

Abstract
Small noncoding RNAs (sRNA/sncRNAs) are generated from different genomic loci and play important roles in biological processes, such as cell proliferation and the regulation of gene expression. Next-generation sequencing (NGS) has provided an unprecedented opportunity to discover and quantify diverse kinds of sncRNA, such as tRFs (tRNA-derived small RNA fragments), phasiRNAs (phased, secondary, small-interfering RNAs), Piwi-interacting RNA (piRNAs) and plant-specific 24-nt short interfering RNAs (siRNAs). However, currently available web-based tools do not provide approaches to comprehensively analyze all of these diverse sncRNAs. This study presents a novel integrated platform, sRNAtools (https://bioinformatics.caf.ac.cn/sRNAtools), that can be used in conjunction with high-throughput sequencing to identify and functionally annotate sncRNAs, including profiling microRNAss, piRNAs, tRNAs, small nuclear RNAs, small nucleolar RNAs and rRNAs and discovering isomiRs, tRFs, phasiRNAs and plant-specific 24-nt siRNAs for up to 21 model organisms. Different modules, including single case, batch case, group case and target case, are developed to provide users with flexible ways of studying sncRNA. In addition, sRNAtools supports different ways of uploading small RNA sequencing data in a very interactive queue system, while local versions based on the program package/Docker/virtureBox are also available. We believe that sRNAtools will greatly benefit the scientific community as an integrated tool for studying sncRNAs.
Keywords: small noncoding RNA, software, phasiRNA, tRF, 24-nt siRNA, wood formation
Key Points
Diverse kinds of sRNA/sncRNA can be comprehensively identified, profiled and functionally annotated by sRNAtools for up to 21 model species.
Different modules, including single case, batch case, group case and target case are developed to provide users with flexible ways of studying sncRNA.
sRNAtools supports different ways of uploading small RNA sequencing data in a very interactive queue system, while local versions are also available.
Very user-friendly and interactive web interfaces are provided by sRNAtools to present the obtained results, which greatly facilitate the study of sncRNAs.
Introduction
In recent years, in addition to microRNAs (miRNAs), many important small noncoding RNAs (sncRNAs) have been identified in animals and plants and shown to play important cellular roles, such as in the regulation of gene expression, RNA processing and cell proliferation. For example, tRFs (tRNA-derived small RNA fragments) are small noncoding RNAs generated from mature or precursor transfer RNAs (tRNAs). They are observed in almost every branch of life [1] and play important roles in the regulation of gene expression [2]. The 24-nucleotide small-interfering RNAs (24-nt siRNAs), which account for a large percentage of the total plant siRNA pool, play crucial roles in guiding plant-specific RNA-directed DNA methylation to transcriptionally silent transposon elements, transgenes, repetitive sequences and some endogenous genes [3, 4]. PhasiRNAs, phased, secondary, small-interfering RNAs, are another type of sncRNA that are common in plants and that have been well described for their ability to function ‘in trans’ to suppress target transcript levels [5].
Next-generation sequencing (NGS) has been widely used in the high-throughput characterization of small noncoding RNA transcriptomes. It has offered an unprecedented opportunity to discover and quantify small RNAs and to identify differentially expressed small RNA transcripts under various conditions. There is a range of publicly available tools for small RNA transcriptome analysis from high-throughput sequencing data, such as miRDeep [6], Mireval [7], miRNAkey [8], miRanalyzer [9], miRTRAP [10], DSAP [11], CAP-miRSeq [12], miRspring [13], tRF2Cancer [14], tDRmapper [15], DARIO [16], ncPRO-seq [17], ShortStack [18], DeAnnIso [19], PhaseTank [20], mirPRo [21], miRge 2.0 [22] and miRquant 2.0 [23]. However, to the best of our knowledge, the focus of most of these available tools is directed toward miRNAs and their functionalities. Although some integrated tools have been developed to study sncRNA, such as CPSS 2.0 [24], mirTools 2.0 [25], the UEA sRNA workbench [26], Oasis 2.0 [27], Unitas [28], sRNAnalyzer [29], sRNAtoolbox [30, 31] and SPAR [32], a comprehensive and convenient web-based tool to identify and analyze diverse kinds of sncRNA and their potential functions in different species is still lacking.
Here, we present sRNAtools (https://bioinformatics.caf.ac.cn/sRNAtools or https://bioinformatics.sc.cn/sRNAtools), which can be used to identify and functionally annotate diverse kinds of sncRNA for up to 21 model species (including human, mouse, Arabidopsis, rice, etc.), including (i) profiling of miRNAs, Piwi-interacting RNA (piRNAs), piRNA-producing loci (piRNA cluster), tRNAs, natsiRNA, small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs) and rRNAs and (ii) discovering isomiRs, novel miRNAs, tRFs, plant-specific 24-nt siRNAs and phasiRNAs. sRNAtools also provides web-based approaches to detect sncRNAs in multiple transcriptomes. Meanwhile, sRNAtools supports the uploading of data of diverse formats, including collapsed FASTA format, FASTQ format and inputs of GSM IDs/SRR IDs and accessible links. sRNAtools uses several JavaScript libraries (such as igv.js and forna.js) to make the web interfaces more interactive and intuitive. We believe that sRNAtools is a very comprehensive and convenient sncRNA analysis web tool and will greatly benefit the study of sncRNAs.
Analysis workflow
Data sources
Precursor and mature tRNA sequences were downloaded from the GtRNAdb database [33]. miRNA precursor sequences and other known small noncoding RNA sequences were retrieved from the latest miRBase database (release v22.0) (http://www.mirbase.org/) and latest RFAM database (release v14.0) [34] (Supplementary Table S1), respectively. mRNA sequences and gene annotations were downloaded from the Phytozome database (v12.1) [35] for plant species and the Ensembl database [36] for other species. The lncRNA sequences were obtained from NONCODE [37] and CANTATAdb [38]. circRNA sequences were retrieved from CIRCpedia v2 [39], CircFunBase [40] and PlantcircBase [41]. Natural antisense transcripts (NATs) were downloaded from RNAcentral [42] and PlantNATsDB [43]. piRNAs and the loci that produce them were retrieved from piRBase [44] and piRNAclusterDB [45], respectively. Plant 24-nt siRNA-producing loci were obtained from Pln24NT [46]. Sequences containing tRNA genes and 100 bp downstream of the 3′-end of such genes were extracted as precursor tRNA genes. For mature tRNA sequences, the introns were removed and ‘CCA’ was added to the 3′-end of the tRNA gene sequences. The fully supported species for different RNA classes (miRNA, tRNA, piRNA/24-nt siRNA, piRNA/24-nt siRNA-producing sites, lncRNA, circRNA, NAT, snRNA, snoRNA and rRNA) include human, mouse, rat, zebrafish, chicken, pig, rhesus, fly, Caenorhabditis elegans, Arabidopsis, rice, maize, soybean and tomato (Supplementary Table S1 and S2). Other sncRNAs, including tRF, isomiR, novel miRNA and phasiRNA, can be detected ab initio based on RNA libraries and genome sequences.
To functionally annotate the sRNA target genes, several popular gene functional datasets were collected, including the Gene Ontology (GO) and KEGG pathways from clusterProfiler package [47], Reactome pathways from ReactomePA packages [48] and Disease Ontology, Network of Cancer Gene and DisGeNET disease genes from DOSE packages [49]. The functional datasets from the Molecular Signatures Database (MSigDB) [50] were also included, which contains chemical and genetic perturbation genes, microRNA target motifs, cancer gene neighborhoods, cancer module genes, oncogenic signature genes and immunological signature genes. Gene functional interaction data were obtained from the STRING database [51].
Overview of sRNAtools workflow
The overall workflow of sRNAtools is shown in Figures 1 and 2. The sRNAtools web server provides four functional modules: single case, batch case, group case and target gene case. The single-case module allows users to identify and profile diverse sncRNAs for a single sample. The batch-case module allows users to submit multiple samples at the same time. The group-case and target-gene-case modules are designed to study potential sncRNA functions by analyzing their differential expression, sncRNA targets and target gene function enrichments. Finally, all of the results are shown in interactive tables and figures on the web page, which are all available for downloading in different forms.
Figure 1.
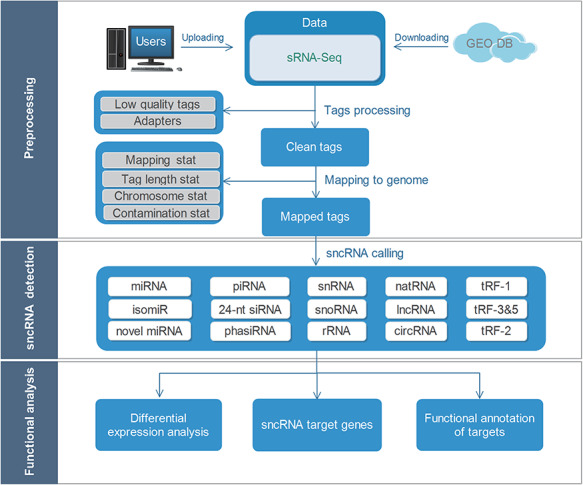
Overall sRNAtools workflow.
Figure 2.
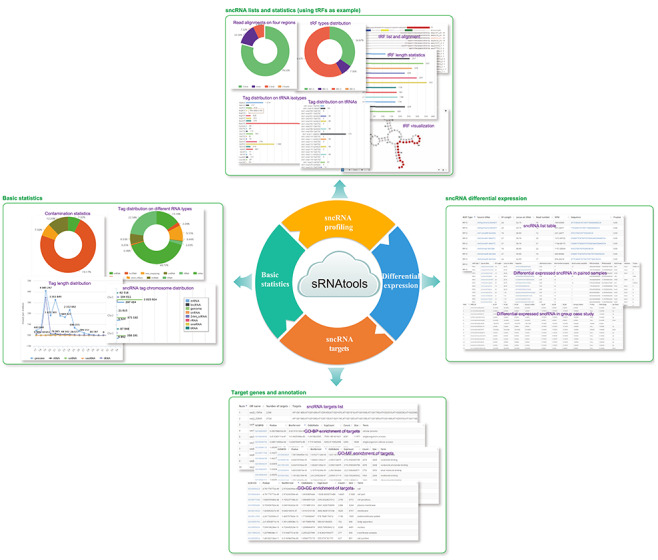
Screenshots of sRNAtools outputs. The outputs typically contain four types of information: the basic mapping statistics of tags, sncRNA list and annotation, differential expression list and annotation and sncRNA target gene list and functional enrichments.
Small RNA-seq data pre-processing
Cutadapt (v1.18) in Python (v2.7) is used to sequentially trim 5′ and 3′ adapters from raw reads of a FASTQ file. FASTX-Toolkit is then utilized to trim and filter reads with low quality (Q20 as the default setting). Users can input the adapters or choose the adapter type from several small RNA sequencing protocols, including smRNA-seq [52], ENCODE microRNA-seq [53], ENCODE small RNA-seq [53] and single-cell small RNA-seq [with unique molecular identifiers (UMI)] [54]. For smRNA-seq, three protocol types are provided: TruSeq small RNA (-a TGGAATTCTCGGGTGCCAAGG), Illumina v1.0 small RNA (-a TCGTATGCCGTCTTCTGCTTG) and Illumina v1.5 small RNA (-a ATCTCGTATGCCGTCTTCTGCTTG). For ENCODE microRNA-seq, the parameter for Cutadapt is ‘-a ACGGGCTAATATTTATCGGTGGAGCATCACGATCTCGTAT -g CAGTCG -g TGACTC -g GCTAGA -g ATCGAT’. For ENCODE small RNA-seq, four different options are provided: ENCODE3 (-a TGGAATTCTC), A_Tailing_No_Barcode (-a AAAAA -e 0), A_Tailing_N3 (-a AAAAA -e 0 --clip_R1 5) and A_Tailing_N4 (-a AAAAA -e 0 --clip_R1 6). For single-cell small RNA-seq, a pipeline (https://github.com/eyay/smallseq) is integrated in sRNAtools to remove UMI and adapters. Furthermore, the web server can estimate the adapters based on highly abundant miRNA sequences in the relevant species and enable the automatic detection of quality type (Phred33 or Phred64). The reads of lengths within the predefined range (18–45 as the default) are kept for downstream analysis. In the quality control step, cross-species contamination checking is performed based on miRTrace [55].
sncRNA identification and expression profiling
After adapter trimming, low-quality filtering and contamination checking [55], the clean sequencing data are mapped to a reference genome and different RNA sequence libraries using Bowtie (v1.2.2) [56] (http://bowtie-bio.sourceforge.net). In this process, a maximum of one mismatch (-v 1) is allowed, and the best alignments of reads with no more than 20 hits (-a -m 20 --best -strata) are reported as the default setting. To measure expression, all small RNA reads/tags reported when mapping against the respective RNA library are selected to calculate reads/tags per million (RPM/TPM) for each RNA. For miRNA, tRNA, piRNA or 24-nt siRNA, a separate multiple mapping threshold can be set by users due to their different constitutions of repeats, with 10, 30 and 50 multiple mapping times as the default thresholds. The mapped tags are then used to identify and profile sncRNAs, including miRNAs, piRNAs, tRNAs, snRNAs, snoRNAs, rRNAs and other sncRNAs, based on the constructed libraries (Figure 1). If the sequencing reads can map to multiple types of RNA libraries, the priority order of miRNA, tRNA, piRNA, snRNA, snoRNA, rRNA, mRNA, lncRNA, circRNA and NAT is used. For tRF identification, the reads aligned to precursor tRNA genes and mature tRNA sequences with the same strand as the source tRNA are used for tRF analysis. To distinguish actual tRFs from random degradation fragments, the binomial test is utilized [14]. The algorithm for identification of the templated and non-templated 5′-isomiRs and 3′-isomiRs is based on isomiR2Function [57]. Plant-specific 24-nt siRNA is identified using the algorithm implemented in Pln24NT [46]. For novel miRNA identification, two commonly used programs, miRDeep [6] and Mireap (http://mireap.sourceforge.net/), are implemented based on genome-mapped reads that cannot be mapped to any RNA libraries. PHASIS (v3.0) [58] is implemented to detect phasiRNA and phasiRNA loci.
Detection of differential expression
To compare differentially expressed sncRNAs between two samples (without replicates), DEseq [59] and edgeR [60] can be used to infer the statistical significance of differential expression [61]. Meanwhile, the difference in expression between two experimental groups with multiple samples/replicates can be analyzed using DEseq [59], DEseq2 [60], edgeR [62], Wilcoxon’s rank-sum test [61] and Student’s t-test. As the default, a sncRNA is considered to be significantly differentially expressed when the P value is ≤0.05 and the fold change is at least 1.5-fold relative to normalized read counts. The read/tag counts of identified sncRNAs are normalized to the total number of small RNA reads that are matched to all mapped reads in each sample [reads/tags per million (RPM/TPM)].
Analysis of target genes
To identify target genes of sncRNAs, sRNAtools implements four widely used miRNA target prediction tools [Tapirhybrid (v1.1) [63] and Targetfinder (v1.0) [64] for plant species, and RNAhybrid (v2.2.1) [65] and miRanda (v1.9) [66] for other species]. The parameters can be set by users for each target prediction tool. Various functional gene enrichments are further used to explore the potential biological function of predicted targeted genes, including GO [67] and KEGG pathway for all supported species, and Reactome pathway, Disease Ontology, Network of Cancer Gene, DisGeNET disease genes and MSigDB functional gene sets for human, mouse, rat, zebrafish, fly and C. elegans (Supplementary Table S1). In this functional enrichment process, the GO annotation terms and pathways of predicted targets are first extracted from the relevant annotation dataset, and then Fisher’s exact test is used to perform enrichment analysis (P value <0.01 as the default). Meanwhile, the gene functional network of target genes can be visualized interactively in netviewer [68].
sRNAtools web service
Data inputs
The single-case and batch-case modules allow users to submit small RNA sequencing data, GSM sample IDs from the GEO database (https://www.ncbi.nlm.nih.gov/geo), SRR sample IDs from the SRA database (https://www.ncbi.nlm.nih.gov/sra) or accessible web links of their data. In the single case, when uploading sequencing data, the uploading of data in FASTQ format is also supported. We added a functionality in the web server to support different small RNA sequencing protocols that use different adapters when constructing the sequencing library [such as smRNA-seq [52], ENCODE short total RNA-seq [53], ENCODE miRNA-seq [53] and single-cell small RNA-seq (with UMI)] [54] when the users upload a raw FASTQ file.
It is recommended that the file be in collapsed FASTA format or a FASTA file in zip or gz compressed format to speed up the uploading. The maximum upload size for a single file is as large as 3 Gb. The header for collapsed FASTA format is in the form of ‘>seq1_160’, where ‘seq1’ is a user-definable unique ID and ‘160’ represents the frequency of tags in seq1. In the group-case module, the single-case analysis job IDs are required as inputs, and each group should contain at least one sample. When a single-case analysis job ID is given, the web server will retrieve the corresponding sncRNA list file automatically. The target analysis module can be used to analyze sncRNA target genes and their potential functions. After the submission of data, the data analysis queue system provides users with a job ID that can be used to retrieve the results once the job is finished.
Users can set a length interval in advance, and only the tag sequences within this interval (18–45 nt as the default) will be considered for sncRNA detection. Meanwhile, sRNAtools provides many other useful parameters, such as the number of allowed mismatches (with the default of a maximum of one mismatch) and the multiple mapping times in tag sequence mapping. Two parameters, P value (<0.05 as the default) and minimum tag abundance (>20 RPM as the default), can be set by users to define high-confidence tRFs from random fragments. To detect sncRNAs that are differentially expressed between samples, the desired statistical significance in terms of P value threshold and fold change in normalized sequence counts can be defined by users. All input web pages are organized with examples to help users achieve correct inputs.
Data outputs
All sRNAtools outputs are presented in intuitive web interfaces, which typically contain the following information: (i) basic mapping statistics of small RNA tags, including cross-species taxon distribution (contamination evaluation), tag length distribution, tag distribution among different RNA types and tag chromosome position distribution; (ii) sncRNA result lists, expression and/or statistical plots of miRNAs, tRFs, tRNAs, rRNAs, snRNAs, snoRNAs, isomiRs, novel miRNAs, 24-nt siRNAs, piRNAs and phasiRNAs; (iii) list of differentially expressed sncRNAs and expression dot-plots; and (iv) sncRNA target genes and target functional annotation (Figure 2). Note that all tags can be shown in addition to the most abundant tags in sncRNA result lists.
Using tRFs as examples, the basic mapping statistics include pie summary charts of tags mapping on tRNAs, pie summary charts of tags mapping on four tRF regions of tRNAs (5′-end, 3′-end, internal and 3′-trailer), distribution charts of small RNA tag lengths, distribution charts of mapped tags on different tRNA isotypes and distribution charts of mapped tags on different tRNAs (Figure 2). Note that, as in miRNA profiling procedures in other miRNA tools (e.g. mirTools [61]), all charts for basic mapping statistics are based on the frequency of unique reads and the expression level of total reads (the number of reads for each tag). The tRF result list provides a detailed annotation for each detected tRF, including tRF type, source tRNA, tRF length, location of tRF on the tRNA, absolute tag count, normalized RPM (reads per million) expression value, tRF sequence and the detection P value. A link to a detailed information page is also included for each tRF, in which the sequence alignments of tRFs aligned to source tRNAs are shown in tabulated format, while the location of tRFs on tRNA secondary structures is also displayed by Forna.js [69].
In group-case study, if there are no replicates, the outputs contain the list of differentially expressed sncRNAs and plots of the correlation of expression between the two samples. The list of differentially expressed sncRNAs contains the expression values of the two groups, the expression fold change, up/down tags and the differential expression P value. If replicates are provided, the sncRNAs differentially expressed between the two groups are listed and the annotation of the group expression list contains the expression value of each sample, the expression fold change, up/down tags and the statistical P value. Furthermore, in pages on the differential expression for a group-case study, target gene analysis can be performed directly based on the differentially expressed sncRNAs. The sncRNA target output contains the list of predicted sncRNA target genes and their functional annotation with enrichments of Gene Ontology, KEGG pathways and other pathways. Meanwhile, interactive virtualization of the interaction network of target genes is also provided in the target output web page.
Comparison with other integrated sRNA tools
Given the small size of sRNA high-throughput sequencing data, there is a range of publicly available tools for integrated small RNA transcriptome analysis from high-throughput sequencing data, such as DARIO [16], ncPRO-seq [17], the UEA sRNA workbench [26], CPSS 2.0 [24], mirTools 2.0 [25], Unitas [28], Oasis 2.0 [27], sRNAnalyzer [29], sRNAtoolbox [30] and SPAR [32] (Supplementary Table S3). However, to the best of our knowledge, the main focus of these available tools is directed toward a limited subset of sRNAs or a limited number of functionalities.
In addition to the most extensively studied microRNAs, many important small noncoding RNAs (sncRNAs) have recently been identified in animals and plants, which play very important cellular roles, such as tRFs, the 24-nucleotide small-interfering RNAs (24-nt siRNAs) and phasiRNAs. A comprehensive and convenient web-based tool to identify and analyze all of these diverse sRNAs and their potential functions in different species is still lacking. sRNAtools can not only profile known RNAs based on different available RNA libraries (Supplementary Table S3), such as of miRNA, piRNA, tRNA, rRNA, snRNA and snoRNA, but also identify other sRNAs ab initio, such as novel miRNAs, isomiRs, tRFs, 24-nt siRNAs and phasiRNAs, which are not easily identified by genomic overlapping methods. In the identification of tRFs, a binomial test is utilized, which can distinguish high-confidence tRFs from random degradation fragments.
Although some tools such as Oasis and sRNAtoolbox with multiple functionalities have been developed, the functional modules are not integrated with each other. In contrast, sRNAtools provides a one-stop analysis function for convenient sRNA-seq data analysis. For the data input, sRNAtools supports a total of four different ways for users to submit their data and provides a very interactive queue system to analyze data. sRNAtools also supports the data uploading in the batch model while local versions are also available, which can help users to analyze the data on their own servers. The outputs of most existing tools are not intuitive and interactive for users, especially users without bioinformatics expertise. sRNAtools integrates many JavaScript libraries, which makes manipulation easier and enables the output results to be shown in a user-friendly way. All of the tables and figures in the results pages can be downloaded in different formats, such as png, jepg, pdf and svg for figures, and xls, txt, csv and pdf for tables. Other specific features of sRNAtools include that (i) it can check the cross-species contamination by miRTrace, especially in studies of clinical and field parasitology and food quality control; (ii) igv.js is used for the first time to show the small RNA tag mapping in the web page; and (iii) Docker is used to construct the local version, which is easily to be installed and configured.
Comparison with comprehensive sncRNA databases for human
To better illustrate the constitution of the data in sRNAtools, we also compared the dataset in sRNAtools with other comprehensive small RNA databases for human (Supplementary Table S4), including DASHR [52], DASHR 2.0 [70] and GENCODE [71]. DASHR 2.0 is an updated version of DASHR, which contains the highest number of RNA records for miRNA, tRNA, snRNA, rRNA and scRNA, while sRNAtools contains the highest number of records for piRNA, snoRNA, lncRNA and circRNA. sRNAtools uses statistical methods to identify tRFs ab initio, enabling many more tRFs to be identified with statistical confidence. Besides, sRNAtools can also identify other sncRNAs for human, such as isomiR and novel miRNA, which is not available in DASHR. GENCODE contains the smallest number of sncRNA records.
Validation using public datasets
miRNAs have been implicated in brain development and neuron functions, and the miRNA malfunction has been revealed in many neurological disorders. In part due to the cellular heterogeneity in neural circuits, the mechanisms of miRNA have been difficult to study. To systematically study miRNAs in neurons, Miao et al. firstly used the Cre-loxP binary system in mice to target cell types. Next, using deep sequencing and qPCR validation, they revealed that several miRNAs show distinct expression profiles in glutamatergic and GABAergic neurons and subtypes of GABAergic neurons in the neocortex and cerebellum [72]. In total, 7, 23 and 10 differentially expressed miRNAs were validated by qPCR in comparisons of Purkinje cells versus whole cerebellum, Camk2a versus Gad2 samples and PV versus SST samples. By analyzing deep-sequencing data of this study (GSE30286), sRNAtools can successfully identify all of the validated differentially expressed miRNAs (Supplementary Table S5).
Transfer RNAs (tRNAs) are subjected to numerous RNA modifications, which can directly control their folding and stability [73]. N7-methylguanosine (m7G) at nucleotide 46 (m7G46) is one of the most prevalent modifications and has important physiological functions in mammals. Deficiency of the m7G tRNA methyltransferase METTL1/WDR4 complex results in neurological disease [74]. A total of 22 m7G modifications were identified in mammalian systems, and knockout of METTL1 was shown to greatly impact the stability of 22 m7G tRNAs [the exception being cysteine (Cys)] [75]. Based on public small RNA sequencing data of Mettl1 knockout and control in mouse embryonic stem cells (mESCs) (GSE112670), tRNA expression profiling by sRNAtools showed that the majority of m7G tRNAs (18/22) showed significant differential expression (Supplementary Table S6). When knocking out Mettl1, most of the m7G tRNAs were downregulated, indicating that the stability of m7G tRNA was indeed affected. The finding that Cys tRNAs were upregulated was unexpected, although this is consistent with the report of Shuibin et al. [75] (Supplementary Table S6).
tRFs have been reported in many diseases, such as epilepsy and developmental disorders [76, 77]. Qing et al. utilized deep sequencing and qPCR to demonstrate that the level of small RNA fragments derived from tRNAs strikingly increased in ischemic rat brain [78]. A variety of tRNA-derived small RNAs were profiled in response to ischemia, among which ValCAC, GlyGCC, HisGTG, ValAAC, GlyCCC, GluTTC and GluCTC showed highly differential expression and ValCAC and GlyGCC were confirmed by qPCR and northern blotting. By using the same deep-sequencing data (GSE70473), sRNAtools successfully showed similar results, indicating the high specificity of sRNAtools in tRF identification (Supplementary Table S7).
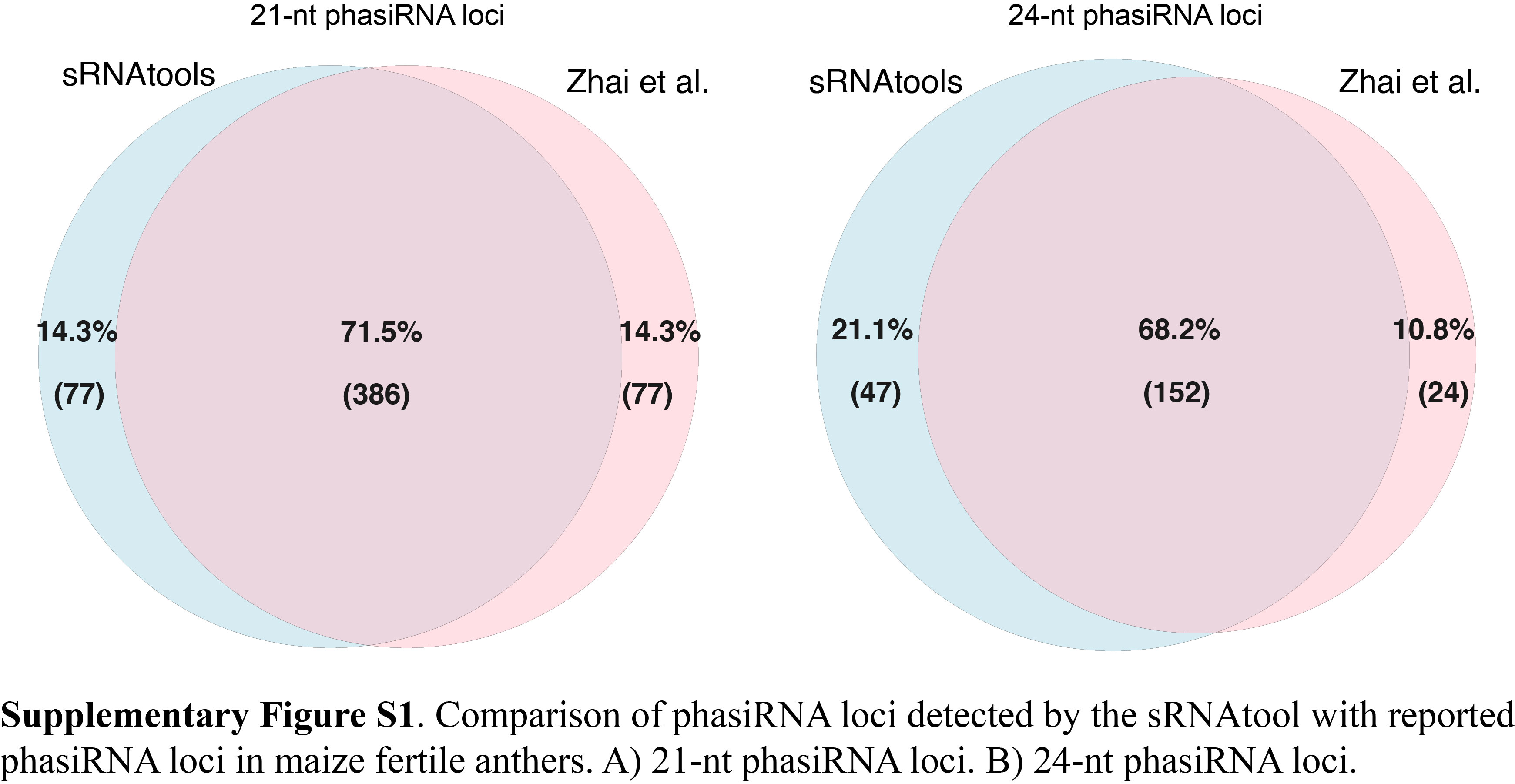
Phased small-interfering RNAs (phasiRNAs), a class of sncRNA that is widespread in the plant kingdom, are derived from cleavage fragments with 21- or 24-nt intervals from precursor RNA transcripts. In Arabidopsis, several genomic loci were reported to generate phasiRNAs [20, 79]. Many more phasiRNA loci have been revealed in monocots, such as 463 21-PHAS and 176 24-PHAS loci in maize fertile anthers [80]. Based on small RNA-seq data from different Arabidopsis tissues obtained from the GEO database, sRNA can successfully detect all of the phasiRNA loci reported (Supplementary Table S8). The results also indicate that the activity of phasiRNA shows different dynamics among different Arabidopsis tissues. In addition, by using the 32 samples of small RNA-seq data (GSE52293), sRNAtools can cover 71.5% of 21-nt phasiRNA loci and 68.2% of 24-nt phasiRNA loci in smRNA-seq data obtained from maize fertile anthers (Supplementary Figure S1).
Data upload speed and analysis speed evaluation
To test the data upload and analysis efficiency, we uploaded the dataset used as described above (Supplementary Table S9) to the HPC server from both China and the USA. The sizes of samples GSE30286 (18 samples), GSE112670 (4 samples), GSE70473 (10 samples), Arabidopsis tissues (14 samples) and GSE52293 (32 samples) were 145, 87, 80, 213 and 925 Mb, respectively. The uploading took 3, 2, 2, 4 and 16 min from China, and 7, 5, 5, 11 and 45 min from the USA, respectively. It is expected that the speed is higher in China due to the server being located in China. The data analysis times were about 38 min, 21 min, 12 min, 56 min and 5 h and 20 min, respectively.
Case studies
Woody plants provide large amounts of biomass, which can serve as raw material for the production of renewable energy and other commercial products. Poplar is a model system to study woody plants. miRNAs play important roles in the formation of poplar wood [81, 82]. To fully understand the characteristics of poplar sncRNAs, especially in wood formation, we used sRNAtools based on sRNA-seq to study sncRNAs in all poplar wood tissues (xylem, phloem and cambium) and leaves sRNA-seq data have been deposited in GEO database under accession number GSE139897. The mapping statistics showed that (i) tRNA tags are relatively more abundant in xylem; (ii) miRNA tags are highly abundant in leaves; and (iii) 24-nt siRNA tags are relatively more abundant in cambium (Figure 3A). Accordingly, the length distribution showed three clear peaks at 19, 21 and 24 nt in xylem, leaf and cambium, respectively (Figure 3B). After careful checking, we found that the 19-nt peaks mainly represent tRFs, indicating the importance of tRFs in xylem development (Figure 4). The miRNA expression revealed that several miRNAs known to be associated with wood development (e.g. mir-167 and miR475b) were expressed at low levels in wood tissue compared with the levels in leaves, indicating their negative regulatory roles in wood formation (Figure 5A). Meanwhile, we also identified significantly more novel miRNAs in cambium (P = 0.023), which is in accordance with its highly dynamic cellular status (Figure 5B). The integrated novel miRNA list and associated detailed information (abundance, target gene, mapping between miRNA and target region, and the secondary structure of the precursor, etc.) are available at https://bioinformatics.caf.ac.cn/sRNAtools/result_ptc_novel.php. The 24-nt siRNAs are also more abundant in cambium, although the P value is not significant. In comparison with other gramineous monocots, poplar contains a small number of phasiRNAs and woody tissues generate lower levels of phasiRNAs than leaves (Figure 5B).
Figure 3.
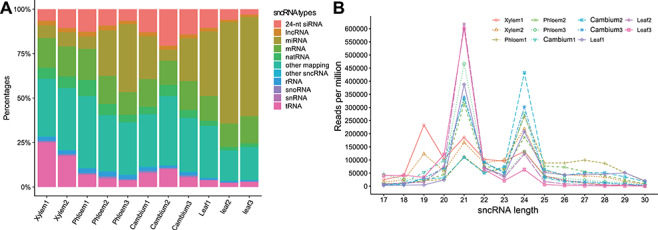
Mapping statistics and length distribution of small RNA tags among poplar xylem, phloem, cambium and leaf tissues. Each tissue has three replicates, except for xylem with two replicates after filtering out a low-quality replicate. (A) Mapping statistics among different RNA types. (B) Length distribution for different sncRNA tags.
Figure 4.
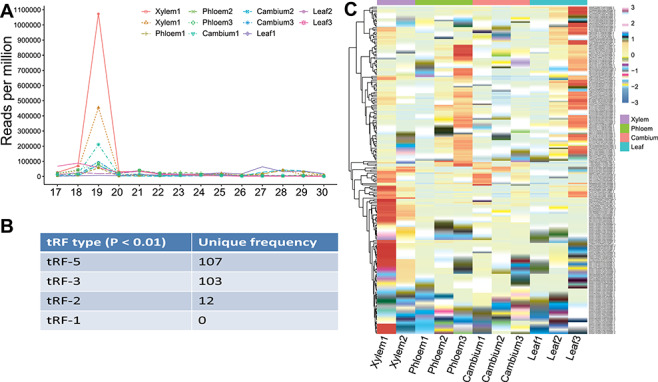
tRNA fragment distribution among different poplar wood tissues and leaves. (A) Length distribution of tRNA fragment (tRF) tags indicating abundant 19-nt tRFs in xylem tissues. (B) High-confidence 19-nt tRFs (P value <0.01) identified among all of the tissues. (C) Expression heatmap of high-confidence 19-nt tRFs revealing high expression dynamics of tRFs in woody tissues.
Figure 5.
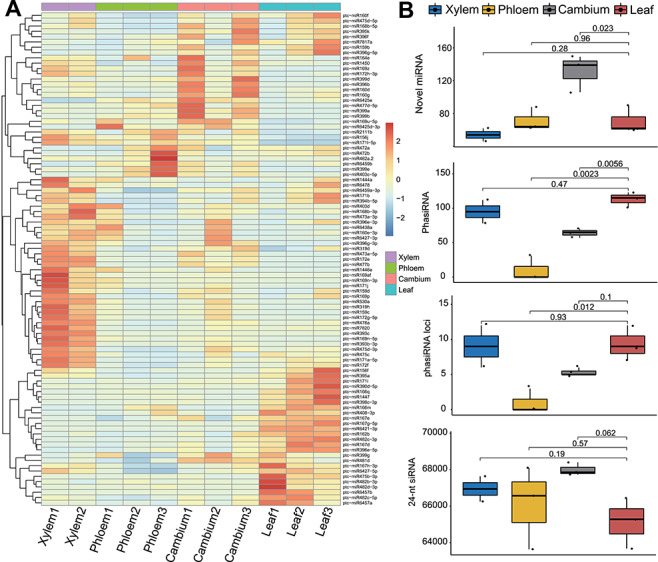
Identification and profiling of other sncRNAs in poplar woody tissues. (A) miRNA expression heatmap. (B) Novel miRNA, phasiRNA and their loci and 24-nt siRNA identified among different tissues.
Implementation
The web server is hosted within a PHP/Apache environment under a Linux system and is equipped with four hexadeca-core (64 cores) Intel Xeon processors (2.1 GHz each) and 512 GB of RAM. The back-end pipeline is implemented in the Python/Perl language, and some plots are drawn by R (http://www.r-project.org). Several JavaScript libraries, including JQuery (https://jquery.com/), DataTable (https://datatables.net), Highchart (https://www.highcharts.com), Fornac.js [69] and igv.js [83], were used to create dynamic and interactive data visualization in web browser interfaces, which provide users with a highly intuitive interface for manipulating the tool and viewing the analysis results.
Perspectives
In addition to miRNAs, other important sncRNA molecules have been discovered in the functional genomics era. High-throughput sequencing greatly facilitates study of the small RNA transcriptome and offers an effective method to comprehensively investigate sncRNAs in genomes. However, the convenient detection and profiling of all of these sncRNAs from large amounts of sequencing data remain a challenge. Therefore, we developed an automated and easy-to-use web service, sRNAtools, for research communities to identify, profile and functionally annotate sncRNAs based on high-throughput sequencing. Currently, sRNAtools supports 21 model reference genomes across vertebrates, insects, nematodes and plants. More species will be supported in the future. The specific RNA annotation for some species, such as piRNA annotation for chimpanzee, is still not available. When the relevant data become available in a public database, they will be added to sRNAtools. Furthermore, the data in sRNAtools will be updated regularly to keep pace with changes in the source databases. We also developed a local pipeline, Docker (https://www.docker.com), and a VirtualBox (https://www.virtualbox.org) version of sRNAtools, which are available to download on the download web page. Recently, more and more RNA modifications have been revealed in sncRNAs. A sncRNA modification identification function will be developed in sRNAtools. Overall, we believe that sRNAtools will greatly benefit the scientific community as an integrated tool for studying sncRNAs.
Author contributions
Q.L. constructed the sRNAtools web server and drafted the manuscript. C.J.D., X.Q.L. and G.G.G. participated in the pipeline development in the web server. X.Q.L. constructed the local pipeline, Docker and a VirtualBox version of sRNAtools. C.J.D. participated in drafting the manuscript. J.F.C. participated in the administration of the HPC server. C.J.D. participated in the small RNA-seq of poplar wood and leaf tissues. X.H.S. was involved in planning of the study and headed the project. All authors read and approved the final manuscript.
Supplementary Material
{kind=link}
Qi Liu was a PhD candidate at the State Key Laboratory of Tree Genetics and Breeding, Research Institute of Forestry, Chinese Academy of Forestry, working on gene functional study based on systems biology and small RNA data integration and visualization.
Changjun Ding is a researcher at the State Key Laboratory of Tree Genetics and Breeding, Research Institute of Forestry, Chinese Academy of Forestry, working on functional study of small RNA in hybrid vigor of forestry trees.
Xiaoqiang Lang is a scientist at the West China Medical Center of Sichuan University, working on bioinformatics big data analysis pipeline development and integration.
Ganggang Guo is a researcher at Institute of Crop Science of Chinese Academy of Agricultural Sciences, working on research on crop germplasm resources, genetic breeding and small RNA molecular biology.
Jiafei Chen is a scientist at the State Key Laboratory of Tree Genetics and Breeding, Research Institute of Forestry, Chinese Academy of Forestry, working on bioinformatics project management and high-performance computer administration.
Xiaohua Su is a researcher and associate director of the State Key Laboratory of Tree Genetics and Breeding, director and chief expert of Forest Tree Breeding, Research Institute of Forestry, Chinese Academy of Forestry. She is also a member of Co-Innovation Center for Sustainable Forestry in Southern China, Nanjing Forestry University. Her team works on bioinformatics data integration, mapping of quantitative trait loci (QTL) and genetic engineering breeding.
Acknowledgements
We thank Jeremy Allen, PhD, from Liwen Bianji, Edanz Group China, for editing the English text of a draft of this manuscript.
Funding
Basic Research Fund of RIF (Grant No. CAFYBB2017ZY006); The National Natural Science Foundation of China (Grant No. 31870662).
References
- 1. Lee YS, Shibata Y, Malhotra A, et al. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 2009;23:2639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kumar P, Kuscu C, Dutta A. Biogenesis and function of transfer RNA-related fragments (tRFs). Trends Biochem Sci 2016;41:679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elvira-Matelot E, Hachet M, Shamandi N, et al. Arabidopsis RNASE THREE LIKE2 modulates the expression of protein-coding genes via 24-nucleotide small interfering RNA-directed DNA methylation. Plant Cell 2016;28:406–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. You C, Cui J, Wang H, et al. Conservation and divergence of small RNA pathways and microRNAs in land plants. Genome Biol 2017;18:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fei Q, Xia R, Meyers BC. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 2013;25:2400–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedlander MR, Chen W, Adamidi C, et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 2008;26:407–15. [DOI] [PubMed] [Google Scholar]
- 7. Gao D, Middleton R, Rasko JE, et al. miREval 2.0: a web tool for simple microRNA prediction in genome sequences. Bioinformatics 2013;29:3225–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ronen R, Gan I, Modai S, et al. miRNAkey: a software for microRNA deep sequencing analysis. Bioinformatics 2010;26:2615–6. [DOI] [PubMed] [Google Scholar]
- 9. Hackenberg M, Rodriguez-Ezpeleta N, Aransay AM. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res 2011;39:W132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hendrix D, Levine M, Shi W. miRTRAP, a computational method for the systematic identification of miRNAs from high throughput sequencing data. Genome Biol 2010;11:R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang PJ, Liu YC, Lee CC, et al. DSAP: deep-sequencing small RNA analysis pipeline. Nucleic Acids Res 2010;38:W385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun Z, Evans J, Bhagwate A, et al. CAP-miRSeq: a comprehensive analysis pipeline for microRNA sequencing data. BMC Genomics 2014;15:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Humphreys DT, Suter CM. miRspring: a compact standalone research tool for analyzing miRNA-seq data. Nucleic Acids Res 2013;41:e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng LL, Xu WL, Liu S, et al. tRF2Cancer: a web server to detect tRNA-derived small RNA fragments (tRFs) and their expression in multiple cancers. Nucleic Acids Res 2016;44:W185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Selitsky SR, Sethupathy P. tDRmapper: challenges and solutions to mapping, naming, and quantifying tRNA-derived RNAs from human small RNA-sequencing data. BMC Bioinformatics 2015;16:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fasold M, Langenberger D, Binder H, et al. DARIO: a ncRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res 2011;39:W112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen CJ, Servant N, Toedling J, et al. ncPRO-seq: a tool for annotation and profiling of ncRNAs in sRNA-seq data. Bioinformatics 2012;28:3147–9. [DOI] [PubMed] [Google Scholar]
- 18. Axtell MJ. ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 2013;19:740–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y, Zang Q, Zhang H, et al. DeAnnIso: a tool for online detection and annotation of isomiRs from small RNA sequencing data. Nucleic Acids Res 2016;44:W166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo Q, Qu X, Jin W. PhaseTank: genome-wide computational identification of phasiRNAs and their regulatory cascades. Bioinformatics 2015;31:284–6. [DOI] [PubMed] [Google Scholar]
- 21. Shi J, Dong M, Li L, et al. mirPRo-a novel standalone program for differential expression and variation analysis of miRNAs. Sci Rep 2015;5:14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu Y, Baras AS, Halushka MK. miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinformatics 2018;19:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kanke M, Baran-Gale J, Villanueva J, et al. miRquant 2.0: an expanded tool for accurate annotation and quantification of microRNAs and their isomiRs from small RNA-sequencing data. J Integr Bioinform 2016;13:307. [DOI] [PubMed] [Google Scholar]
- 24. Wan C, Gao J, Zhang H, et al. CPSS 2.0: a computational platform update for the analysis of small RNA sequencing data. Bioinformatics 2017;33:3289–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu J, Liu Q, Wang X, et al. mirTools 2.0 for non-coding RNA discovery, profiling, and functional annotation based on high-throughput sequencing. RNA Biol 2013;10:1087–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stocks MB, Mohorianu I, Beckers M, et al. The UEA sRNA Workbench (version 4.4): a comprehensive suite of tools for analyzing miRNAs and sRNAs. Bioinformatics 2018;34:3382–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rahman RU, Gautam A, Bethune J, et al. Oasis 2: improved online analysis of small RNA-seq data. BMC Bioinformatics 2018;19:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gebert D, Hewel C, Rosenkranz D. Unitas: the universal tool for annotation of small RNAs. BMC Genomics 2017;18:644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu X, Kim TK, Baxter D, et al. sRNAnalyzer-a flexible and customizable small RNA sequencing data analysis pipeline. Nucleic Acids Res 2017;45:12140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rueda A, Barturen G, Lebron R, et al. sRNAtoolbox: an integrated collection of small RNA research tools. Nucleic Acids Res 2015;43:W467–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aparicio-Puerta E, Lebron R, Rueda A, et al. sRNAbench and sRNAtoolbox 2019: intuitive fast small RNA profiling and differential expression. Nucleic Acids Res 2019;47:W530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuksa PP, Amlie-Wolf A, Katanic Z, et al. SPAR: small RNA-seq portal for analysis of sequencing experiments. Nucleic Acids Res 2018;46:W36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chan PP, Lowe TM. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 2016;44:D184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kalvari I, Argasinska J, Quinones-Olvera N, et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res 2018;46:D335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goodstein DM, Shu S, Howson R, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 2012;40:D1178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hubbard T, Barker D, Birney E, et al. The Ensembl genome database project. Nucleic Acids Res 2002;30:38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao Y, Li H, Fang S, et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res 2016;44:D203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szczesniak MW, Rosikiewicz W, Makalowska I. CANTATAdb: a collection of plant long non-coding RNAs. Plant Cell Physiol 2016;57:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong R, Ma XK, Li GW, et al. CIRCpedia v2: an updated database for comprehensive circular RNA annotation and expression comparison. Genomics Proteomics Bioinformatics 2018;16:226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meng X, Hu D, Zhang P, et al. CircFunBase: a database for functional circular RNAs. Database (Oxford) 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chu Q, Zhang X, Zhu X, et al. PlantcircBase: a database for plant circular RNAs. Mol Plant 2017;10:1126–8. [DOI] [PubMed] [Google Scholar]
- 42. Consortium TR. RNAcentral: a hub of information for non-coding RNA sequences. Nucleic Acids Res 2019;47:D1250–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen D, Yuan C, Zhang J, et al. PlantNATsDB: a comprehensive database of plant natural antisense transcripts. Nucleic Acids Res 2012;40:D1187–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J, Zhang P, Lu Y, et al. piRBase: a comprehensive database of piRNA sequences. Nucleic Acids Res 2019;47:D175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rosenkranz D. piRNA cluster database: a web resource for piRNA producing loci. Nucleic Acids Res 2016;44:D223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Q, Ding C, Chu Y, et al. Pln24NT: a web resource for plant 24-NT siRNA producing loci. Bioinformatics 2017;33:2065–7. [DOI] [PubMed] [Google Scholar]
- 47. Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu G, He QY. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst 2016;12:477–9. [DOI] [PubMed] [Google Scholar]
- 49. Yu G, Wang LG, Yan GR, et al. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015;31:608–9. [DOI] [PubMed] [Google Scholar]
- 50. Liberzon A, Birger C, Thorvaldsdottir H, et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leung YY, Kuksa PP, Amlie-Wolf A, et al. DASHR: database of small human noncoding RNAs. Nucleic Acids Res 2016;44:D216–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davis CA, Hitz BC, Sloan CA, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 2018;46:D794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Faridani OR, Abdullayev I, Hagemann-Jensen M, et al. Single-cell sequencing of the small-RNA transcriptome. Nat Biotechnol 2016;34:1264–6. [DOI] [PubMed] [Google Scholar]
- 55. Kang W, Eldfjell Y, Fromm B, et al. miRTrace reveals the organismal origins of microRNA sequencing data. Genome Biol 2018;19:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang K, Sablok G, Qiao G, et al. isomiR2Function: an integrated workflow for identifying microRNA variants in plants. Front Plant Sci 2017;8:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kakrana A, Li P, Patel P, et al. . PHASIS: a computational suite for de novo discovery and characterization of phased, siRNA-generating loci and their miRNA triggers. bioRxiv 2017:158832.
- 59. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhu E, Zhao F, Xu G, et al. mirTools: microRNA profiling and discovery based on high-throughput sequencing. Nucleic Acids Res 2010;38:W392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 2012;40:4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bonnet E, He Y, Billiau K, et al. TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 2010;26:1566–8. [DOI] [PubMed] [Google Scholar]
- 64. Fahlgren N, Jogdeo S, Kasschau KD, et al. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 2010;22:1074–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res 2006;34:W451–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Betel D, Koppal A, Agius P, et al. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 2010;11:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources Nucleic Acids Res 2017;45:D331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu Q, Ding C, Chu Y, et al. PoplarGene: poplar gene network and resource for mining functional information for genes from woody plants. Sci Rep 2016;6:31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kerpedjiev P, Hammer S, Hofacker IL. Forna (force-directed RNA): simple and effective online RNA secondary structure diagrams. Bioinformatics 2015;31:3377–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kuksa PP, Amlie-Wolf A, Katanic Z, et al. DASHR 2.0: integrated database of human small non-coding RNA genes and mature products. Bioinformatics 2019;35:1033–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012;22:1760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. He M, Liu Y, Wang X, et al. Cell-type-based analysis of microRNA profiles in the mouse brain. Neuron 2012;73:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pan T. Modifications and functional genomics of human transfer RNA. Cell Res 2018;28:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shaheen R, Abdel-Salam GM, Guy MP, et al. Mutation in WDR4 impairs tRNA m(7)G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol 2015;16:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lin S, Liu Q, Lelyveld VS, et al. Mettl1/Wdr4-mediated m(7) G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol Cell 2018;71:244–255 e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Blanco S, Dietmann S, Flores JV, et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J 2014;33:2020–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hogg MC, Raoof R, El Naggar H, et al. Elevation in plasma tRNA fragments precede seizures in human epilepsy. J Clin Invest 2019;129:2946–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li Q, Hu B, Hu GW, et al. tRNA-derived small non-coding RNAs in response to ischemia inhibit angiogenesis. Sci Rep 2016;6:20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chen HM, Li YH, Wu SH. Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc Natl Acad Sci U S A 2007;104:3318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhai J, Zhang H, Arikit S, et al. Spatiotemporally dynamic, cell-type-dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc Natl Acad Sci U S A 2015;112:3146–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Quan M, Xiao L, Lu W, et al. Association genetics in Populus reveal the allelic interactions of Pto-MIR167a and its targets in wood formation. Front Plant Sci 2018;9:744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xiao L, Quan M, Du Q, et al. Allelic interactions among Pto-MIR475b and its four target genes potentially affect growth and wood properties in Populus. Front Plant Sci 2017;8:1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.