

Abstract
There is increasing evidence that widespread cortical cerebral blood flow deficits occur early in the course of Parkinson’s disease. Although cerebral blood flow measurement has been suggested as a potential biomarker for early diagnosis of Parkinson’s disease, as well as a means for tracking response to treatment, the relationship of cerebral blood flow to α-synucleinopathy, a major pathological hallmark of Parkinson’s disease, remains unclear. Therefore, we performed arterial spin-labeling magnetic resonance imaging and diffusion tensor imaging on transgenic mice overexpressing human wild-type α-synuclein and age-matched controls to measure cerebral blood flow and degenerative changes. As reported for early-stage Parkinson’s disease, α-synuclein mice exhibited a significant reduction in cortical cerebral blood flow, which was accompanied by motor coordination deficits and olfactory dysfunction. Although no overt degenerative changes were apparent in diffusion tensor imaging images, magnetic resonance imaging volumetric analysis revealed a significant reduction in olfactory bulb volume, similar to that seen in Parkinson’s disease patients. Our data, representing the first report of cerebral blood flow deficit in an animal model of Parkinson’s disease, suggest a causative role for α-synucleinopathy in cerebral blood flow deficits in Parkinson’s disease. Thus, α-synuclein transgenic mice comprise a promising model to study Parkinson’s disease-related mechanisms of cerebral blood flow deficits and to investigate further its utility as a potential biomarker for Parkinson’s disease.
Keywords: Neurodegeneration, Parkinson’s disease, olfactory dysfunction, dopamine, magnetic resonance imaging
Introduction
Of the challenges facing Parkinson’s disease (PD) research, few are more pressing than the ability to detect the disease before disabling clinical symptoms appear. Although PD has long been viewed as a motor system disorder caused by degeneration of dopaminergic neurons in the substantia nigra, the disease is accompanied by numerous non-motor manifestations that add significantly to the overall disease burden.1,2 In fact, many non-motor symptoms – including olfactory dysfunction, sleep disorders, autonomic dysfunctions, anxiety, and depression – can precede motor symptoms by years and even decades.3,4 Since PD’s motor and non-motor symptoms are likely caused by the same protein aggregation pathologic mechanism, research into early non-motor manifestations may provide clues to the onset and progression of motor symptoms and point the way toward early interventions.
Reduction in cortical cerebral blood flow (CBF) and dysregulation of glucose metabolism represent early manifestations of PD.5 Although the human brain comprises only 2% of an individual’s total body weight, a healthy brain accounts for approximately 20% of the body’s energy consumption. This high energy demand is accommodated by neurovascular and neurometabolic coupling (an activity-coupled increase in local CBF and glucose oxidation). Arterial spin-labeling (ASL) magnetic resonance imaging (MRI) studies have demonstrated significant cortical CBF deficits affecting frontal, parietal, and occipital areas in the brains of PD patients.6–10 In one study, the magnitude of CBF decrease among various cortical regions ranged from 20 to 40%, relative to the control group.7 Although there is variability in regional CBF among these studies, reduced regional CBF has been reported in PD patients both with8,10,11 and without6–9 dementia. A recent report9 described spatial co-localization of parietal cortical thinning and CBF reduction in non-demented patients with relatively mild PD, indicating that CBF changes occur early on in PD. Moreover, this pattern of cortical thinning and CBF reduction was negatively associated with motor symptoms. Because adequate CBF is essential for neuronal function, decreases lead to brain functional decline and potentially contribute to neurodegeneration. As a result, reduced CBF has been suggested as a possible early PD biomarker.7,10
Nevertheless, the mechanism behind CBF alteration in PD remains poorly understood, including its relationship to α-synucleinopathy, a major pathological hallmark of PD. α-Synuclein pathology likely develops years – possibly decades – before clinical symptoms and is present in virtually all sporadic and familial PD patients.12 In addition, its distribution correlates with clinical symptoms.12 Thus, understanding the mechanism of CBF alteration in PD, including its relationship with α-synuclein pathology, may help elucidate the causes and progression of both motor and non-motor symptoms and point the way toward early interventions. We hypothesized that if α-synuclein pathology is associated with deficits in CBF, α-synuclein transgenic mice that overexpress human wild-type α-synuclein should exhibit reduced CBF. Thus, we performed ASL MRI on 18-month-old α-synuclein and age-matched non-transgenic control mice to measure CBF. To assess whether CBF alterations are accompanied by overt neurodegenerative changes, we also studied apparent diffusion coefficient (ADC) and fractional anisotropy (FA) using diffusion tensor imaging (DTI).
Materials and methods
Mice
Male transgenic mice overexpressing human wild-type α-synuclein under the direction of a mouse Thy-1 promoter [C57BL/6N-Tg(Thy1-SNCA)15Mjff/J; stock number 017682] and non-transgenic male controls matched for age (C57BL/6NJ; stock number 005304) were acquired at seven to eight weeks of age from The Jackson Laboratory under an agreement with the Michael J. Fox Foundation. Two of the most commonly used rodent models of PD are MPTP and α-synuclein-overexpressing transgenic mice. Generally, only the males of these models are used as variability and/or diminished PD phenotype is inherent in the models’ females.13,14 Thus, to enable comparison with existing literature, only male mice were chosen for the current study. The mice were group-housed and maintained in a 12/12 h light/dark cycle and at 24°C room temperature until 18 months of age. Animal husbandry was in accordance with the ARRIVE guidelines, National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the Society for Neuroscience Policies on the Use of Animals and Humans in Neuroscience Research. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the South Texas Veterans Health Care System, Audie L. Murphy division and the University of Texas Health Science Center, San Antonio. A total of seven each α-synuclein and non-transgenic control mice were used. All behavioral tests were performed during the light cycle, and 20–40 lux light intensity was maintained throughout the behavioral tests. Proper righting reflex was assessed before the behavioral tests, and the mice were acclimatized to the testing room for at least 1 h prior to the session. The smallest possible number of mice was used (based on power calculations) and all efforts were made to minimize suffering. All experiments were randomized using GraphPad QuickCalcs and performed blind coded.
Behavioral testing
Home cage activity
General activity in the home cage was recorded using the ActiMot2 infrared light beam activity monitoring system (25 beams each on the X and Z directions and 16 beams on the Y direction; TSE Systems, Inc., Chesterfield, MO). The home activity of the mice was recorded at a sampling rate of 100 Hz with a recording interval of 10 min for 24 h continuously. Ambulatory and fine movements, speed, total distance travelled, and rearing, as well as the number of sleep bouts during the light and dark phases were analyzed and compared between the groups.
Open field
The ActiMot2 open field apparatus was equipped with 32 infrared sensors each, spaced 1.5 cm apart, on the X, Y, and Z directions. The open field arena, which is made of plexiglass (51.5 × 51.5 cm), was illuminated at 20 lux light intensity. For each test, mice were placed individually into a corner of the arena and activity was recorded using the ActiMot2 infrared light beam activity monitoring system for 10 min to assess the reactivity of the mouse to a novel environment. The parameters included ambulatory and fine movements, speed, total distance travelled, rearing, and visits to the center of the arena. Between each test, the open field was wiped with ethanol solution and dried to remove odor trails.
Static bar test
The test was performed as described before.15 In brief, a 9 mm diameter wooden rod (60 cm long) was fixed by a G-clamp to a laboratory shelf, such that the rod protrudes horizontally. The mouse was placed at the distal end of the rod facing outward and performance was recorded with a video camera as the mouse turned and moved along the beam. Later, the videos were analyzed for the following metrics: (i) time to reorient 180° from the starting position (T-turn), (ii) time to travel to the shelf end (T-travel), (iii) total time to traverse the beam (T total = T-turn + T-travel), and (iv) the number of errors/slips as the mouse was moving forward.
Horizontal bar test
The horizontal bar test measures both motor coordination and strength. In this test, the mice were allowed to grasp the center point of a 2 or 6 mm diameter copper bar (38 cm long, held 49 cm above the floor) with their forepaws only, and the time was recorded until either falling from the bar or reaching one end of the bar within the maximum test time of 30 s (completion of the task).15 Scoring was as follows: falling within 1–5 s = 1; falling within 6–10 s = 2; falling within 11–20 s = 3; falling within 21–30 s = 4; falling after 30 s = 5; completing the task = 5.
Hindlimb extension reflex
Extension reflex of the hindlimb was tested by suspending the mice by tail in midair and observing the pattern of hindlimb extension reflex for about 5 s. The pattern was scored as follows: absence of hindlimb extension or the mouse gripping its tail = 0; extension of only one hindlimb or extension of both hindlimbs without splayed toes = 1; extension of both hindlimbs with splaying of toes = 2.16
Nesting behavior
Nest building was assessed as described previously.17 Briefly, the mice were individually housed in clean plastic cages with approximately 2 cm of bedding lining each floor. One hour prior to the onset of the dark phase of the lighting cycle, a piece of pre-weighed 51 mm square × 5 mm thick Nestlet™ cotton pad (Nestlets™, Ancare Corp., Belmont, New York) was placed on the floor of each cage. The nests were assessed 24 h later on a rating scale of 1–5.17
Test for general anosmia
General anosmia was assessed using the buried food retrieval paradigm.18 The mice were placed in a testing cage, in which a sweetened cereal pellet (Cap’n Crunch; Quaker Oats Company) was buried 0.5 cm below the bedding, so that it was not visible. The testing cage consisted of a clean mouse cage that was filled ∼3 cm with fresh bedding. The retrieval time was recorded from the instant the mouse was released in the center of the testing cage until the cereal pellet was found (maximum 15 min). Retrieval time for an unscented glass marble and time to find an exposed (vs. hidden) cereal pellet were determined similarly to assess any potential confounding of the buried cereal test by anxiety-related digging behaviors or deficits in motor coordination, respectively. Similarly, since food was used as a cue, body weight and food intake were monitored prior to the test to assess any potential confounding of the buried cereal test by differences in food intake between the groups.
Magnetic resonance imaging
MRI was performed on an 11.7 T magnet with 740 mT/m gradients (Bruker, Billerica, MA, USAs) and with a 1 cm transmit/receive surface coil. At 18 months of age, seven each male α-synuclein and age-matched non-transgenic control mice were subjected to MRI scans. Animals were initially anesthetized with 2% isoflurane and then secured in a custom-made head holder with ear and tooth bars. After preparation, animals were moved into the MRI bore and 1–1.5% isoflurane was maintained under spontaneous breathing conditions. A vacuum line placed next to the head of the animal was always open to prevent buildup of isoflurane in the scanner bore. A transceiver surface coil was placed on the top of the head for imaging, and a labeling coil was placed under the heart for ASL.19 Respiration rate was monitored with an MR-compatible small animal monitoring and gating system (SA Instruments, Inc., Stony Brook, NY). The rectal temperature was maintained at 37 ± 0.5°C with a feedback-regulated circulating warm water pad. Heart rate and arterial oxygen saturation were monitored (MouseOx, STARR Life Science Corp., Oakmont, PA) and maintained within normal physiological ranges. The olfactory bulbs and whole brains were scanned separately.
Cerebral blood flow
CBF was measured using continuous ASL with a 2.36 s labeling pulse to the labeling coil in the presence of a 20 mT/m gradient and a 350 ms post-label delay.20 Paired images with and without labeling were acquired in an interleaved fashion. MRI parameters were: spin-echo, echo planar imaging (EPI), labeling duration 2.36 s, nine 1-mm thick coronal images for the brain and five 0.5-mm thick coronal images for the olfactory bulb, field of view = 12.8 × 12.8 mm, data matrix 64 × 64, TR = 3.0 s, and TE = 17.6 ms. Two hundred repetitions were acquired.
Diffusion MRI
DTI was obtained with a single low b value (10 s/mm2) and 30 directions with 1200 s/mm2. MRI parameters are: spin-echo, EPI, 11 1-mm coronal images for the brain and 9 0.5-mm coronal images for the olfactory bulb, field of view = 12.8 × 12.8 mm, data matrix = 64 × 64, TR = 3 s, TE = 28.7 ms, and two repetitions.
Transverse relaxation time (T2*)
The T2* maps were calculated from a 3D multiple echo gradient echo sequence with a spectral width of 59 kHz. Five echoes were acquired with the first TE = 2.2 ms and an inter-echo time of 2.2 ms, and only echoes during the positive gradient lobes were acquired. The TR was 40 ms. The field of view for the brain scans was 12.8 × 12.8 × 9 mm with an acquired matrix size of 86 × 86×45, reconstructed to 128 × 128×90, whereas the field of view for the olfactory bulb scans was 12.8 × 12.8 × 4 mm with an acquired matrix size of 86 × 86×40, reconstructed to 128 × 128×40. Five averages were acquired.
Maps of CBF (ml blood/g tissue/min), FA, and ADC (mm2/s) values were calculated using Matlab (MathWorks, Natick, MA).21–25 Volume of olfactory bulb and brain were measured separately from T2*-weighted images. Bilateral region of interest (ROI) analysis was performed by manually drawing ROI of various brain regions involved in olfactory and motor/executive function using the coronal Allen Mouse Brain Atlas (Allen Institute for Brain Science; available from: http://mouse.brain-map.org).26
Statistical analysis
Sample size was calculated by power analysis using GPower 3.1.7 software (Universität Düsseldorf, Germany). Existing literature and our own published work20,27 served as the basis for calculating effect size. Assuming an effect size of 1.9 (power 90% and alpha 0.05), the sample size for behavioral tests was estimated to be 7, whereas with an effect size of 2.1, the sample size for MRI was estimated to be 6. Although the sample size for MRI was estimated to be 6, we performed scans on all seven mice used in the behavioral tests. However, one of the α-synuclein mice was excluded from the analysis due to localized artifacts. Statistical analyses of the data were performed in R version 3+ (Vienna, Austria) or GraphPad InStat (GraphPad Software, Inc.). For the MRI data, we used two-way ANOVA with the Tukey-Kramer method of correction for multiple comparisons due to unequal sample sizes (seven control vs. six α-synuclein). However, for the data on the time to turn and travel, the static bar, the horizontal bar test, and home cage activity, as well as for open field ambulatory movement, fine movement, rearing, and center vs. periphery activity, we used two-way ANOVA with the Tukey HSD method of correction for multiple comparisons due to equal sample sizes (7 each/strain). For all other group comparisons between α-synuclein and non-transgenic control mice, we used an unpaired t-test with Welch correction for continuous variables, and the Mann-Whitney test for data that were by design not normally distributed (Hindlimb reflex score and Nesting score). All sets of continuous data were tested for normality using the Shapiro-Wilk test, and fewer than 5% of the tests concluded that the set was non-normal at the 0.05 significance level, confirming that the data sets met the assumption of a normal distribution. Results were expressed as mean ± SD and differences in mean were considered significant at P < 0.05.
Results
Magnetic resonance imaging
At 18 months of age α-synuclein transgenic mice exhibited a significant decrease in CBF (Figures 1 and 2). Compared with age- and sex-matched non-transgenic controls, there was a 36.6% reduction (P = 0.0283) in cortical CBF in α-synuclein mice (Figure 2). Although the mean value of subcortical CBF in α-synuclein mice was 20.2% less than the mean value in control mice in our study, the difference was not statistically significant (P > 0.05), possibly due to the amount of variation inherent in measurement of CBF for animals of this small size. To get more insight into the relevance of reduced CBF to PD, we performed regional analysis in those areas of the brain involved in movement and in olfaction, an early functional loss in PD and other neurodegenerative disorders such as Alzheimer’s disease. For areas involved in movement, we measured CBF in the substantia nigra, striatum, motor cortex, somatosensory cortex, and cingulate cortex; and for areas involved in olfaction, the olfactory bulb, olfactory tubercle, and piriform cortex. Except for the olfactory bulb, all cortical areas showed significant decrease in CBF, with somatosensory and motor cortices having the largest percent decrease in CBF (Figure 3(a)). The difference in olfactory bulb CBF was not statistically significant (P > 0.05). We have not attempted to measure CBF in other areas of the olfactory-limbic archicortex and basal ganglia, since these nuclei are very small and MRI does not provide sufficient contrast and spatial resolution to delineate them clearly in the mouse brain.
Figure 1.

Representative CBF maps illustrated on coronal views of arterial spin-labeling magnetic resonance images taken from 18-month-old α-synuclein (n = 6) and age-matched non-transgenic control mice (n = 7). A reduction in CBF can be clearly seen in the widespread brain areas of the α-synuclein mice compared with the control mice. The color-coded dotted lines indicate various ROIs used for quantitative analysis of the imaging data represented in Figure 3. Although only unilateral ROIs are shown, bilateral ROIs were used for quantitative analysis.
CC: cingulate cortex; MC: motor cortex; OT: olfactory tubercle; PC: piriform cortex; SN: substantia nigra; SSC: somatosensory cortex; ST: striatum.
Figure 2.
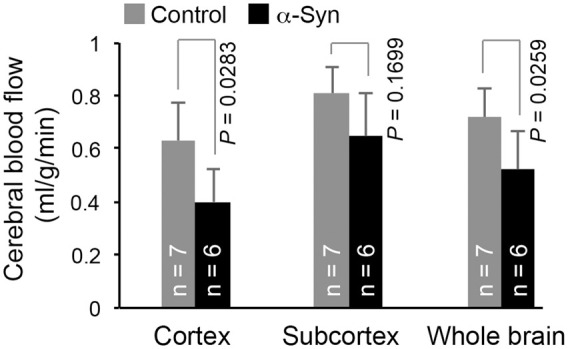
Compared with control mice, about 37% and 28% reductions in CBF were observed in the cortex and whole brain (cortex + subcortex), respectively, of α-synuclein mice (α-Syn). The reduction in subcortical CBF in α-synuclein mice was not statistically significant (P > 0.05). Data represent mean ± SD. Two-way ANOVA with Tukey-Kramer method of correction for multiple comparisons. The whole brain was excluded from the two-way ANOVA due to it being a composite variable (cortex + subcortex = whole brain). Whole brain: unpaired t-test with Welch correction.
Figure 3.

Plots of quantitative data illustrating changes in CBF (a), ADC (b), and FA (c) in various regions of interest (ROI). All cortical ROI of α-synuclein mice showed significant reductions (P < 0.05) in CBF. [AQ: Please check edit: ‘However, the CBF reduction in the olfactory bulb and the two subcortical ROIs…’]However, the CBF reduction in the olfactory bulb and the two subcortical ROIs (striatum and substantia nigra) was not significant (P > 0.05). None of the ROIs showed a significant change in either ADC or FA, except the substantia nigra, in which the ADC value of α-synuclein mice was significantly reduced (P < 0.05). Data represent mean ± SD. Two-way ANOVA with Tukey-Kramer method of correction for multiple comparisons. The olfactory bulb CBF was excluded from the two-way ANOVA due to being confounded by the use of isoflurane during MRI. Olfactory bulb CBF: unpaired t-test with Welch correction.
To assess potential neurodegenerative changes associated with α-synuclein overexpression, we also measured ADC and FA using DTI and performed volumetric analysis of olfactory bulb and brain. As for the CBF, only those regions that were big enough to draw a clear ROI were included in the analysis. The ADC and FA values of these regions are summarized in Figure 3(b) and (c). No significant changes in either ADC or FA were observed in these regions, except for substantia nigra, which showed a significant decrease in ADC values, indicative of a microstructural alteration in the tissue. Volumetric analysis revealed no significant changes in the brains of α-synuclein mice, compared with controls (Figure 4(a)). However, consistent with what is reported for PD patients,28 we observed a modest but significant (P = 0.0128) reduction in olfactory bulb volume in α-synuclein mice compared with non-transgenic controls (Figure 4(b)).
Figure 4.

Histogram plots of MRI volumetric analysis of brain (a) and olfactory bulb (b) of α-synuclein and control mice. Compared with control mice, a modest but statistically significant (P < 0.05) reduction in olfactory bulb volume was observed in α-synuclein mice. Data represent mean ± SD. Unpaired t-test with Welch correction.
Behavioral testing
To quantify the Parkinsonian phenotype, the mice were subjected to both motor and olfactory behavioral tests. The α-synuclein mice showed significant impairment in motor coordination, as assessed by the static rod test (Figure 5(a)), as mice took more time to turn (P = 0.0078) and made more errors/slips (P = 0.0049) while traversing the rod (Figure 5(b)). The time α-synuclein mice took to traverse the rod (Figure 5(a)) was not statistically different from the time for control mice (P > 0.05); however, there was a dramatic difference in the way α-synuclein mice crossed the rod. Control mice traversed the rod with a normal stride – their legs holding them up such that there was visible space between the rod and their torsos (Figure 5(c)). The legs of the α-synuclein mice struggled to hold them up, as they ran their torsos dragged along the rod (Figure 5(d)), and their hind paws frequently slipped off. Similar results were obtained with the horizontal bar test, which measures both motor coordination and strength. The α-synuclein mice scored very poorly compared with controls (Figure 5(e)). As shown in Figure 5(f) to (i), α-synuclein mice also scored poorly in hindlimb extension reflex and nesting behavior, deficits in both of which have been shown to correlate with loss of brain dopamine levels. Body weights were measured prior to the motor coordination tests. Since α-synuclein mice weighed only 10% more than control mice (Figure 5(j)), it is unlikely that the results of the motor coordination tests were confounded by any difference in body weight. Lastly, we assessed general activity levels of the mice in their home cages as well as in a novel environment (open field). As shown in Figure 6, none of the parameters tested was statistically different between α-synuclein and control mice, including ambulatory and fine movement, speed, total distance travelled, rearing, and number of sleep bouts during the light and dark phase of the day. Thus, the data on motor coordination and general activity indicate that, as seen in early-stage PD, α-synuclein mice exhibit significant deficits in motor coordination but not in overall activity levels.
Figure 5.

α-Synuclein mice performed poorly in various tests for motor coordination. In the static bar test, α-synuclein mice took significantly more time to turn (T-turn; a) and made a significantly greater number of errors/slips (b) while traversing a 9 mm rod. T-total = T-turn + T-travel. (c and d) Representative photographs of control (c) and α-synuclein mice (d) while performing the static bar test. Note how the legs of the α-synuclein mice struggled to hold them up on the rod. (e) Result of horizontal bar test. Scores for latency to fall from horizontal bars of 2 mm and 4 mm diameter were significantly reduced in α-synuclein mice. Compared with control animals, α-synuclein mice also scored poorly on the hindlimb extension reflex test (f) and nest building (g). (h and i) Representative images of nest construction. Deacon’s nesting scoring scale: 1 = Nestlet not noticeably touched; 2 = Nestlet partially shredded; 3 = Nestlet mostly shredded but no identifiable nest site; 4 = an identifiable but flat nest; 5 = a perfect nest with wall surrounding mouse body. (j) Body weight of α-synuclein and control mice. Data represent mean ± SD. Static bar test (a) and Horizontal bar test (e): two-way ANOVA with Tukey HSD method of correction for multiple comparisons. The T-total (a) was excluded from the two-way ANOVA due to it being a composite variable (T-turn + T-travel = T-total). Hindlimb extension reflex test and nest building: Mann-Whitney test. All other data: unpaired t-test with Welch correction.
Figure 6.

General activity in home cage (a–f) and novel environment (g–l). Overall activity levels were similar among α-synuclein and control mice (P > 0.05). Various parameters tested in home cage activity are represented separately for the dark (12 h) and light (12 h) phases of the lighting cycle. Data represent mean ± SD. Home cage activity (a–f), and open field ambulatory movement (g), fine movement (h), and rearing (j): two-way ANOVA with Tukey HSD method of correction for multiple comparisons. All other data: Unpaired t-test with Welch correction.
Since olfactory dysfunction is an early warning sign of PD, and PD-related α-synuclein pathology may potentially begin in the olfactory system,29–31 we tested the mice for general anosmia using a buried food retrieval paradigm. The α-synuclein mice took significantly more time than control mice to retrieve the hidden cereal (Figure 7; P = 0.0077). Since the retrieval time for an unscented glass marble or time to find an exposed (vs. hidden) piece of cereal was not significantly different between α-synuclein and control mice (data not shown), it is unlikely that the results of the buried food retrieval were confounded by motor coordination deficits.
Figure 7.
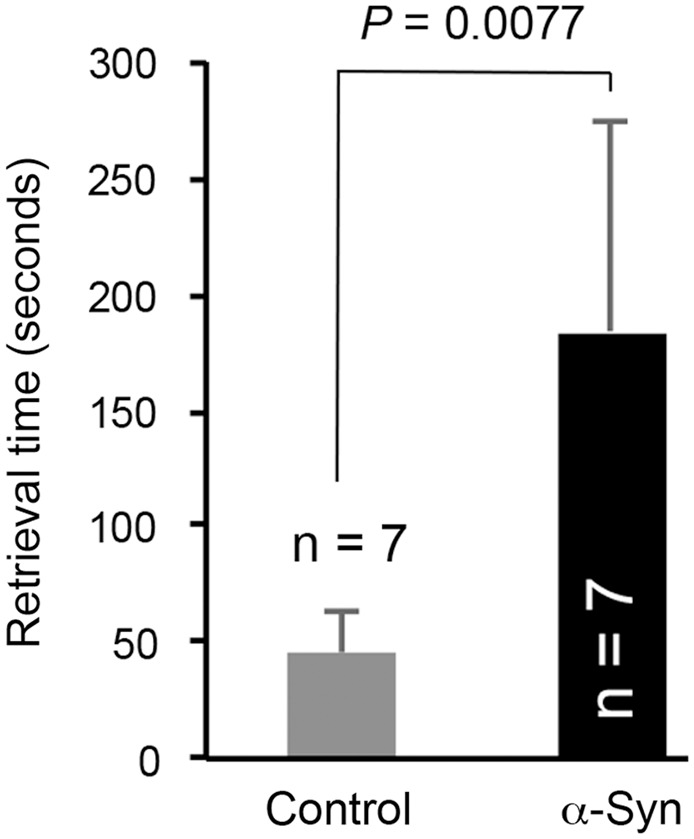
α-Synuclein mice took significantly more time than control mice to retrieve hidden cereal, a measure of olfactory dysfunction. Data represent mean ± SD. Unpaired t-test with Welch correction.
Discussion
ASL MRI analysis of transgenic mice overexpressing wild-type human α-synuclein revealed significant cortical CBF deficits, suggesting a causal relationship between α-synuclein pathology and CBF deficits in PD. We observed a 36.6% reduction in cortical CBF in α-synuclein mice. In early to moderate stage PD, the magnitude of regional cortical CBF ranges from 20 to 40% relative to age-matched controls.7,10 Thus, the extent of perfusion decrease observed in α-synuclein mice is similar to that reported for PD patients.
In the current study, using the ASL method, the average cortical CBF in control mice under 1–1.5% isoflurane anesthesia was 0.631 ± 0.055 ml/g/min. This is comparable to the results of a recent study using positron emission tomography (PET), in which the CBF in mice under 1.5% isoflurane anesthesia was found to be 0.62 ± 0.05 ml/g/min.32 However, the CBF values in anesthetized mice in both studies were slightly higher than that reported for awake mice using the autoradiographic method, which was 0.5 ml/g/min.33 It is likely that the anesthetic (isoflurane) contributed to the slightly higher CBF reported in our study. Indeed, isoflurane has a cerebral vasodilatory effect,34 which leads to increased CBF.35 It is also worth noting that a study of macaque monkeys found a significant increase in CBF only with high doses (2%) of isoflurane.35 At the low doses (1–1.5%) that we used in the current study, the increase in CBF was only modest.35
α-Synuclein pathology appears early on in virtually all sporadic and familial PD patients, its distribution correlates with clinical symptoms,12 and widespread cortical hypoperfusion and reduced glucose metabolism appear early on in PD. Nonetheless, the relationship between α-synuclein pathology and CBF dysfunction has not yet been defined. Cerebral blood flow is tightly linked to tissue metabolism through neurovascular and neurometabolic coupling. We hypothesize that the reduced CBF in α-synuclein mice and in PD patients arises from abnormal modulation of neurovascular coupling and CBF autoregulation by α-synuclein overexpression. The renin-angiotensin system plays a critical role in the control of CBF by affecting neurovascular coupling and CBF autoregulation. All known components of the renin-angiotensin system are reported in the brain36 as well as the kidney, and it has been shown that a counter-regulatory mechanism exists between the renin-angiotensin system and dopamine, also in both the brain and the kidney. In animal studies, depletion of dopamine induces compensatory overactivation of the renin-angiotensin system.37 α-Synuclein is found in axon terminals, and it plays an active role in the downregulation of dopamine secretion.38–40 Together, these studies indicate that overexpression of α-synuclein might lead to dysregulation of the counter-regulatory mechanism between the renin-angiotensin system and dopamine, leading to abnormal upregulation of the local renin-angiotensin system and deficits in CBF. Of note, overexpression of normal α-synuclein, rather than mutant forms, correlates with disease pathology in PD.41–43 Moreover, excess activation of the local renin-angiotensin system activates NOX family NADPH oxidases, the second most important source of reactive oxygen species after mitochondria. NADPH oxidase complexes induce oxidative stress and exacerbate inflammation. Indeed, oxidative stress and neuroinflammation are very prominent features in the PD brain.37
In an alternative hypothesis, reduced CBF in α-synuclein mice could also arise from abnormal neurometabolic coupling due to α-synuclein overexpression. By strategic positioning between blood vessels and neurons, astrocytes play a critical role in the local supply of energy substrate to neurons through the modulation of blood flow, as well as through glucose uptake and metabolism.44 Indeed, PD features widespread energetic defects. Analysis of 6.8 million raw data points from nine genome-wide expression studies and 185 laser-captured human midbrain dopamine neurons revealed PD patients underexpress 10 gene sets that are associated with mitochondrial electron transport, glucose utilization, and glucose sensing.45 So far, no data exist on the potential interplay between α-synuclein and neurometabolic coupling in CBF deficits. However, it is worth noting that patients with diabetes but without PD had lower striatal dopamine transporter binding and higher α-synuclein levels in their cerebrospinal fluid compared with healthy controls.46 Moreover, PD patients with diabetes mellitus had a 4.5-fold increased risk of motor progression and 9.3-fold increased risk of cognitive decline compared with patients with PD alone.46 In sum, although it does not reveal the mechanism for deficits in CBF, our study opens up new avenues for detailed mechanistic studies on the role of α-synuclein, a protein with largely unknown function, in CBF regulation in health and disease.
The inability to detect a significant reduction in CBF of the olfactory bulb might be explained by confounding due to isoflurane, an inhalant anesthetic, during MRI. Isoflurane has a marked odor that might have caused overactivation of the olfactory bulb, leading to activity-dependent changes in olfactory bulb CBF. Evidence of this possibility comes from the larger standard deviations for CBF in the olfactory bulb area of the brain compared with other brain areas. Although the change in olfactory bulb CBF was not significant, MRI volumetric analysis revealed a significant reduction (11%) in the volume of the olfactory bulb in α-synuclein mice, which is consistent with reports from human PD patients showing a 13% reduction in this metric.28 Change in olfactory bulb volume has been shown to affect olfactory performance47 and, in fact, a correlation between odor detection threshold and olfactory bulb volume has been reported in PD patients.28 In line with these reports, we found that α-synuclein mice took significantly more time to retrieve buried cereal, indicative of an increase in odor detection threshold. It is plausible to hypothesize that the reduction in olfactory bulb volume is a consequence of a decrease in the number and/or size of olfactory glomeruli, which form the anatomical basis of the olfactory sensory map that enables odor detection. This notion receives support from a recent study using post-mortem human olfactory bulb, which revealed that global glomerular voxel volume of olfactory bulb in PD patients with 9 to 20 years of disease duration was approximately 47% that of controls.48 The same study further showed that the higher α-synuclein load in the olfactory bulb from PD cases, the lower the global glomerular voxel volume, suggesting a causal relationship between α-synuclein pathology and olfactory bulb volume. Although olfactory bulb volume is significantly reduced in α-synuclein mice, no volumetric change was observed in rest of the brain. It is unclear what makes olfactory bulb more susceptible to α-synuclein overexpression-related atrophy, compared with other areas of the brain. The total energy requirement for the detection and discrimination of odorants is quite large. It has been shown that the energetically demanding odor-evoked oxidative metabolism is tightly correlated to high capillary density in the olfactory bulb.49 Not surprisingly, the highest brain insulin-binding affinities and insulin receptor density and kinase activity were localized to the olfactory bulb, compared with those found in all other brain regions.50,51 Interestingly, dysregulation of glucose metabolism52 and olfactory dysfunction are early events in the course of sporadic PD. The olfactory bulb is a metabolic sensor, playing a role in controlling energy homeostasis in response to sensory and hormonal signals. Since α-synuclein is a novel player in the regulation of glucose uptake and utilization,53 and since α-synucleinopathy first appears in the olfactory bulb,29 understanding the specific mechanism of olfactory bulb atrophy and olfactory dysfunction in α-synuclein transgenic mice may provide clues to early pathogenic mechanisms in PD.
Olfactory bulb α-synucleinopathy has a high sensitivity (95%) and specificity (91%) for PD, and it accurately predicts the presence of α-synuclein deposits in other brain regions.29 Based on these studies, as well as the fact that α-synucleinopathy in the olfactory system precedes its development in the nigrostriatal pathway, it has been proposed that olfactory bulb biopsies might be used to confirm early diagnosis of PD. However, the high risk/benefit ratio of this invasive procedure has precluded its adoption. Understanding the exact relationship between α-synucleinopathy and CBF deficits in PD would potentially allow us to use CBF deficits as a surrogate non-invasive marker for α-synucleinopathy, and in combination with olfactory bulb volumetric analysis and olfactory testing, it may provide a valuable biomarker set for identifying individuals at high risk for developing PD.
The CBF deficit and olfactory dysfunction in α-synuclein mice was accompanied by significant impairment in motor coordination tests, including nest building and the hindlimb extension reflex test, which are sensitive to brain dopamine levels.54,55 However, overall activity levels in both the home cage and open field were not affected, indicating that motor impairments seen in α-synuclein mice are similar to those observed during prodromal/early-stage PD, where dopaminergic dysfunction likely precedes overt neurodegenerative changes. Indeed, MRI studies have revealed only slight or no gray matter atrophy in early to moderate stage PD patients,7,56 although cortical CBF was decreased.7 Although we did not observe significant changes in brain volume, ADC, or FA between α-synuclein and control mice, we did find, as reported for patients with PD,57 a significant reduction in the ADC value in the substantia nigra of α-synuclein mice, highlighting the selective vulnerability of this region. Together, previous reports and the current data suggest that functional changes precede overt structural changes in PD. Therefore, CBF measurement would likely be a more sensitive biomarker than gray matter loss in early stage PD.
In summary, this study reports significant CBF deficits accompanied by motor coordination impairment, olfactory dysfunction, and olfactory bulb atrophy in transgenic mice overexpressing human wild-type α-synuclein. Cerebral blood flow is tightly coupled to tissue metabolism, and PD is characterized by widespread cortical hypoperfusion and deficits in energy homeostasis. However, the mechanism of cortical hypoperfusion and its relationship to α-synucleinopathy remains unknown. As far as we are aware, our study is the first to report a CBF deficit in any animal model of PD and suggests a role for α-synucleinopathy in cortical hypoperfusion in PD, setting the stage for detailed mechanistic analysis of the relationship between α-synucleinopathy and cortical hypoperfusion and its potential role in the onset and progression of various motor and non-motor manifestations of PD. Since decreased CBF is a common feature of aging, an over-arching PD risk factor, its measurement by non-invasive imaging technology has promise as a biomarker for early diagnosis, and in association with olfactory testing, could assist in tracking disease progression and treatment response.
Footnotes
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a Merit Review Grant from the Veterans Health Administration (1 101 BX 003157) and a grant from the William and Ella Owens Medical Research Foundation, both awarded to RAC, as well as pilot and career development grants from the Perry & Ruby Stevens Parkinson’s Disease Center of Excellence, awarded to KCB.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions: RAC and KCB conceived and designed the study. QS performed the MRI scans and data analysis, assisted by KCB. KCB and ETH performed the behavioral tests. MJM and ETH designed and performed the statistical analysis. All authors were involved in overall data analysis. KCB wrote the initial draft of the article and RAC edited it. All authors commented on the article and approved the final version.
ORCID iD: KC Biju https://orcid.org/0000-0002-8409-6374
References
- 1.Marras C, Chaudhuri KR.Nonmotor features of Parkinson's disease subtypes. Mov Disord 2016; 31: 1095–1102. [DOI] [PubMed] [Google Scholar]
- 2.Cumming P, Borghammer P.Molecular imaging and the neuropathologies of Parkinson's disease. Curr Top Behav Neurosci 2012; 11: 117–148. [DOI] [PubMed] [Google Scholar]
- 3.Pfeiffer RF.Non-motor symptoms in Parkinson's disease. Parkinsonism Relat Disord 2016; 22Suppl 1: S119–S122. [DOI] [PubMed] [Google Scholar]
- 4.Schapira AHV, Chaudhuri KR, Jenner P.Non-motor features of Parkinson disease. Nat Rev Neurosci 2017; 18: 509. [DOI] [PubMed] [Google Scholar]
- 5.Borghammer P, Chakravarty M, Jonsdottir KY, et al. Cortical hypometabolism and hypoperfusion in Parkinson's disease is extensive: probably even at early disease stages. Brain Struct Funct 2010; 214: 303–317. [DOI] [PubMed] [Google Scholar]
- 6.Barzgari A, Sojkova J, Maritza Dowling N, et al. Arterial spin labeling reveals relationships between resting cerebral perfusion and motor learning in Parkinson's disease. Brain Imaging Behav 2019; 13: 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Seara MA, Mengual E, Vidorreta M, et al. Cortical hypoperfusion in Parkinson's disease assessed using arterial spin labeled perfusion MRI. Neuroimage 2012; 59: 2743–2750. [DOI] [PubMed] [Google Scholar]
- 8.Kamagata K, Motoi Y, Hori M, et al. Posterior hypoperfusion in Parkinson's disease with and without dementia measured with arterial spin labeling MRI. J Magn Reson Imaging 2011; 33: 803–807. [DOI] [PubMed] [Google Scholar]
- 9.Madhyastha TM, Askren MK, Boord P, et al. Cerebral perfusion and cortical thickness indicate cortical involvement in mild Parkinson's disease. Mov Disord 2015; 30: 1893–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melzer TR, Watts R, MacAskill MR, et al. Arterial spin labelling reveals an abnormal cerebral perfusion pattern in Parkinson's disease. Brain 2011; 134(Pt 3): 845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin WC, Chen PC, Huang YC, et al. Dopaminergic therapy modulates cortical perfusion in Parkinson disease with and without dementia according to arterial spin labeled perfusion magnetic resonance imaging. Medicine (Baltimore) 2016; 95: e2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luk KC, Lee VM.Modeling Lewy pathology propagation in Parkinson's disease. Parkinsonism Relat Disord 2014; 20Suppl 1: S85–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grant LM, Richter F, Miller JE, et al. Vocalization deficits in mice over-expressing alpha-synuclein, a model of pre-manifest Parkinson's disease. Behav Neurosci 2014; 128: 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller DB, Ali SF, O'Callaghan JP, et al. The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann N Y Acad Sci 1998; 844: 153–165. [PubMed] [Google Scholar]
- 15.Deacon RM.Measuring motor coordination in mice. J Vis Exp 2013; 75: e2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaworski DM, Soloway P, Caterina J, et al. Tissue inhibitor of metalloproteinase-2(TIMP-2)-deficient mice display motor deficits. J Neurobiol 2006; 66: 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deacon RM.Assessing nest building in mice. Nat Protoc 2006; 1: 1117–1119. [DOI] [PubMed] [Google Scholar]
- 18.Lehmkuhl AM, Dirr ER, Fleming SM.Olfactory assays for mouse models of neurodegenerative disease. J Vis Exp 2014; 90: e51804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muir ER, Shen Q, Duong TQ.Cerebral blood flow MRI in mice using the cardiac-spin-labeling technique. Magn Reson Med 2008; 60: 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cong L, Muir ER, Chen C, et al. Multimodal MRI evaluation of the MitoPark mouse model of Parkinson's disease. PLoS One 2016; 11: e0151884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Q, Meng X, Fisher M, et al. Pixel-by-pixel spatiotemporal progression of focal ischemia derived using quantitative perfusion and diffusion imaging. J Cereb Blood Flow Metab 2003; 23: 1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Q, Ren H, Cheng H, et al. Functional, perfusion and diffusion MRI of acute focal ischemic brain injury. J Cereb Blood Flow Metab 2005; 25: 1265–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen Q, Ren H, Fisher M, et al. Dynamic tracking of acute ischemic tissue fates using improved unsupervised ISODATA analysis of high-resolution quantitative perfusion and diffusion data. J Cereb Blood Flow Metab 2004; 24: 887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Q, Ren H, Fisher M, et al. Statistical prediction of tissue fate in acute ischemic brain injury. J Cereb Blood Flow Metab 2005; 25: 1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen Q, Huang S, Duong TQ.Ultra-high spatial resolution basal and evoked cerebral blood flow MRI of the rat brain. Brain Res 2015; 1599: 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007; 445: 168–176. [DOI] [PubMed] [Google Scholar]
- 27.Biju KC, Evans RC, Shrestha K, et al. Methylene blue ameliorates olfactory dysfunction and motor deficits in a chronic MPTP/probenecid mouse model of Parkinson's disease. Neuroscience 2018; 380: 111–122. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, You H, Liu JF, et al. Association of olfactory bulb volume and olfactory sulcus depth with olfactory function in patients with Parkinson disease. AJNR Am J Neuroradiol 2011; 32: 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beach TG, White CL, III, Hladik CL, et al. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 2009; 117: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berendse HW, Ponsen MM.Detection of preclinical Parkinson's disease along the olfactory trac(t). J Neural Transm Suppl 2006; 70: 321–325. [DOI] [PubMed] [Google Scholar]
- 31.Del Tredici K, Rub U, De Vos RA, et al. Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 2002; 61: 413–426. [DOI] [PubMed] [Google Scholar]
- 32.Temma T, Yamazaki M, Miyanohara J, et al. Sequential PET estimation of cerebral oxygen metabolism with spontaneous respiration of (15)O-gas in mice with bilateral common carotid artery stenosis. J Cereb Blood Flow Metab 2017; 37: 3334–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welsh FA, Sims RE, McKee AE.Effect of glucose on recovery of energy metabolism following hypoxia-oligemia in mouse brain: dose-dependence and carbohydrate specificity. J Cereb Blood Flow Metab 1983; 3: 486–492. [DOI] [PubMed] [Google Scholar]
- 34.Iida H, Ohata H, Iida M, et al. Isoflurane and sevoflurane induce vasodilation of cerebral vessels via ATP-sensitive K+ channel activation. Anesthesiology 1998; 89: 954–960. [DOI] [PubMed] [Google Scholar]
- 35.Li CX, Patel S, Wang DJ, et al. Effect of high dose isoflurane on cerebral blood flow in macaque monkeys. Magn Reson Imaging 2014; 32: 956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKinley MJ, Albiston AL, Allen AM, et al. The brain renin-angiotensin system: location and physiological roles. Int J Biochem Cell Biol 2003; 35: 901–918.[12676175] [DOI] [PubMed] [Google Scholar]
- 37.Labandeira-Garcia JL, Rodriguez-Pallares J, Dominguez-Meijide A, et al. Dopamine-angiotensin interactions in the basal ganglia and their relevance for Parkinson's disease. Mov Disord 2013; 28: 1337–1342. [DOI] [PubMed] [Google Scholar]
- 38.Nemani VM, Lu W, Berge V, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010; 65: 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senior SL, Ninkina N, Deacon R, et al. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur J Neurosci 2008; 27: 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venda LL, Cragg SJ, Buchman VL, et al. Alpha-synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci 2010; 33: 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahn TB, Kim SY, Kim JY, et al. alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 2008; 70: 43–49. [DOI] [PubMed] [Google Scholar]
- 42.Pinotsi D, Michel CH, Buell AK, et al. Nanoscopic insights into seeding mechanisms and toxicity of alpha-synuclein species in neurons. Proc Natl Acad Sci USA 2016; 113: 3815–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singleton AB, Farrer M, Johnson J, et al. Alpha-synuclein locus triplication causes Parkinson's disease. Science 2003; 302: 841. [DOI] [PubMed] [Google Scholar]
- 44.Escartin C, Rouach N.Astroglial networking contributes to neurometabolic coupling. Front Neuroenergetics 2013; 5: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng B, Liao Z, Locascio JJ, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson's disease. Sci. Transl. Med 2010; 2: 52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pagano G, Polychronis S, Wilson H, et al. Diabetes mellitus and Parkinson disease. Neurology 2018; 90: e1654–e1662. [DOI] [PubMed] [Google Scholar]
- 47.Haehner A, Rodewald A, Gerber JC, et al. Correlation of olfactory function with changes in the volume of the human olfactory bulb. Arch Otolaryngol Head Neck Surg 2008; 134: 621–624. [DOI] [PubMed] [Google Scholar]
- 48.Zapiec B, Dieriks BV, Tan S, et al. A ventral glomerular deficit in Parkinson's disease revealed by whole olfactory bulb reconstruction. Brain 2017; 140: 2722–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lecoq J, Tiret P, Najac M, et al. Odor-evoked oxygen consumption by action potential and synaptic transmission in the olfactory bulb. J Neurosci 2009; 29: 1424–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baskin DG, Porte D, Jr, Guest K, et al. Regional concentrations of insulin in the rat brain. Endocrinology 1983; 112: 898–903. [DOI] [PubMed] [Google Scholar]
- 51.Hill JM, Lesniak MA, Pert CB, et al. Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience 1986; 17: 1127–1138. [DOI] [PubMed] [Google Scholar]
- 52.Dunn L, Allen GF, Mamais A, et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol Aging 2014; 35: 1111–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez-Araujo G, Nakagami H, Hayashi H, et al. Alpha-synuclein elicits glucose uptake and utilization in adipocytes through the Gab1/PI3K/Akt transduction pathway. Cell Mol Life Sci 2013; 70: 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lieu CA, Chinta SJ, Rane A, et al. Age-related behavioral phenotype of an astrocytic monoamine oxidase-B transgenic mouse model of Parkinson's disease. PLoS One 2013; 8: e54200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sager TN, Kirchhoff J, Mork A, et al. Nest building performance following MPTP toxicity in mice. Behav Brain Res 2010; 208: 444–449. [DOI] [PubMed] [Google Scholar]
- 56.Burton EJ, McKeith IG, Burn DJ, et al. Cerebral atrophy in Parkinson's disease with and without dementia: a comparison with Alzheimer's disease, dementia with Lewy bodies and controls. Brain 2004; 127(Pt 4): 791–800. [DOI] [PubMed] [Google Scholar]
- 57.Zhong Z, Merkitch D, Karaman MM, et al. High-spatial-resolution diffusion MRI in Parkinson disease: lateral asymmetry of the substantia nigra. Radiology 2019; 291: 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]