

Abstract
Ribonucleic acid sequencing (RNA-seq) identifies and quantifies RNA molecules from a biological sample. Transformation from raw sequencing data to meaningful gene or isoform counts requires an in silico bioinformatics pipeline. Such pipelines are modular in nature, built using selected software and biological references. Software is usually chosen and parameterized according to the sequencing protocol and biological question. However, while biological and technical noise is alleviated through replicates, biases due to the pipeline and choice of biological references are often overlooked. Here, we show that the current standard practice prevents reproducibility in RNA-seq studies by failing to specify required methodological information. Peer-reviewed articles are intended to apply currently accepted scientific and methodological standards. Inasmuch as the bias-less and optimal RNA-seq pipeline is not perfectly defined, methodological information holds a meaningful role in defining the results. This work illustrates the need for a standardized and explicit display of methodological information in RNA-seq experiments.
Keywords: RNA-sequencing, reproducibility, computational workflow
Introduction
Ribonucleic acid sequencing (RNA-seq) enables the identification and quantification of RNA molecules from a biological sample. Microarrays, long considered the state of the art for large-scale RNA quantification, need a known genome annotation for probe design, prior to the actual experiment [1]. In contrast, in an RNA-seq experiment, a genome annotation is introduced after the sequencing step, permitting one to reanalyze the same dataset using different software and references [2], maintaining the relevance of datasets after genome and genomic annotation updates. In the past decade, RNA-seq has been rapidly democratized due to a dramatic lowering of the cost of sequencing. This has led to the diversification and multiplication of sequencing analysis applications, and the generation of large numbers of datasets to analyze. Hundreds of different software applications have been published to fulfill this need in a very modular fashion [3], allowing the usage of custom in silico pipelines defined by user-selected software and references for each analytical step.
Quantification results in RNA-seq are subject to different types of noise which are usually categorized as either technical or biological in nature [4]. Technical noise represents variations due to the laboratory manipulations, from RNA extraction to sequencing. Biological noise includes a broader array of sources, depending on the experimental design. This covers differences that can go from non-genetic individuality of cells exposed to homogeneous conditions [5], all the way to inter-individual genetic variations. Biological and technical sources of noise are stochastic in nature, and thus perfect reproducibility is impossible to achieve. Replicates can be used to quantify the variations and alleviate their impacts [6]. Several studies have already investigated the number of replicates needed for reproducible results [7–9]. However, another source of discrepancy seems to have often been overlooked. The optimal RNA-seq in silico processing pipeline, from raw sequencing files to meaningful gene or isoform counts, has not yet been (and perhaps will never be) defined. Thus, the same data can be processed in a multitude of ways, using different combinations of modular software and references. The distribution of a gene’s quantification across all the different RNA-seq pipelines (used or possible) is what we have dubbed the ‘in silico design noise’. Theoretically, such noise is deterministic in nature. While some software does have stochastic processes, these are undesirable when they cause random variability in the results. Given the exact same inputs (i.e. same dataset, software and parameters), it is preferable to always obtain the same results. Therefore, this ‘in silico design noise’ is actually a function of the software and parameter selection. One can reduce the impact of such noise and ensure the reproducibility of the analysis by explicitly specifying every information related to data transformations used to process the RNA-seq datasets. In this survey, we scrutinize the exhaustivity of the scientific literature regarding reported methodology of RNA-seq experiments. We find that only a minority of articles (25%) describe all essential computational steps and fewer still specify all parameter values to ensure complete reproducibility. From these analyses, we stress that a better disclosure of methodological information by users, developers and editors will beget more reproducible scientific literature.
Standard steps in an RNA-seq computational analysis pipeline
While RNA-seq experiments can be analyzed in different ways, in organisms with annotated genomes, RNA-seq computational pipelines typically all follow the same series of steps to obtain a quantification of transcripts from a raw read file (Figure 1), following which, diverse further analyses are possible [10]. Here, we focus on the first steps that are common to all RNA-seq pipelines, from read file to transcript quantification, consisting of several design choices of tools and references that are essential to use in order to compute quantification and to specify in order to ensure reproducibility of the results (Figure 1). The first such element is the description of the source of the raw read file, whether the RNA-seq data were generated in the context of the current study or whether the data were obtained from a repository such as the Gene Expression Omnibus. It is common to preprocess the data by verifying their quality, trimming reads of lower quality and removing non-biological sequences (i.e. sequencing adapters and indexes) before performing the alignment. We note however that current quality values per nucleotide are such that a preprocessing step might increasingly be considered unnecessary. While every other step is essential in RNA-seq, preprocessing is the only step that produces the same file type as it uses. This means that it does not, in a computational manner, need to be run. The alignment type (whether reads are aligned on the genome or the transcriptome) and the annotation file used to define the genomic features considered are also essential elements of methodological information. Finally, once the source of the reads and the identity of the genome/transcriptome are defined, one must also indicate the tools used to align the reads to the genome/transcriptome and to quantify the abundance of the different genomic features annotated. Each of these steps requires specification of parameter values to entirely describe how the step was carried out (Figure 1).
Figure 1.
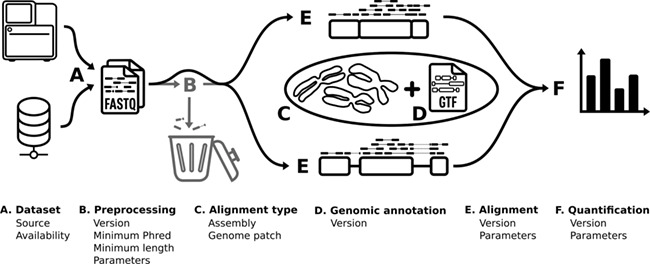
RNA-seq bioinformatics pipeline. Schematic of the RNA-seq bioinformatics methodology. The pipeline is divided into six steps (A–F). Each step is specified using a series of parameters displayed on the figure.
RNA-seq pipelines are diverse, consisting of many different software tools and references
To investigate methodology reporting practices in RNA-seq computational analysis pipelines, 1000 randomly chosen articles performing RNA-seq were analyzed by two independent reviewers as described in the Methods (Supplementary data). To ensure only consideration of articles with comparable pipelines, we kept only articles that included Methods starting from a raw read file and obtaining transcript or gene quantification. A single species study was necessary due to species-specific references used in RNA-seq. Homo sapiens was chosen due to the large availability of studies and references. The article exclusion criteria are described in Supplementary Table 1 and following their application, 465 articles remained. From these articles, information about the RNA-seq pipeline, from FASTQ files to gene or isoform count matrices, was extracted to determine which tools and parameters are used in the RNA-seq literature.
As shown in Figure 2, many references and tools are commonly used in RNA-seq computational pipelines. Most RNA-seq analyses align reads against a genome rather than a transcriptome (Figure 2C), but the choice of reference annotation is distributed among several sources (Figure 2D). For the three steps involving tool choices (Figure 2 panels B, E and F representing the preprocessing, alignment and quantification steps, respectively), no tool is reported to be used in more than 25% of the articles and thus several different tools are common in RNA-seq pipelines. We note however that most commonly used alignment tools are part of the Tuxedo suite.
Figure 2.
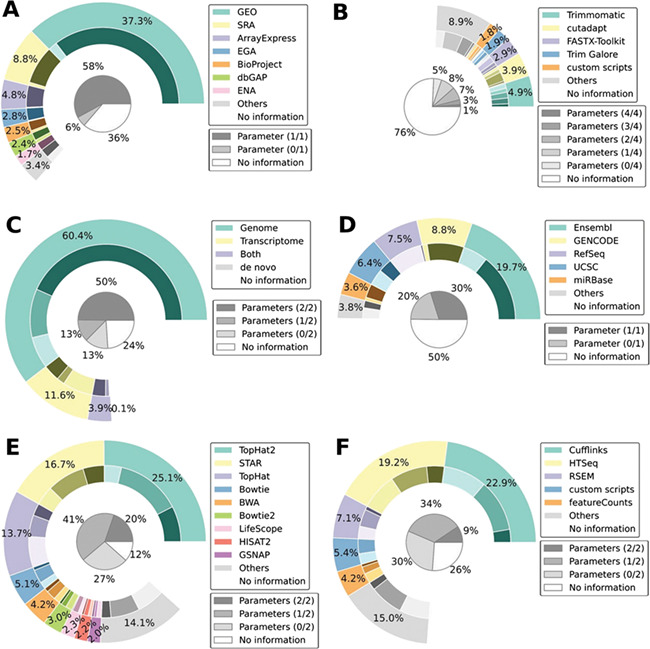
RNA-seq reported methodology is incomplete. Distribution of software and reference usage for the six methodological steps of an RNA-seq experiment (A. dataset, B. preprocessing tool, C. alignment type, D. genomic annotation, E. alignment tool and F. quantification tool). The outer donut chart illustrates the distribution of the primary criterion for each step. The inner donut chart illustrates the degree of parameter specification: the darker the shade, the more complete the information. The inner pie chart is the summation of all shades from the inner donut. Complete results are available as Supplementary data.
RNA-seq is a young and quickly evolving field. The commonly used sequencing parameters, in terms of read length, depth and read pairing, have changed in the past years, and new software is being developed to better use this more informative data [10, 11]. But we observed latency in tool usage in the literature. While older tools need to be readily available for comparative studies, users generating new results should be encouraged to modernize their pipelines and move to updated or newer tools. We have highlighted the use of the Tuxedo suite family (TopHat, TopHat2, HISAT and HISAT2) [12–14] to observe that TopHat, while being three times reimplemented (Figure 3), is still being used as an aligner for newly published studies despite indications in the last TopHat2 release that HISAT2 should now be preferred (in TopHat 2.1.1 release 23 February 2016). Ever increasing numbers of computational resources are becoming available, and newer software is usually more computationally efficient. Efforts to reanalyze studies with state-of-the-art pipelines should be put forward. The fast pace nature of RNA-seq technology requires one to be agile for timely contributions. If updating software to newer, benchmarked and better-performing software has an impact on the results, then one can argue that the implementation was useful.
Figure 3.
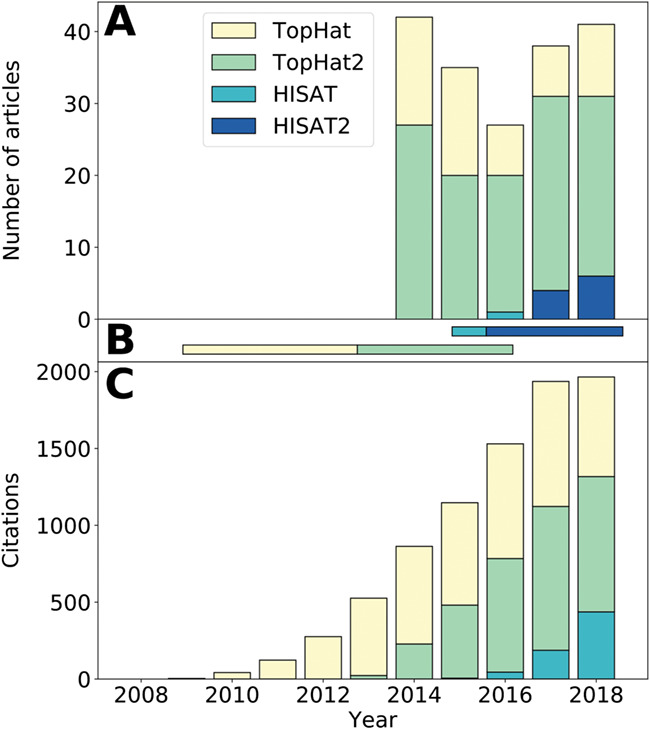
Observed latency in tool usage—a TopHat–HISAT case study. A illustrates the distribution of articles using tools from the TopHat–HISAT family found in our methodological literature review. B presents the recommended usage period for each tool. Dates were extracted from the TopHat and HISAT pages, using official release dates and notices given by the authors. C represents the distribution of new citations per year for each software original publication. The citation count was extracted from Scopus in January 2019. HISAT and HISAT2 share the same color considering that HISAT2 was never published independently of HISAT. While A only includes articles using RNA-seq with Homo sapiens, C includes all articles citing one of the tools.
RNA-seq methodology is incompletely reported with some steps much better described than others
Because several different references and software tools are used for each of the different RNA-seq analysis steps, the number of resulting pipelines is combinatorially large, increasing the importance of ensuring the complete description of all steps. Unfortunately, among the 465 articles considered, many failed to even mention any information regarding an RNA-seq analysis step (no information shares in Figure 2). An interesting observation is the non-uniform distribution of this lacking information. If the distribution could be explained by a random phenomenon or the presence of a bimodal distribution of articles ranked by a global ‘reporting quality’ metric, one would observe approximately the same share missing from each of the steps described in Figure 2, which is not the case. This non-uniform distribution could be interpreted as mirroring the community-conceived importance of the different steps. For example, the alignment tool is specified in 88% of the articles, whereas the preprocessing tool is indicated in only 24%. This could mean that the community of RNA-seq users believes that the alignment is far more important than the preprocessing in an RNA-seq experiment. The vast differences highlighted for preprocessing could also be explained by RNA-seq workflows that do not include a preprocessing step, or by prior preprocessing by the sequencing facility. In either case, the manuscript should contain all relevant information about preprocessing. The field currently lacks a meaningful quantitative assessment comparing the inherent bias of the different steps to the biological background. With such information, it would be possible to rank the importance of each decision on the final quantification, and appreciate if those are in relationship with the methodological results shown here. In any case, display of information for all analysis steps of an RNA-seq experiment is an important goal that could be achieved through awareness and enforced publication guidelines.
We also note that even when a step is described in an article, information is often missing regarding versions or parameter values, which are necessary to ensure reproducibility. Such is the case, for example, for the step specifying the genomic annotation, for which one can observe a non-uniform distribution in the proportion of articles giving details about the version number (Figure 2D, differences in the inner donut chart shading). In particular, most articles using Ensembl [15] and GENCODE [16] annotations specify the version number while few articles using RefSeq [17] do. A possible explanation for this is the availability of such information at the moment users download the genome annotation. A clear display of the annotation version for the different species by RefSeq on their website could help it reach the same level of version specification as Ensembl and GENCODE. In general, an upfront display of version information is crucial for any database, which could be subject to modifications. Any information that is easily accessible seems to bear more importance than information that is harder to find. One also has to be able to access previous versions of a reference to keep older datasets relevant. While Git is not currently designed to support large data files, Git-like features would answer these goals, by providing the possibility to navigate across the different versions of a reference, explicitly versioning data files and their differences. Current scientific data hosting platforms (e.g. figshare, Git LFS, Open Science Framework, Quilt, Zenodo) do not support either versioning or diffing of files, which is necessary for efficient data tracking.
Another possible cause for the unreliable distribution of information is the availability of alternative sources for the same references. Genome sequences and annotations can be readily downloaded from their official maintainer’s website, but can also be found on other file transfer protocol (FTP) servers or websites. Duplication of information only increases the risk of out-of-date data and versioning errors.
To summarize the findings, Figure 4 presents how many essential steps were correctly specified per article. This provides an idea of how the lack of information is distributed in the literature. While Figure 4A, showing the distribution of articles according to the number of steps they described, offers an optimistic view of article methodological quality, Figure 4B, displaying the distribution of articles classified by the number of steps they completely describe, represents the actual reproducibility potential. It is worth noting that some articles display absolutely no information other than the fact they have performed RNA-seq. Supplementary Figure 1 illustrates the completeness of each essential criterion per year. We do not note any obvious improvement of the situation in recent years.
Figure 4.
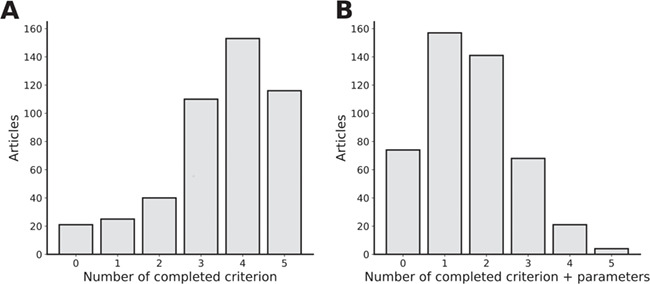
Article distribution by completeness. A. Distribution of articles by the number of essential criteria that have been specified in the methodology. Essential criteria are considered to be the dataset, alignment type, genomic annotation, alignment tool and quantification tool. B. A criterion needed to have every parameter specified to be accepted.
Measures that could be undertaken to increase methodological reporting of RNA-seq computational pipelines
Guidelines for adequate information presentation in RNA-seq already exist. The MINSEQE, minimum information about a high-throughput sequencing experiment, analogous to the former minimum information about a microarray experiment [18], describes the required information ‘to enable the unambiguous interpretation and facilitate reproduction of the results of the experiment’. Results of the methodological review suggest that such guidelines are routinely not being followed. A more reliable way to enforce a desired behavior would be at the publishing level, with editors requiring standardized minimal information about an RNA-seq experiment. Nevertheless, it would not be enough. The problem is also due to the way information is presented. Textually describing the experiments is a potential source of loss of clarity. The bias-less way to report an RNA-seq in silico experiment is probably a direct access to the code used to generate the results. If this code is housed in a collaborative version control system (e.g. Git), one would also be able to update the code for the peer-reviewing step or additional corrections. In fact, Git is already used for software version control and even scientific paper writing and peer-reviewing [19]. But this solution suffers from potential code readability and computing infrastructure issues. In lieu of an in silico physical preservation of a workflow, a semantical one offers workflow reproducibility, with less infrastructure dependency [20]. A schematic and semantic view of the data transformation pipeline, as already proposed in other areas of scientific computing [21], would help to better illustrate every data and software linkage. In this view, we advocate the use of workflow management systems, such as Nextflow [22] and Snakemake [23]. These tools promote reproducibility by explicitly defining and compartmentalizing the different pipeline steps, and enabling a scalable execution of the pipeline in dedicated containers and virtual environments. Users can therefore publish more readily readable and reproducible code, all while following previously described rules for reproducible computational research [24].
Conclusion
In summary, we illustrate the lack of information, the unreliable distribution of references and the latency in software usage in the RNA-seq literature by the means of a methodological review of the literature. The current state of the literature prohibits meaningful meta-analysis of the literature and large-scale reproducibility studies. We believe this situation will be improved by acknowledging the issue, clearly displaying the technical requirements for RNA-seq methodological reproducibility and with scientific publishers demanding standardized, high-quality methodology [25].
Supplementary Material
Joël Simoneau is an MSc student in the Department of Biochemistry of the Université de Sherbrooke. His research focuses on the role of the computational workflow in RNA-seq.
Simon Dumontier is an undergraduate student in bioinformatics/pharmacology with an interest in determining how to choose the most appropriate RNA-seq pipeline.
Ryan Gosselin, PhD, is an associate professor at the Department of Chemical & Biotechnological Engineering of the Université de Sherbrooke. His research focuses on the optimisation of complex data acquisition technologies using multivariate analyses.
Michelle S. Scott, PhD, is an associate professor in the Department of Biochemistry of the Université de Sherbrooke. Her research focuses on elaborating RNA-seq workflows for the accurate quantification of whole transcriptomes.
Key Points
RNA-seq pipelines are diverse, consisting of many different software tools and references.
RNA-seq methodology is incompletely reported with some steps much better described than others.
Clearly displaying the technical requirements and demanding standardized methodology will improve RNA-seq reproducibility.
Funding
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC; RGPIN-2018-05412 to M.S.S.). J.S. was supported by a Masters scholarship from NSERC. M.S.S. holds a Fonds de Recherche du Québec—Santé Research Scholar Junior 2 Career Award. M.S.S. is a member of the Centre de Recherche du Centre Hospitalier de l’Université de Sherbrooke.
References
- 1. Schena M, Shalon D, Davis RW, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995;270:467–70. [DOI] [PubMed] [Google Scholar]
- 2. Zhao S, Fung-Leung WP, Bittner A, et al. Comparison of RNA-seq and microarray in transcriptome profiling of activated T cells. PLoS One 2014;9:e78644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ison J, Rapacki K, Ménager H, et al. Tools and data services registry: a community effort to document bioinformatics resources. Nucleic Acids Res 2016;44:D38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hansen KD, Wu Z, Irizarry RA, et al. Sequencing technology does not eliminate biological variability. Nat Biotechnol 2011;29:572–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spudich JL, Koshland DE. Non-genetic individuality: chance in the single cell. Nature 1976;262:467–71. [DOI] [PubMed] [Google Scholar]
- 6. Auer PL, Doerge RW. Statistical design and analysis of RNA sequencing data. Genetics 2010;185:405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marioni JC, Mason CE, Mane SM, et al. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 2008;18:1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Busby MA, Stewart C, Miller CA, et al. Scotty: a web tool for designing RNA-seq experiments to measure differential gene expression. Bioinformatics 2013;29:656–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schurch NJ, Schofield P, Gierliński M, et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016;22:839–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Conesa A, Madrigal P, Tarazona S, et al. A survey of best practices for RNA-seq data analysis. Genome Biol 2016;17:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Z, Gerstein M, Snyder M. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res 2018;46:D754–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frankish A, Diekhans M, Ferreira A-M, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019;47:D766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O’Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 2016;44:D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brazma A. Minimum information about a microarray experiment (MIAME)—successes, failures, challenges. ScientificWorldJournal 2009;9:420–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Katz DS, Niemeyer KE, Smith AM. Publish your software: introducing the journal of open source software (JOSS). Comput Sci Eng 2018;20:84–8. [Google Scholar]
- 20. Santana-Perez I, Ferreira da Silva R, Rynge M, et al. Reproducibility of execution environments in computational science using semantics and clouds. Future Gener Comput Syst 2017;67:354–67. [Google Scholar]
- 21. Gil Y, Deelman E, Ellisman M, et al. Examining the challenges of scientific workflows. Computer 2007;40:24–32. [Google Scholar]
- 22. Di Tommaso P, Chatzou M, Floden EW, et al. Nextflow enables reproducible computational workflows. Nat Biotechnol 2017;35:316–9. [DOI] [PubMed] [Google Scholar]
- 23. Koster J, Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 2012;28:2520–2. [DOI] [PubMed] [Google Scholar]
- 24. Sandve GK, Nekrutenko A, Taylor J, et al. Ten simple rules for reproducible computational research. PLoS Comput Biol 2013;9:e1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simoneau J, Scott MS. In silico analysis of RNA-seq requires a more complete description of methodology. Nat Rev Mol Cell Biol 2019;20:451–2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.