

Abstract
Homeobox B4 (HOXB4), which belongs to the homeobox (HOX) family, possesses transcription factor activity and has a crucial role in stem cell self-renewal and tumorigenesis. However, its biological function and exact mechanism in cervical cancer remain unknown. Here, we found that HOXB4 was markedly downregulated in cervical cancer. We demonstrated that HOXB4 obviously suppressed cervical cancer cell proliferation and tumorigenic potential in nude mice. Additionally, HOXB4-induced cell cycle arrest at the transition from the G0/G1 phase to the S phase. Conversely, loss of HOXB4 promoted cervical cancer cell growth both in vitro and in vivo. Bioinformatics analyses and mechanistic studies revealed that HOXB4 inhibited the activity of the Wnt/β-catenin signaling pathway by direct transcriptional repression of β-catenin. Furthermore, β-catenin re-expression rescued HOXB4-induced cervical cancer cell defects. Taken together, these findings suggested that HOXB4 directly transcriptional repressed β-catenin and subsequently inactivated the Wnt/β-catenin signaling pathway, leading to significant inhibition of cervical cancer cell growth and tumor formation.
Subject terms: Cancer, Cell growth
Introduction
Cervical cancer, which is now the fourth most commonly diagnosed type of cancer and the fourth leading cause of cancer-related death among females, causes an estimated 570,000 cases and 311,000 deaths in 2018 worldwide1. Notably, it remains to be the second leading cause of cancer-related death in women aged 20–39 years after breast cancer2. Human papillomavirus (HPV) infection is clearly a necessary but not a sufficient cause of cervical cancer1,3. However, the underlying pathogenesis is still unclear.
The WNT signal transduction cascade orchestrates embryonic development, tissue homeostasis, and regeneration4. However, dysregulation of the Wnt/β-catenin signaling pathway is closely associated with various diseases, including cancer5–8. Without Wnt signaling, β-catenin can be phosphorylated by a destruction complex (including Axin, APC, PP2A, GSK3, and CK1α), and subsequently degraded by the ubiquitin–proteasome system8. Conversely, Wnt binds to its receptors and recruits Axin to the plasma membrane, where it associates with phosphorylated LRP8. Subsequently, the destruction complex disrupts, and β-catenin is stabilized8. Then β-catenin access to the nucleus, binds to its nuclear partner TCF and upregulates the expression of target genes8. Accumulating evidence suggests that the canonical Wnt/β-catenin signaling is involved in the occurrence and progression of cervical cancer9–16.
Homeobox (HOX) gene, a subgroup of transcription factors containing a highly conserved homeodomain, has a pivotal role in development17. Aberrations of HOX gene expression have been found in cancers including cervical cancer18–20. HOXB4, the positive regulator implicated in the self-renewal of hematopoietic stem cells (HSCs)21, has been recognized as an oncogene or a tumor suppressor gene, depending on the specific type of cancer. HOXB4 was overexpressed in many cancers, including ovarian cancer22,23, cervical cancer24–27, lung cancer28, renal cancer29, mesothelioma30, and leukemia31–34, which promoted tumor growth and metastasis23. Moreover, increased HOXB4 expression resulted in drug resistance31,35. In addition, high HOXB4 expression was associated with a significantly shorter overall survival30. However, some studies have shown that HOXB4 was downregulated in cancer tissues36–40 and reduced in vitro cell proliferation and migration41. Other studies focused on the epigenetic alterations, showing hypermethylation of the HOXB4 promoter in cancers37–40. Although recent studies have demonstrated that HOXB4 was involved in cervical cancer, its contribution to cervical cancer remains largely unknown.
In this study, HOXB4 was determined to be downregulated in cervical squamous cell carcinoma. Functional studies showed that HOXB4 had a growth-inhibition role in cervical cancer cells. Bioinformatics analyses and mechanistic studies revealed that HOXB4 inhibited cervical tumorigenesis through direct transcriptional repression of β-catenin, which subsequently suppressed the activity of the Wnt/β-catenin signaling pathway.
Materials and methods
Tissue specimens
The study was approved by the First Affiliated Hospital of Xi’an Jiaotong University, and all patients were informed of the application of the surgical samples. A total of 97 specimens (including 41 normal cervix, 11 HSIL, and 45 cervical squamous cell cancer) were collected for this study. The histological classification of all primary tissues was confirmed by the International Federation of Gynecology and Obstetrics (FIGO) classification. Fresh specimens obtained during surgery were routinely fixed in formalin and embedded in paraffin or quickly frozen in liquid nitrogen for future RNA isolation and protein extraction.
Xenograft mouse model
This study was conducted following the guidelines of the Animal Care and Use Committee of the Medical School of Xi’an Jiaotong University. Six- to eight-week-old female BALB/c nude mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. Under specific pathogen-free conditions, mice were allowed to eat and drink freely and raised at a constant temperature on a 12-h day-night cycle. Mice were randomly divided into two groups and subcutaneously injected with 1 × 106 cervical cancer cells. At the end of the experiment, mice were euthanized by cervical dislocation. Tumors were harvested and quantified with tumor weight and tumor volume (0.5 × length × width × width).
Cell culture
All cell lines were purchased from American Type Culture Collection (ATCC) and tested for mycoplasma contamination. Cells were maintained in the recommended media containing 10% fetal bovine serum and 1% penicillin–streptomycin at 37 °C and 5% CO2. HeLa, SiHa, C-33A, and 293T cells were cultured in Dulbecco’s modified Eagle medium (Sigma-Aldrich), whereas CaSki cells were cultured in RPMI1640 medium (Sigma-Aldrich). To establish cell lines with stable expression genes or shRNAs, each cell line was transfected with the indicated plasmid using Lipofectamine 2000 reagent (Invitrogen) and selected by G418 (Calbiochem) for 3 weeks. For transient transfection, cells were transfected with plasmids and collected 48 h after being transfected.
PCR and qRT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen). Briefly, the sample was treated with 20% chloroform, vortexed, and centrifuged for 15 min. The upper aqueous phase (containing RNA) was collected and an equal volume of isopropanol was added, followed by a centrifugation step for 15 min. The RNA pellet was washed with 75% ethanol, air-dried, and then recovered in RNase-free water. The RNA template was converted into a cDNA using PrimeScript RT Reagent Kit (TaKaRa). The cDNA was then used as a template for PCR amplification using PrimeSTAR HS DNA Polymerase (TaKaRa) or qRT-PCR using SYBR Green Master Mix (Life). Primer sequences were listed in Supplementary Table 4.
Plasmid construction
All gene expression plasmids were generated by PCR amplification of the cDNA library and cloned into pIRES2-AcGFP-Neo (Clontech) expression plasmids. Short hairpin RNA (shRNA) was used to silence target gene expression and purchased from Shanghai GenePharma Co., Ltd. For the gene promoter dual-luciferase reporter vector, PCR amplification was performed from genomic DNA and then inserted into the pGL3-Basic luciferase vector (Promega). The mutated promoter dual-luciferase reporter vector was established by site-directed mutagenesis. Briefly, the plasmid template was amplified in a mutagenesis reaction with two overlapping primers containing the target mutation site. Then a digestion step by DpnI (NEB) was required to eliminate the original plasmid template. Finally, the recombination reaction and then transformation into E. coli competent cells were performed to isolate mutant plasmid. Detailed information regarding primers and oligonucleotide sequences was provided in Supplementary Table 4.
Immunohistochemistry and immunocytochemistry
Immunohistochemical staining of 5-μm-thick sections was performed using formalin-fixed, paraffin-embedded, and tissue specimens or xenograft tumor samples, according to standard protocols. Briefly, sections were deparaffinized, rehydrated, heated for antigen-retrieval, and pretreated with 3% H2O2 for 10 min. Sections were preincubated with 10% goat serum to block non-specific binding, incubated with primary antibodies overnight, and then added biotinylated secondary antibody. DAB was used as a chromogen, and hematoxylin was used for counterstaining. The IHC score was calculated by multiplying the staining grade (+0 unstained, +1 weak, +2 moderate, and +3 strong) with the staining ratio of cells (+0 <5%, +1 5–25%, +2 25–50%, +3 50–75% and +4 >75%). A score <3 was negative, while a score ≥3 was positive. For immunocytochemistry, cells grown on coverslips were fixed with 4% paraformaldehyde. After permeabilizing with 0.1% Triton X-100 and blocking with 10% goat serum, antibodies were applied to immunostaining cells. Detailed antibody information was provided in Supplementary Table 5.
Western blot
Cells were lysed on ice with RIPA lysis buffer pre-added with protease inhibitor Cocktail (Sigma-Aldrich) for 30 min, and then centrifuged at 4 °C for 15 min. Supernatants were measured by protein concentration assay (BCA, ThermoScientific) and then denatured by a 5×SDS loading buffer at 95 °C for 5 min. Nucleoproteins were extracted using Nuclear and Cytoplasmic Protein Extraction Kit (ThermoScientific). Proteins in the cell lysate were separated by SDS–PAGE gel electrophoresis and then transferred to a PVDF membrane. After blocking in 5% non-fat milk for 1 h, the membrane was incubated with primary antibodies under gentle agitation at 4 °C overnight. The membrane was then exposed to HRP-conjugated secondary antibody at room temperature for 1 h and subjected to chemiluminescence using Pierce ECL Western Blotting Substrate (ThermoScientific). Detailed antibody information was provided in Supplementary Table 5.
Oncomine database analysis
Using the Oncomine (www.oncomine.org) database, the gene expression of HOXB4 in cancer vs. normal tissue was analyzed (P < 0.001; fold change > 2).
RNA-Seq
Sequencing was carried out using the BGISEQ-500 platform at BGI Genomics Co., Ltd. The mRNA was enriched with oligo(dT)-attached magnetic beads. After purification, mRNA was cut into small fragments, and reverse transcription of cDNA was initiated by random hexamers. The A-Tailing Mix and RNA Index Adapters were ligated to the ends of cDNA fragments. The ligation products were then purified and amplified by PCR and validated for quality control. Reads were generated on BGIseq500 platform (BGI) after heating denaturation of PCR products. Using HISAT (http://ccb.jhu.edu/software/hisat), RNA-Seq reads were mapped to the reference genome (human assembly GRCh38.p11). The gene expression level was calculated by RSEM (http://deweylab.biostat.wisc.edu/RSEM). Differently expressed genes can be identified if its expression differed between any two groups with a fold change > 1 and the FDR < 0.01.
GSEA analysis
TCGA clinical data were derived from The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/), containing 306 cervical squamous cell carcinoma (CESC) patients. Normalized TCGA expression data and RNA-seq gene expression data were analyzed using GSEA software provided by the Broad Institute (http://www.broad.mit.edu/gsea). The enrichment score (ES) was the main result of gene set enrichment analysis, which reflected the degree to which a gene set was over-represented at the top or the bottom of a ranked list of genes. A positive ES indicated that the gene set was enriched at the top of the ranked list and correlated with the first phenotype; a negative ES indicated that the gene set was enriched at the bottom of the ranked list and correlated with the second phenotype.
Dual-luciferase reporter assay
Dual-luciferase reporter assay was performed using the Dual-Luciferase Reporter (DLR) Assay System (Promega). Briefly, 5 × 104 cells were inoculated into a 24-well culture plate and transiently transfected with 1 mg of the luciferase reporter plasmid and 0.1 mg of the Renilla luciferase plasmid (internal control). Cells were collected 48 h after transfection and the luciferase activity was evaluated at 420 nm. Results were shown as the fold change of the experimental group relative to the control group.
EMSA
Electrophoretic mobility shift assay (EMSA) was carried out using LightShift® Chemiluminescent EMSA Kit (ThermoScientific). Briefly, biotin 5′ end-labeled DNA was heated to 95 °C for 5 min and subsequently with 1 °C min-1 dropped to 4 °C. Typical binding reactions included 20 fmol dsDNA with different concentrations of HOXB4 nuclear protein extracts or 4 pmol unlabeled DNA and incubated at room temperature for 20 min. Reactions were loaded into a pre-run 6% native polyacrylamide gel, electrophoresed in 0.5× TBE buffer, transferred to a nylon membrane, and then cross-linked at a UV light. The bands were detected using chemiluminescence.
ChIP-qPCR
The chromatin immunoprecipitation (ChIP) assay was performed using the EZ-Magna ChIP Assay kit (Millipore) according to the manufacturer’s instructions. Briefly, cells were cross-linked with 1% formaldehyde for 10 min, quenched with 1× glycine for 5 min, washed with cold PBS, and incubated with a lysis buffer containing protease inhibitors for 15 min on ice. After centrifugation and discarding the supernatant, the cell pellet was collected and then sheared into DNA fragments by sonication. After centrifugation, the supernatant was immunoprecipitated using 5 μg antibodies against HOXB4 (Santa) or IgG (negative control) overnight at 4 °C and pulled down using fully re-suspended protein A/G magnetic beads. Finally, immunoprecipitation was collected, washed, and treated with proteinase K and RNase to purify DNA. The extracted DNA fragments were used as templates for qPCR analysis, and data were normalized with 5% input, respectively. Primers were presented in Supplementary Table 4.
Cell proliferation and cell cycle assays
Cell proliferation was measured by cell counting, MTT, and colony formation assays. For the cell counting assay, a total of 1 × 104 cells were inoculated in a 6-well culture plate for 7 days and counted every 2 days. For the MTT assay, cells were seeded in a 96-well culture plate at a density of 1000 cells, and 3-(4,5-dimethylthiazolyl)-2,5-diphenyl tetrazolium bromide (Sigma-Aldrich) was added to each well for 7 days. The OD value was measured at 490 nm every 2 days. For the colony formation assay, cells were inoculated in a 6-well culture plate with 1000 cells per well and collected on the 14th day. After washing twice with PBS, cells were fixed with 4% paraformaldehyde for 10 min and then stained with 0.1% crystal violet for 20 min. Visible colonies were counted and photographed. For cell cycle analysis, after synchronizing by serum starvation for 24 h, cells were inoculated in a 6-well culture plate at a density of 6 × 105 per well. Cells were harvested, fixed with 75% cold ethanol, stained with propidium iodide (Sigma-Aldrich) for 30 min, and then run on a FACS Calibur flow cytometer (BD Biosciences) using CellQuest software. Results were analyzed with FlowJo software.
Statistical analysis
SPSS 20.0 and GraphPad Prism 8.0 software were used for all statistical analyses. Before statistical analysis, data were analyzed for normal distribution and presented as mean ± s.d. Student’s t-test was used for comparisons between the two groups unless stated otherwise. P < 0.05 was considered significant and shown in the figure. All experiments were performed three or more times with similar results, independently under identical or similar conditions, except stated otherwise.
Results
HOXB4 was downregulated in cervical cancer
HOXB4, a member of the Antp homeobox family, encodes a protein with a homeobox DNA-binding domain, and functions as a sequence-specific transcription factor (Fig. 1a). Oncomine analysis (http://Oncomine.org) showed that HOXB4 was upregulated in breast cancer and downregulated in colorectal cancer, indicating that HOXB4 may have a dual role in cancer pathogenesis (Fig. 1b). To identify the potential role of HOXB4 in cervical cancer, we first performed immunohistochemistry (IHC) in 41 cases of the normal cervix (NC), 11 cases of HSIL, and 45 cases of squamous cell carcinoma (SCC), and tested the expression of HOXB4. The results were classified as negative and positive. Positive HOXB4 staining was observed in the cytoplasm and/or nucleus (Fig. 1c). The percentage of positive HOXB4 staining was 65.9% in NC (27/41), 45.5% in HSIL (5/11), and 31.1% in SCC (14/45) (Fig. 1d and Supplementary Table 1). Moreover, the average HOXB4 IHC score of SCC was much lower than that of NC (Fig. 1e). Additionally, we verified that HOXB4 was downregulated in randomly selected SCC patient tissue samples compared with normal specimens by western blot, which used GAPDH for normalization (Fig. 1f). Moreover, qRT-PCR showed that compared with the normal cervix, the expression of HOXB4 mRNA in randomly selected SCC patient tissues was reduced (Fig. 1g). These data indicated that compared with the normal cervix, the HOXB4 expression was downregulated in cervical cancer.
Fig. 1. HOXB4 was downregulated in cervical cancer.

a Illustration of the HOXB4 domains. The relative position of the hexapeptide domain mediating cofactor (PBX) binding and the homeodomain mediating DNA binding were shown. b Oncomine analysis of HOXB4 expression in human cancers compared with normal tissues (www.oncomine.org). c–e Expression of HOXB4 in normal cervix and cervical cancer. Histopathology analysis (IHC staining) of HOXB4 was performed in 41 normal cervix (NC), 11 high-grade squamous intraepithelial lesion (HSIL), and 45 squamous cervical cancer (SCC). Representative images (c) with various levels of staining were shown. Scale bar: 10 μm. HOXB4 IHC staining was classified into two categories (negative or positive). Numbers of positive cases were shown (d). The relative IHC score (e) of HOXB4 was quantified as the product of the grade of staining intensity and the percentage of positively stained cells. One-way ANOVA was used to analyze the data. f Expression of HOXB4 in human tissues. Proteins were extracted from eight normal cervix tissues and eight SCC tumors and assessed by WB, with GAPDH normalization (f, left). The relative protein expression (f, right) of HOXB4 was normalized to that of GAPDH using ImageJ software. g HOXB4 mRNA expression levels in ten normal cervix tissues and fourteen SCC tumors and assessed by qRT-PCR. Data were normalized to GAPDH.
HOXB4 suppressed tumorigenesis of cervical cancer cells in vivo
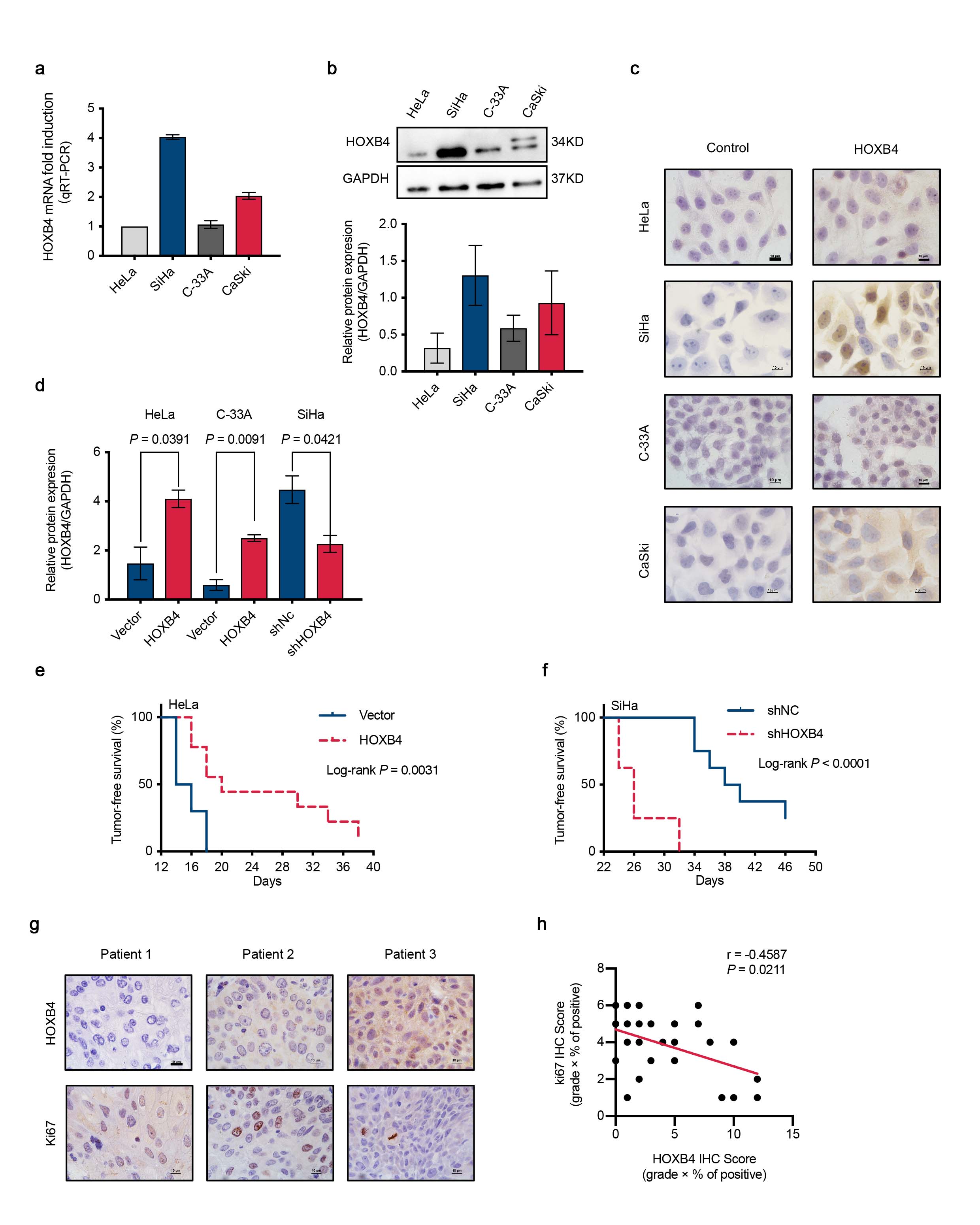
To elucidate the role of aberrant HOXB4 expression in cervical cancer, we detected HOXB4 mRNA and protein levels in a group of cervical cancer cell lines. Whereas SiHa cells showed abundant expression, HOXB4 was moderately expressed in HeLa, C-33A, and CaSki cell lines (Supplementary Fig. 1a–c). Next, both loss-of-function and gain-of-function of HOXB4 were performed in cervical cancer cell lines. We generated HeLa and C-33A cell lines stably overexpressing HOXB4 or a SiHa cell line with stably knocking down of HOXB4 by two independently targeted short hairpin RNAs (shRNAs) (Fig. 2a and Supplementary Fig. 1d).
Fig. 2. HOXB4 suppressed cervical tumorigenesis in vivo.

a HOXB4 expression in modified cervical cancer cell lines. HOXB4 protein expression in HeLa (a, left) and C-33A (a, middle) cells transfected with either empty vector (Vector) or HOXB4 overexpression vector (HOXB4) plasmid. SiHa (a, right) cells were transfected with either control (shNC) or shRNAs targeting the HOXB4 (shHOXB4) plasmid and detected using WB. GAPDH was used as a loading control. b–g Suppression of tumorigenesis by HOXB4. Each mouse (n = 8 biologically independent samples) was subcutaneously injected with 1 × 106 HeLa cells into the left (b black arrows, Vector) and right (b white arrows, HOXB4). SiHa cells were also injected (e left: black arrows, shNC; right: white arrows, shHOXB4). At the end of the experiment, mice were killed and tumors were harvested. Representative images of mice and tumors were shown (b, e). Tumor volumes (mm3) (c, f) and tumor weight (g) (d, g) were plotted. h–i Expression of Ki67. Immunohistochemical staining was performed with HOXB4 and Ki67 (h) from mouse xenograft tumors and quantification by IHC scores (i). Representative images (h) were shown. Scale bar: 10 μm.
To investigate the function of HOXB4 in cervical cancer cells in vivo, nude mice were injected HOXB4-overexpressed HeLa cells and HOXB4-depleted SiHa cells subcutaneously and analyzed for tumor formation. Mice engrafted with HOXB4-overexpressed cancer cells had smaller tumor volumes throughout the experiment than mice engrafted with control cells (Fig. 2b). At 38 days after tumor cell implantation, we observed a 94.4% decrease in the size and a 96.5% decrease in the weight of the tumors formed by HOXB4-overexpressed HeLa cells compared with those of the control tumors (Fig. 2c, d). Notably, HOXB4 overexpression significantly extended the tumor-free survival of animal (Supplementary Fig. 1e). On day 48, compared to the control mice, mice implanted with HOXB4-depleted SiHa cells showed larger tumor volumes, with a 17.1-fold increase in tumor size and a 5.8-fold increase in tumor weight; these mice also had shortened tumor-free survival (Fig. 2e–g and Supplementary Fig. 1f). In addition, the results showed that the IHC score for Ki67 staining was significantly decreased in the tumors stably overexpressing HOXB4, but markedly increased in the tumors with stable knockdown of HOXB4, compared with control tumors (Fig. 2h, i), demonstrating that HOXB4 suppressed tumorigenesis by inhibiting cervical cancer cell proliferation in vivo. Consistent with our observation of in vivo experiments, IHC staining of primary tumors indicated that HOXB4 expression was negatively correlated with Ki67 in cervical cancer patients (Supplementary Fig. 1g, h).
HOXB4 inhibited the proliferation of cervical cancer cells and arrested the cell cycle in vitro
To further verify whether HOXB4 inhibited cervical cancer cell growth in vitro, we performed cell counting, MTT, and colony formation experiments. Compared with the control, the overexpression of HOXB4 in the HeLa cell line significantly reduced cell proliferation and decreased colony formation (Fig. 3a–c). The inhibitory effect of HOXB4 on cell proliferation was further confirmed in the C-33A cell line (Fig. 3d–f). Conversely, HOXB4 knockdown in the SiHa cell line markedly increased cell growth and colony formation relative to control shRNA (Fig. 3g–i). Taken together, these data indicated that HOXB4 inhibited the growth of cervical cancer cells in vitro.
Fig. 3. HOXB4 inhibited cell proliferation and arrested the cell cycle in vitro.

a–i Inhibition of cell proliferation by HOXB4. Cell counting (a), MTT (b), and colony formation (c) assays were performed in HOXB4-overexpressing HeLa cells and control cells. Representative images of the colony formation assay with crystal violet staining (c, right) were shown, and colonies were quantified by counting (c, left). These proliferation experiments (cell counting, MTT, and colony formation assays) were also performed in HOXB4-overexpressing C-33A cells (d–f) and HOXB4-depleted SiHa cells (g–i). j–o Cell cycle distribution in modified transfected cervical cancer cell lines. Cell cycle profiles were obtained by FACS analysis of propidium iodide-stained cells and analyzed by flow cytometry in HOXB4-overexpressing HeLa (j, k) and C-33A (l, m) cells, as well as in HOXB4-depleted SiHa (n, o) cells.
To explore the mechanism of HOXB4 inhibiting the proliferation of cervical cancer cells, flow cytometry was used to analyze the cell cycle of cells stained with propidium iodide. The data showed that, compared with control cells, HeLa cells with continuous upregulation of HOXB4 caused an increase in the percentage of cells in the G0/G1 phase and a decrease in the percentage of cells in the S phase (Fig. 3j, k). Similar results were also observed in C-33A cells (Fig. 3l, m). However, down-regulation of HOXB4 accelerated the G1/S transition, reduced the ratio of G0/G1 phase SiHa cells, and increased the ratio of S phase cells (Fig. 3n, o). Collectively, the cell cycle profiles suggested that HOXB4 inhibited the G1/S transition.
HOXB4 downregulated the activity of the Wnt/β-catenin signaling pathway
To explore the molecular mechanism of HOXB4 in cervical cancer cells, RNA sequencing (RNA-seq) analysis was performed using three HeLa cell lines with stable overexpression of HOXB4 and three control cell lines. A total of 17,894 genes were detected. Gene set enrichment analysis (GSEA) results of six samples revealed that 28/50 gene sets were upregulated by phenotype Group_Vector and 22/50 gene sets were upregulated by phenotype Group_HOXB4 (Supplementary Table 2). Of these gene sets, changes in the Wnt/β-catenin signaling pathway were identified after modification of HOXB4. CTNNB1 and MYC are two major molecules in the Wnt/β-catenin signaling pathway4, which were significantly decreased due to the overexpression of HOXB4, in the HeLa cell line, as shown in our RNA-seq results (Fig. 4a and Supplementary Table 3).
Fig. 4. HOXB4 inactivated the Wnt/β-catenin signaling pathway.

a Heat map of HOXB4 RNA-seq results. All enriched features of the enriched gene sets, which were filtered and analyzed by GSEA analysis of HOXB4 RNA-seq data, were shown between the vector control (Vector) and HOXB4-overexpressing (HOXB4) HeLa cells. Gene expression values were represented as colors, where the range of colors (red, pink, light blue, dark blue) showed the range of expression values (high, moderate, low, lowest). n = 3 per group. b–g Expression of β-catenin and c-Myc. The mRNA levels of HOXB4, β-catenin, and c-Myc were detected by qRT-PCR in HOXB4-overexpressing HeLa (b) and C-33A (d) cells, as well as in HOXB4-depleted SiHa (f) cells. Data were normalized to GAPDH. The protein expression of HOXB4, total β-catenin, nuclear β-catenin, and c-Myc was assessed in HOXB4-modified HeLa (c), C-33A (e), and SiHa (g) cells by western blot and normalized to GAPDH and histone H3 (H3) respectively. h–j Inactivation of the Wnt/β-catenin reporter luciferase by HOXB4. HeLa (h), C-33A (i), or SiHa (j) cells transfected with the indicated plasmids were analyzed by β-catenin reporter luciferase assays (TOPFlash (TOP) and FOPFlash (FOP)).
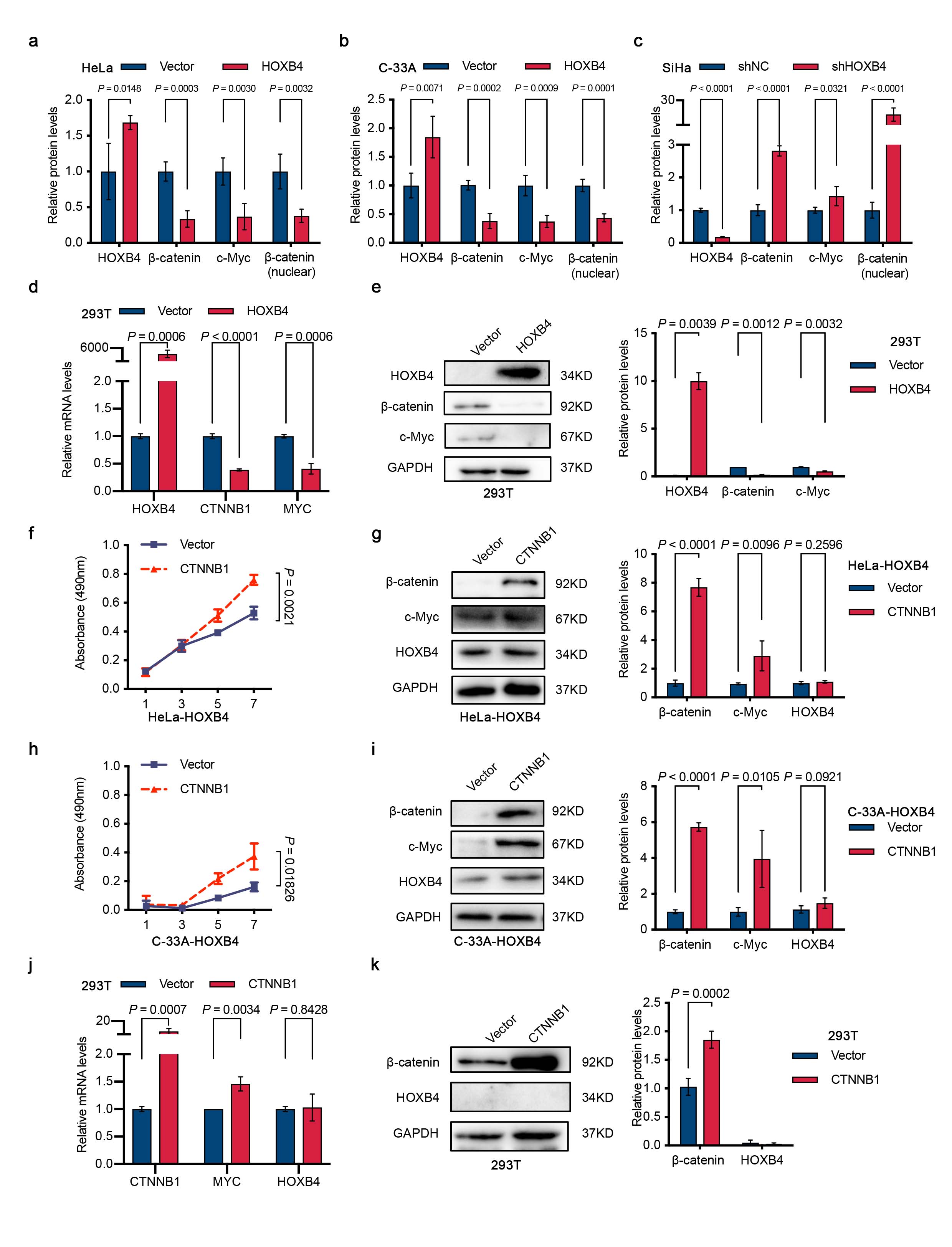
To further verify the result indicated by the bioinformatics analysis of RNA-seq, quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and western blot experiments confirmed that HOXB4 overexpression reduced the mRNA and protein levels of β-catenin and the target gene c-Myc, in HeLa and C-33A cells (Fig. 4b–e and Supplementary Fig. 2a, b). Consistent with the down-regulation of total β-catenin levels, the nucleoprotein level also decreased in the HeLa and C-33A cell lines where HOXB4 was overexpressed (Fig. 4c, e and Supplementary Fig. 2a, b). In contrast, knocking down of HOXB4 in the SiHa cell line resulted in β-catenin accumulation and increased c-Myc expression, both at the mRNA and protein levels (Fig. 4f, g and Supplementary Fig. 2c). Likewise, the β-catenin level in the nuclear fraction of SiHa cells knocked down by HOXB4 was increased compared to control cells (Fig. 4g and Supplementary Fig. 2c). In addition, in 293T cells, overexpression of HOXB4 suppressed the expression levels of β-catenin and c-Myc (Supplementary Fig. 2d, e).
The TOP/FOP flash luciferase reporter assay was used to detect the activity of the Wnt/β-catenin signaling pathway, showing an approximately 71.6% reduction and 56.0% reduction in the activity of the β-catenin reporter (TOP flash) in HeLa and C-33A cells stably overexpressing HOXB4, respectively, compared with the control cells (Fig. 4h, i). In contrast, HOXB4 knockdown in SiHa cells resulted in a 2.4-fold increase in β-catenin reporter activity (Fig. 4j). Collectively, these results indicated that HOXB4 downregulated Wnt/β-catenin signaling pathway activity.
In addition, the rescue experiment, in which we restored the expression of β-catenin in the HeLa and C-33A cell lines overexpressing HOXB4, reversed the growth inhibitory effect of HOXB4 in vitro and upregulated the expression of the downstream gene c-Myc, without changing HOXB4 (Supplementary Fig. 2f–i). Similarly, transiently transfected 293T cells with β-catenin did not affect either the mRNA level or the protein levels of HOXB4, compared with cells transfection with empty vector control (Supplementary Fig. 2j, k). These data confirmed that HOXB4 inhibited the activity of the Wnt/β-catenin signaling pathway by downregulating β-catenin expression; however, the Wnt/β-catenin signaling pathway had no effect on HOXB4 in cervical cancer cells.
HOXB4 bound directly to the promoter of β-catenin
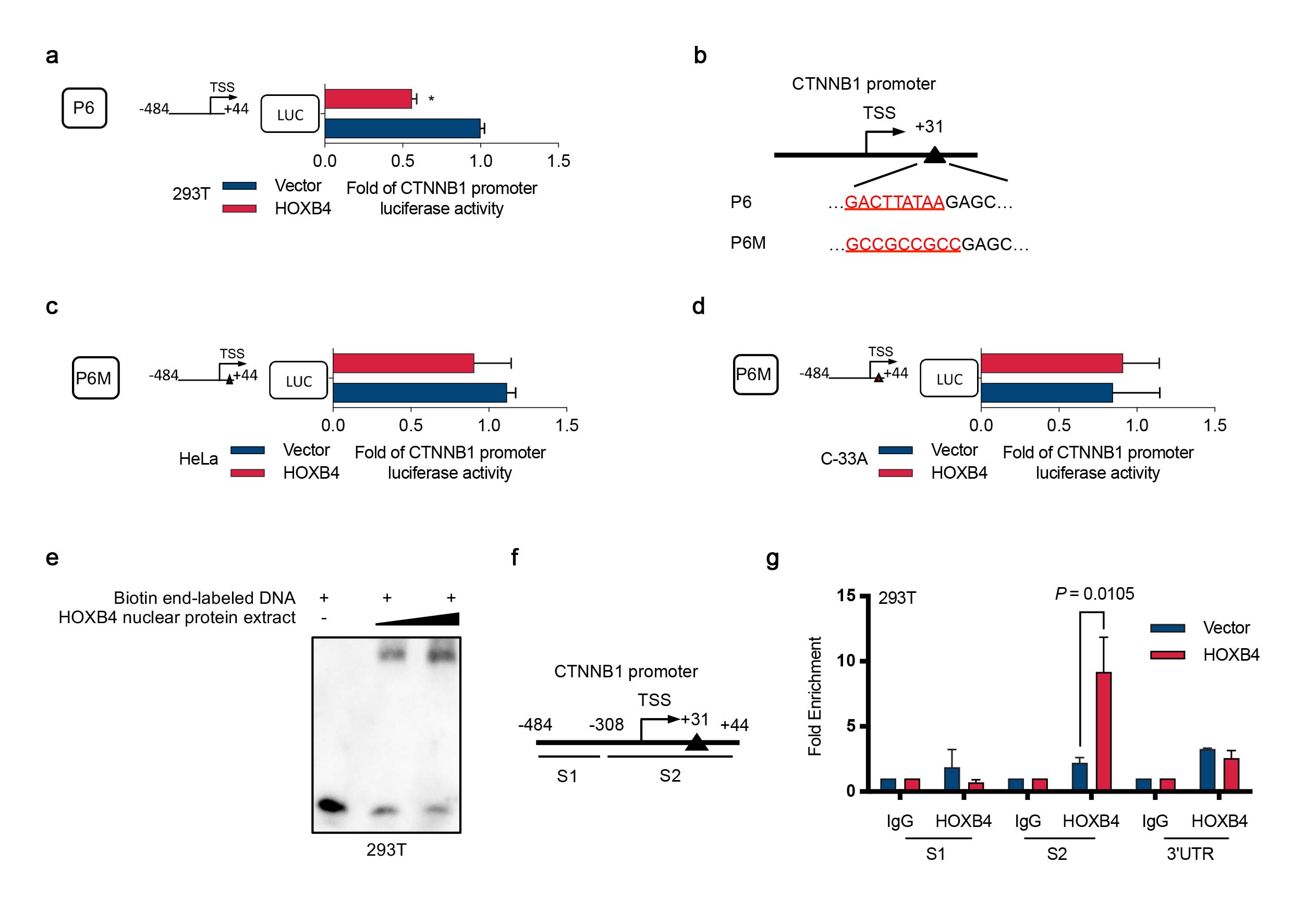
Analysis of the binding site of HOXB4 using the JASPAR database (http://jaspar.genereg.net/) revealed that the HOXB4 DNA-binding motif contained 5′-TC/AATTA-3′ (Fig. 5a). In addition, the transcription factor binding site search and visualization integrated web tool, the LASAGNA-Search 2.0 (https://biogrid-lasagna.engr.uconn.edu/) showed that HOXB4 bound to the CTNNB1 gene. To test whether HOXB4 transcriptionally regulated β-catenin expression, we searched the β-catenin promoter region of approximately 2 kb (−2000 to +44 bp, relative to the transcription start site [TSS]) in the UCSC genome browser (http://genome.ucsc.edu/), and constructed six promoter reporters (P1-P6) containing different fragments (Fig. 5b). In HeLa and C-33A cells overexpressing HOXB4, the luciferase activity of P6 (−484 to +44 bp) was decreased, whereas in SiHa cells knocking down HOXB4 increased P6 luciferase activity (Fig. 5b–d). In agreement with these findings, overexpression of HOXB4 in 293T cells downregulated the P6 luciferase reporter activity (Supplementary Fig. 3a). Based on the above data, we searched the β-catenin promoter region P6, found the putative HOXB4 binding site, and constructed a mutation of the P6 promoter-reporter (P6M) (Supplementary Fig. 3b). Notably, the mutation of the binding site abolished the HOXB4-mediated reduction in promoter activity (Supplementary Fig. 3c, d). These data suggested that HOXB4 transcription suppressed the expression of β-catenin.
Fig. 5. HOXB4 bound to the promoter of β-catenin directly.

a Transcription factor binding sites of HOXB4. b–d Luciferase assays assessing the promoter of β-catenin. β-catenin promoter fragments were constructed, and luciferase activity relative to the control was measured in HOXB4-modified HeLa (b), C-33A (c), and SiHa (d) cells. e, f EMSA assay for the promoter of β-catenin. EMSA was used to detect the direct association of nuclear HOXB4 protein with its binding sites on the promoter regions of β-catenin in HOXB4-overexpressing HeLa (e) and C-33A (f) cells. g, h ChIP assays for the promoter of β-catenin. ChIP-qPCR assay in HOXB4-overexpressing HeLa (g) and C-33A (h) cells were performed with HOXB4 antibody and IgG antibody (negative control).
Electrophoretic mobility shift assay (EMSA) was used to determine whether the HOXB4 nuclear protein interacted directly with the putative HOXB4 binding site in HOXB4-overexpressed HeLa and C-33A cells, which was confirmed by the “shift” in migration of the labeled DNA band (Fig. 5e, f). Consistent with our findings, there was a direct interaction between HOXB4 protein and the HOXB4 binding site, 5′-ATAA-3′, in HOXB4-overexpressing 293T cells (Supplementary Fig. 3e). Also, Chromatin immunoprecipitation (ChIP)-qPCR with HOXB4 antibody and IgG (control) indicated enrichment of the β-catenin promoter region S2 (−308 to +44) with the HOXB4 antibody, confirming that HOXB4 inhibited β-catenin transcription by binding to its promoter directly (Fig. 5g, h and Supplementary Fig. 3f). Similar results were observed in 293T cells (Supplementary Fig. 3g). Thus, these data showed that HOXB4 inhibited β-catenin transcription by binding to its promoter at the HOXB4 binding site, 5′-ATAA-3′, directly.
HOXB4 was negatively correlated with Wnt/β-catenin signaling activity in cervical cancer tissues
To further confirm the growth inhibitory effect of HOXB4 was related to the activity of the Wnt/β-catenin signaling pathway in vivo, we performed immunohistochemical staining of xenograft tumors with the indicated antibodies. Compared with the control group, the overexpression of HOXB4 significantly reduced the expression of β-catenin and c-Myc (Fig. 6a, b). Conversely, HOXB4 knockdown upregulated the expression of β-catenin and c-Myc, relative to the control group (Fig. 6c, d).
Fig. 6. HOXB4 was negatively correlated with β-catenin.

a–d Expression of HOXB4, β-catenin, and c-Myc in vivo. The expression of HOXB4, β-catenin, and c-Myc was measured in the xenograft tumors formed by HOXB4-modified HeLa (a, b) and SiHa (c, d) cells using IHC staining. Representative images were shown. Scale bar: 10 μm. e–g Expression of HOXB4/β-catenin/c-Myc in human cervical cancer. IHC analysis of HOXB4, β-catenin, and c-Myc (e) and quantification of IHC scores (f, g) of human cervical cancer samples. Representative images (e) were shown. Scale bar: 10 μm. Correlation between HOXB4 and β-catenin (f) or c-Myc (g) was calculated. n = 30, Pearson chi-square test.
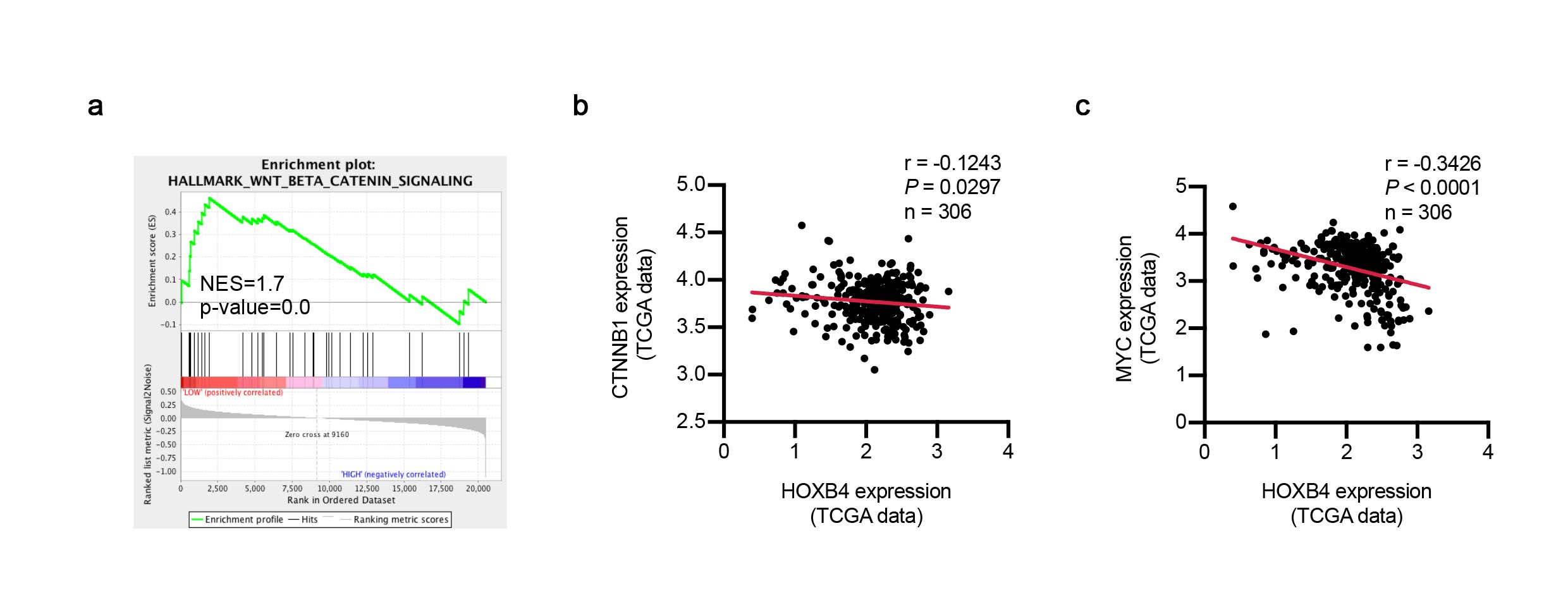
To investigate the relevance of our findings to human cervical cancer, we searched the cervical cancer database from The Cancer Genome Atlas (TCGA) and performed GSEA analysis. We found that LOW group (low expression of HOXB4) was enriched the Wnt/catenin signaling gene set (Supplementary Fig. 4a). Moreover, TCGA data revealed that the expression of HOXB4, β-catenin, and c-Myc showed a significant negative correlation in cervical cancer (Supplementary Fig. 4b, c). The immunohistochemical staining of cervical cancer patient samples further verified the correlation between HOXB4, β-catenin, and c-Myc (Fig. 6e). The IHC score also showed that HOXB4 expression was negatively correlated with β-catenin and c-Myc expression (Fig. 6f, g).
Collectively, in cervical cancer, the absence of HOXB4 upregulated β-catenin and its downstream gene c-Myc, which contributed to the enhancement of the cell cycle and promoted tumor progression. In HOXB4-overexpressed cervical cancer cells, HOXB4 was accessed to the nucleus and transcriptionally repressed β-catenin expression, leading to impaired nuclear accumulation of β-catenin, which subsequently contributed to the inhibition of tumorigenesis (Fig. 7).
Fig. 7. A model of the HOXB4/β-catenin/c-Myc in cervical cancer.

In cervical cancer, loss of HOXB4 upregulated β-catenin and its downstream gene c-Myc, which contributed to the enhancement of the cell cycle and promoted tumor progression. In HOXB4-overexpressing cervical cancer cells, HOXB4 was accessed to the nucleus and transcriptionally repressed β-catenin expression, leading to impaired nuclear accumulation of β-catenin, which subsequently contributed to the inhibition of tumor progression.
Discussion
The HOX gene family encodes a class of transcription factors, which have a crucial role in embryonic development. Studies have also shown that their aberrant expression is related to cancers18,42–44. HOXB4, a member of the Antp homeobox protein family, regulates embryonic development and participates in tumor progression as an oncogene or tumor suppressor gene. In ovarian cancer, the expression of HOXB4 was closely related to poor prognosis and transcriptionally activated DHDDS to stimulate the proliferation and invasion of tumor cells23. Other studies also revealed that HOXB4 was upregulated in ovarian cancer cells and drug-resistant cells35. Down-regulation of HOXB4 enhanced the cytotoxic effects of Taxol and DDP on ovarian cancer cells35. Therefore, in ovarian cancer, HOXB4 may act as an oncogene. However, the primary role of HOXB4 in breast cancer seems to be tumor suppression. HOXB4 inhibited cell migration and EMT by regulating the STARD13-related signaling pathway36. In addition, HOXB4 was downregulated in most breast cancer tissues36,45, and its overexpression enhanced sensitivity to doxorubicin36. Hence, HOXB4 may serve as a tumor suppressor gene or an oncogene, depending on the specific cancers.
In the present study, we sought to investigate the role and molecular mechanism of HOXB4 in cervical cancer. First of all, our research demonstrated that compared with the normal cervix, HOXB4 was significantly downregulated in cervical cancer, which was inconsistent with previous studies24–26. Barba-de detected the expression of HOXB4 in paraffin-embedded tissue microarray in cervical tissues by immunostaining24. HOXB4 immunoreactivity was observed in almost all cervical cancer tissues (69/70), but not in normal tissues (0/3)24. However, their study only detected HOXB4 expression in three normal cervical epithelium tissues, and the sample size was too small to draw any conclusions. HOXB4 staining was seen in the cytoplasm and/or nucleus of positive cells in normal cervical tissues (Fig. 1c). In normal tissues, the negative staining rate of HOXB4 was 34.1% (14/41) and the positive staining rate was 65.9% (27/41) (Fig. 1d and Supplementary Table 1). In addition, they did not consider the expression of HOXB4 in adjacent tissues, which were positive in all samples (19/19)24. Moreover, it is inappropriate to evaluate immunohistochemistry results only by negative and positive staining. In our study, according to the average IHC score (combined staining intensity and positive cell ratio), HOXB4 staining was divided into two categories, negative and positive. The HOXB4 immunoreactivity score revealed that HOXB4 was expressed at different levels and significantly higher than that in cervical cancer (Fig. 1e). Lopez found that HOXB4 was only expressed in 3/11 normal cervical tissues but in the most cervical carcinoma samples (16/17) using RT-PCR25. A similar result showed the absence of HOXB4 gene expression in 14 normal cervical tissues26. These data were inconsistent with ours. The HOXB4 mRNA expression in 10 normal cervical specimens was 30.6 (9.5–52.7) detected by qRT-PCR, which was higher than the level of 14 specimens of squamous cervical cancer [7.7 (3.3–12.2)] (95% CI, P = 0.0155) (Fig. 1g). Furthermore, western blot confirmed HOXB4 was upregulated in normal cervix compared with cervical cancer (Fig. 1f). Secondly, we conducted a series of in vitro and in vivo experiments using cervical cancer cell lines with gain or loss of function of HOXB4, and identified that HOXB4 inhibited cell proliferation and arrested the cell cycle at the transition from G0/G1 phase to S phase. Thus, our findings clearly proved that HOXB4 had as a tumor growth-inhibitor in cervical cancer.
Mechanism studies showed that HOXB4 negatively regulated Wnt/β-catenin signaling pathway. Mutations in the Wnt pathway and its components are responsible for a variety of growth-related pathologies and cancers. Transcription factors of the HOX family have an important role in numerous cellular processes, including cell growth by regulating various signaling pathways, such as the Wnt/β-catenin signaling pathway46. Studies on human non-small cell lung cancer (NSCLC) have shown that knocking down HOXB5 significantly inhibited cell proliferation by inhibiting β-catenin and the downstream targets c-Myc and cyclin D147. In the cutaneous squamous cell carcinoma (CSCC) cells, HOXB7 bound to β-catenin, and HOXB7 knockdown reduced cell viability and tumor growth by inhibiting the Wnt/β-catenin signaling pathway48. HOXA9 transcriptional activation of WNT6 ultimately activated the canonical WNT/β-catenin pathway in glioblastoma (GBM)49. HOX proteins not only promote the development of cancer but also inhibit tumorigenesis by downregulating the Wnt/β-catenin signaling. In lung cancer, HOXA4 was downregulated and inhibited the cell growth by promoting the transcription of GSK3β, thereby inhibiting Wnt signaling50. In prostate cancers, HOXB13 downregulated TCF4 and its responsive genes c-Myc and cyclin D1, subsequently inhibiting cell growth51. Similarly, in colorectal cancer, HOXB13 downregulated the protein expression of TCF4, thereby hindering the cell growth52. Our results showed that HOXB4 directly bound to the promoter of β-catenin at 5′-ATAA-3′, transcriptionally inhibited its expression and subsequently suppressed the activity of the Wnt/β-catenin signaling pathway, which was consistent with previous studies42 that the homeodomain (evolutionally conserved 60-amino-acid long DNA-binding domain) only recognized and bound to four base-pair sequences (TAAT/ATTA/ TTAT/ATAA).
GESA analysis of the data from TCGA-CESC confirmed that HOXB4 inhibited the activity of the Wnt/β-catenin signaling pathway and correlation analysis showed that in cervical cancer, the expression of HOXB4, β-catenin, and c-myc genes were significantly negatively correlated (Supplementary Fig. 4). However, the clinical correlation between HOXB4 and β-catenin gene expression in TCGA-CESC was low (r = −0.1243, P < 0.05). There may be two explanations. First, the patient’s gene expression was affected by many factors, such as pathological type, pathological stage, age, drug therapy, basic gene expression level, etc. These factors will affect the expression of mRNA. Second, the pathological tissue of the patient we obtained may include multiple cell types, such as cervical cancer cells, stromal cells, immune cells, etc., which may affect the correlation between the two genes in cancer tissues.
In summary, we found that HOXB4 was markedly downregulated in cervical cancer. A series of in vitro and in vivo experiments proved that HOXB4 functioned as a growth-inhibition role in cervical cancer. Mechanistically, HOXB4 inhibited cervical tumorigenesis by repressing the transcription of β-catenin, thereby inhibiting Wnt/β-catenin signaling pathway activity.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Lu Li for the help in plasmid construction experiments and Nan Cui for the help in the experimental design. We thank all members of the Peng-Sheng Zheng laboratory for their critical reading and suggestions. This work was supported by a grant to Prof. Peng-Sheng Zheng from the National Natural Science Foundation of China (Nos. 81472728 and 81672910).
Author contributions
D.L. designed and performed the experiments, analyzed the data, and wrote the manuscript. W.T.Y. provided advice, helped with data analysis, and edited the manuscript. P.S.Z. supervised the overall study, provided funding support, designed experiments, analyzed data, and wrote the manuscript. All authors read and approved the final paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University with informed consent from patients.
Footnotes
Edited by G. Blandino
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information The online version contains supplementary material available at 10.1038/s41419-021-03411-6.
References
- 1.Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2020;28:394. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 3.Walboomers JMM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 4.Huang P, et al. Activating Wnt/beta-catenin signaling pathway for disease therapy: challenges and opportunities. Pharmacol. Therap. 2019;196:79–90. doi: 10.1016/j.pharmthera.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Ring A, Kim Y-M, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. Rep. 2014;10:512–525. doi: 10.1007/s12015-014-9515-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duchartre Y, Kim Y-M, Kahn M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 2016;99:141–149. doi: 10.1016/j.critrevonc.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nusse R, Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Cao H-Z, Zheng P-S. LGR5 promotes the proliferation and tumor formation of cervical cancer cells through the Wnt/beta-catenin signaling pathway. Oncotarget. 2014;5:9092–9105. doi: 10.18632/oncotarget.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui N, Yang W-T, Zheng P-S. Slug inhibits the proliferation and tumor formation of human cervical cancer cells by up-regulating the p21/p27 proteins and down-regulating the activity of the Wnt/beta-catenin signaling pathway via the trans-suppression Akt1/p-Akt1 expression. Oncotarget. 2016;7:26152–26167. doi: 10.18632/oncotarget.8434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Zheng P-S, Yang W-T. EZH2-mediated repression of GSK-3 beta and TP53 promotes Wnt/beta-catenin signaling-dependent cell expansion in cervical carcinoma. Oncotarget. 2016;7:36115–36129. doi: 10.18632/oncotarget.8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao H-Z, Liu X-F, Yang W-T, Chen Q, Zheng P-S. LGR5 promotes cancer stem cell traits and chemoresistance in cervical cancer. Cell Death Dis. 2017;8:e3039. doi: 10.1038/cddis.2017.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X-F, Li X-Y, Zheng P-S, Yang W-T. DAX1 promotes cervical cancer cell growth and tumorigenicity through activation of Wnt/beta-catenin pathway via GSK3 beta. Cell Death Dis. 2018;9:339. doi: 10.1038/s41419-018-0359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Yang W-T, Zheng P-S, Liu X-F. SOX17 restrains proliferation and tumor formation by down-regulating activity of the Wnt/ss-catenin signaling pathway via trans-suppressing ss-catenin in cervical cancer. Cell Death Dis. 2018;9:741. doi: 10.1038/s41419-018-0782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen M, Li L, Zheng P-S. SALL4 promotes the tumorigenicity of cervical cancer cells through activation of the Wnt/beta-catenin pathway via CTNNB1. Cancer Sci. 2019;110:2794–2805. doi: 10.1111/cas.14140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burk RD, et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378. doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abate-Shen C. Deregulated homeobox gene expression in cancer: Cause or consequence? Nat. Rev. Cancer. 2002;2:777–785. doi: 10.1038/nrc907. [DOI] [PubMed] [Google Scholar]
- 18.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer. 2010;10:361–371. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- 19.Zhai Y, et al. Gene expression analysis of Preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007;67:10163–10172. doi: 10.1158/0008-5472.CAN-07-2056. [DOI] [PubMed] [Google Scholar]
- 20.Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J. Mol. Med. 2014;92:811–823. doi: 10.1007/s00109-014-1181-y. [DOI] [PubMed] [Google Scholar]
- 21.Antonchuk J, Sauvageau G, Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109:39–45. doi: 10.1016/S0092-8674(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 22.Hong JH, et al. Expression pattern of the class I homeobox genes in ovarian carcinoma. J. Gynecol. Oncol. 2010;21:29–37. doi: 10.3802/jgo.2010.21.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N, Gou J-H, Xiong J, You J-J, Li Z-Y. HOXB4 promotes the malignant progression of ovarian cancer via DHDDS. BMC Cancer. 2020;20:222. doi: 10.1186/s12885-020-06725-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barba-de la Rosa AP, et al. Hox B4 as potential marker of non-differentiated cells in human cervical cancer cells. J. Cancer Res. Clin. Oncol. 2012;138:293–300. doi: 10.1007/s00432-011-1081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez R, et al. HOXB homeobox gene expression in cervical carcinoma. Int. J. Gynecol. Cancer. 2006;16:329–335. doi: 10.1111/j.1525-1438.2006.00350.x. [DOI] [PubMed] [Google Scholar]
- 26.Hung YC, et al. Homeobox gene expression and mutation in cervical carcinoma cells. Cancer Sci. 2003;94:437–441. doi: 10.1111/j.1349-7006.2003.tb01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Romero R, et al. The role of developmental HOX genes in cervical cancer. Rev. Med. del. Inst. Mexicano del. Seguro Soc. 2015;53 Suppl 2:S188–S193. [PubMed] [Google Scholar]
- 28.Bodey B, Bodey B, Groger AM, Siegel SE, Kaiser HE. Immunocytochemical detection of homeobox B3, B4, and C6 gene product expression in lung carcinomas. Anticancer Res. 2000;20:2711–2716. [PubMed] [Google Scholar]
- 29.Shears L, Plowright L, Harrington K, Pandha HS, Morgan R. Disrupting the interaction between HOX and PBX causes necrotic and apoptotic cell death in the renal cancer lines CaKi-2 and 769-P. J. Urol. 2008;180:2196–2201. doi: 10.1016/j.juro.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 30.Morgan R, et al. HOX transcription factors are potential targets and markers in malignant mesothelioma. BMC Cancer. 2016;16:85. doi: 10.1186/s12885-016-2106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, et al. HOXB4 knockdown reverses multidrug resistance of human myelogenous leukemia K562/ADM cells by downregulating P-gp, MRP1 and BCRP expression via PI3K/Akt signaling pathway. Int. J. Oncol. 2016;49:2529–2537. doi: 10.3892/ijo.2016.3738. [DOI] [PubMed] [Google Scholar]
- 32.Dumas P-Y, et al. MiR-10a and HOXB4 are overexpressed in atypical myeloproliferative neoplasms. BMC Cancer. 2018;18:1098. doi: 10.1186/s12885-018-4993-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, et al. Expression of HOXB4, PRDM16 and HOXA9 in patients with acute myeloid leukemia and its clinical significance. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2016;24:326–331. doi: 10.7534/j.issn.1009-2137.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Umeda S, et al. Prognostic significance of HOXB4 in de novo acute myeloid leukemia. Hematology. 2012;17:125–131. doi: 10.1179/102453312X13376952196250. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Sun J, Gao S, Hu H, Xie P. HOXB4 knockdown enhances the cytotoxic effect of paclitaxel and cisplatin by downregulating ABC transporters in ovarian cancer cells. Gene. 2018;663:9–16. doi: 10.1016/j.gene.2018.04.033. [DOI] [PubMed] [Google Scholar]
- 36.Zhou G, Liu X, Xiong B, Sun Y. Homeobox B4 inhibits breast cancer cell migration by directly binding to StAR-related lipid transfer domain protein 13. Oncol. Lett. 2017;14:4625–4632. doi: 10.3892/ol.2017.6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benezeder T, et al. Multigene methylation analysis of enriched circulating tumor cells associates with poor progression-free survival in metastatic breast cancer patients. Oncotarget. 2017;8:92483–92496. doi: 10.18632/oncotarget.21426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daugaard I, et al. Identification and validation of candidate epigenetic biomarkers in lung adenocarcinoma. Sci. Rep. 2016;6:35807. doi: 10.1038/srep35807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aquino Xavier FC, de Souza Setubal Destro MF, Esteves Duarte CM, Nunes FD. Epigenetic repression of HOXB cluster in oral cancer cell lines. Arch. Oral. Biol. 2014;59:783–789. doi: 10.1016/j.archoralbio.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Shen S, et al. Seven-CpG-based prognostic signature coupled with gene expression predicts survival of oral squamous cell carcinoma. Clin. Epigenet. 2017;9:88. doi: 10.1186/s13148-017-0392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonfim-Silva R, et al. Functional analysis of HOXA10 and HOXB4 in human medulloblastoma cell lines. Int. J. Oncol. 2017;51:1929–1940. doi: 10.3892/ijo.2017.4151. [DOI] [PubMed] [Google Scholar]
- 42.de Bessa Garcia SA, Araujo M, Pereira T, Mouta J, Freitas R. HOX genes function in breast cancer development. Biochim. Biophys. Acta. 2020;1873:188358. doi: 10.1016/j.bbcan.2020.188358. [DOI] [PubMed] [Google Scholar]
- 43.Kuo T-L, Cheng K-H, Chen L-T, Hung W-C. Deciphering the potential role of Hox genes in pancreatic cancer. Cancers. 2019;11:734. doi: 10.3390/cancers11050734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu K-Y, Wang L-T, Hsu S-H, Wang S-N. Homeobox genes and hepatocellular carcinoma. Cancers. 2019;11:621. doi: 10.3390/cancers11050621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cantile M, et al. In vivo expression of the whole HOX gene network in human breast cancer. Eur. J. Cancer. 2003;39:257–264. doi: 10.1016/S0959-8049(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 46.Yu M, Zhan J, Zhang H. HOX family transcription factors: related signaling pathways and post-translational modifications in cancer. Cell Signal. 2020;66:109469. doi: 10.1016/j.cellsig.2019.109469. [DOI] [PubMed] [Google Scholar]
- 47.Zhang B, Li N, Zhang H. Knockdown of Homeobox B5 (HOXB5) inhibits cell proliferation, migration, and invasion in non-small cell lung cancer cells through inactivation of the Wnt/beta-catenin pathway. Oncol. Res. 2018;26:37–44. doi: 10.3727/096504017X14900530835262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao D, Chen H-Q. Specific knockdown of HOXB7 inhibits cutaneous squamous cell carcinoma cell migration and invasion while inducing apoptosis via the Wnt/beta-catenin signaling pathway. Am. J. Physiol. 2018;315:C675–C686. doi: 10.1152/ajpcell.00291.2017. [DOI] [PubMed] [Google Scholar]
- 49.Goncalves CS, et al. A novel molecular link between HOXA9 and WNT6 in glioblastoma identifies a subgroup of patients with particular poor prognosis. Mol. Oncol. 2020;14:1224–1241. doi: 10.1002/1878-0261.12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng S, et al. HOXA4, down-regulated in lung cancer, inhibits the growth, motility and invasion of lung cancer cells. Cell Death Dis. 2018;9:465. doi: 10.1038/s41419-018-0497-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung CY, Kim RS, Lee SJ, Wang CH, Jeng MH. HOXB13 homeodomain protein suppresses the growth of prostate cancer cells by the negative regulation of T-cell factor 4. Cancer Res. 2004;64:3046–3051. doi: 10.1158/0008-5472.CAN-03-2614. [DOI] [PubMed] [Google Scholar]
- 52.Jung C, et al. HOXB13 is downregulated in colorectal cancer to confer TCF4-mediated transactivation. Br. J. Cancer. 2005;92:2233–2239. doi: 10.1038/sj.bjc.6602631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.