 ms pulse interval, five pulses, 40% attenuation rate, and (+/−) polarity. Ten micrograms of Flp recombinase expression vector were electroporated into T-iPSCs to eliminate the neomycin resistance gene from the modified 21HAC2. Twenty micrograms of pCART2.2-Ubc-hKO1 and 10 μg pEF1a-PhiC31 integrase were electroporated into T-iPSCs carrying the basal-HAC.
T cell differentiation from T-iPSCs
T-iPSCs (TkT3v1-7) containing the basal-HAC with KO1 were differentiated into T cells as described previously with a slight modification.25,26 Briefly, the iPSCs were cultured on a feeder of mouse embryonic fibroblasts before differentiation. Two feeder cell lines, C3H10T1/2 feeder and OP9-DL1 cells (both obtained from RIKEN BRC), were maintained in basal Eagle’s medium (Life Technologies) containing 1% fetal calf serum (FCS) and 1% L-glutamine-penicillin-streptomycin solution (PSG, Sigma) on 0.1% gelatin-coated dishes and alpha-MEM (Gibco) supplemented with 15% FBS and 1% PSG, respectively. To obtain hematopoietic progenitor cells (HPCs), the iPSCs were seeded onto 10T1/2 cells in Iscove’s modified Dulbecco’s medium (Sigma) supplemented with 15% FCS, 1% PSG, Insulin-Transferrin-Selenium solution (ITS-G, GIBCO), 0.45 mM a-monothioglycerol (Nacalai Tesque), 50 μg/mL L(+)-ascorbic acid (PAA, Nacalai Tesque), 20 ng/mL VEGF (R&D Systems) and incubated at 37°C with 5% O2. Seven days later, 30 ng/mL SCF and 10 ng/mL FLT-3L (PeproTech) were added, and the cells were cultured for 7 days at 37°C with 20% O2. Cells on 10T1/2 feeders were seeded on OP9-DL1 cells in alpha-MEM supplemented with 15% FBS, 1% PSG, PAA, 1% ITS-G, 10 ng/mL IL-7 (PeproTech), and 10 ng/mL FLT-3L and cultured for 21 days. Media were changed every 2–4 days, and cells on 10T1/2 and OP9-DL1 feeder cells were reseeded onto fresh feeders every 7 days. The obtained cells were stimulated by anti-CD3 antibody OKT3 (Thermo Fisher Scientific) in the presence of 10 ng/mL IL-7 and 10 nM dexamethasone (Dexart, Fuji Pharma, Japan) for 7 days to induce mature CD8 T cells.26
FCM analyses
FCM analyses using fluorescein isothiocyanate (FITC) mouse anti-human CD46 antibody, PE mouse anti-human CD9 antibody, and PE mouse anti-human CD71 antibody (BD Pharmingen) in DMD-iPSCs were performed on FACSAriaI (BD Biosciences) as described previously.22,23,46
To characterize HIDEMs, the cells were stained with PE mouse anti-human CD13 (BD Biosciences), PE mouse anti-human CD146 (BioCytex), FITC mouse anti-human CD44 (BD Biosciences), FITC mouse anti-human alkaline phosphatase (Santa Cruz Biotechnology), PE mouse anti-human CD49b (BioLegend), PE mouse anti-CD45 (BD Biosciences), FITC anti-human CD31 (Immunostep), FITC anti-human CD56 (BioLegend), PE rat anti-Flk1 (BD Biosciences), and PE mouse anti-SSEA4 (BD Biosciences) antibodies. The cells (1 × 106 cells) were stained with the recommended concentration of each antibody in PBS(−) containing 1% FBS. FCM was performed using a Gallios (Beckman Coulter). Generated fcs files were analyzed by Kaluza software (Beckman Coulter).
T-iPSCs were harvested in PBS containing trypsin (Life Technologies), CaCl2 (Nacalai Tesque), and KSR (Life Technologies) and stained with SSEA4-VioBlue (Miltenyi Biotech) or TRA1-60-Alexa 647 (BD Biosciences) antibodies. Trypsinized sacs were stained with CD34-Pacific Blue and CD43-APC antibodies (both from BD Biosciences). Cells cultured on OP9-DL1 feeder cells for 21 days were harvested by pipetting, stained with a CD3-APC antibody (BioLegend), washed, and resuspended in PBS containing 2% FCS and propidium iodide. FCM data were acquired on BD LSRFortessa or BD FACSAriaII (BD Biosciences). Generated fcs files were analyzed using FlowJo 9 software (BD Biosciences).
Neural induction
For neural differentiation, we used the quick aggregation method for serum-free embryoid body formation (SFEBq). Undifferentiated hiPSCs were maintained in StemFit medium (Ajinomoto) under feeder-free conditions, dissociated with 0.5 × TrypLE Select (Thermo Fisher Scientific), and seeded in 96-well low-cell-adhesion plates (LIPIDURE-Coat Plate A-V96, NOF) at 9,000 cells/well. The cells were cultured for 14 days in SFEBq medium, DMEM/F12 containing 20% KSR, 2 mM Glutamax, 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol, and 1% penicillin/streptomycin as well as 2 μM XAV939 (Stemgent) for Wnt signal inhibition and 100 nM LDN193189 (Stemgent) and 10 μM SB431542 (WAKO) for dual SMAD inhibition. The medium contained 30 μM Y-27632 (WAKO) for the first 6 days.
On day 15, to induce neuronal differentiation, cell aggregates were transferred to 4-well culture slides (Lab-Tek II, Thermo Fisher Scientific) coated with poly-L-ornithine and fibronectin and cultured in Neurobasal Plus Medium with B-27 Plus Supplement (Thermo Fisher Scientific) and 2 mM Glutamax for 2 weeks.
Acknowledgments
This study was supported in part by the Regional Innovation Strategy Support Program of Ministry of Education, Culture, Sports, Science and Technology (MEXT) (Y.K. and M.O.), Japan Science and Technology Agency (JST) Core Research for Evolutional Science and Technology (CREST) grant number JPMJCR18S4 (Y.K.), and Centers for Clinical Application Research on Specific Disease/Organ (Type C) from Japan Agency for Medical Research and Development (AMED) under grant number JP20bm1004001 (S.K., Y.K., and K.T.). We thank Dr. Takafumi Nakamura at Tottori University and Dr. Gene Kurosawa at Fujita Health University for providing MV-H (wild type), chimeric MV-H of anti-CD9, anti-CD71ScFv, and MV-F expression vector. We thank Toko Kurosaki, Yukako Sumida, Masami Morimura, Kei Yoshida, Eri Kaneda, Yutaro Akakura, Takuro Suematsu, Dr. Kazuomi Nakamura, Dr. Yuji Nakayama, and Tomoko Ashiba at Tottori University and Kazuyo Takaki at Kyowa Kirin Co., Ltd. for their technical assistance. We also thank Dr. Hidetoshi Hoshiya and Dr. Sara Benedetti at University College London, Dr. Hiroyuki Kugoh, Dr. Hiroyuki Satofuka, Dr. Takashi Moriwaki, Dr. Tetsuya Ohbayashi, and Dr. Takahito Ohira at Tottori University for critical discussions. This research was partly performed at the Tottori Bio Frontier managed by Tottori prefecture. Work in the Tedesco laboratory was supported by Muscular Dystrophy UK, the European Research Council, and the National Institute for Health Research (NIHR; the views expressed are those of the authors and not necessarily those of the National Health Service, NIHR, or Department of Health). We also thank Mitchell Arico from Edanz Group (https://en-author-services.edanz.com/) for editing a draft of this manuscript.
Author contributions
Y.K. and N.U. participated in all aspects of this work and prepared the manuscript; M. Osaki performed teratoma formation experiments; A.O. and S.N. performed plasmid construction; Y.K., N.U., S.A., N.K., K.K., Y.Y., C.S., S. Takata, M.S., and Y.N. performed cell culture and MMCT experiments; K.K., S. Takata, and M. Oshimura performed cytogenetic analyses; M.H. performed neural differentiation experiments; G.F. and F.S.T. discussed data, revised the manuscript, and assisted N.U. to generate HIDEMs; N.K., C.S., and Y.Y. performed expression analyses; K.T. and M. Oshimura supervised the study; S.N., K.F., and S. Takayanagi performed T cell experiments, and A.K. and S.K. designed the T cell experiments.
Declaration of interests
K.F., S. Takayanagi, and A.K. are employed by Kirin Holdings, Co., Ltd. S.N. is employed by Kyowa Kirin, Co., Ltd. The other authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.12.012.
Supplemental information
References
- 1.Kazuki Y., Oshimura M. Human artificial chromosomes for gene delivery and the development of animal models. Mol. Ther. 2011;19:1591–1601. doi: 10.1038/mt.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oshimura M., Uno N., Kazuki Y., Katoh M., Inoue T. A pathway from chromosome transfer to engineering resulting in human and mouse artificial chromosomes for a variety of applications to bio-medical challenges. Chromosome Res. 2015;23:111–133. doi: 10.1007/s10577-014-9459-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kouprina N., Earnshaw W.C., Masumoto H., Larionov V. A new generation of human artificial chromosomes for functional genomics and gene therapy. Cell. Mol. Life Sci. 2013;70:1135–1148. doi: 10.1007/s00018-012-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kazuki Y., Hoshiya H., Takiguchi M., Abe S., Iida Y., Osaki M., Katoh M., Hiratsuka M., Shirayoshi Y., Hiramatsu K. Refined human artificial chromosome vectors for gene therapy and animal transgenesis. Gene Ther. 2011;18:384–393. doi: 10.1038/gt.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kazuki Y., Hiratsuka M., Takiguchi M., Osaki M., Kajitani N., Hoshiya H., Hiramatsu K., Yoshino T., Kazuki K., Ishihara C. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol. Ther. 2010;18:386–393. doi: 10.1038/mt.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomizuka K., Shinohara T., Yoshida H., Uejima H., Ohguma A., Tanaka S., Sato K., Oshimura M., Ishida I. Double trans-chromosomic mice: maintenance of two individual human chromosome fragments containing Ig heavy and kappa loci and expression of fully human antibodies. Proc. Natl. Acad. Sci. USA. 2000;97:722–727. doi: 10.1073/pnas.97.2.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuroiwa Y., Kasinathan P., Choi Y.J., Naeem R., Tomizuka K., Sullivan E.J., Knott J.G., Duteau A., Goldsby R.A., Osborne B.A. Cloned transchromosomic calves producing human immunoglobulin. Nat. Biotechnol. 2002;20:889–894. doi: 10.1038/nbt727. [DOI] [PubMed] [Google Scholar]
- 8.Kazuki Y., Kobayashi K., Aueviriyavit S., Oshima T., Kuroiwa Y., Tsukazaki Y., Senda N., Kawakami H., Ohtsuki S., Abe S. Trans-chromosomic mice containing a human CYP3A cluster for prediction of xenobiotic metabolism in humans. Hum. Mol. Genet. 2013;22:578–592. doi: 10.1093/hmg/dds468. [DOI] [PubMed] [Google Scholar]
- 9.Tedesco F.S., Gerli M.F., Perani L., Benedetti S., Ungaro F., Cassano M., Antonini S., Tagliafico E., Artusi V., Longa E. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 2012;4:140ra89. doi: 10.1126/scitranslmed.3003541. [DOI] [PubMed] [Google Scholar]
- 10.Benedetti S., Uno N., Hoshiya H., Ragazzi M., Ferrari G., Kazuki Y., Moyle L.A., Tonlorenzi R., Lombardo A., Chaouch S. Reversible immortalisation enables genetic correction of human muscle progenitors and engineering of next-generation human artificial chromosomes for Duchenne muscular dystrophy. EMBO Mol. Med. 2018;10:254–275. doi: 10.15252/emmm.201607284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi S., Kazuki Y., Nakayama Y., Nanba E., Oshimura M., Ohbayashi T. A method for producing transgenic cells using a multi-integrase system on a human artificial chromosome vector. PLoS ONE. 2011;6:e17267. doi: 10.1371/journal.pone.0017267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki T., Kazuki Y., Oshimura M., Hara T. A novel system for simultaneous or sequential integration of multiple gene-loading vectors into a defined site of a human artificial chromosome. PLoS ONE. 2014;9:e110404. doi: 10.1371/journal.pone.0110404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honma K., Abe S., Endo T., Uno N., Oshimura M., Ohbayashi T., Kazuki Y. Development of a multiple-gene-loading method by combining multi-integration system-equipped mouse artificial chromosome vector and CRISPR-Cas9. PLoS ONE. 2018;13:e0193642. doi: 10.1371/journal.pone.0193642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karagiannis P., Takahashi K., Saito M., Yoshida Y., Okita K., Watanabe A., Inoue H., Yamashita J.K., Todani M., Nakagawa M. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019;99:79–114. doi: 10.1152/physrev.00039.2017. [DOI] [PubMed] [Google Scholar]
- 15.Pawlowski M., Ortmann D., Bertero A., Tavares J.M., Pedersen R.A., Vallier L., Kotter M.R.N. Inducible and Deterministic Forward Programming of Human Pluripotent Stem Cells into Neurons, Skeletal Myocytes, and Oligodendrocytes. Stem Cell Reports. 2017;8:803–812. doi: 10.1016/j.stemcr.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papapetrou E.P., Schambach A. Gene Insertion Into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 2016;24:678–684. doi: 10.1038/mt.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki T., Kazuki Y., Hara T., Oshimura M. Current advances in microcell-mediated chromosome transfer technology and its applications. Exp. Cell Res. 2020;390:111915. doi: 10.1016/j.yexcr.2020.111915. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki T., Kazuki Y., Oshimura M., Hara T. Highly Efficient Transfer of Chromosomes to a Broad Range of Target Cells Using Chinese Hamster Ovary Cells Expressing Murine Leukemia Virus-Derived Envelope Proteins. PLoS ONE. 2016;11:e0157187. doi: 10.1371/journal.pone.0157187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinenko S.A., Skvortsova E.V., Liskovykh M.A., Ponomartsev S.V., Kuzmin A.A., Khudiakov A.A., Malashicheva A.B., Alenina N., Larionov V., Kouprina N., Tomilin A.N. Transfer of Synthetic Human Chromosome into Human Induced Pluripotent Stem Cells for Biomedical Applications. Cells. 2018;7:261. doi: 10.3390/cells7120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liskovykh M., Lee N.C., Larionov V., Kouprina N. Moving toward a higher efficiency of microcell-mediated chromosome transfer. Mol. Ther. Methods Clin. Dev. 2016;3:16043. doi: 10.1038/mtm.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasegawa Y., Ikeno M., Suzuki N., Nakayama M., Ohara O. Improving the efficiency of gene insertion in a human artificial chromosome vector and its transfer in human-induced pluripotent stem cells. Biol. Methods Protoc. 2018;3:bpy013. doi: 10.1093/biomethods/bpy013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katoh M., Kazuki Y., Kazuki K., Kajitani N., Takiguchi M., Nakayama Y., Nakamura T., Oshimura M. Exploitation of the interaction of measles virus fusogenic envelope proteins with the surface receptor CD46 on human cells for microcell-mediated chromosome transfer. BMC Biotechnol. 2010;10:37. doi: 10.1186/1472-6750-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiratsuka M., Ueda K., Uno N., Uno K., Fukuhara S., Kurosaki H., Takehara S., Osaki M., Kazuki Y., Kurosawa Y. Retargeting of microcell fusion towards recipient cell-oriented transfer of human artificial chromosome. BMC Biotechnol. 2015;15:58. doi: 10.1186/s12896-015-0142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maffioletti S.M., Gerli M.F., Ragazzi M., Dastidar S., Benedetti S., Loperfido M., VandenDriessche T., Chuah M.K., Tedesco F.S. Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human ES and patient-specific iPS cells. Nat. Protoc. 2015;10:941–958. doi: 10.1038/nprot.2015.057. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura T., Kaneko S., Kawana-Tachikawa A., Tajima Y., Goto H., Zhu D., Nakayama-Hosoya K., Iriguchi S., Uemura Y., Shimizu T. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Minagawa A., Yoshikawa T., Yasukawa M., Hotta A., Kunitomo M., Iriguchi S., Takiguchi M., Kassai Y., Imai E., Yasui Y. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell. 2018;23:850–858.e4. doi: 10.1016/j.stem.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy M., Awong G., Sturgeon C.M., Ditadi A., LaMotte-Mohs R., Zúñiga-Pflücker J.C., Keller G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Eiraku M., Watanabe K., Matsuo-Takasaki M., Kawada M., Yonemura S., Matsumura M., Wataya T., Nishiyama A., Muguruma K., Sasai Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell. 2008;3:519–532. doi: 10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Maroof A.M., Keros S., Tyson J.A., Ying S.W., Ganat Y.M., Merkle F.T., Liu B., Goulburn A., Stanley E.G., Elefanty A.G. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weick J.P., Held D.L., Bonadurer G.F., 3rd, Doers M.E., Liu Y., Maguire C., Clark A., Knackert J.A., Molinarolo K., Musser M. Deficits in human trisomy 21 iPSCs and neurons. Proc. Natl. Acad. Sci. USA. 2013;110:9962–9967. doi: 10.1073/pnas.1216575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tedesco F.S. Human artificial chromosomes for Duchenne muscular dystrophy and beyond: challenges and hopes. Chromosome Res. 2015;23:135–141. doi: 10.1007/s10577-014-9460-6. [DOI] [PubMed] [Google Scholar]
- 33.Zatti S., Martewicz S., Serena E., Uno N., Giobbe G., Kazuki Y. Complete restoration of multiple dystrophin isoforms in genetically corrected Duchenne muscular dystrophy patient-derived cardiomyocytes. Mol. Ther. Methods Clin. Dev. 2014;1 doi: 10.1038/mtm.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi I.Y., Lim H., Estrellas K., Mula J., Cohen T.V., Zhang Y., Donnelly C.J., Richard J.P., Kim Y.J., Kim H. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived using a Human iPSC-Based Model. Cell Rep. 2016;15:2301–2312. doi: 10.1016/j.celrep.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 35.Tedesco F.S., Hoshiya H., D’Antona G., Gerli M.F., Messina G., Antonini S., Tonlorenzi R., Benedetti S., Berghella L., Torrente Y. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011;3:96ra78. doi: 10.1126/scitranslmed.3002342. [DOI] [PubMed] [Google Scholar]
- 36.Tomizuka K., Yoshida H., Uejima H., Kugoh H., Sato K., Ohguma A., Hayasaka M., Hanaoka K., Oshimura M., Ishida I. Functional expression and germline transmission of a human chromosome fragment in chimaeric mice. Nat. Genet. 1997;16:133–143. doi: 10.1038/ng0697-133. [DOI] [PubMed] [Google Scholar]
- 37.Inoue J., Mitsuya K., Maegawa S., Kugoh H., Kadota M., Okamura D., Shinohara T., Nishihara S., Takehara S., Yamauchi K. Construction of 700 human/mouse A9 monochromosomal hybrids and analysis of imprinted genes on human chromosome 6. J. Hum. Genet. 2001;46:137–145. doi: 10.1007/s100380170101. [DOI] [PubMed] [Google Scholar]
- 38.Matano M., Date S., Shimokawa M., Takano A., Fujii M., Ohta Y., Watanabe T., Kanai T., Sato T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015;21:256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 39.Drost J., van Jaarsveld R.H., Ponsioen B., Zimberlin C., van Boxtel R., Buijs A., Sachs N., Overmeer R.M., Offerhaus G.J., Begthel H. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521:43–47. doi: 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 40.Paulis M., Susani L., Castelli A., Suzuki T., Hara T., Straniero L., Duga S., Strina D., Mantero S., Caldana E. Chromosome Transplantation: A Possible Approach to Treat Human X-linked Disorders. Mol. Ther. Methods Clin. Dev. 2020;17:369–377. doi: 10.1016/j.omtm.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fusaki N., Ban H., Nishiyama A., Saeki K., Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kazuki Y., Kimura M., Nishigaki R., Kai Y., Abe S., Okita C., Shirayoshi Y., Schulz T.C., Tomizuka K., Hanaoka K. Human chromosome 21q22.2-qter carries a gene(s) responsible for downregulation of mlc2a and PEBP in Down syndrome model mice. Biochem. Biophys. Res. Commun. 2004;317:491–499. doi: 10.1016/j.bbrc.2004.03.069. [DOI] [PubMed] [Google Scholar]
- 43.Ikeno M., Masumoto H., Okazaki T. Distribution of CENP-B boxes reflected in CREST centromere antigenic sites on long-range alpha-satellite DNA arrays of human chromosome 21. Hum. Mol. Genet. 1994;3:1245–1257. doi: 10.1093/hmg/3.8.1245. [DOI] [PubMed] [Google Scholar]
- 44.Shinohara T., Tomizuka K., Miyabara S., Takehara S., Kazuki Y., Inoue J., Katoh M., Nakane H., Iino A., Ohguma A. Mice containing a human chromosome 21 model behavioral impairment and cardiac anomalies of Down’s syndrome. Hum. Mol. Genet. 2001;10:1163–1175. doi: 10.1093/hmg/10.11.1163. [DOI] [PubMed] [Google Scholar]
- 45.Hiratsuka M., Uno N., Ueda K., Kurosaki H., Imaoka N., Kazuki K., Ueno E., Akakura Y., Katoh M., Osaki M. Integration-free iPS cells engineered using human artificial chromosome vectors. PLoS ONE. 2011;6:e25961. doi: 10.1371/journal.pone.0025961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uno N., Fujimoto T., Komoto S., Kurosawa G., Sawa M., Suzuki T., Kazuki Y., Oshimura M. A luciferase complementation assay system using transferable mouse artificial chromosomes to monitor protein-protein interactions mediated by G protein-coupled receptors. Cytotechnology. 2018;70:1499–1508. doi: 10.1007/s10616-018-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.