

Abstract
Primary biliary cholangitis (PBC) causes chronic and persistent cholestasis in the liver, eventually resulting in cirrhosis and hepatic failure without appropriate treatment. PBC mainly develops in middle-aged women, but it is also common in young women and men. PBC is considered a model of autoimmune disease because of the presence of disease-specific autoantibodies, that is, antimitochondrial antibodies (AMAs), intense infiltration of mononuclear cells into the bile ducts, and a high prevalence of autoimmune diseases such as comorbidities. Histologically, PBC is characterized by degeneration and necrosis of intrahepatic biliary epithelial cells surrounded by a dense infiltration of mononuclear cells, coined as chronic non-suppurative destructive cholangitis, which leads to destructive changes and the disappearance of small- or medium-sized bile ducts. Since 1990, early diagnosis with the detection of AMAs and introduction of ursodeoxycholic acid as first-line treatment has greatly altered the clinical course of PBC, and liver transplantation-free survival of patients with PBC is now comparable to that of the general population.
Keywords: Anti-mitochondrial antibody, Epidemiology, Bezafibrate
INTRODUCTION
Primary biliary cholangitis (PBC), formally named primary biliary cirrhosis until 2016, is a chronic cholestatic liver disease. The first patient presenting with symptoms resembling PBC was described in the literature in 1851 [1]. When the term “primary biliary cirrhosis” initially appeared in the title of an article published in 1949 by Dauphinee and Sinclair [2], most early descriptions of PBC involved patients at the cirrhotic stage, with jaundice, ascites, and variceal bleeding; therefore, the nomenclature “primary biliary cirrhosis” was correct at that time. However, the use of biochemical and immunological tests in clinical settings has enabled the diagnosis of PBC at earlier stages. Furthermore, the establishment of ursodeoxycholic acid (UDCA) as a first-line treatment drug remarkably reduced disease progression to cirrhosis. In fact, while half of patients diagnosed as PBC had one or more symptoms at diagnosis in the 1980’s, 75–80% of patients were asymptomatic without any complaint of symptom at diagnosis after 2000 in Japan. The serious gap between the disease manifestation and its misnomer became wider, and the term “cirrhosis” became not merely an inaccuracy but an active stigma for patients, motivated by the change of nomenclature of the disease, from cirrhosis to cholangitis [3-10]. The Asian Pacific Association for the Study of Liver (APASL) also officially approved this decision, and the new nomenclature “primary biliary cholangitis” is currently used in the official journal of the APASL [11].
EPIDEMIOLOGY
The prevalence and incidence of PBC vary considerably worldwide (Fig. 1). This discrepancy can be attributed to the true epidemiological difference between regions or study periods, to the variation in study designs for case finding or ascertainment, or difference in the diagnosis of PBC among physicians. Notably, awareness of PBC may still not be satisfactory in some Asian and African countries where epidemiological studies are scarce, and the sample size in some studies is very small. PBC is believed to be a rare disease in the Asia-Pacific region, and both the prevalence and incidence seemed to be lower in the Asia-Pacific region, as indicated by recent epidemiological studies in South Korea or Hong Kong (Fig. 1) [12,13]. In contrast, a 2016 study in Japan reported that the point prevalence of PBC was 33.8 per 100,000 in the Japanese population [14], which was comparable to that in European countries, the United States, and Canada. A recent review from South Korea defined a “rare disease” by either the prevalence (1,500–2,500 per 100,000 population) or patient number (<20,000) [15]. According to this definition, PBC is a rare disease, but interestingly, there is a substantial difference in the prevalence between Japan and South Korea or Hong Kong.
Figure 1.
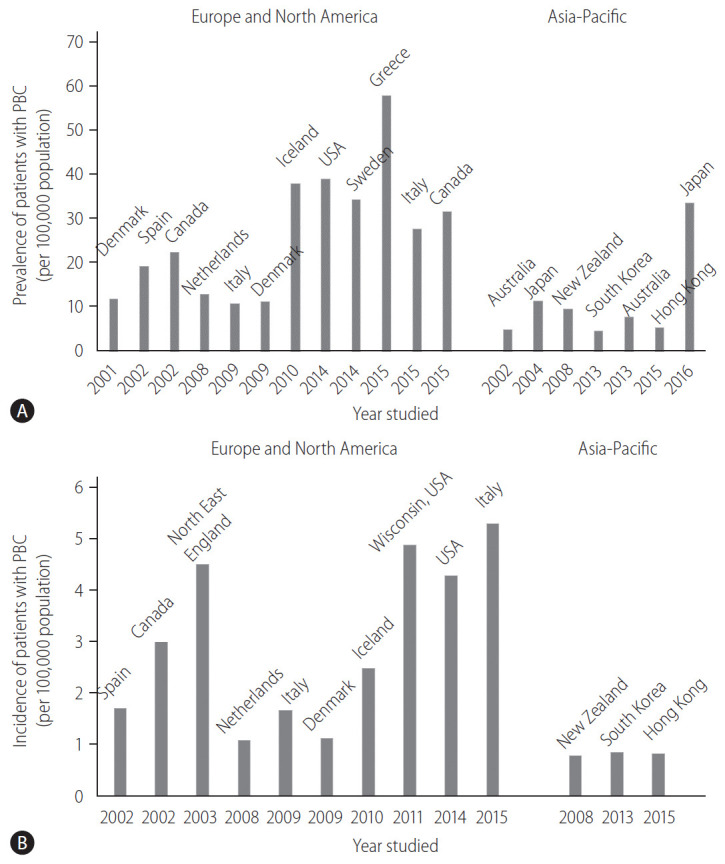
Epidemiological data of PBC over time and in different geographical regions. (A) The prevalence (per 100,000 population) and (B) incidence (per 100,000 population) of PBC. PBC, primary biliary cholangitis.
Other studies also indicated an increasing trend in the prevalence of PBC over time (Fig. 1A) [16]. Longitudinal studies in identical regions consistently showed an increase in the prevalence of PBC [14,17,18]. Since sequential studies demonstrated an increasing incidence of PBC (1.67 in 2009 and 5.31 in 2015, in Italy [19,20]) or a relatively stable incidence (2.6 in Sweden [18]), it is unclear whether a true increase or improved overall survival has contributed to the increasing prevalence of PBC.
One of the signatures of PBC is its female preponderance. Retrospective analysis indicated that the female:male ratio was 9:1 [21,22] in the 1990s and early 2000s, and this overt female predisposition has provided researchers a clue in clarifying the etiology of PBC. Notably, female predominance is still clearly observed today; however, it is less pronounced; the female:male ratio was less than 5:1 in most recent epidemiological studies and even 2.1:1 [19]. The reason for the relative increase in the number of male patients with PBC remains unclear, but a better recognition and a true increase in the incidence of PBC are likely responsible.
ETIOLOGY
Tolerance breakdown against autoantigens
PBC is a multifactorial and enigmatic disease; it remains unknown how and why PBC develops. Autoimmune attack targeted at biliary epithelial cells (BECs) through tolerance breakdown triggers disease onset.
The hallmark of PBC is anti-mitochondrial antibody (AMA), which are detected in 90–95% of patients with PBC [23,24], and the most disease-specific autoantibodies in human immunopathology. The high specificity of AMAs for PBC suggests that AMAs are not simple serological markers for diagnosis but are important in the immunopathology of PBC. AMAs recognize a family of enzymes located in the inner membrane of the mitochondria, named the 2-oxo-acid dehydrogenase complex (2-OADC), which mainly includes the pyruvate dehydrogenase complex E2 subunit (PDC-E2), the branched-chain 2-OADC E2 subunit (BCOADC-E2), the 2-oxoglutaric acid dehydrogenase complex E2 subunit (OGDC-E2), and dihydrolipoamide dehydrogenase-binding protein (E3BP) [25]. AMAs and autoreactive CD4+ and CD8+ T cell epitopes are confined within a shared peptide sequence of the inner lipoyl domain of human PDC-E2 [26].
BECs and hepatocytes of patients with PBC express large amounts of human leukocyte antigen (HLA) classes I and II molecules [27,28]. In patients with PBC, BECs acts as nonprofessional antigen-presenting cells, and the interplay of BECs and T cells may, to some extent, account for bile duct loss. Indeed, BECs expresses adhesion molecules, cytokines, and chemokines, and recruit mononuclear cells in the biliary tract of the liver. One example is fractalkine (CX3CL1), a chemokine with both chemoattractant and cell-adhesive functions [29]. T helper type 1-cytokine predominance and lipopolysaccharide in the microenvironment of injured bile ducts induce the upregulation of fractalkine expression in BECs, followed by the chemoattraction of mononuclear cells expressing its receptor (CX3CR1), including CD4+ and CD8+ T cells [30,31]. Serum fractalkine levels in PBC are high in patients with marked cholangitis activity (CA) at early stages, and they decrease in response to treatment [32].
PDC-E2 is a ubiquitous protein located in nearly all nucleated cells in the human body, and it remains unclear why autoreactive T cells specific for PDC-E2 elicit cytotoxicity only against BECs in the liver. In this regard, it should be noted that PBC recurs even after liver transplantation (LT), indicating that the immunopathological susceptibility of BECs in PBC is not major histocompatibility complex (MHC)-specific but a general feature shared with autologous BECs. The hypothesis to solve this enigma is that human intrahepatic BECs could maintain PDC-E2 immunologically intact within apoptotic blebs (apotopes) during apoptosis [33]. Interestingly, a unique triad consisting of BEC apotopes, macrophages from patients with PBC, and AMAs could lead to rigorous production of inflammatory cytokine production [34].
Genetic predisposition
Accumulating evidence also suggests that a combination of genetic predisposition and environmental triggering factors plays a crucial role in tolerance breakdown.
Genetic predisposition is believed to be a major contributing factor in the development of PBC [35].
A recent study in Iceland, which took advantage of the unique local genealogical database, demonstrated that the familial risk of PBC was present not only in first-degree relatives but also in second- and third-degree relatives of patients with PBC, with increased relative risk ratios (RRs) of 9.13 (95% confidence interval [CI], 4.17–16.76), 3.61 (1.48–8.92), and 2.59 (1.35–4.67), respectively [36]. Furthermore, the increased risk of PBC tended toward significance even in fourth- and fifth-degree relatives with RRs of 1.66 (1.00–3.02) and 1.42 (0.99–2.20), respectively. These findings clearly emphasize the importance of genetic risk in the pathogenesis of PBC.
Recent genome-wide association study (GWAS) analyses from North America, European countries, Japan, and China identified other HLA alleles that are strongly associated with susceptibility to PBC and revealed more than 40 non-HLA alleles contributing to PBC susceptibility (Table 1) [37-48]. Although risk alleles differ among studies and populations, they primarily belong to genes and pathways involved in antigen presentation and production of interleukin (IL)-12 (IRF5, SOCS1, TNFAIP3, NF-B, and IL-12A), activation of T cells, and interferon (IFN)-γ production (TNFSF15, IL12R, TYK2, STAT4, SOCS1, NF-κB, and TNFAIP3) as well as activation of B cells and production of immunoglobulins (POU2AF1, SPIB, PRKCB, IKZF3, and ARID3A). These immune pathways could be important in the pathogenesis of PBC.
Table 1.
Major gene loci associated with susceptibility of PBC, and other autoimmune diseases*
| Chromosome No. | Gene loci | PBC (Europe/North America) | PBC (Japan/China) | RA | IBD | MS | SLE |
|---|---|---|---|---|---|---|---|
| 1 | CD58 | Yes | ✓ | ✓ | |||
| 1 | MMEL1, TNFRSF14 | Yes | ✓ | ✓ | ✓ | ||
| 1 | IL12RB2 | Yes | |||||
| 1 | DENND1B | Yes | ✓ | ||||
| 2 | IL1RL2/IL1RL1 | Yes | ✓ | ||||
| 2 | STAT4 | Yes | Yes | ✓ | ✓ | ||
| 2 | CD28/CTLA4/ICOS | Yes | ✓ | ✓ | |||
| 2 | CCL20 (LARC) | Yes | ✓ | ||||
| 2 | BCL2L11 | ||||||
| 2 | GPR35 | ✓ | |||||
| 3 | PLCL2 | Yes | ✓ | ||||
| 3 | CD80 | Yes | Yes | ✓ | |||
| 3 | IL12A, SCHIP1 | Yes | Yes | ✓ | |||
| 3 | FOXP1 | ||||||
| 3 | MST1 | ✓ | |||||
| 4 | DGK Q | Yes | ✓ | ✓ | |||
| 4 | NF-kB1 | Yes | Yes | ✓ | |||
| 4 | IL21 | Yes | ✓ | ||||
| 5 | IL7R | Yes | Yes | ✓ | ✓ | ||
| 5 | PAM/C5orf30 | Yes | ✓ | ||||
| 5 | LOC285626/IL12B | Yes | ✓ | ✓ | |||
| 6 | TNFAIP3 | Yes | ✓ | ✓ | ✓ | ✓ | |
| 6 | BACH2 | ✓ | |||||
| 7 | ELMO1 | Yes | ✓ | ✓ | ✓ | ||
| 7 | IRF5 | Yes | ✓ | ✓ | ✓ | ||
| 9 | TNFSF15 | Yes | ✓ | ||||
| 10 | IL2RA | ✓ | ✓ | ✓ | |||
| 11 | RPS6KA4 | Yes | ✓ | ✓ | |||
| 11 | CXCR5 | Yes | Yes | ✓ | ✓ | ||
| 11 | POU2AF1 | Yes | |||||
| 11 | CCDC88B | Yes | ✓ | ||||
| 11 | SIK2 | ||||||
| 12 | TNFRSF1A | Yes | Yes | ✓ | |||
| 12 | SH2B3 | Yes | ✓ | ||||
| 12 | HDAC7 | ✓ | |||||
| 12 | RFX4, RIC8B | ||||||
| 13 | TNFSF11 (RANKL) | Yes | ✓ | ||||
| 14 | RAD51L1 | Yes | |||||
| 14 | TNFAIP2 | Yes | |||||
| 15 | IL16 | Yes | |||||
| 16 | IL21R | Yes | |||||
| 16 | PRKCB | Yes | ✓ | ||||
| 16 | CLEC16A, SOCS1 | Yes | ✓ | ✓ | ✓ | ||
| 16 | CSNK2A2, CCDC113 | Yes | |||||
| 16 | IRF8 | Yes | ✓ | ✓ | ✓ | ||
| 17 | IKZF3-ORMDL3 | Yes | Yes | ✓ | ✓ | ||
| 17 | MAPT, CRHR1 | Yes | |||||
| 18 | TYK2 | Yes | ✓ | ✓ | ✓ | ||
| 18 | ARID3A | Yes | |||||
| 18 | SPIB | Yes | |||||
| 18 | TCF4 | ||||||
| 18 | CD226 | ✓ | ✓ | ||||
| 19 | PRKD2, STRN4 | ||||||
| 21 | PSMG1 | ✓ | |||||
| 21 | UBASH3A | ✓ | |||||
| 22 | MAP3K7IP1/RPl3, SYNGR1 | Yes | Yes | ✓ |
PBC, primary biliary cholangitis; RA, rheumatoid arthritis; IBD, inflammatory bowel disease; MS, multiple sclerosis; SLE, systemic lupus erythematosus; IL, interleukin; RANKL, receptor activator of nuclear factor-kappaB ligand.
Environmental triggers
As environmental triggers, large-scale case-control studies have consistently found an association between urinary tract infections and cigarette smoking with PBC [49-52]. Bacterial infection may have an impact on the etiology of PBC because PDC-E2 has a molecular mimic between human PDC-E2 and Escherichia coli (E. coli) PDC-E2, and thus, E. coli infection may trigger the breaking of immunological tolerance against human PDC-E2. Case-control studies also illustrate that xenobiotic modification of PDC-E2 with chemicals abundantly found in daily life, such as lipsticks, hair dyes, and nail polish, has a role in generating immunogenic neoantigens and breaking tolerance in PBC [51,53-57]. Finally, dysbiosis of the gut microbiota was found in patients with PBC, and interestingly, it was partially resolved with UDCA treatment [58].
Microbiota and bile acids
Early environmental exposures can influence microbiome development. Evidence for the role of the microbiome in the etiology of PBC includes the effect of diet and the higher prevalence of PBC in Western nations in the Northern hemisphere [59]. Diet is the primary external factor that can affect the microbiome, and changes in the microbiome can be affected by bile acids [60]. It is known that not all commensal bacteria are equally susceptible to bile acids [61], with E. coli and Helicobacter spp. being particularly resistant. Bile acids act as signaling molecules involved in glucose-lipid metabolism. The main bile acids in humans include cholic and chenodoxycholic acids and are synthesized in the liver [62]. Bile acids have several other effects that maintain healthy liver tissue, including the regulation of glucose tolerance and insulin sensitivity. Bile acids are also known to induce sterol regulatory element-binding protein-1c production, which regulates the biosynthesis of cholesterol, and also regulates the expression of lipogenesis genes such as glycerol-3-phosphate acyltransferase, acetyl-CoA carboxylase, and fatty acid synthase, thus offering protection against hyperlipidemia [63]. Besides their effects on liver homeostasis, bile acids may alter the microbiome and hepatic regeneration, and thereby play a role in the pathogenesis of PBC [64]. The chronic cholestasis seen in PBC that results from the microbiota affected by bile acids in turn can lead to aberrant expression of bile acid transporters and nuclear receptors, which can in turn cause liver damage, leading to a vicious cycle for chronic liver damage with bile acids as the pivotal determinant [65]. It is clear that the role of bile acids and bile salts in the pathogenesis of PBC is extremely complicated, and is influenced by a number of interacting mechanisms.
Studies on changing the microbiome to be more consistent with healthy controls can be achieved through the use of UDCA treatment, but the effects of generating a healthier “microbiome” on the development of autoimmune liver disease are still unclear. It is known that a healthy microbiome tends to be beneficial for products such as single chain fatty acids [66] and less proinflammatory cytokines such as TNFα and IFNγ [67].
DIAGNOSIS
A diagnosis of PBC is made when 2 or 3 following items are met: the 1) consistent elevation of cholestatic enzymes, 2) detection of AMA, and 3) typical liver histology [58,68,69]. Figure 2 shows a diagnostic flowchart. Because of the very high sensitivity and specificity of AMA in the diagnosis of PBC, detectable AMA and elevation of alkaline phosphatase (ALP) levels are adequate for the diagnosis of PBC, and liver biopsy is not mandatory in many cases. Nevertheless, since histopathological stage at baseline is an independent prognostic marker of survival, as demonstrated by a recent large-scale retrospective study [70], liver biopsy is recommended for predicting long-term outcome but is not necessary in making a diagnosis. A histological examination is required in atypical cases, including suspicious AMA-negative PBC and autoimmune hepatitis/PBC overlap.
Figure 2.
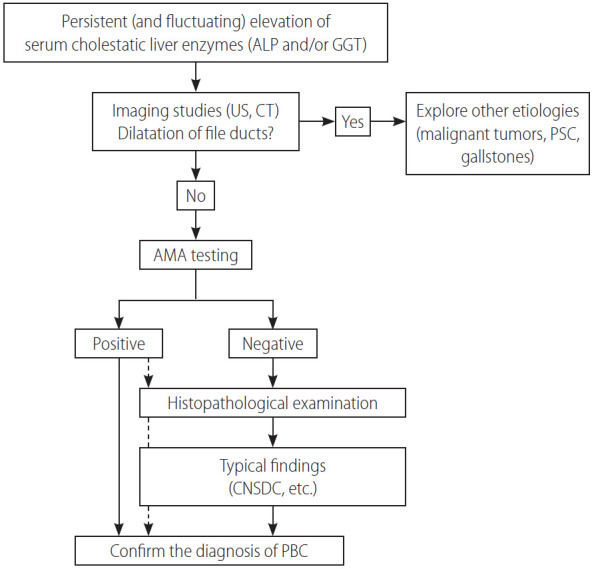
A diagnostic flowchart of patients with PBC. ALP, alkaline phosphatase; GGT, gamma glutamyl transferase; US, ultrasonography; CT, computed tomography; PSC, primary sclerosing cholangitis; AMA, anti-mitochondrial antibody; CNSDC, chronic non-suppurative destructive cholangitis; PBC, primary biliary cholangitis.
In clinical settings, AMA is often determined by enzyme-linked immunosorbent assay (ELISA) or immunofluorescence. Traditionally, at least nine AMA subtypes have been identified, and four of them (M2, M4, M8, and M9) seemed to be associated with PBC [71]. Since anti-M2 antibody was directed to 2-OADC and the most specific antibody to PBC, the M2 subtype of AMA (AMA-M2) is currently used as AMA in daily clinical practice. In ELISA, a combination of three recombinant mitochondrial proteins (PDC-E2, BCOADC-E2, and OGDC-E2) was used as the antigen. The titer of AMAs is not associated with disease progression or the patient’s clinical course. AMAs are occasionally detected in less than 1% of healthy individuals with normal liver test results [72,73]. Individuals who are AMA-positive are at higher risk of developing PBC and require close follow-up, although the risk does not appear to be high, as previously believed. A large-scale cohort study in France demonstrated that the prevalence of AMA-positive patients without evidence of PBC was 16.1 per 100,000 population, and one in six patients with AMA positivity and a normal ALP level developed PBC within 5 years [74]. Conversely, a recent study from China demonstrated that more than 80% of patients with AMA without elevation of serum ALP levels also developed histological characteristics of PBC, suggesting the presence of undiagnosed PBC patients among those with normal ALP levels and AMA positivity [75]. It remains unclear whether these individuals will progress to advanced disease, as in typical PBC, and how they should be clinically treated.
Among several anti-nuclear antibodies, sp100 and gp210 are frequently found in the sera of patients with PBC, and, thus, aid in diagnosing patients with probable PBC but undetectable AMA positivity. A combination of three mitochondrial antigens, sp100, and gp210, (“PBC screen”) had a sensitivity of 83.8% and specificity of 94.7% for diagnosing PBC and was considered appropriate as the first-line screening test [76]. Molecular mimicry between mitochondrial antigens and sp100/gp210 was reported [77]. The detection of gp210 may be associated with progression of the disease in UDCA-treated patients [78], but this observation requires further validation.
Histopathologically, the pathology of PBC is exclusively located in the intrahepatic small- or middle-sized bile ducts. A dense infiltration of mononuclear cells around the intrahepatic small bile ducts (interlobular bile ducts), coined as chronic non-suppurative destructive cholangitis (CNSDC), and granuloma formation are characteristic findings. Eventually, intrahepatic small bile ducts disappear from the liver, and chronic cholestatic features gradually develop. Hepatitis activity (HA) and chronic CA contribute to progressive hepatocellular damage and fibrosis, resulting in liver cirrhosis and hepatic failure.
The Scheuer’s [79] or Ludwig et al.’s [80] classifications has been used for a long time as the classification system in histological staging of PBC pathology. In Scheuer’s classification, florid duct lesions (CNSDC), ductular proliferation, scarring, and nodular cirrhosis are representative findings of stages 1, 2, 3, and 4, respectively (Fig. 3). However, as described in the original report by Scheuer, there is considerable overlap of findings between these stages; CNSDC can be observed even in the liver with nodular cirrhosis. Additionally, the pathology of PBC is not always distributed evenly in the liver; hence, sampling error can occur when determining the stages with these systems.
Figure 3.

Histopathological findings in PBC characterizing stage 1, 2, 3, and 4 of the Scheruer’s classification. (A) Chronic non-suppurative destructive cholangitis (arrow, hematoxylin, and eosin staining). Black bar indicates 400 mm. (B) Ductular proliferation (hematoxylin and eosin staining). Black bar, 400 mm. (C) Scarring (silver impregnation staining). Black bar, 100 mm. (D) Nodular cirrhosis (hematoxylin and eosin staining). Black bar, 200 mm. All these figures were kindly provided by Professor Kenichi Harada (Kanazawa University, Kanazawa, Japan).
In order to overcome these limitations, Nakanuma et al. proposed a new histological staging and grading system for PBC (Tables 2-4) [81]. In Nakanuma’s classification, the scores for fibrosis, bile duct loss, and deposition of orcein-positive granules were used for staging, whereas CA and HA were used for grading. CA is determined by the presence of chronic cholangitis or CNSDC, and HA is defined by the presence of interface hepatitis or lobular hepatitis. Overall survival was stratified better with Nakanuma’s classification than with the classic system [82].
Table 2.
Nakanuma’s classification: staging of PBC [81]
| Stage 1 (no progression): score 0* |
| Stage 2 (mild progression): score 1–3 |
| Stage 3 (moderate progression): score 4–6 |
| Stage 4 (advanced progression): score 7–9 |
PBC, primary biliary cholangitis.
The score for staging is the sum of the scores for fibrosis, bile duct loss, and deposition of orcein-positive granules, as shown below.
Table 3.
Nakanuma’s classification: scoring of PBC [81]
| Fibrosis | Bile duct loss | Deposition of orcein-positive granules | |
|---|---|---|---|
| Score 0 | No or limited portal fibrosis | No | No deposition |
| Score 1 | Portal fibrosis | Yes, in <1/3 of the portal tracts | Deposition in several periportal hepatocytes in <1/3 of the portal tracts |
| Score 2 | Bridging fibrosis | Yes, in 1/3–2/3 of the portal tracts | Deposition in variable periportal hepatocytes in 1/3–2/3 of the portal tracts |
| Score 3 | Cirrhosis | Yes, in >2/3 of the portal tracts | Deposition in many periportal hepatocytes in >2/3 of the portal tracts |
Table 4.
Nakanuma’s classification: grading of necroinflammatory activities of PBC [81]
| Cholangitis activity | |
|---|---|
| CA0 (no activity) | No cholangitis but mild duct epithelial damage may be present |
| CA1 (mild activity) | 1 bile duct with evident chronic cholangitis |
| CA2 (moderate activity) | ≥2 bile ducts with evident chronic cholangitis |
| CA3 (marked activity) | ≥1 bile duct with CNSDC |
| Hepatitis activity | |
| HA0 (no activity) | No interface hepatitis and no or minimal lobular hepatitis |
| HA1 (mild activity) | Interface hepatitis affecting ≥10 continuous hepatocytes in 1 portal tract or fibrous septum, and mild-moderate lobular hepatitis |
| HA2 (moderate activity) | Interface hepatitis affecting ≥10 continuous hepatocytes in ≥2 portal tracts or fibrous septa, and mild-moderate lobular hepatitis |
| HA3 (marked activity) | Interface hepatitis affecting ≥20 continuous hepatocytes in ≥1/2 of the portal tracts, and moderate lobular hepatitis or bridging or zonal necrosis |
PBC, primary biliary cholangitis; CA, cholangitis activity; CNSDC, chronic non-suppurative destructive cholangitis; HA, hepatitis activity.
TREATMENT
UDCA
Since the first report demonstrating its efficacy for PBC [83], UDCA has dramatically altered the natural course of PBC and has been approved as a first-line therapy for PBC worldwide [58,69,84]. UDCA is used at a dose of 13–15 mg/kg/day, and is recommended for all patients with PBC with elevated liver biochemistry levels.
UDCA improves serum biochemical abnormalities, delays the histological progression and development of varices, and prolongs LT-free survival. Interestingly, a recent retrospective study on a large cohort demonstrated that the LT-free survival of UDCA-treated PBC patients was significantly improved compared to those that received no treatment and also in a population with incomplete biochemical response to UDCA [85].
Approximately 20–30% of patients with PBC exhibited incomplete biochemical responses to UDCA. The outcomes of these patients were significantly worse than those with complete responses to UDCA [85]. It is strongly recommended that patients with incomplete responses to UDCA commence second-line treatment in addition to UDCA. For this purpose, various criteria employing combinations of biochemical markers at 1 year after commencement of UDCA treatment have been proposed (Table 5) [86-95]. Therefore, the “UDCA response score,” which predicts treatment response before starting UDCA treatment with baseline clinical variables, has been proposed [96]. Very recently, the validity of the UDCA response score was demonstrated in a Japanere cohort [97].
Table 5.
Criteria defining biochemical responses to UDCA
| Criteria | Number of patients | Duration | Definition |
|---|---|---|---|
| Qualitative definition | |||
| Barcelona [95] | 192 | 1 year | Normal ALP level or reduction in the ALP level by >40% |
| Paris-I [88] | 292 | 1 year | ALP level <3×ULN, AST level <2×ULN, normal bilirubin level |
| Rotterdam [90] | 375 | 1 year | Normal bilirubin level, normal albumin level |
| Toronto [91] | 69 | 2 year | ALP level ≤1.67×ULN |
| Ehime [86] | 83 | 6 months | Normal GGT level or reduction in the GGT level by ≥70% |
| Paris-II [89] | 165 | 1 year | ALP level <1.5×ULN, AST level <1.5×ULN, normal bilirubin level |
| Rochester [94] | 73 | 1 year | ALP level ≤1.67×ULN, bilirubin level ≤1 mg/dL |
| International (Global PBC) [93] | 4,845 | 1 year | ALP level <2×ULN, normal bilirubin level |
| Quantitative scores | |||
| GLOBE score [92] | 4,119 | 1 year | Bilirubin level, ALP level, albumin level, and platelet count at 1 year, age at baseline |
| UK-PBC score [87] | 3,165 | 1 year | ALP level, AST/ALT level, and bilirubin level at 1 year, albumin level and platelet count at baseline |
UDCA, ursodeoxycholic acid; ALP, alkaline phosphatase; ULN, upper limit of normal; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma glutamyl transferase.
Obeticholic acid (OCA)
OCA is a selective ligand of the farnesoid X receptor (FXR). In an international cohort, prospective, randomized, placebo-controlled trial (POISE trial), 217 patients with PBC who showed an incomplete response (serum ALP level >1.67×upper limit of normal [ULN]) or an abnormal total bilirubin level (<2×ULN), or were intolerant to UDCA were enrolled and received 5–10 mg of OCA, 10 mg of OCA, and a placebo for 1 year. The primary end point was an ALP level <1.67×ULN with >15% reduction from the baseline and normal bilirubin levels. Among those patients, 46–47% achieved the primary end point [98]. Consequently, OCA received accelerated approval from the United States Food and Drug Administration (FDA) approval on May 27, 2016.
Although OCA has become the long-awaited second-line drug officially approved for PBC, it is still unsatisfactory for several reasons. First, the response rate to OCA was at most 50%, which means that up to 50% of the patients did not respond to OCA. Second, pruritus, a symptom frequently experienced by patients with PBC, appeared as an adverse effect in 56–68% of those treated with OCA. Third, the appropriate treatment duration for OCA should be continued for patients refractory to UDCA; thus, OCA may be prescribed lifelong along with UDCA. Based on the high cost of OCA ($69,350 per year), its never-ending prescription places substantial economic burdens on patients and societies; thus, it is not cost-effective [99]. Finally, it has not yet been confirmed whether the primary end points (ALP level <1.67×ULN with >15% reduction from the baseline and normal bilirubin level) are associated with improvement of long-term outcomes. Therefore, follow-up studies of the POISE trial are required by the FDA; a phase 3 study is currently ongoing (COBALT, NCT02308111), and the interim results at 3-year treatment suggested biochemical efficacy and safety [100] as well as histological improvement [101] of OCA. Regardless, the safety concerns of OCA should not be underestimated as the FDA released a warning in September 2017 stating that the use of OCA in PBC patients with decompensated cirrhosis (Child-Pugh-Turcotte class B and C) was associated with clinical worsening or even death.
Fibrates
Fibrates are peroxisome proliferator-activated receptor-α and pregnane X receptor agonists and result in the reduction of de novo bile acid synthesis and upregulation of bile acid transporters [102]. Fibrates (fenofibrate and bezafibrate) were originally indicated for dyslipidemia and are used to decrease serum cholesterol and triglycerides. Bezafibrate was first reported as biologically effective for patients with PBC who were refractory to UDCA in 1999 [103]. A prospective, randomized, placebo-controlled study of bezafibrate in PBC patients with incomplete responses to UDCA from France demonstrated that an add-on of bezafibrate to UDCA for 2 years significantly improved liver biochemistry levels and liver stiffness [104]. In another study, additional evidence from Japan showed that the observed LT-free survival of patients treated with combination therapy of UDCA and bezafibrate was significantly superior to the expected LT-free survival of those treated with UDCA monotherapy according to the GLOBE and UK-PBC scores [105]. In Japan, bezafibrate has been used as a de facto second line treatment for patients who exhibited an incomplete response to UDCA. The proportion of patients treated with UDCA and bezafibrate and the LT-free survival stratified by the diagnosis year are shown in Figure 4. Until 1990, only 56% and 2% of patients were treated with UDCA and bezafibrate, respectively, and the LT-free 5-year survival rate of patients with PBC was 59%. By contrast, 91% and 16–17% of patients were treated with UDCA and bezafibrate after 2000, and the LT-free 5-year survival rate was significanltly improved to 93–94% (P<0.001). Bezafibrate may also improve pruritus of PBC [106]. A prospective clinical trial on this topic is ongoing (FITCH trial, NCT02701166).
Figure 4.

A change of treatment and outcome over time in Japan. (A) The proportion of patients treated with UDCA (light gray bars) and bezafibrate (dark gray bars) in Japan stratified by the diagnosis year. (B) The LT-free survival rate of patients with PBC in Japan, stratified by the diagnosis year. LT, liver transplantation; UDCA, ursodeoxycholic acid; PBC, primary biliary cholangitis.
Fenofibrate was reported to decrease serum ALP levels in studies from Japan and China [107,108], whereas the adjunct use of fenofibrate with UDCA showed no association with decreased serum ALP levels in a United Kingdom study [109]. Participants were recruited for a prospective randomized study in China (clinical trial ID: NCT02965911). However, these two prospective clinical trials showed notable improvements in liver enzyme levels at 12 or 24 months as the primary end point. Even after these trials were terminated, it is still unknown whether long-term outcomes are improved with additional treatment with fibrates, so follow-up studies of these trials are needed.
LT
Despite improvements in medical treatment for PBC, LT is the only treatment option for patients with decompensating events or intolerable pruritus. A recent study utilizing the European Liver Transplant Registry demonstrated a significant decrease in LT in PBC over the last 30 years after the introduction of UDCA in clinical settings [110]. The proportion of LT for PBC decreased from 20% of all LT cases in 1986 to 4% in 2015 (P<0.001). The absolute number of transplants was the highest in 1994 (n=279), which decreased to an average of 200 in the last decade. This decrease is striking in contrast to the substantial increase in the prevalence of PBC at the same time [16].
While the long-term outcome after LT for PBC is excellent overall [111-113], recurrence of PBC after LT is not uncommon. The reported incidence of recurrent PBC differs widely between 11% and 42% [114-129]. Although several studies have reported risk factors associated with recurrence of PBC, most consistently demonstrated that the use of tacrolimus is associated with an increased risk of recurrence [115,116,123,126-128]. For example, a recent study of 785 patients with PBC from North America and Europe who underwent LT between February 1983 and June 2016 indicated that tacrolimus was linked to the recurrence of PBC. Although the use of cyclosporine was protective, the 5-year probabilities of recurrence of PBC were reported to be 28% and 11% in patients receiving tacrolimus and cyclosporine, respectively (P<0.001) [125]. On the other hand, the role of tacrolimus as an increasing agent of recurrent PBC does not seem to be the case in other ethnicities. Recently, two cohort studies from Japan demonstrated that the increased frequency of recurrent PBC is associated with initial treatment with cyclosporine after LT [117,122].
Although it is believed that recurrence of PBC does not have a significant impact on long-term outcomes, such as overall survival [123,130], a recent study of 785 PBC patients from 13 centers in North America and Europe who received LT with a median follow-up of 6.9 years (interquartile range, 6.1–7.9) reported opposite and unexpected results that disease recurrence was found in 240 patients (31%). Importantly, graft and patient survival rates were significantly impaired in those with recurrent PBC (P=0.004 and 0.001, respectively) [125]. It is imperative to determine whether recurrent PBC has a clinically significant impact on patient and graft survival. Furthermore, since UDCA treatment seems to be effective in improving markers of cholestasis, it does not effectively reduce the frequency and risk of recurrence of PBC after LT [130]. Therefore, effective strategies to halt the recurrence of PBC are urgently needed.
Treatment flowchart
In Figure 5, a proposed treatment flowchart for patients with PBC is shown. UDCA treatment (13–15 mg/kg/day) is recommended for all patients diagnosed with PBC, with elevated cholestatic liver enzymes. Treatment response should be judged at 1 year of UDCA treatment with the biochemical criteria shown in Table 5. Since most newly diagnosed PBC patients are at an early stage in daily clinical practice, the Paris II criteria designed for early stage patients may be suitable and easy to use. If the treatment response is complete, UDCA monotherapy should be continued and no additional treatment is required. In patients with incomplete response, add-on of the second-line treatment is definitely necessary; otherwise, the long-term outcome would deteriorate. The evidence-based choice of second-line medication is very difficult because no drug has been demonstrated to improve long-term outcomes. In Japan, a de facto use of bezafibrate as a second-line treatment appears to contribute to the improvement of long-term outcome, and it is expected to demonstrate in a large scale cohort a significant association of a combination of UDCA and bezafibrate with reduced mortality or need for LT.
Figure 5.
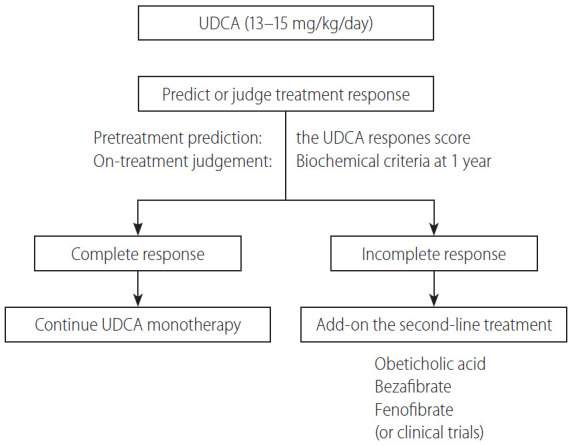
A treatment flowchart of patients with PBC. UDCA, ursodeoxycholic acid; PBC, primary biliary cholangitis.
Alternatively, response to UDCA can be predicted before commencement of UDCA therapy using the UDCA Response Score, and initiation of treatment with a combination of UDCA and bezafibrate would be a choice in cases with prediction of poor response.
MANAGEMENT OF SYMPTOMS, EXTRAHEPATIC MANIFESTATIONS, AND HEPATOCELLULAR CARCINOMA (HCC)
Patients with PBC frequently present with numerous symptoms. The most dominant clinical symptoms in the early stage of PBC are fatigue and pruritus, followed by jaundice. Since these symptoms can significantly deteriorate the quality of life of patients with PBC, it is strongly recommended to carefully monitor these symptoms with objective and reproducible measures such as the PBC-40 questionnaire [131]. In Table 6, symptoms in PBC and corresponding recommended or emerging treatment options are summarized. In addition, along with an improvement in the long-term outcome of patients with PBC, the development of HCC is not a rare event.
Table 6.
Symptoms in PBC and corresponding treatment option
| Symptom | Treatment | Description |
|---|---|---|
| Fatigue | Modafinil | RCT failed to show efficacy |
| Pruritus | Anion-exchange resins (cholestyramine) | The first-line treatment, despite limitations |
| Rifampicin | The second-line treatment | |
| µ-opioid receptor antagonists (naloxone or naltrexone) | The second-line treatment | |
| κ-opioid receptor agonist (nalfurafine hydrochloride) | Approved only in Japan | |
| Ileal bile acid transporter inhibitor | Linerixibat; efficacy was shown in the Phase 2a; now global phase 2b (NCT02966834) | |
| Maralixibat; failed to show efficacy | ||
| Bezafibrate | Now being investigated (NCT02701166) | |
| Sicca syndrome | Artificial tears and saliva | Should be initially used |
| Pilocarpine or cevimeline | May be helpful in refractory cases |
PBC, primary biliary cholangitis; RCT, randomized clinical trial.
Fatigue
Fatigue is the most common and debilitating symptom in PBC, experienced by approximately 50% of patients (ranging from 20% to 80% depending on each study) [132-134]. Although it is difficult to define the cutoff clearly according to the presence of fatigue, it has been repeatedly shown that fatigue has a great impact on the impairment of the quality of life of patients with PBC [133,135]. Fatigue is not associated with disease severity and staging, but may be related to age at onset and gender [136].
The cause of fatigue remains unknown but seems to be complex in origin, probably multifactorial in most patients and associated with depression, autonomic dysfunction, and sleep disturbance [135]. Recent studies with magnetic resonance imaging revealed that neuroimaging changes in the brain can be detected even in the early stage of PBC [137]. Fatigue cannot be treated with UDCA, and a recent systemic review failed to define any established treatment for fatigue in PBC [138]. Fatigue may be improved by LT, but it can also persist in a substantial portion of patients even after LT, making the role of LT as a therapeutic option for severe fatigue questionable [139]. Modafinil, which is officially approved by the FDA for wakefulness disorders, has been used, but a recent randomized, placebo-controlled clinical trial failed to indicate beneficial effects of this drug in reducing fatigue in patients with PBC [140].
Pruritus
Pruritus is another common symptom in PBC, affecting 20–80% of patients. Pruritus can occur locally or diffusely, and its presence and severity change throughout the clinical course of PBC. It tends to become more pronounced with the progression of PBC but can be present even in the very early stage. Pruritus can be highly bothersome and intolerable to patients, such as causing sleep disturbance, and is an important indication for LT. Similar to fatigue, the severity of pruritus is objectively assessable with the PBC-40 questionnaire [131]. The cause of pruritus remains unknown, although several substances are hypothesized to be related to pruritus in cholestatic liver diseases [141]. Most notably, lysophosphatidic acid (LPA) may be a potential candidate for initiating pruritus [142], and the activity of serum autotaxin, which converts lysophosphatidylcholine into LPA, and is related to the severity of pruritus and responds to therapeutic interventions [143,144]. Hence, LPA-autotaxin is an important candidate as a therapeutic target, yet clinically unavailable.
Although several lines of treatment options are recommended, severe pruritus is frequently intractable. In daily clinical practice, anion-exchange resins such as cholestyramine, rifampsin, and opiate antagonists are used [145]. Anion-exchange resins are recommended as the first-line therapy in the American Association for the Study of Liver Diseases and European Association for the Study of the Liver guidelines, despite its limited efficacy [68,146]. Cholestyramine is prescribed as 4 g per dose to a maximum of 16 g/day, and it is important to be administered 1 hour after or 4 hours before other medications (especially UDCA) to avoid inhibiting their absorption [68]. Rifampicin is a pregnane X receptor agonist that has been used for pruritus at 150–300 mg twice daily. Opiate antangonists interefere with increased endogenous opioid levels in patients with cholestatic pruritus [145]. While m-opioid receptor antagonists, such as naloxone or naltrexone, have been used, nalfurafine hydrochloride, a selective κ-opioid receptor agonist, is currently used in Japan for cholestatic pruritus and exhibits substantial efficacy [147]. As an emerging novel therapy, an ileal bile acid transporter (IBAT) inhibitor compound (linerixibat) that inhibits reabsorption of bile acids in the ileum effectively decreased pruritus in patients with PBC in a phase 2a study [148]. A global phase 2b study (clinical trial ID: NCT02966834) investigating the efficacy of linerixibat is almost complete. A phase 2 trial of another IBAT inhibitor, maralixibat, failed to demonstrate a significant anti-pruritic effect against placebo, presumably because of a significant placebo effect [149]. As described earlier, bezafibrate is now being investigated in a clinical trial for the treatment of cholestatic pruritus (clinical trial ID: NCT02701166) [106].
Sicca syndrome
The sicca complex is frequently present in patients with PBC, manifesting as dry eyes and/or dry mouth. External glands, including the lachrymal or salivary glands, are also affected in PBC. A current retrospective study revealed the prevalence of Sjögren’s syndrome in up to 56% of patients with PBC [150]; however, the sicca complex affects patients with PBC who do not meet the criteria of Sjögren’s syndrome. Patients with sicca syndrome may experience many symptoms, including burning, itching, irritated eyes, blepharitis, dysphagia, stomatitis, dental caries, and dry cough, resulting in severe impairment of their quality of life. Early recognition of sicca symptoms and consultations with ophthalmologists or dentists are suggested. Artifical tears and saliva are often helpful, and pilocarpine or cevimeline may alleviate the symptoms in refractory cases.
Osteopenia and osteoporosis
Osteopenic bone disease, including osteopenia and osteoporosis, is a common disorder in PBC that mainly affects middle-aged women and is associated with an increased risk for fragility fractures. The decrease in bone mineral density found in PBC is multifactorial. Chronic cholestasis leads to malabsorption and deficiency of vitamin D, which is essential for bone metabolism. Other factors associated with bone diseases include age, sex, low body mass index, history of fragility fracture, and advanced stage of PBC [151,152]. Intervention with bisphosphonates for patients with osteoporosis and those with a history of fragility fracture is safe and improves bone mineral density [153]; nonetheless, it remains unclear whether bisphosphonate use is associated with a decrease in fragility fractures. Recently, denosumab, a fully human monoclonal antibody against the receptor activator of nuclear factor-kappaB ligand (RANKL), was demonstrated to be effective for treating osteoporosis in patients with PBC [154]. Since RANK-RANKL signaling might be implicated in the pathogenesis of PBC [155], treatment with denosumab could be used to target both osteoporosis and PBC.
Hyperlipidemia and metabolic syndrome
Chronic cholestasis is a main feature of PBC, and consequently, hyperlipidemia is common and affects up to 80% of patients [156]. Several prospective studies have indicated that an increase in serum lipid levels is not associated with a higher risk for cardiovascular diseases related to atherosclerosis, and treatment for hyperlipidemia per se is not necessary. Notably, these studies were conducted in the 1990s when metabolic syndrome was relatively rare in patients with PBC. A recent study from Italy demonstrated that cardiovascular events developed more frequently in patients with metabolic syndrome [157], indicating the importance of treatment intervention for patients with hyperlipidemia if metabolic syndrome exists.
HCC
HCC is occasionally encountered in patients with PBC. The reported incidences and risk factors for developing HCC from several large-scale retrospective cohorts are summarized in Table 7. Surprisingly, the incidence rates (cases per 1,000 patient-years) of HCC in all patients with PBC are similar across different regions: 3.6 in Barcelona, Spain; 3.7 in Padova, Italy [158]; 3.6 in a nationwide study in Japan [159]; and 3.4 in an international cohort [160]. The incidence rate was exceptionally high (6.6) in a cohort from Beijing, China [161], presumably because of the high rate of individuals with previous hepatitis B virus (HBV) infection. A history of HBV infection was identified as an independent risk factor for HCC in this study. The incidence rate was higher in men than in women. In those studies, male sex and advanced histological stage independently contributed to the development of HCC [158-161]. Treatment response was included among possible risk factors only in the international cohort study, and biochemical non-response at 1 year of UDCA treatment (Paris-I not fulfilled) significantly increased the future risk of HCC (adjusted hazard ratio, 3.44; 95% CI, 1.65–7.14; P<0.0001) [160]. Taken together, close monitoring of HCC is strongly recommended for high-risk patients with PBC, such as male patients, those with advanced-stage disease, and non-responders to UDCA. The mean survival of patients who developed HCC was 36 months after diagnosis, and another cohort indicated 5- and 10-year survival rates of 49.5% and 31.7%, respectively [162].
Table 7.
Incidence and risk factors for HCC in patients with PBC
| Country/region | Number |
Incidence* |
Risk factor | |||
|---|---|---|---|---|---|---|
| Total | HCC | All | Male patients | Female patients | ||
| Barcelona, Spain [158] | 389 | 13 | 3.6 | NA | NA | Advanced histological stage |
| Padova, Italy [158] | 327 | 11 | 3.7 | NA | NA | Advanced histological stage (all), male sex |
| Japan [171] | 2,946 | 71 | 3.6 | 9.5 | 2.9 | Male sex, advanced histological stage (in female patients) |
| International [160] | 4,565 | 123 | 3.4 | 6.7 | 2.6 | Advanced age, male sex, thrombocytopenia at 12 months, biochemical non-response |
| Beijing, China [161] | 1,865 | 70 | 6.6 | NA | NA | Advanced age, male sex, co-existence of diabetes, History of HBV infection |
HCC, hepatocellular carcinoma; PBC, primary biliary cholangitis; NA, not applicable; HBV, hepatitis B virus.
Cases per 1,000 patient-years.
FUTURE PERSPECTIVE
Although the long-term outcome of PBC has been significantly improved, there remain several unmet needs in PBC. UDCA and other second-line treatments have little efficacy for patients with advanced disease, for whom LT is the only therapeutic option. No “cure” is achieved with current treatment, and patients are required to take medications for life long. A variety of symptoms, including fatigue and pruritus, are very difficult to manage and easily reduce the health-related quality of life.
To overcome these unmet needs in this field, rigorous efforts should be directed at improving our understanding of the environmental etiology and genetic basis of PBC, molecular mechanisms of disease progression, and gender bias to identify critical pathways for therapeutic interventions. Relevant animal models that recapitulate human PBC should be established for preclinical studies with designer drugs guided by this new knowledge. Our goal is achieving the “cure” for PBC.
Acknowledgments
We deeply appreciate the secretarial assistance of Ms. Kayono Unno. This work was supported by a Health and Labor Sciences Research Grant (research on intractable hepatobiliary disease) issued by the Ministry of Health, Labor, and Welfare of Japan. We would like to thank Editage (www.editage.jp) for English language editing.
Abbreviations
- 2-OADC
2-oxo-acid dehydrogenase complex
- ALP
alkaline phosphatase
- AMA
anti-mitochondrial antibody
- APASL
Asian Pacific Association for the Study of the Liver
- BCOADC-E2
branched chain 2-oxoacid dehydrogenase complex E2 subunit
- BECs
biliary epithelial cells
- CA
cholangitis activity
- CI
confidence interval
- CNSDC
chronic non-suppurative destructive cholangitis
- E. coli
Escherichia coli
- E3BP
dihydrolipoamide dehydrogenase-binding protein
- ELISA
enzyme-linked immunosorbent assay
- FDA
Food and Drug Administration
- FXR
farnesoid X receptor
- GWAS
genome-wide association study
- HA
hepatitis activity
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HLA
human leukocyte antigen
- IBAT
ileal bile acid transporter
- IFN
interferon
- IL
interleukin
- LPA
lysophosphatidic acid
- LT
liver transplantation
- MHC
major histocompatibility complex
- OCA
obeticholic acid
- OGDC-E2
2-oxo-glutamic acid dehydrogenase complex E2 subunit
- PBC
primary biliary cholangitis
- PDC-E2
pyruvate dehydrogenase complex E2 subunit
- RANKL
receptor activator of nuclear factor-kappaB ligand
- RRs
risk ratios
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Footnotes
Conflicts of Interest: Atsushi Tanaka received consultant fees from EA Pharma, GlaxoSmithKline, and Gilead Sciences.
REFERENCES
- 1.Addison T, Gull W. On a certain affection of the skin-vitiligoidea-alpha-plana, B-tuberosa. Guys Hosp Rep. 1851;7:265–276. [Google Scholar]
- 2.Dauphinee JA, Sinclair JC. Primary biliary cirrhosis. Can Med Assoc J. 1949;61:1–6. [PMC free article] [PubMed] [Google Scholar]
- 3.Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. Clin Res Hepatol Gastroenterol. 2015;39:e57–e59. doi: 10.1016/j.clinre.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. Dig Liver Dis. 2015;47:924–926. doi: 10.1016/j.dld.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. Am J Gastroenterol. 2015;110:1536–1538. doi: 10.1038/ajg.2015.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. Clin Gastroenterol Hepatol. 2015;13:1867–1869. doi: 10.1016/j.cgh.2015.08.025. [DOI] [PubMed] [Google Scholar]
- 7.Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. J Hepatol. 2015;63:1285–1287. doi: 10.1016/j.jhep.2015.06.031. [DOI] [PubMed] [Google Scholar]
- 8.Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. Gastroenterology. 2015;149:1627–1629. doi: 10.1053/j.gastro.2015.08.031. [DOI] [PubMed] [Google Scholar]
- 9.Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. Gut. 2015;64:1671–1672. doi: 10.1136/gutjnl-2015-310593. [DOI] [PubMed] [Google Scholar]
- 10.Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: From ‘cirrhosis’ to ‘cholangitis’. Hepatology. 2015;62:1620–1622. doi: 10.1002/hep.28140. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka A, Ma X, Yokosuka O, Weltman M, You H, Amarapurkar DN, et al. Autoimmune liver diseases in the Asia-Pacific region: proceedings of APASL symposium on AIH and PBC 2016. Hepatol Int. 2016;10:909–915. doi: 10.1007/s12072-016-9767-9. [DOI] [PubMed] [Google Scholar]
- 12.Cheung KS, Seto WK, Fung J, Lai CL, Yuen MF. Epidemiology and natural history of primary biliary cholangitis in the Chinese: a territory-based study in Hong Kong between 2000 and 2015. Clin Transl Gastroenterol. 2017;8:e116. doi: 10.1038/ctg.2017.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim KA, Ki M, Choi HY, Kim BH, Jang ES, Jeong SH. Population-based epidemiology of primary biliary cirrhosis in South Korea. Aliment Pharmacol Ther. 2016;43:154–162. doi: 10.1111/apt.13448. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka A, Mori M, Matsumoto K, Ohira H, Tazuma S, Takikawa H. Increase trend in the prevalence and male-to-female ratio of primary biliary cholangitis, autoimmune hepatitis, and primary sclerosing cholangitis in Japan. Hepatol Res. 2019;49:881–889. doi: 10.1111/hepr.13342. [DOI] [PubMed] [Google Scholar]
- 15.Jeong SH. Current epidemiology and clinical characteristics of autoimmune liver diseases in South Korea. Clin Mol Hepatol. 2018;24:10–19. doi: 10.3350/cmh.2017.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol. 2012;56:1181–1188. doi: 10.1016/j.jhep.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 17.French J, van der Mei I, Simpson S, Jr, Ng J, Angus P, Lubel J, et al. Increasing prevalence of primary biliary cholangitis in Victoria, Australia. J Gastroenterol Hepatol. 2020;35:673–679. doi: 10.1111/jgh.14924. [DOI] [PubMed] [Google Scholar]
- 18.Marschall HU, Henriksson I, Lindberg S, Söderdahl F, Thuresson M, Wahlin S, et al. Incidence, prevalence, and outcome of primary biliary cholangitis in a nationwide Swedish population-based cohort. Sci Rep. 2019;9:11525. doi: 10.1038/s41598-019-47890-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lleo A, Jepsen P, Morenghi E, Carbone M, Moroni L, Battezzati PM, et al. Evolving trends in female to male incidence and male mortality of primary biliary cholangitis. Sci Rep. 2016;6:25906. doi: 10.1038/srep25906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marzioni M, Bassanelli C, Ripellino C, Urbinati D, Alvaro D. Epidemiology of primary biliary cholangitis in Italy: evidence from a real-world database. Dig Liver Dis. 2019;51:724–729. doi: 10.1016/j.dld.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Neuberger J, Lombard M, Galbraith R. Primary biliary cirrhosis. Gut. 1991;Suppl:S73–S78. doi: 10.1136/gut.32.suppl.s73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talwalkar J, Lindor K. Primary biliary cirrhosis. Lancet. 2003;362:53–61. doi: 10.1016/S0140-6736(03)13808-1. [DOI] [PubMed] [Google Scholar]
- 23.Van de Water J, Gershwin ME, Leung P, Ansari A, Coppel RL. The autoepitope of the 74-kD mitochondrial autoantigen of primary biliary cirrhosis corresponds to the functional site of dihydrolipoamide acetyltransferase. J Exp Med. 1988;167:1791–1799. doi: 10.1084/jem.167.6.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oertelt S, Rieger R, Selmi C, Invernizzi P, Ansari AA, Coppel RL, et al. A sensitive bead assay for antimitochondrial antibodies: chipping away at AMA-negative primary biliary cirrhosis. Hepatology. 2007;45:659–665. doi: 10.1002/hep.21583. [DOI] [PubMed] [Google Scholar]
- 25.Dubel L, Tanaka A, Leung PS, Van de Water J, Coppel R, Roche T, et al. Autoepitope mapping and reactivity of autoantibodies to the dihydrolipoamide dehydrogenase-binding protein (E3BP) and the glycine cleavage proteins in primary biliary cirrhosis. Hepatology. 1999;29:1013–1018. doi: 10.1002/hep.510290403. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka A, Leung PSC, Gershwin ME. The genetics and epigenetics of primary biliary cholangitis. Clin Liver Dis. 2018;22:443–455. doi: 10.1016/j.cld.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Björkland A, Festin R, Mendel-Hartvig I, Nyberg A, Lööf L, Tötterman TH. Blood and liver-infiltrating lymphocytes in primary biliary cirrhosis: increase in activated T and natural killer cells and recruitment of primed memory T cells. Hepatology. 1991;13:1106–1111. doi: 10.1002/hep.1840130617. [DOI] [PubMed] [Google Scholar]
- 28.Krams SM, Van de Water J, Coppel RL, Esquivel C, Roberts J, Ansari A, et al. Analysis of hepatic T lymphocyte and immunoglobulin deposits in patients with primary biliary cirrhosis. Hepatology. 1990;12:306–313. doi: 10.1002/hep.1840120219. [DOI] [PubMed] [Google Scholar]
- 29.Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 30.Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, et al. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. 2005;41:506–516. doi: 10.1002/hep.20582. [DOI] [PubMed] [Google Scholar]
- 31.Shimoda S, Harada K, Niiro H, Taketomi A, Maehara Y, Tsuneyama K, et al. CX3CL1 (fractalkine): a signpost for biliary inflammation in primary biliary cirrhosis. Hepatology. 2010;51:567–575. doi: 10.1002/hep.23318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harada K, Kakuda Y, Nakamura M, Shimoda S, Nakanuma Y. Clinicopathological significance of serum fractalkine in primary biliary cirrhosis. Dig Dis Sci. 2013;58:3037–3043. doi: 10.1007/s10620-013-2734-6. [DOI] [PubMed] [Google Scholar]
- 33.Lleo A, Selmi C, Invernizzi P, Podda M, Coppel RL, Mackay IR, et al. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology. 2009;49:871–879. doi: 10.1002/hep.22736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lleo A, Bowlus CL, Yang GX, Invernizzi P, Podda M, Van de Water J, et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology. 2010;52:987–998. doi: 10.1002/hep.23783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka A, Leung PSC, Gershwin ME. The genetics of primary biliary cholangitis. Curr Opin Gastroenterol. 2019;35:93–98. doi: 10.1097/MOG.0000000000000507. [DOI] [PubMed] [Google Scholar]
- 36.Örnolfsson KT, Olafsson S, Bergmann OM, Gershwin ME, Björnsson ES. Using the Icelandic genealogical database to define the familial risk of primary biliary cholangitis. Hepatology. 2018;68:166–171. doi: 10.1002/hep.29675. [DOI] [PubMed] [Google Scholar]
- 37.Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat Commun. 2015;6:8019. doi: 10.1038/ncomms9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirschfield GM, Liu X, Han Y, Gorlov IP, Lu Y, Xu C, et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet. 2010;42:655–657. doi: 10.1038/ng.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. 2009;360:2544–2555. doi: 10.1056/NEJMoa0810440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirschfield GM, Xie G, Lu E, Sun Y, Juran BD, Chellappa V, et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun. 2012;13:328–335. doi: 10.1038/gene.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juran BD, Hirschfield GM, Invernizzi P, Atkinson EJ, Li Y, Xie G, et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum Mol Genet. 2012;21:5209–5221. doi: 10.1093/hmg/dds359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawashima M, Hitomi Y, Aiba Y, Nishida N, Kojima K, Kawai Y, et al. Genome-wide association studies identify PRKCB as a novel genetic susceptibility locus for primary biliary cholangitis in the Japanese population. Hum Mol Genet. 2017;26:650–659. doi: 10.1093/hmg/ddw406. [DOI] [PubMed] [Google Scholar]
- 43.Liu JZ, Almarri MA, Gaffney DJ, Mells GF, Jostins L, Cordell HJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2012;44:1137–1141. doi: 10.1038/ng.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X, Invernizzi P, Lu Y, Kosoy R, Lu Y, Bianchi I, et al. Genomewide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet. 2010;42:658–660. doi: 10.1038/ng.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43:329–332. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet. 2012;91:721–728. doi: 10.1016/j.ajhg.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu F, Tang R, Zuo X, Shi X, Wei Y, Zheng X, et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat Commun. 2017;8:14828. doi: 10.1038/ncomms14828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong M, Li J, Tang R, Zhu P, Qiu F, Wang C, et al. Multiple genetic variants associated with primary biliary cirrhosis in a Han Chinese population. Clin Rev Allergy Immunol. 2015;48:316–321. doi: 10.1007/s12016-015-8472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burroughs AK, Rosenstein IJ, Epstein O, Hamilton-Miller JM, Brumfitt W, Sherlock S. Bacteriuria and primary biliary cirrhosis. Gut. 1984;25:133–137. doi: 10.1136/gut.25.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corpechot C, Chrétien Y, Chazouillères O, Poupon R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. J Hepatol. 2010;53:162–169. doi: 10.1016/j.jhep.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 51.Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–1202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Howel D, Fischbacher CM, Bhopal RS, Gray J, Metcalf JV, James OF. An exploratory population-based case-control study of primary biliary cirrhosis. Hepatology. 2000;31:1055–1060. doi: 10.1053/he.2000.7050. [DOI] [PubMed] [Google Scholar]
- 53.Ala A, Stanca CM, Bu-Ghanim M, Ahmado I, Branch AD, Schiano TD, et al. Increased prevalence of primary biliary cirrhosis near superfund toxic waste sites. Hepatology. 2006;43:525–531. doi: 10.1002/hep.21076. [DOI] [PubMed] [Google Scholar]
- 54.Amano K, Leung PS, Rieger R, Quan C, Wang X, Marik J, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol. 2005;174:5874–5883. doi: 10.4049/jimmunol.174.9.5874. [DOI] [PubMed] [Google Scholar]
- 55.McNally RJ, James PW, Ducker S, Norman PD, James OF. No rise in incidence but geographical heterogeneity in the occurrence of primary biliary cirrhosis in North East England. Am J Epidemiol. 2014;179:492–498. doi: 10.1093/aje/kwt308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prince MI, Chetwynd A, Diggle P, Jarner M, Metcalf JV, James OF. The geographical distribution of primary biliary cirrhosis in a well-defined cohort. Hepatology. 2001;34:1083–1088. doi: 10.1053/jhep.2001.29760. [DOI] [PubMed] [Google Scholar]
- 57.Rieger R, Leung PS, Jeddeloh MR, Kurth MJ, Nantz MH, Lam KS, et al. Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J Autoimmun. 2006;27:7–16. doi: 10.1016/j.jaut.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 58.Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut. 2018;67:534–541. doi: 10.1136/gutjnl-2016-313332. [DOI] [PubMed] [Google Scholar]
- 59.Ma HD, Zhao ZB, Ma WT, Liu QZ, Gao CY, Li L, et al. Gut microbiota translocation promotes autoimmune cholangitis. J Autoimmun. 2018;95:47–57. doi: 10.1016/j.jaut.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ryan PM, Stanton C, Caplice NM. Bile acids at the cross-roads of gut microbiome-host cardiometabolic interactions. Diabetol Metab Syndr. 2017;9:102. doi: 10.1186/s13098-017-0299-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Staley C, Weingarden AR, Khoruts A, Sadowsky MJ. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl Microbiol Biotechnol. 2017;101:47–64. doi: 10.1007/s00253-016-8006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hofmann AF, Hagey LR, Krasowski MD. Bile salts of vertebrates: structural variation and possible evolutionary significance. J Lipid Res. 2010;51:226–246. doi: 10.1194/jlr.R000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen ML, Takeda K, Sundrud MS. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol. 2019;12:851–861. doi: 10.1038/s41385-019-0162-4. [DOI] [PubMed] [Google Scholar]
- 64.Yang H, Duan Z. Bile acids and the potential role in primary biliary cirrhosis. Digestion. 2016;94:145–153. doi: 10.1159/000452300. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Tang R, Leung PSC, Gershwin ME, Ma X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun Rev. 2017;16:885–896. doi: 10.1016/j.autrev.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 66.Melbye P, Olsson A, Hansen TH, Søndergaard HB, Bang Oturai A. Short-chain fatty acids and gut microbiota in multiple sclerosis. Acta Neurol Scand. 2019;139:208–219. doi: 10.1111/ane.13045. [DOI] [PubMed] [Google Scholar]
- 67.Jones-Hall YL, Nakatsu CH. The intersection of TNF, IBD and the microbiome. Gut Microbes. 2016;7:58–62. doi: 10.1080/19490976.2015.1121364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69:394–419. doi: 10.1002/hep.30145. [DOI] [PubMed] [Google Scholar]
- 69.Working Subgroup for Clinical Practice Guidelines for Primary Biliary Cirrhosis Guidelines for the management of primary biliary cirrhosis: the intractable hepatobiliary disease study group supported by the Ministry of Health, Labour and Welfare of Japan. Hepatol Res. 2014;44 Suppl S1:71–90. doi: 10.1111/hepr.12270. [DOI] [PubMed] [Google Scholar]
- 70.Murillo Perez CF, Hirschfield GM, Corpechot C, Floreani A, Mayo MJ, van der Meer A, et al. Fibrosis stage is an independent predictor of outcome in primary biliary cholangitis despite biochemical treatment response. Aliment Pharmacol Ther. 2019;50:1127–1136. doi: 10.1111/apt.15533. [DOI] [PubMed] [Google Scholar]
- 71.Joshi S, Cauch-Dudek K, Heathcote EJ, Lindor K, Jorgensen R, Klein R. Antimitochondrial antibody profiles: are they valid prognostic indicators in primary biliary cirrhosis? Am J Gastroenterol. 2002;97:999–1002. doi: 10.1111/j.1572-0241.2002.05620.x. [DOI] [PubMed] [Google Scholar]
- 72.Mattalia A, Quaranta S, Leung PS, Bauducci M, Van de Water J, Calvo PL, et al. Characterization of antimitochondrial antibodies in health adults. Hepatology. 1998;27:656–661. doi: 10.1002/hep.510270303. [DOI] [PubMed] [Google Scholar]
- 73.Shibata M, Onozuka Y, Morizane T, Koizumi H, Kawaguchi N, Miyakawa H, et al. Prevalence of antimitochondrial antibody in Japanese corporate workers in Kanagawa prefecture. J Gastroenterol. 2004;39:255–259. doi: 10.1007/s00535-003-1285-6. [DOI] [PubMed] [Google Scholar]
- 74.Dahlqvist G, Gaouar F, Carrat F, Meurisse S, Chazouillères O, Poupon R, et al. Large-scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis. Hepatology. 2017;65:152–163. doi: 10.1002/hep.28859. [DOI] [PubMed] [Google Scholar]
- 75.Sun C, Xiao X, Yan L, Sheng L, Wang Q, Jiang P, et al. Histologically proven AMA positive primary biliary cholangitis but normal serum alkaline phosphatase: is alkaline phosphatase truly a surrogate marker? J Autoimmun. 2019;99:33–38. doi: 10.1016/j.jaut.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 76.Liu H, Norman GL, Shums Z, Worman HJ, Krawitt EL, Bizzaro N, et al. PBC screen: an IgG/IgA dual isotype ELISA detecting multiple mitochondrial and nuclear autoantibodies specific for primary biliary cirrhosis. J Autoimmun. 2010;35:436–442. doi: 10.1016/j.jaut.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 77.Shimoda S, Nakamura M, Ishibashi H, Kawano A, Kamihira T, Sakamoto N, et al. Molecular mimicry of mitochondrial and nuclear autoantigens in primary biliary cirrhosis. Gastroenterology. 2003;124:1915–1925. doi: 10.1016/s0016-5085(03)00387-1. [DOI] [PubMed] [Google Scholar]
- 78.Nakamura M, Kondo H, Mori T, Komori A, Matsuyama M, Ito M, et al. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis. Hepatology. 2007;45:118–127. doi: 10.1002/hep.21472. [DOI] [PubMed] [Google Scholar]
- 79.Scheuer P. Primary biliary cirrhosis. Proc R Soc Med. 1967;60:1257–1260. doi: 10.1177/003591576706001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis) Virchows Arch A Pathol Anat Histol. 1978;379:103–112. doi: 10.1007/BF00432479. [DOI] [PubMed] [Google Scholar]
- 81.Nakanuma Y, Zen Y, Harada K, Sasaki M, Nonomura A, Uehara T, et al. Application of a new histological staging and grading system for primary biliary cirrhosis to liver biopsy specimens: interobserver agreement. Pathol Int. 2010;60:167–174. doi: 10.1111/j.1440-1827.2009.02500.x. [DOI] [PubMed] [Google Scholar]
- 82.Harada K, Hsu M, Ikeda H, Zeniya M, Nakanuma Y. Application and validation of a new histologic staging and grading system for primary biliary cirrhosis. J Clin Gastroenterol. 2013;47:174–181. doi: 10.1097/MCG.0b013e31827234e4. [DOI] [PubMed] [Google Scholar]
- 83.Poupon R, Chrétien Y, Poupon RE, Ballet F, Calmus Y, Darnis F. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet. 1987;1:834–836. doi: 10.1016/s0140-6736(87)91610-2. [DOI] [PubMed] [Google Scholar]
- 84.Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ, et al. Primary biliary cirrhosis. Hepatology. 2009;50:291–308. doi: 10.1002/hep.22906. [DOI] [PubMed] [Google Scholar]
- 85.Harms MH, van Buuren HR, Corpechot C, Thorburn D, Janssen HLA, Lindor KD, et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol. 2019;71:357–365. doi: 10.1016/j.jhep.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 86.Azemoto N, Abe M, Murata Y, Hiasa Y, Hamada M, Matsuura B, et al. Early biochemical response to ursodeoxycholic acid predicts symptom development in patients with asymptomatic primary biliary cirrhosis. J Gastroenterol. 2009;44:630–634. doi: 10.1007/s00535-009-0051-9. [DOI] [PubMed] [Google Scholar]
- 87.Carbone M, Sharp SJ, Flack S, Paximadas D, Spiess K, Adgey C, et al. The UK-PBC risk scores: derivation and validation of a scoring system for long-term prediction of end-stage liver disease in primary biliary cholangitis. Hepatology. 2016;63:930–950. doi: 10.1002/hep.28017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Corpechot C, Abenavoli L, Rabahi N, Chrétien Y, Andréani T, Johanet C, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48:871–877. doi: 10.1002/hep.22428. [DOI] [PubMed] [Google Scholar]
- 89.Corpechot C, Chazouillères O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long-term outcome. J Hepatol. 2011;55:1361–1367. doi: 10.1016/j.jhep.2011.02.031. [DOI] [PubMed] [Google Scholar]
- 90.Kuiper EM, Hansen BE, de Vries RA, den Ouden-Muller JW, van Ditzhuijsen TJ, Haagsma EB, et al. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology. 2009;136:1281–1287. doi: 10.1053/j.gastro.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 91.Kumagi T, Guindi M, Fischer SE, Arenovich T, Abdalian R, Coltescu C, et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am J Gastroenterol. 2010;105:2186–2194. doi: 10.1038/ajg.2010.216. [DOI] [PubMed] [Google Scholar]
- 92.Lammers WJ, Hirschfield GM, Corpechot C, Nevens F, Lindor KD, Janssen HL, et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology. 2015;149:1804–1812. doi: 10.1053/j.gastro.2015.07.061. e4. [DOI] [PubMed] [Google Scholar]
- 93.Lammers WJ, van Buuren HR, Hirschfield GM, Janssen HL, Invernizzi P, Mason AL, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147:1338–1349. doi: 10.1053/j.gastro.2014.08.029. e5; quiz e15. [DOI] [PubMed] [Google Scholar]
- 94.Momah N, Silveira MG, Jorgensen R, Sinakos E, Lindor KD. Optimizing biochemical markers as endpoints for clinical trials in primary biliary cirrhosis. Liver Int. 2012;32:790–795. doi: 10.1111/j.1478-3231.2011.02678.x. [DOI] [PubMed] [Google Scholar]
- 95.Parés A, Caballería L, Rodés J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. 2006;130:715–720. doi: 10.1053/j.gastro.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 96.Carbone M, Nardi A, Flack S, Carpino G, Varvaropoulou N, Gavrila C, et al. Pretreatment prediction of response to ursodeoxycholic acid in primary biliary cholangitis: development and validation of the UDCA response score. Lancet Gastroenterol Hepatol. 2018;3:626–634. doi: 10.1016/S2468-1253(18)30163-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yagi M, Matsumoto K, Komori A, Abe M, Hashimoto N, Inao M, et al. A validation study of the ursodeoxycholic acid response score in Japanese patients with primary biliary cholangitis. Liver Int. 2020;40:1926–1933. doi: 10.1111/liv.14534. [DOI] [PubMed] [Google Scholar]
- 98.Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375:631–643. doi: 10.1056/NEJMoa1509840. [DOI] [PubMed] [Google Scholar]
- 99.Samur S, Klebanoff M, Banken R, Pratt DS, Chapman R, Ollendorf DA, et al. Long-term clinical impact and cost-effectiveness of obeticholic acid for the treatment of primary biliary cholangitis. Hepatology. 2017;65:920–928. doi: 10.1002/hep.28932. [DOI] [PubMed] [Google Scholar]
- 100.Trauner M, Nevens F, Shiffman ML, Drenth JPH, Bowlus CL, Vargas V, et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol Hepatol. 2019;4:445–453. doi: 10.1016/S2468-1253(19)30094-9. [DOI] [PubMed] [Google Scholar]
- 101.Bowlus CL, Pockros PJ, Kremer AE, Parés A, Forman LM, Drenth JPH, et al. Long-term obeticholic acid therapy improves histological endpoints in patients with primary biliary cholangitis. Clin Gastroenterol Hepatol. 2020;18:1170–1178. doi: 10.1016/j.cgh.2019.09.050. e6. [DOI] [PubMed] [Google Scholar]
- 102.Honda A, Ikegami T, Nakamuta M, Miyazaki T, Iwamoto J, Hirayama T, et al. Anticholestatic effects of bezafibrate in patients with primary biliary cirrhosis treated with ursodeoxycholic acid. Hepatology. 2013;57:1931–1941. doi: 10.1002/hep.26018. [DOI] [PubMed] [Google Scholar]
- 103.Iwasaki S, Tsuda K, Ueta H, Aono R, Ono M, Saibara T, et al. Bezafibrate may have a beneficial effect in pre-cirrhotic primary biliary cirrhosis. Hepatology Research. 1999;16:12–18. [Google Scholar]
- 104.Corpechot C, Chazouillères O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A placebo-controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med. 2018;378:2171–2181. doi: 10.1056/NEJMoa1714519. [DOI] [PubMed] [Google Scholar]
- 105.Honda A, Tanaka A, Kaneko T, Komori A, Abe M, Inao M, et al. Bezafibrate improves GLOBE and UK-PBC scores and long-term outcomes in patients with primary biliary cholangitis. Hepatology. 2019;70:2035–2046. doi: 10.1002/hep.30552. [DOI] [PubMed] [Google Scholar]
- 106.Bolier R, de Vries ES, Parés A, Helder J, Kemper EM, Zwinderman K, et al. Fibrates for the treatment of cholestatic itch (FITCH): study protocol for a randomized controlled trial. Trials. 2017;18:230. doi: 10.1186/s13063-017-1966-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cheung AC, Lapointe-Shaw L, Kowgier M, Meza-Cardona J, Hirschfield GM, Janssen HL, et al. Combined ursodeoxycholic acid (UDCA) and fenofibrate in primary biliary cholangitis patients with incomplete UDCA response may improve outcomes. Aliment Pharmacol Ther. 2016;43:283–293. doi: 10.1111/apt.13465. [DOI] [PubMed] [Google Scholar]
- 108.Dohmen K, Mizuta T, Nakamuta M, Shimohashi N, Ishibashi H, Yamamoto K. Fenofibrate for patients with asymptomatic primary biliary cirrhosis. World J Gastroenterol. 2004;10:894–898. doi: 10.3748/wjg.v10.i6.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hegade VS, Khanna A, Walker LJ, Wong LL, Dyson JK, Jones DEJ. Long-term fenofibrate treatment in primary biliary cholangitis improves biochemistry but not the UK-PBC risk score. Dig Dis Sci. 2016;61:3037–3044. doi: 10.1007/s10620-016-4250-y. [DOI] [PubMed] [Google Scholar]
- 110.Harms MH, Janssen QP, Adam R, Duvoux C, Mirza D, Hidalgo E, et al. Trends in liver transplantation for primary biliary cholangitis in Europe over the past three decades. Aliment Pharmacol Ther. 2019;49:285–295. doi: 10.1111/apt.15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Adam R, Karam V, Delvart V, O’Grady J, Mirza D, Klempnauer J, et al. Evolution of indications and results of liver transplantation in Europe. A report from the European Liver Transplant Registry (ELTR) J Hepatol. 2012;57:675–688. doi: 10.1016/j.jhep.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 112.Singal AK, Guturu P, Hmoud B, Kuo YF, Salameh H, Wiesner RH. Evolving frequency and outcomes of liver transplantation based on etiology of liver disease. Transplantation. 2013;95:755–760. doi: 10.1097/TP.0b013e31827afb3a. [DOI] [PubMed] [Google Scholar]
- 113.The Japanese Liver Transplantation Society Liver transplantation in Japan. -Registry by the Japanese Liver Transplantation Society- Jpn J Transplant. 2015;52:134–147. [Google Scholar]
- 114.Bhanji RA, Mason AL, Girgis S, Montano-Loza AJ. Liver transplantation for overlap syndromes of autoimmune liver diseases. Liver Int. 2013;33:210–219. doi: 10.1111/liv.12027. [DOI] [PubMed] [Google Scholar]
- 115.Carbone M, Mells GF, Alexander GJ, Westbrook RH, Heneghan MA, Sandford RN, et al. Calcineurin inhibitors and the IL12A locus influence risk of recurrent primary biliary cirrhosis after liver transplantation. Am J Transplant. 2013;13:1110–1111. doi: 10.1111/ajt.12132. [DOI] [PubMed] [Google Scholar]
- 116.Charatcharoenwitthaya P, Pimentel S, Talwalkar JA, Enders FT, Lindor KD, Krom RA, et al. Long-term survival and impact of ursodeoxycholic acid treatment for recurrent primary biliary cirrhosis after liver transplantation. Liver Transpl. 2007;13:1236–1245. doi: 10.1002/lt.21124. [DOI] [PubMed] [Google Scholar]
- 117.Egawa H, Sakisaka S, Teramukai S, Sakabayashi S, Yamamoto M, Umeshita K, et al. Long-term outcomes of living-donor liver transplantation for primary biliary cirrhosis: a Japanese multicenter study. Am J Transplant. 2016;16:1248–1257. doi: 10.1111/ajt.13583. [DOI] [PubMed] [Google Scholar]
- 118.Guy JE, Qian P, Lowell JA, Peters MG. Recurrent primary biliary cirrhosis: peritransplant factors and ursodeoxycholic acid treatment post-liver transplant. Liver Transpl. 2005;11:1252–1257. doi: 10.1002/lt.20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hytiroglou P, Gutierrez JA, Freni M, Odin JA, Stanca CM, Merati S, et al. Recurrence of primary biliary cirrhosis and development of autoimmune hepatitis after liver transplant: a blind histologic study. Hepatol Res. 2009;39:577–584. doi: 10.1111/j.1872-034X.2008.00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jacob DA, Neumann UP, Bahra M, Klupp J, Puhl G, Neuhaus R, et al. Long-term follow-up after recurrence of primary biliary cirrhosis after liver transplantation in 100 patients. Clin Transplant. 2006;20:211–220. doi: 10.1111/j.1399-0012.2005.00471.x. [DOI] [PubMed] [Google Scholar]
- 121.Khettry U, Anand N, Faul PN, Lewis WD, Pomfret EA, Pomposelli J, et al. Liver transplantation for primary biliary cirrhosis: a long-term pathologic study. Liver Transpl. 2003;9:87–96. doi: 10.1053/jlts.2003.36392. [DOI] [PubMed] [Google Scholar]
- 122.Kogiso T, Egawa H, Teramukai S, Taniai M, Hashimoto E, Tokushige K, et al. Risk factors for recurrence of primary biliary cholangitis after liver transplantation in female patients: a Japanese multicenter retrospective study. Hepatol Commun. 2017;1:394–405. doi: 10.1002/hep4.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liermann Garcia RF, Evangelista Garcia C, McMaster P, Neuberger J. Transplantation for primary biliary cirrhosis: retrospective analysis of 400 patients in a single center. Hepatology. 2001;33:22–27. doi: 10.1053/jhep.2001.20894. [DOI] [PubMed] [Google Scholar]
- 124.Manousou P, Arvaniti V, Tsochatzis E, Isgro G, Jones K, Shirling G, et al. Primary biliary cirrhosis after liver transplantation: influence of immunosuppression and human leukocyte antigen locus disparity. Liver Transpl. 2010;16:64–73. doi: 10.1002/lt.21960. [DOI] [PubMed] [Google Scholar]
- 125.Montano-Loza AJ, Hansen BE, Corpechot C, Roccarina D, Thorburn D, Trivedi P, et al. Factors associated with recurrence of primary biliary cholangitis after liver transplantation and effects on graft and patient survival. Gastroenterology. 2019;156:96–107. doi: 10.1053/j.gastro.2018.10.001. e1. [DOI] [PubMed] [Google Scholar]
- 126.Montano-Loza AJ, Wasilenko S, Bintner J, Mason AL. Cyclosporine A protects against primary biliary cirrhosis recurrence after liver transplantation. Am J Transplant. 2010;10:852–858. doi: 10.1111/j.1600-6143.2009.03006.x. [DOI] [PubMed] [Google Scholar]
- 127.Neuberger J, Gunson B, Hubscher S, Nightingale P. Immunosuppression affects the rate of recurrent primary biliary cirrhosis after liver transplantation. Liver Transpl. 2004;10:488–491. doi: 10.1002/lt.20123. [DOI] [PubMed] [Google Scholar]
- 128.Sanchez EQ, Levy MF, Goldstein RM, Fasola CG, Tillery GW, Netto GJ, et al. The changing clinical presentation of recurrent primary biliary cirrhosis after liver transplantation. Transplantation. 2003;76:1583–1588. doi: 10.1097/01.TP.0000090867.83666.F7. [DOI] [PubMed] [Google Scholar]
- 129.Sylvestre PB, Batts KP, Burgart LJ, Poterucha JJ, Wiesner RH. Recurrence of primary biliary cirrhosis after liver transplantation: histologic estimate of incidence and natural history. Liver Transpl. 2003;9:1086–1093. doi: 10.1053/jlts.2003.50213. [DOI] [PubMed] [Google Scholar]
- 130.Bosch A, Dumortier J, Maucort-Boulch D, Scoazec JY, Wendum D, Conti F, et al. Preventive administration of UDCA after liver transplantation for primary biliary cirrhosis is associated with a lower risk of disease recurrence. J Hepatol. 2015;63:1449–1458. doi: 10.1016/j.jhep.2015.07.038. [DOI] [PubMed] [Google Scholar]
- 131.Jacoby A, Rannard A, Buck D, Bhala N, Newton JL, James OF, et al. Development, validation, and evaluation of the PBC-40, a disease specific health related quality of life measure for primary biliary cirrhosis. Gut. 2005;54:1622–1629. doi: 10.1136/gut.2005.065862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huet PM, Deslauriers J, Tran A, Faucher C, Charbonneau J. Impact of fatigue on the quality of life of patients with primary biliary cirrhosis. Am J Gastroenterol. 2000;95:760–767. doi: 10.1111/j.1572-0241.2000.01857.x. [DOI] [PubMed] [Google Scholar]
- 133.Mells GF, Pells G, Newton JL, Bathgate AJ, Burroughs AK, Heneghan MA, et al. Impact of primary biliary cirrhosis on perceived quality of life: the UK-PBC national study. Hepatology. 2013;58:273–283. doi: 10.1002/hep.26365. [DOI] [PubMed] [Google Scholar]
- 134.Prince M, Chetwynd A, Newman W, Metcalf JV, James OF. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: follow-up for up to 28 years. Gastroenterology. 2002;123:1044–1051. doi: 10.1053/gast.2002.36027. [DOI] [PubMed] [Google Scholar]
- 135.Jopson L, Jones DE. Fatigue in primary biliary cirrhosis: prevalence, pathogenesis and management. Dig Dis. 2015;33 Suppl 2:109–114. doi: 10.1159/000440757. [DOI] [PubMed] [Google Scholar]
- 136.Carbone M, Mells GF, Pells G, Dawwas MF, Newton JL, Heneghan MA, et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology. 2013;144:560–569. doi: 10.1053/j.gastro.2012.12.005. e7; quiz e13-e14. [DOI] [PubMed] [Google Scholar]
- 137.Grover VP, Southern L, Dyson JK, Kim JU, Crossey MM, Wylezinska-Arridge M, et al. Early primary biliary cholangitis is characterised by brain abnormalities on cerebral magnetic resonance imaging. Aliment Pharmacol Ther. 2016;44:936–945. doi: 10.1111/apt.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee JY, Danford CJ, Trivedi HD, Tapper EB, Patwardhan VR, Bonder A. Treatment of fatigue in primary biliary cholangitis: a systematic review and meta-analysis. Dig Dis Sci. 2019;64:2338–2350. doi: 10.1007/s10620-019-5457-5. [DOI] [PubMed] [Google Scholar]
- 139.Carbone M, Bufton S, Monaco A, Griffiths L, Jones DE, Neuberger JM. The effect of liver transplantation on fatigue in patients with primary biliary cirrhosis: a prospective study. J Hepatol. 2013;59:490–494. doi: 10.1016/j.jhep.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 140.Silveira MG, Gossard AA, Stahler AC, Jorgensen RA, Petz JL, Ali AH, et al. A randomized, placebo-controlled clinical trial of efficacy and safety: modafinil in the treatment of fatigue in patients with primary biliary cirrhosis. Am J Ther. 2017;24:e167–e176. doi: 10.1097/MJT.0000000000000387. [DOI] [PubMed] [Google Scholar]
- 141.Beuers U, Kremer AE, Bolier R, Elferink RP. Pruritus in cholestasis: facts and fiction. Hepatology. 2014;60:399–407. doi: 10.1002/hep.26909. [DOI] [PubMed] [Google Scholar]
- 142.Kremer AE, Martens JJ, Kulik W, Ruëff F, Kuiper EM, van Buuren HR, et al. Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology. 2010;139:1008–1018. doi: 10.1053/j.gastro.2010.05.009. e1. [DOI] [PubMed] [Google Scholar]
- 143.Kremer AE, Martens J, Kulik W, Williamson C, Moolenaar WH, Kondrackienė J, et al. Autotaxin but not bile salts correlate with itch intensity in cholestasis. J Hepatol. 2010;52:S1. [Google Scholar]
- 144.Kremer AE, van Dijk R, Leckie P, Schaap FG, Kuiper EM, Mettang T, et al. Serum autotaxin is increased in pruritus of cholestasis, but not of other origin, and responds to therapeutic interventions. Hepatology. 2012;56:1391–1400. doi: 10.1002/hep.25748. [DOI] [PubMed] [Google Scholar]
- 145.Tandon P, Rowe BH, Vandermeer B, Bain VG. The efficacy and safety of bile acid binding agents, opioid antagonists, or rifampin in the treatment of cholestasis-associated pruritus. Am J Gastroenterol. 2007;102:1528–1536. doi: 10.1111/j.1572-0241.2007.01200.x. [DOI] [PubMed] [Google Scholar]
- 146.European Association for the Study of the Liver EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67:145–172. doi: 10.1016/j.jhep.2017.03.022. [DOI] [PubMed] [Google Scholar]
- 147.Yagi M, Tanaka A, Namisaki T, Takahashi A, Abe M, Honda A, et al. Is patient-reported outcome improved by nalfurafine hydrochloride in patients with primary biliary cholangitis and refractory pruritus? A post-marketing, single-arm, prospective study. J Gastroenterol. 2018;53:1151–1158. doi: 10.1007/s00535-018-1465-z. [DOI] [PubMed] [Google Scholar]
- 148.Hegade VS, Kendrick SF, Dobbins RL, Miller SR, Thompson D, Richards D, et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet. 2017;389:1114–1123. doi: 10.1016/S0140-6736(17)30319-7. [DOI] [PubMed] [Google Scholar]
- 149.Mayo MJ, Pockros PJ, Jones D, Bowlus CL, Levy C, Patanwala I, et al. A randomized, controlled, phase 2 study of maralixibat in the treatment of itching associated with primary biliary cholangitis. Hepatol Commun. 2019;3:365–381. doi: 10.1002/hep4.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Floreani A, Franceschet I, Cazzagon N, Spinazzè A, Buja A, Furlan P, et al. Extrahepatic autoimmune conditions associated with primary biliary cirrhosis. Clin Rev Allergy Immunol. 2015;48:192–197. doi: 10.1007/s12016-014-8427-x. [DOI] [PubMed] [Google Scholar]
- 151.Guañabens N, Cerdá D, Monegal A, Pons F, Caballería L, Peris P, et al. Low bone mass and severity of cholestasis affect fracture risk in patients with primary biliary cirrhosis. Gastroenterology. 2010;138:2348–2356. doi: 10.1053/j.gastro.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 152.Guañabens N, Parés A, Ros I, Caballería L, Pons F, Vidal S, et al. Severity of cholestasis and advanced histological stage but not menopausal status are the major risk factors for osteoporosis in primary biliary cirrhosis. J Hepatol. 2005;42:573–577. doi: 10.1016/j.jhep.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 153.Guañabens N, Monegal A, Cerdá D, Muxí Á, Gifre L, Peris P, et al. Randomized trial comparing monthly ibandronate and weekly alendronate for osteoporosis in patients with primary biliary cirrhosis. Hepatology. 2013;58:2070–2078. doi: 10.1002/hep.26466. [DOI] [PubMed] [Google Scholar]
- 154.Arase Y, Tsuruya K, Hirose S, Ogiwara N, Yokota M, Anzai K, et al. Efficacy and safety of 3-year denosumab therapy for osteoporosis in patients with autoimmune liver diseases. Hepatology. 2020;71:757–759. doi: 10.1002/hep.30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lleo A, Bian Z, Zhang H, Miao Q, Yang F, Peng Y, et al. Quantitation of the rank-rankl axis in primary biliary cholangitis. PLoS One. 2016;11:e0159612. doi: 10.1371/journal.pone.0159612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sorokin A, Brown JL, Thompson PD. Primary biliary cirrhosis, hyperlipidemia, and atherosclerotic risk: a systematic review. Atherosclerosis. 2007;194:293–299. doi: 10.1016/j.atherosclerosis.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 157.Floreani A, Cazzagon N, Franceschet I, Canesso F, Salmaso L, Baldo V. Metabolic syndrome associated with primary biliary cirrhosis. J Clin Gastroenterol. 2015;49:57–60. doi: 10.1097/MCG.0000000000000029. [DOI] [PubMed] [Google Scholar]
- 158.Cavazza A, Caballería L, Floreani A, Farinati F, Bruguera M, Caroli D, et al. Incidence, risk factors, and survival of hepatocellular carcinoma in primary biliary cirrhosis: comparative analysis from two centers. Hepatology. 2009;50:1162–1168. doi: 10.1002/hep.23095. [DOI] [PubMed] [Google Scholar]
- 159.Harada K, Hirohara J, Ueno Y, Nakano T, Kakuda Y, Tsubouchi H, et al. Incidence of and risk factors for hepatocellular carcinoma in primary biliary cirrhosis: national data from Japan. Hepatology. 2013;57:1942–1949. doi: 10.1002/hep.26176. [DOI] [PubMed] [Google Scholar]
- 160.Trivedi PJ, Lammers WJ, van Buuren HR, Parés A, Floreani A, Janssen HL, et al. Stratification of hepatocellular carcinoma risk in primary biliary cirrhosis: a multicentre international study. Gut. 2016;65:321–329. doi: 10.1136/gutjnl-2014-308351. [DOI] [PubMed] [Google Scholar]
- 161.Rong G, Wang H, Bowlus CL, Wang C, Lu Y, Zeng Z, et al. Incidence and risk factors for hepatocellular carcinoma in primary biliary cirrhosis. Clin Rev Allergy Immunol. 2015;48:132–141. doi: 10.1007/s12016-015-8483-x. [DOI] [PubMed] [Google Scholar]
- 162.Imam MH, Silveira MG, Sinakos E, Gossard AA, Jorgensen R, Keach J, et al. Long-term outcomes of patients with primary biliary cirrhosis and hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2012;10:182–185. doi: 10.1016/j.cgh.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 163.Ellinghaus D, Folseraas T, Holm K, Ellinghaus E, Melum E, Balschun T, et al. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology. 2013;58:1074–1083. doi: 10.1002/hep.25977. [DOI] [PubMed] [Google Scholar]
- 164.Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet. 2016;48:510–518. doi: 10.1038/ng.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Folseraas T, Melum E, Rausch P, Juran BD, Ellinghaus E, Shiryaev A, et al. Extended analysis of a genome-wide association study in primary sclerosing cholangitis detects multiple novel risk loci. J Hepatol. 2012;57:366–375. doi: 10.1016/j.jhep.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet. 2017;49:269–273. doi: 10.1038/ng.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Karlsen TH, Franke A, Melum E, Kaser A, Hov JR, Balschun T, et al. Genome-wide association analysis in primary sclerosing cholangitis. Gastroenterology. 2010;138:1102–1111. doi: 10.1053/j.gastro.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 168.Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet. 2013;45:670–675. doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Melum E, Franke A, Schramm C, Weismüller TJ, Gotthardt DN, Offner FA, et al. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nat Genet. 2011;43:17–19. doi: 10.1038/ng.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Srivastava B, Mells GF, Cordell HJ, Muriithi A, Brown M, Ellinghaus E, et al. Fine mapping and replication of genetic risk loci in primary sclerosing cholangitis. Scand J Gastroenterol. 2012;47:820–826. doi: 10.3109/00365521.2012.682090. [DOI] [PubMed] [Google Scholar]
- 171.Harada K, Nakanuma Y. Prevalence and risk factors of hepatocellular carcinoma in Japanese patients with primary biliary cirrhosis. Hepatol Res. 2014;44:133–140. doi: 10.1111/hepr.12242. [DOI] [PubMed] [Google Scholar]