

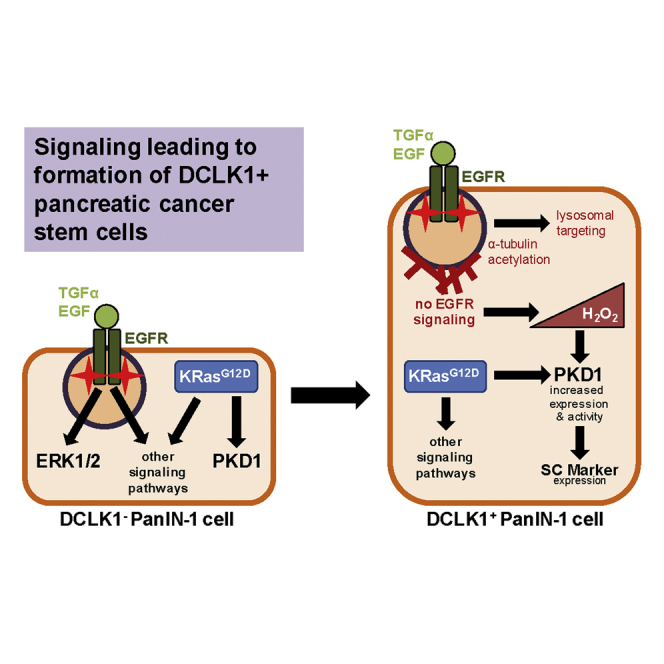
Summary
Doublecortin-like kinase 1 (DCLK1)-positive pancreatic cancer stem cells develop at a precancerous stage and may contribute to the lack of efficacy of pancreatic cancer therapy. Although PanIN cells express oncogenic KRas and have an increased activity of epidermal growth factor receptor (EGFR), we demonstrate that, in DCLK1+ PanIN cells, EGFR signaling is not propagated to the nucleus. Mimicking blockage of EGFR with erlotinib in PanIN organoid culture or in p48cre;KrasG12D mice led to a significant increase in DCLK1+ PanIN cells. As a mechanism of how EGFR inhibition leads to formation of DCLK1+ cells, we identify an increase in hydrogen peroxide contributing to activation of Protein Kinase D1 (PKD1). Active PKD1 then drives stemness and abundance of DCLK1+ cells in lesions. Our data suggest a signaling mechanism that leads to the development of DCLK1+ pancreatic cancer stem cells, which can be exploited to target this population in potential therapeutic approaches.
Subject areas: Cell Biology, Stem Cell Research, Cancer
Graphical Abstract
For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.isci.2020.102019.
Highlights
-
•
DCLK1+ PanIN cells have abrogated EGFR signaling and increased oxidative stress
-
•
Inhibition of EGFR signaling increases DCLK1+ cells, oxidative stress, and PKD1
-
•
PKD1 contributes to the abundance of DCLK1+ stem cells in PanIN lesions
-
•
Stem cell markers, including OCT4 and CD133, are increased by PKD1
Cell Biology; Stem Cell Research; Cancer
Introduction
Pancreatic ductal adenocarcinoma (PDA) has been difficult to target, with a 5-year survival rate of only 9% (Siegel et al., 2020). A key factor in this dismal rate is the presence of cancer stem cells (CSCs), which occur at early stages of tumorigenesis (Qu et al., 2015; Rhim et al., 2012), are resistant to chemotherapy (Gzil et al., 2019; Hermann et al., 2007; Hong et al., 2009), and contribute to tumor recurrence (Vinogradov and Wei, 2012). CSCs have the ability to self-renew and initiate tumors with relatively few cells (Bailey et al., 2014; Gzil et al., 2019; Li et al., 2007), making them essential targets for novel therapeutic approaches.
We here focus on a population of cells with cancer stem cell properties in the pancreas, which is characterized by the expression of Doublecortin-like kinase 1 (DCLK1), a microtubule-associated protein (Lin et al., 2000). These CSCs express, in addition to DCLK1, acetylated α-tubulin (Bailey et al., 2014), β-endorphin (Delgiorno et al., 2014), and the stemness markers CD133 (Bailey et al., 2014), CD24/CD44/ESA (Bailey et al., 2014), and Notch1 (Sureban et al., 2011). Furthermore, DCLK1 in these cells upregulates PD-L1 through the Hippo pathway (Yan et al., 2020), which is implicated in stem cell maintenance (Harvey et al., 2013). Another feature of these cells is the presence of Y1068-phosphorylated epidermal growth factor receptor (EGFR), clustered in an apical region (Delgiorno et al., 2014).
In mouse models for PDA the DCLK1+ population is most abundant in acinar-to-ductal metaplasia (ADM) and pancreatic intraepithelial neoplasia 1A/1B (PanIN-1A/1B) lesions (Bailey et al., 2014; Delgiorno et al., 2014) but is also increased in the serum of patients with stage I and II pancreatic cancer (Qu et al., 2015). Although formed during early tumorigenesis, DCLK1+ lesion cells show bona fide CSC properties. For example, they have enhanced clonogenic ability and can initiate tumor formation (Bailey et al., 2014). Moreover, they disseminate early in the KPC (KrasG12D;p53R172H;Pdx1cre) mouse model for PDA (Qu et al., 2015). Consequently, DCLK1 expression in patient tumors is associated with expression of the cancer stem cell markers CD44/CD24/EpCAM and goes along with shorter median overall survival time, more frequent relapse, and shorter relapse-free survival (Nishio et al., 2017). Thus, understanding the mechanisms driving this malignant cell type is critical to be able to combat pancreatic cancer.
Of the three members of the Protein Kinase D (PKD) family of serine-threonine kinases, Protein Kinase D1 (PKD1) has been identified as a driver of pancreatic cancer initiation (Liou et al., 2015). In KC (p48cre;KrasG12D) mice, PKD1 drives ADM and progression to PanINs (Liou et al., 2015). In these transformation processes PKD1 acts downstream of oncogenic KRas, which initiates its activation through increasing the generation of reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) (Liou et al., 2016). An increase in H2O2 mediates phosphorylation of PKD1 at tyrosine residue 95 (Y95), which is a specific marker for ROS-activated PKD1 (Doppler and Storz, 2007). PKD1 has been shown to activate the transcription factors Notch1 and nuclear factor-kB (NF-kB) to drive early metaplasia (Liou et al., 2015, 2016). However, it is unclear if (or how) PKD1 affects the DCLK1+ stem cell population, which has been shown by lineage tracing experiments to develop from KrasG12D-expressing ADM or PanIN cells (Bailey et al., 2014).
We here show that, in DCLK1+ PanIN cells, although EGFR is phosphorylated at Y1068, owing to the high presence of acetylated α-tubulin and apical clustering, signaling is not propagated to the nucleus. Inhibition of EGFR signaling, either through this mechanism or through inhibition with erlotinib, leads to a significant increase in DCLK1+ PanIN cells. An increase in hydrogen peroxide as a result of EGFR inhibition contributes to activation of PKD1, which contributes to both occurrence and stemness of DCLK1+ cells. In summary, we here identified key signaling events in DCLK1+ pancreatic cancer stem cells that can be exploited to target this population in potential therapeutic approaches.
Results
EGFR, although autophosphorylated, does not signal downstream to ERK1/2 in DCLK1+ PanIN cells
Expression of DCLK1, acetylated α-tubulin, and β-endorphin characterizes a population of pancreatic cancer stem cells that are formed in low-grade lesions early during tumor development (Bailey et al., 2014; Delgiorno et al., 2014). Lineage tracing experiments showed that these cells develop from KrasG12D-expressing ADM or PanIN cells and are distinct to the DCLK1+ Tuft cells that originate in the intestine, but they can also be detected in the pancreas (Bailey et al., 2014; Westphalen et al., 2016). We confirmed previous reports (Bailey et al., 2014; Delgiorno et al., 2014) that, in low-grade pancreatic lesions of p48cre;LSL-KrasG12D (KC) mice, DCLK1, acetylated α-tubulin, and β-endorphin mark the same population of cells (Figures S1A–S1D), and also found that these cells are negative for the intestine-derived Tuft cell marker POU2F3 (Figures S1E and S1F).
Y1068-phosphorylated EGFR is localized to the membrane in most PanIN cells (Figure 1A), but in DCLK1+ PanIN cells it is primarily clustered in apical areas where it co-localizes with acetylated alpha-tubulin (Figures 1B and 1C). EGFR signaling is terminated by lysosomal downregulation (Brankatschk et al., 2012; Sousa et al., 2012; Vieira et al., 1996), which was functionally linked to acetylation of α-tubulin (Gao et al., 2010; Liu et al., 2012). Analyses of acetylated α-tubulin+ and DCLK1+ cells showed an increased expression of the lysosomal marker LAMP-1 (Figure 1D), and this correlated with a decrease in downstream signaling to ERK1/2 in DCLK1+ cells (analyses of pT202/pY204-ERK1/2; Figures 1E and 1F). This suggests that EGFR, although capable of autophosphorylation at Y1068, is targeted to the lysosomes, which prevents downstream signaling to MAPK in these cells.
Figure 1.

EGFR, although autophosphorylated, does not signal downstream to ERK1/2 in DCLK1+ PanIN cells
(A) IHC-IF for pY1068-EGFR (green) and DAPI nuclear stain in p48cre;LSL-KrasG12D (KC) mouse pancreas tissue. Scale bars indicate 50 μm.
(B) IHC-IF for pY1068-EGFR (green) and DCLK1 (pink; top) or acetylated α-tubulin (red; bottom) with DAPI nuclear stain in KC mouse pancreas tissue. Scale bars indicate 50 μm.
(C) Quantification of pY1068-EGFR signal in acetylated α-tubulin+ PanIN cells (∗ indicates statistical significance; t test p value < 0.0001; n = 550 cells). Error bars indicate standard deviation.
(D) IHC-IF for LAMP-1 (green) and either acetylated α-tubulin (red; top) or DCLK1 (pink; bottom) with DAPI nuclear stain in KC mouse pancreas tissue. Scale bars indicate 10 μm.
(E) IHC-IF for pT202/pY204-ERK1/2 (labeled pERK1/2; green) and DCLK1 (pink) in KC mouse pancreas tissue. Scale bars indicate 50 μm.
(F) Quantification of pERK1/2 in DCLK1- or DCLK1+ PanIN-1 lesion cells. Three mice were used to obtain the average fluorescent signal for pERK1/2 in DCLK1- or DCLK1+ cells in ImageJ, represented as fold difference in the graph. Statistical significance, indicated by ∗, was determined using the paired t test; p value = 0.0064. Error bars represent the standard deviation.
Chemical inhibition of EGFR signaling increases the occurrence of DCLK1+ cells in vitro and in vivo
In light of the above data indicating dysfunctional EGFR signaling in DCLK1+ cells, we next tested if inhibition of EGFR signaling could lead to formation of these cells. Therefore, we treated KC mice at an age of 4 weeks with control or the EGFR inhibitor erlotinib over a time span of 8 weeks (Figure S2A). At the endpoint we did not observe significant changes in the abnormal tissue area (including lesions and stroma) (Figure S2B) or apoptotic cell death within lesion areas (Figure S2C). However, as expected, we observed a significant decrease in active (Y1068-phosphorylated) EGFR (Figure S2D), which correlated with a significant increase in DCLK1+ cells within ADM/PanIN lesions (Figures 2A and 2B).
Figure 2.

Chemical inhibition of EGFR signaling increases the occurrence of DCLK1+ cells in vitro and in vivo
(A) IHC for DCLK1 in KC mice treated with vehicle or Erlotinib for 8 weeks. Scale bars indicate 50 μm.
(B) Quantification of the number of DCLK1+ cells per ADM/PanIN lesion area, shown as fold change. Counting was done manually, and area measurements were done using Aperio ImageScope. Pancreata of n = 5 mice per treatment group were analyzed. Statistical significance, indicated by ∗, was determined using the t test; p value = 0.0071. Error bars indicate standard deviation.
(C) Imaging of brightfield, DAPI nuclear stain, and DCLK1 (green) immunofluorescence in SM3 cell PanIN organoids. Scale bars indicate 50 μm.
(D) Quantification of flow cytometry analysis of DCLK1- and DCLK1+ SM3 cells, where the bars indicate the average of three experiments. Error bars indicate standard deviation.
(E) Representative plot of flow cytometry analysis of DCLK1 in control or erlotinib-treated SM3 cells.
(F) Quantification of flow cytometry in control or erlotinib-treated SM3 cells, where the bars indicate the average and ∗ indicates statistical signficance (t test; p value = 0.05; n = 3). Error bars represent the standard deviation.
(G) Flow cytometry results from three independent experiments, using the Cell Meter Intracellular Fluorimetric Hydrogen Peroxide Assay Kit to measure intracellular hydrogen peroxide resulting from treatment of SM3 cells (n = 3 per group) with control or erlotinib (1 μM) for 2 days. Statistical significance, indicated by ∗, was determined using the paired t test; p value = 0.0224. Error bars represent the standard deviation.
In order to test if inhibition of EGFR can indeed lead to an increase in the DCLK1+ cell population within PanIN lesion cells, we used an organoid system. SM3 are non-transformed cells from duct-like PanIN cells derived from Pdx1cre;KrasG12D mice (Agbunag et al., 2006) that can be propagated for 25–30 passages. SM3 cells in organoid culture form PanIN-like structures (Liou et al., 2017) that contain DCLK1+ cells (Figure 2C). Flow cytometric separation of DCLK1- and DCLK1+ cells in SM3 PanIN cells showed that the DCLK1+ population (8.75% ± 3.24% of cells in n = 3 replicates; Figure 2D) is also positive for the in vivo markers acetylated α-tubulin and β-endorphin (Figure S2E). Treatment of SM3 PanIN cells with erlotinib decreased EGFR activity, as measured by the presence of pERK1/2 (Figure S3A). Moreover, although fluorescence-activated cell sorting (FACS) analyses of SM3 PanIN cells treated with vehicle or erlotinib for 2 days did not significantly alter proliferation of DCLK1- or DCLK1+ populations (Figure S3B), erlotinib did significantly increase the presence of DCLK1+ cells (Figures 2E and 2F). Taken together, the data obtained with erlotinib support our finding that DCLK1+ cells can develop owing to dysfunctional EGFR signaling.
DCLK1+ pancreatic lesion cells show increased hydrogen peroxide levels
Inhibition of EGFR signaling previously has been shown to increase oxidative stress (Sobhakumari et al., 2013), and after treatment of SM3 PanIN cells with erlotinib we detected an approximately 3-fold increase in intracellular hydrogen peroxide (H2O2) (Figure 2G). Therefore, we next tested if oxidative stress levels are increased in the pancreatic DCLK1+ and acetylated α-tubulin+ cell population in vivo. Immunofluorescence-immunohistochemistry (IF-IHC) analyses of pancreatic lesion areas in KC mice indicated a significant increase in 4-HNE, a marker for intracellular oxidative stress, in the acetylated α-tubulin-positive cell population (Figures 3A and 3B). Moreover, specific probing for hydrogen peroxide (using FBBBE as a probe) showed a significant, over 3-fold increase in H2O2 in DCLK1+ PanIN cells when compared with neighboring DCLK1- PanIN cells (Figures 3C and 3D).
Figure 3.

DCLK1+ pancreatic lesion cells show increased hydrogen peroxide levels
(A) IHC-IF for acetylated α-tubulin (red) and 4-HNE (green) with DAPI nuclear stain in KC pancreas lesions. Scale bars indicate 50 μm.
(B) Quantification of 4-HNE in acetylated α-tubulin+ and acetylated α-tubulin- lesion cells. Signal quantified in ImageJ and displayed as fold change (t test p value < 0.0001; n = 100 cells). ∗ indicates statistical significance, and error bars represent standard deviation.
(C) Representative images of the fluorescent hydrogen peroxide sensor FBBBE (green) combined with DCLK1 (red) IHC-IF on fresh frozen KC mouse tissue. Scale bars indicate 50 μm.
(D) Quantification of the fluorescent FBBBE signal, indicating intracellular hydrogen peroxide, in DCLK1- and DCLK1+ cells from pancreata of n = 3 mice. Signal quantified in ImageJ and displayed as fold change. Statistical significance, indicated by ∗, was determined using the t test; p value <0.0001. Error bars represent standard deviation.
PKD1 is highly expressed in DCLK1-positive pancreatic lesion cells and contributes to their abundance within lesions
Protein Kinase D1 is a kinase that is responsive to hydrogen peroxide (Doppler and Storz, 2007, 2017) and drives pancreatic lesion formation downstream of oncogenic KRas-induced oxidative stress (Liou et al., 2015, 2016). We therefore evaluated its expression and activity in the DCLK1+ stem cell population. First, we found that PKD1 is highly expressed in DCLK1+ cells of low-grade pancreatic lesions in KC mice (Figure 4A). Such a positive correlation of PKD1 and DCLK1 expression can also be detected in the QCMG patient dataset (Bailey et al., 2016), which contains mRNA expression data of 96 pancreatic adenocarcinoma samples (Figure 4B). Since DCLK1+ cells show high levels of oxidative stress, we next probed for PKD1 phosphorylation at Y95, a residue whose phosphorylation was specifically linked to PKD activation by hydrogen peroxide (Doppler and Storz, 2007). We found that DCLK1+ lesion cells showed an increase of Y95-phosphorylation of PKD1 (Figures 4C–4E), and this was confirmed by co-staining of pY95-PKD with acetylated α-tubulin+ (Figure S4A).
Figure 4.

PKD1 is highly expressed in DCLK1-positive pancreatic lesion cells
(A) IHC-IF for DCLK1 (green) and PKD1 (red), with DAPI nuclear stain in KC mouse pancreas. The scale bars indicate 50 μm.
(B) PKD1 and DCLK1 mRNA expression data with regression line from https://www.cbioportal.org/ using the QCMG dataset containing 96 pancreatic adenocarcinoma samples.
(C) IHC for pY95-phosphorylated PKD in lesions of KC mice.
(D) IHC-IF for pY95-phosphorylated PKD (green) and DCLK1 (pink) in lesions of a KC mouse with DAPI nuclear stain. Scale bars represent 20 μm.
(E) Quantification of the percentage of DCLK1+ cells with or without pY95-PKD (t test p value < 0.0001; n = 4 mice). ∗ indicates statistical significance, and error bars represent standard deviation.
We next analyzed our erlotinib-treated KC mice for expression of PKD1 and for ROS-activated PKD1 (as indicated by PKD phosphorylation at pY95). Both PKD1 expression and its ROS-induced tyrosine phosphorylation were significantly increased when mice were treated with erlotinib (Figures 5A–5C). A similar upregulation of PKD1 expression by erlotinib has been found in SM3 cells (Figure S4B). Overall, this supports our model that abrogation of proper EGFR signaling leads to increased expression and activity of PKD1.
Figure 5.

PKD1 contributes to the abundance of DCLK1-positive pancreatic lesion cells
(A) IHC-IF for pY95-PKD (green; top) or PKD1 (red; bottom), combined with DAPI nuclear stain in vehicle or erlotinib-treated KC mice. Scale bars indicate 50 μm.
(B) Quantification of pY95-PKD+ cells in vehicle or erlotinib treated KC mice. The number of pY95-PKD+ cells per lesion were counted and expressed as fold change (t test p value = 0.0079; n = 4 mice per group). ∗ indicates statistical significance, and error bars represent standard deviation.
(C) Quantification of PKD1+ cells in vehicle or erlotinib-treated KC mice. The number of PKD1+ cells per lesion were counted and expressed as fold change (t test p value = 0.0256; n = 4 mice per group). ∗ indicates statistical significance, and error bars represent standard deviation.
(D) Representative images of IHC for DCLK1 in KC and KC;PKD1−/− mice at 8 weeks of age. Scale bars indicate 50 μm.
(E) Quantification of DCLK1 IHC in PanIN-1 lesions of KC or KC;PKD1−/− mice (n = 4 pancreata per genotype). Statistical significance, indicated by ∗, was determined using the t test, p value <0.05. Error bars indicate standard deviation.
(F) Quantification of pY1068-EGFR in KC or KC;PKD1−/− mice. Signal was quantified in ImageJ and expressed as fold change (t test p value = 0.5206; n = 3 mice per group). Error bars represent standard deviation.
Eventually, we tested if oxidative stress-activated PKD1 is not only a marker for DCLK1+ cells but also a driving factor for the presence of these cells. Therefore, we compared KC mice with KC mice in which PKD1 is conditionally knocked out (p48cre;KrasG12D;PKD1−/−) in pancreatic epithelial cells (described in Liou et al., 2015). We detected significantly fewer DCLK1+ cells in PanIN lesions from KC;PKD1−/− mice, when compared with PanIN lesions of KC mice (Figures 5D and 5E). As a control, demonstrating that PKD1 does not affect EGFR signaling, KC and KC;PKD−/− mice were analyzed for EGFR signaling. As expected, we found EGFR signaling (as determined by EGFR Y-1068-phosphorylation) unchanged between samples (Figure 5F).
DCLK1+/acetylated α-tubulin+ lesion cells express stem cell markers dependent on the presence of PKD1
The DCLK1+ cell population previously has been discussed to have cancer stem cell properties (Bailey et al., 2014; Delgiorno et al., 2014). In order to verify that DCLK1+ and PKD1+ cells in low-grade lesions show stem cell characteristics, we sorted primary pancreatic cells from KC mice for DCLK1. As expected, the isolated DCLK1+ cells expressed not only increased levels of DCLK1 and PKD1 (Figures S5A and S5B) but also increased levels of typical stem cell markers such as OCT4 and CD133 (Figure 6A). Moreover, DCLK1+ cells formed PanIN-like structures when seeded in 3D culture, whereas DCLK1- cells, when seeded at the same number, were incapable of forming larger duct-like structures (Figure 6B). All PanIN-like ductal structures formed by DCLK1+ cells were also positive for PKD1 (Figure S5C).
Figure 6.

DCLK1+/acetylated α-tubulin+ lesion cells express stem cell markers dependent on the presence of PKD1
(A and B) Primary cells from KC mouse pancreas were isolated and sorted for DCLK1. Sorted cells were analyzed via qPCR, where the graphs represent three replicates and the error bars represent the standard deviation. Statistical significance is indicated by ∗; the p value for Oct4 is 0.0005 and the p value for CD133 is 0.0014 (A). Sorted cells (10,000 per condition) were plated on top of Matrigel. Images represent day 5, and the scale bars indicate 200 μm (B).
(C) IHC-IF analysis of OCT4 (green) or CD133 (green) and acetylated α-tubulin (red) in KC mouse lesions. Scale bars indicate 50 μm (top panel, OCT4) and 20 μm (bottom panel, CD133).
(D) Quantification of OCT4 in lesions of 8-week-old KC (n = 4) and KC;PKD1−/− (n = 5) mice. Statistical significance , indicated by ∗, was determined by the t test; p value <0.0001.Error bars represent standard deviation.
(E) Quantification of CD133 in lesions of 8-week-old KC (n = 4) and KC;PKD1−/− (n = 4) mice. Statistical significance, indicated by ∗, was determined by the t test; p value = 0.0073. Error bars represent standard deviation.
We then confirmed that the acetylated α-tubulin+/DCLK1+ pancreatic cancer cells in lesions of KC mice express these stem cell markers in vivo. Both OCT4 and CD133 were expressed in acetylated α-tubulin+ cells (Figure 6C). Moreover, we detected a significantly decreased expression of both OCT4 and CD133 in PanIN lesions from KC;PKD1−/− mice, when compared with PanIN lesions of KC mice (Figures 6D, 6E, S5D, and S5E). Together this indicates that DCLK1+ lesion cells express stem cell markers dependent on the presence of PKD1.
Ectopic expression of active PKD1 in PanIN cells increases the DCLK1+ stem cell population
We next tested if ectopic expression of active PKD1 can increase the occurrence of DCLK1+ stem cells. Flow cytometry analyses of SM3 PanIN cells indicated that all DCLK1+ cells (5.98% for the experiment shown; 8.9% ± 4.3% in n = 5 replicates) were also positive for PKD1 (Figure 7A). This was confirmed by IF-IHC analyses of SM3 PanIN organoids, which also were positive for both markers (Figure 7B).
Figure 7.

Ectopic expression of active PKD1 in PanIN cells increases the DCLK1+ stem cell population
(A) Representative plot of flow cytometry analysis for cells positive for DCLK1 and PKD1 in SM3 PanIN cells (left); quantification of the percentage of SM3 cells that are DCLK1+ and PKD1+ (right). “Other” indicates single positive cells or double-negative cells. Error bars indicate the standard deviation.
(B) Immunofluorescence for DCLK1 (green in overlay) and PKD1 (pink in overlay) in SM3 organoids with DAPI nuclear stain. Scale bars indicate 10 μm.
(C and D) Human constitutively active PKD1 (PKD1.CA) was overexpressed in mouse SM3 PanIN cells and flow cytometry analyses of cells for DCLK1 were performed. C shows representative flow cytometry plots. D shows quantification of flow cytometry analyses, where the graph indicates the fold change in DCLK1+ cells from five independent experiments. Statistical significance, indicated by ∗, was determined by the paired t test; p value = 0.0205. Error bars represent the standard deviation.
(E) Quantitative PCR analysis for huPRKD1, to indicate overexpression of human constitutively active PKD1. Statistical significance is represented by ∗ (t test; p value = 0.0003). Error bars indicate the standard deviation.
(F) Quantitative PCR analysis for expression of indicated genes and Gapdh (normalization) in SM3 cells overexpressing PKD1.CA versus control (n = 3 per condition). Statistical significance, indicated by ∗, was determined by the t test; DCLK1, p value = 0.0043; Pdx1, p value = 0.0003; Notch1, p value = 0.0002; Oct4, p value = 0.0279; Satb2, p value = 0.0040; Hes1, p value = 0.0135; CD133, p value = 0.0015. Error bars indicate the standard deviation.
We then infected SM3 cells with a constitutively active version of human PKD1 (PKD1.S738E.S742E; PKD1.CA) and found that this increased the abundance of DCLK1+ cells within the SM3 cell population (Figures 7C–7E). Analyses of a series of stem cell markers indicated that expression of PKD1.CA increased the expression of DCLK1, Pdx1, Notch1, Oct4, Satb2, Hes1, and CD133 stem cell population (Figure 7F). Together these data indicate that active PKD1 can be a driver of formation of DCLK1+ stem cells in SM3 PanIN organoids, which supports the in vivo data seen in KC mice.
Discussion
Pancreatic ductal adenocarcinoma is notoriously difficult to treat since it often is detected at a metastatic stage. Recent data suggest that cells that form metastases are seeded from low-grade dysplasia (ADM/PanIN) prior to the progression to cancer (Rhim et al., 2012). Expression of DCLK1 marks a subpopulation of PanIN cells that have been discussed as possible tumor and metastases initiating cells (Westphalen et al., 2016). DCLK1 is widely expressed on circulating tumor cells in serum from patients with stages I and II PDA (Qu et al., 2015), and DCLK1 expression was associated with a higher TNM stage and a higher rate of lymph node metastasis (Zhou et al., 2017). Considering evidence that the DCLK1+ cell population plays a direct role in the aggressiveness of PDA, elucidating differences of these cells to other PanIN cells is key to finding targeted therapeutics.
Mouse models of PDA indicate that DCLK1+ cells are most abundant in low-grade dysplasia (Bailey et al., 2014) but are distinct to Tuft cells, which also express DCLK1 but orchestrate inflammatory responses in glandular tissues (Gerbe et al., 2009; Gerbe and Jay, 2016; Howitt et al., 2016). Lineage tracing indicates that DCLK1+ cells develop from PanIN cells by so far unidentified processes (Bailey et al., 2014). Here we show that DCLK1+ cells have dysfunctional EGFR signaling and that this may act as a driver of formation of this cell population. In DCLK1- PanIN cells activated EGFR was detected at the cell membrane (Figure 1A), whereas in DCLK1+ cells the activated receptor is clustered in the same apical region where acetylation of α-tubulin occurs (Figure 1B). Tubulin acetylation has been implicated in the regulation of dynamic transport processes within cells. For example, it promotes secretory vesicle flux and is required for efficient anterograde cargo transport (Dompierre et al., 2007; Reed et al., 2006). However, tubulin acetylation also was linked to premature delivery of EGFR vesicles to the degradative compartment, resulting in accelerated receptor degradation and downregulated signaling (Gao et al., 2010). Specifically, mitogenic signaling through ERK1/2 by the EGFR is attenuated by endocytosis and lysosomal degradation (Sousa et al., 2012). Both disturbance of vesicle trafficking and more rapid lysosomal progression may occur in DCLK1+ cells, with the net effect of blocking efficient EGFR downstream signaling toward ERK1/2 (Figures 1E and 1F).
A downregulation of EGFR signaling as a driver of formation of DCLK1+ PanIN cells is further supported by our data using erlotinib as a chemical inhibitor of EGFR. We indeed found that the DCLK1+ cell population increased in response to erlotinib both in vitro (Figures 2E and 2F) and in vivo (Figures 2A and 2B), thereby providing further evidence that disruption of EGFR signaling is important in generating these PDA stem cells. Although erlotinib is approved for use with gemcitabine in patients with advanced pancreatic cancer owing to the increased 1-year survival rate of 23% (compared with 17% for gemcitabine + placebo) (Moore et al., 2007), patients with primarily resectable PDA gain no benefit from erlotinib (Sinn et al., 2017). Considering that the DCLK1+ population is associated with poorer outcome (Nishio et al., 2017), and disrupting EGFR signaling increases the DCLK1+ population, future studies could stratify patients to determine if erlotinib has different effectiveness in DCLK1-high versus DCLK1-low tumors.
Our data identify an increase in hydrogen peroxide as a potential driver of formation of DCLK1+ PanIN cells (Figures 2G and 3A–3D). This increase may be due to dysfunctional EGFR signaling. It previously has been shown that inhibition of EGFR increases oxidative stress (Orcutt et al., 2011). Similarly, we found that erlotinib increased the formation of hydrogen peroxide in SM3 PanIN cells (Figure 2G). Moreover, increased levels of hydrogen peroxide have been shown to perturb signal transduction via endosomes (Kano et al., 2011), suggesting a potentiating effect on the downregulation of EGFR signaling. As a major target for increased levels of hydrogen peroxide we identified the oxidative stress-responsive kinase PKD1. In mouse pancreas PKD1 is not expressed in normal tissue but upregulated during the ADM process and during the formation of PanIN lesions (Liou et al., 2015) and, as we show here, highly expressed in DCLK1+ cells (Figures 4A–4E). Moreover, comparison of low-grade dysplasia in KC mice to KC mice with a PKD1 knockout indicated that PKD1 may be a driver of occurrence of DCLK1+ cells (Figures 5D and 5E).
We found that the expression of PKD1 in DCLK1+ cells regulates the presence of stem cell markers such as CD133 and Oct4 (Figures 6D, 6E, and 7F). Notch, which regulates CD133 and CD133+ stem cell growth and metastasis (Kumar et al., 2016), previously has been implicated downstream of PKD1 (Liou et al., 2015). In addition, Oct4 together with Klf4 and Sox2 co-occupies the promoters of pluripotency-related genes (Evans et al., 2007; Wei et al., 2013). Thus, an increased expression of PKD1 and upregulation of its activity may be a key regulator of generation of DCLK1+ stem cells. A role for another PKD isoform, PKD3, in tumor stemness recently has been shown for breast cancer (Lieb et al., 2020), further underlining a potential function for this kinase family in regulating tumor cell stemness.
In summary, the DCLK1+ stem cell population plays a major role in PDA and understanding the differences between this cell type and other lesion cells can shed light on potential therapeutic approaches. With our findings that EGFR signaling is dysfunctional and not propagated to the nucleus and that oxidative stress/PKD1 signaling is increased, we identified key signaling events driving the formation of DCLK1+ lesion cells. In future studies this knowledge can be exploited to target this pancreatic cancer stem cell population and to develop potential therapeutic approaches.
Limitations of the study
Although our study shows that disruption of EGFR signaling, increased oxidative stress, and PKD1 contribute to the occurrence of DCLK1+ cells in PanIN lesions, the study was often limited to murine tissue and primary cell culture. With the exception of Figure 4B, in which DCLK1 and PKD1 were shown to be correlated in PDA patient tissue, experiments were conducted in a precancerous murine model harboring a KrasG12D mutation. In addition, although this signaling is important in precancerous lesions, further studies are needed to understand the relevance to therapeutic approaches in patients. For example, erlotinib treatment in patients provided a modest increase in survival (Moore et al., 2007). In light of our study, combining erlotinib with DCLK1 inhibition may be efficacious. However, to address this, further studies are needed utilizing tumor models.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Storz (Storz.Peter@mayo.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate unique datasets or code.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
The authors would like to thank Dr. Nabeel Bardeesy (Massachusetts General Hospital) for providing SM3 cells. We would like to acknowledge the support of Dr. Laura Lewis-Tuffin and the Cell and Tissue Analysis Shared Resource facility at Mayo Clinic in Florida for assistance with FACS analyses and our colleagues in the Storz laboratory for helpful discussions. We also thank the Champions for Hope (Funk-Zitiello Foundation) and Gary Chartrand Foundation for their continuous support. This work was supported by NIH grant CA200572 and the Mayo Clinic Cancer Center GI Cancer program (to P.S. and T.P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
Conceived and designed the experiments: A.K.F.M., G.-Y.L., P.S. Performed the experiments: A.K.F.M., H.R.D., L.I.B., G.-Y.L., B.E. Analyzed the data: A.K.F.M., G.-Y.L., H.R.D., L.I.B., P.S. Contributed reagents and tools: M.L., T.P. Wrote the paper: A.K.F.M., P.S.
Declaration of interests
The authors declare no competing interests.
Published: January 22, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.102019.
Supplemental information
References
- Agbunag C., Lee K.E., Buontempo S., Bar-Sagi D. Pancreatic duct epithelial cell isolation and cultivation in two-dimensional and three-dimensional culture systems. Methods Enzymol. 2006;407:703–710. doi: 10.1016/S0076-6879(05)07055-2. [DOI] [PubMed] [Google Scholar]
- Bailey J.M., Alsina J., Rasheed Z.A., McAllister F.M., Fu Y.Y., Plentz R., Zhang H., Pasricha P.J., Bardeesy N., Matsui W. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology. 2014;146:245–256. doi: 10.1053/j.gastro.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P., Chang D.K., Nones K., Johns A.L., Patch A.M., Gingras M.C., Miller D.K., Christ A.N., Bruxner T.J., Quinn M.C. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- Brankatschk B., Wichert S.P., Johnson S.D., Schaad O., Rossner M.J., Gruenberg J. Regulation of the EGF transcriptional response by endocytic sorting. Sci. Signal. 2012;5:ra21. doi: 10.1126/scisignal.2002351. [DOI] [PubMed] [Google Scholar]
- Delgiorno K.E., Hall J.C., Takeuchi K.K., Pan F.C., Halbrook C.J., Washington M.K., Olive K.P., Spence J.R., Sipos B., Wright C.V. Identification and manipulation of biliary metaplasia in pancreatic tumors. Gastroenterology. 2014;146:233–244.e5. doi: 10.1053/j.gastro.2013.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompierre J.P., Godin J.D., Charrin B.C., Cordelieres F.P., King S.J., Humbert S., Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler H., Storz P. A novel tyrosine phosphorylation site in protein kinase D contributes to oxidative stress-mediated activation. J. Biol. Chem. 2007;282:31873–31881. doi: 10.1074/jbc.M703584200. [DOI] [PubMed] [Google Scholar]
- Doppler H., Storz P. Mitochondrial and oxidative stress-mediated activation of protein kinase D1 and its importance in pancreatic cancer. Front. Oncol. 2017;7:41. doi: 10.3389/fonc.2017.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P.M., Zhang W., Chen X., Yang J., Bhakat K.K., Liu C. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J. Biol. Chem. 2007;282:33994–34002. doi: 10.1074/jbc.M701847200. [DOI] [PubMed] [Google Scholar]
- Gao Y.S., Hubbert C.C., Yao T.P. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem. 2010;285:11219–11226. doi: 10.1074/jbc.M109.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbe F., Brulin B., Makrini L., Legraverend C., Jay P. DCAMKL-1 expression identifies Tuft cells rather than stem cells in the adult mouse intestinal epithelium. Gastroenterology. 2009;137:2179–2180. doi: 10.1053/j.gastro.2009.06.072. author reply 2180-2181. [DOI] [PubMed] [Google Scholar]
- Gerbe F., Jay P. Intestinal tuft cells: epithelial sentinels linking luminal cues to the immune system. Mucosal Immunol. 2016;9:1353–1359. doi: 10.1038/mi.2016.68. [DOI] [PubMed] [Google Scholar]
- Gzil A., Zarebska I., Bursiewicz W., Antosik P., Grzanka D., Szylberg L. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol. Biol. Rep. 2019;46:6629–6645. doi: 10.1007/s11033-019-05058-1. [DOI] [PubMed] [Google Scholar]
- Harvey K.F., Zhang X., Thomas D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- Hermann P.C., Huber S.L., Herrler T., Aicher A., Ellwart J.W., Guba M., Bruns C.J., Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Hong S.P., Wen J., Bang S., Park S., Song S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer. 2009;125:2323–2331. doi: 10.1002/ijc.24573. [DOI] [PubMed] [Google Scholar]
- Howitt M.R., Lavoie S., Michaud M., Blum A.M., Tran S.V., Weinstock J.V., Gallini C.A., Redding K., Margolskee R.F., Osborne L.C. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science. 2016;351:1329–1333. doi: 10.1126/science.aaf1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano F., Arai T., Matsuto M., Hayashi H., Sato M., Murata M. Hydrogen peroxide depletes phosphatidylinositol-3-phosphate from endosomes in a p38 MAPK-dependent manner and perturbs endocytosis. Biochim. Biophys. Acta. 2011;1813:784–801. doi: 10.1016/j.bbamcr.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Kumar D., Kumar S., Gorain M., Tomar D., Patil H.S., Radharani N.N.V., Kumar T.V.S., Patil T.V., Thulasiram H.V., Kundu G.C. Notch1-MAPK signaling axis regulates CD133(+) cancer stem cell-mediated melanoma growth and angiogenesis. J. Invest. Dermatol. 2016;136:2462–2474. doi: 10.1016/j.jid.2016.07.024. [DOI] [PubMed] [Google Scholar]
- Li C., Heidt D.G., Dalerba P., Burant C.F., Zhang L., Adsay V., Wicha M., Clarke M.F., Simeone D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- Lieb W.S., Lungu C., Tamas R., Berreth H., Rathert P., Storz P., Olayioye M.A., Hausser A. The GEF-H1/PKD3 signaling pathway promotes the maintenance of triple-negative breast cancer stem cells. Int. J. Cancer. 2020;146:3423–3434. doi: 10.1002/ijc.32798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P.T., Gleeson J.G., Corbo J.C., Flanagan L., Walsh C.A. DCAMKL1 encodes a protein kinase with homology to doublecortin that regulates microtubule polymerization. J. Neurosci. 2000;20:9152–9161. doi: 10.1523/JNEUROSCI.20-24-09152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou G.Y., Bastea L., Fleming A., Doppler H., Edenfield B.H., Dawson D.W., Zhang L., Bardeesy N., Storz P. The presence of interleukin-13 at pancreatic ADM/PanIN lesions alters macrophage populations and mediates pancreatic tumorigenesis. Cell Rep. 2017;19:1322–1333. doi: 10.1016/j.celrep.2017.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou G.Y., Doppler H., Braun U.B., Panayiotou R., Scotti Buzhardt M., Radisky D.C., Crawford H.C., Fields A.P., Murray N.R., Wang Q.J. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 2015;6:6200. doi: 10.1038/ncomms7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou G.Y., Doppler H., DelGiorno K.E., Zhang L., Leitges M., Crawford H.C., Murphy M.P., Storz P. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016;14:2325–2336. doi: 10.1016/j.celrep.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Fan L.X., Zhou X., Sweeney W.E., Jr., Avner E.D., Li X. HDAC6 regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation in renal epithelial cells. PLoS One. 2012;7:e49418. doi: 10.1371/journal.pone.0049418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.J., Goldstein D., Hamm J., Figer A., Hecht J.R., Gallinger S., Au H.J., Murawa P., Walde D., Wolff R.A. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- Nishio K., Kimura K., Amano R., Nakata B., Yamazoe S., Ohira G., Miura K., Kametani N., Tanaka H., Muguruma K. Doublecortin and CaM kinase-like-1 as an independent prognostic factor in patients with resected pancreatic carcinoma. World J. Gastroenterol. 2017;23:5764–5772. doi: 10.3748/wjg.v23.i31.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orcutt K.P., Parsons A.D., Sibenaller Z.A., Scarbrough P.M., Zhu Y., Sobhakumari A., Wilke W.W., Kalen A.L., Goswami P., Miller F.J., Jr. Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res. 2011;71:3932–3940. doi: 10.1158/0008-5472.CAN-10-3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D., Johnson J., Chandrakesan P., Weygant N., May R., Aiello N., Rhim A., Zhao L., Zheng W., Lightfoot S. Doublecortin-like kinase 1 is elevated serologically in pancreatic ductal adenocarcinoma and widely expressed on circulating tumor cells. PLoS One. 2015;10:e0118933. doi: 10.1371/journal.pone.0118933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed N.A., Cai D., Blasius T.L., Jih G.T., Meyhofer E., Gaertig J., Verhey K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Rhim A.D., Mirek E.T., Aiello N.M., Maitra A., Bailey J.M., McAllister F., Reichert M., Beatty G.L., Rustgi A.K., Vonderheide R.H. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Sinn M., Bahra M., Liersch T., Gellert K., Messmann H., Bechstein W., Waldschmidt D., Jacobasch L., Wilhelm M., Rau B.M. CONKO-005: adjuvant chemotherapy with gemcitabine plus erlotinib versus gemcitabine alone in patients after R0 resection of pancreatic cancer: a multicenter randomized phase III trial. J. Clin. Oncol. 2017;35:3330–3337. doi: 10.1200/JCO.2017.72.6463. [DOI] [PubMed] [Google Scholar]
- Sobhakumari A., Schickling B.M., Love-Homan L., Raeburn A., Fletcher E.V., Case A.J., Domann F.E., Miller F.J., Jr., Simons A.L. NOX4 mediates cytoprotective autophagy induced by the EGFR inhibitor erlotinib in head and neck cancer cells. Toxicol. Appl. Pharmacol. 2013;272:736–745. doi: 10.1016/j.taap.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa L.P., Lax I., Shen H., Ferguson S.M., De Camilli P., Schlessinger J. Suppression of EGFR endocytosis by dynamin depletion reveals that EGFR signaling occurs primarily at the plasma membrane. Proc. Natl. Acad. Sci. U S A. 2012;109:4419–4424. doi: 10.1073/pnas.1200164109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureban S.M., May R., Lightfoot S.A., Hoskins A.B., Lerner M., Brackett D.J., Postier R.G., Ramanujam R., Mohammed A., Rao C.V. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011;71:2328–2338. doi: 10.1158/0008-5472.CAN-10-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira A.V., Lamaze C., Schmid S.L. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- Vinogradov S., Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond.) 2012;7:597–615. doi: 10.2217/nnm.12.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z., Gao F., Kim S., Yang H., Lyu J., An W., Wang K., Lu W. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell. 2013;13:36–47. doi: 10.1016/j.stem.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Westphalen C.B., Takemoto Y., Tanaka T., Macchini M., Jiang Z., Renz B.W., Chen X., Ormanns S., Nagar K., Tailor Y. Dclk1 defines quiescent pancreatic progenitors that promote injury-induced regeneration and tumorigenesis. Cell Stem Cell. 2016;18:441–455. doi: 10.1016/j.stem.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Li J., Zhou Y., Yao L., Sun R., Xu Y., Ge Y., An G. Inhibition of DCLK1 down-regulates PD-L1 expression through Hippo pathway in human pancreatic cancer. Life Sci. 2020;241:117150. doi: 10.1016/j.lfs.2019.117150. [DOI] [PubMed] [Google Scholar]
- Zhou B., Sun C., Hu X., Zhan H., Zou H., Feng Y., Qiu F., Zhang S., Wu L., Zhang B. MicroRNA-195 suppresses the progression of pancreatic cancer by targeting DCLK1. Cell. Physiol. Biochem. 2017;44:1867–1881. doi: 10.1159/000485876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate unique datasets or code.