

Abstract
Empowered by the advancement of high-throughput bio technologies, recent research on body-fluid proteomes has led to the discoveries of numerous novel disease biomarkers and therapeutic drugs. In the meantime, a tremendous progress in disclosing the body-fluid proteomes was made, resulting in a collection of over 15 000 different proteins detected in major human body fluids. However, common challenges remain with current proteomics technologies about how to effectively handle the large variety of protein modifications in those fluids. To this end, computational effort utilizing statistical and machine-learning approaches has shown early successes in identifying biomarker proteins in specific human diseases. In this article, we first summarized the experimental progresses using a combination of conventional and high-throughput technologies, along with the major discoveries, and focused on current research status of 16 types of body-fluid proteins. Next, the emerging computational work on protein prediction based on support vector machine, ranking algorithm, and protein–protein interaction network were also surveyed, followed by algorithm and application discussion. At last, we discuss additional critical concerns about these topics and close the review by providing future perspectives especially toward the realization of clinical disease biomarker discovery.
Keywords: body-fluid proteome, protein prediction, clinical application, biomarker discovery
Introduction
Human body fluids are biological fluids that are either excreted or secreted from the bodies of living people [1]. They include, but not limited to, plasma/serum, saliva, urine, cerebrospinal fluid, seminal fluid, amniotic fluid, tear fluid, bronchoalveolar lavage fluid, milk, synovial fluid, nipple aspirate fluid, cervicovaginal fluid, pleural effusion, sputum, exhaled breath condensate and pancreatic juice. It has been widely accepted that human body fluids contain disease-associated proteins that are secreted or leaked from pathological tissues across the body and are often easily obtainable through noninvasive procedures [2]. To date, over 15 000 different proteins have been identified in major human body fluids.
For decades, proteomic applications have spanned across different fields in biomedical and biochemistry research [3] and considered body fluids as the easy and attractive targets to profile [4]. Since the first research on serum globulin separation in 1937 [5], numerous reports on human body-fluid proteomes have been documented. Especially after the use of two-dimensional gel electrophoresis (2-DE) [6], several instrumental milestones appear. For example, in 1970, Freeman and Smith resolved 60 protein components in plasma using conventional gel filtration [7], which clearly demonstrated the complex composition of plasma and the feasibility of profiling blood proteins using those techniques. However, despite its popularity, 2D electrophoresis was known to have limits in terms of its very low efficiency in the analysis of hydrophobic proteins and high sensitivity to the dynamic range and quantitative distribution [8]. Such drawbacks necessitate high-power analytical techniques. In the early 1900s, Sir J.J. Thomson developed mass spectrometry (MS) technique when he obtained mass spectra of small gaseous ions [9]. Since then, MS has become the method of choice for analyzing complex protein samples [10]. Since early 1980s, MS moved from an analytical technique applicable only to small volatile compounds to applications on large biomolecules [11] and has been widely used for comprehensive profiling of human body-fluids proteomes. For examples, the Human Plasma Proteome Project initiated by international Human Proteome Organization has involved a collaboration of many laboratories using MS technology and compiled a core dataset of 3521 distinct proteins in human plasma [12]. In addition, the Sys-BodyFluid database published in 2009 contained 11 kinds of body-fluid proteomes and over 10 000 proteins [13]. Many recent studies concentrated on the discovery of protein biomarkers in pathologic conditions such as cancers, metabolic disease and brain disease [14]. In this regard, it is well-known that the major bottleneck challenges in biomarker discovery lie in the quantitative analysis of highly specific proteins [15]. For example, diseases such as Sjögren’s syndrome, bacterial and viral infectious diseases, and oral cancer all cause alterations of salivary protein expression [16]. Similarly, urine drains from the urinary tract and is therefore particularly enriched in proteins deriving from the kidney, bladder and prostate [17]. Many hereditary glomerular disease proteins have been identified in urine, such as podocin, alpha-actinin-4, CD2-associated protein, myosin-9, myosin 1E, integrin alpha 3 and cubilin, for which the quantitative measure is key to the applications [18].
The ability of MS to identify and to increasingly precisely quantify thousands of proteins from complex fluids has a broad impact on biomedical research [19]. However, regardless the evolving technology, protein identification is still considered as a challenging topic simply because a large amount of proteins are subject to a variety of modifications in body fluids, making the proteome composition highly complex. To facilitate such research, a few computational pipelines have been developed to characterize molecular features of various types of secreted proteins and provide new predictions using statistical and machine-learning methodologies. In 2008, Cui et al. [20] firstly proposed a machine-learning strategy to predict if a protein is likely to enter into bloodstream using support vector machine (SVM) classifier. Soon after that, several related studies were reported to identify secreted proteins associated with different body fluids, including blood [21], urine [22,23], saliva [24,25] and others [26]. In addition to protein identification, those predictors can be used to identify potential biomarkers for specific human diseases based on the context-dependent genomics data [20]. Figure 1 shows the major event nodes related to body-fluid proteome research.
Figure 1.
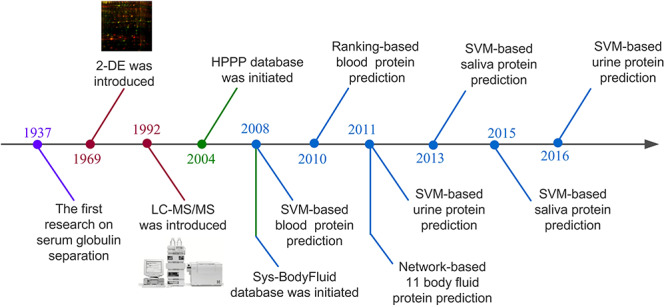
Major events related to proteomics technology development and body-fluid proteome research.
In the following sections, we first review the major techniques and discoveries in protein identification and then focused on the computational work in this field in terms of the methodologies and applications. The discussion will be centered around critical issues related to future application in human fluid proteomics research.
Major methodological strategies for body-fluids protein profiling
Modern proteomic tools have provided different technical frameworks for handling proteome complexity in human body fluids [27]. Several previous works have addressed important issues related to the standardization of sample collection, separation and processing [28,29]. As a summary, Figure 2 shows the currently used analytical workflows, including technologies used to fractionate and analyze proteome in either qualitative or quantitative manner [30].
Figure 2.
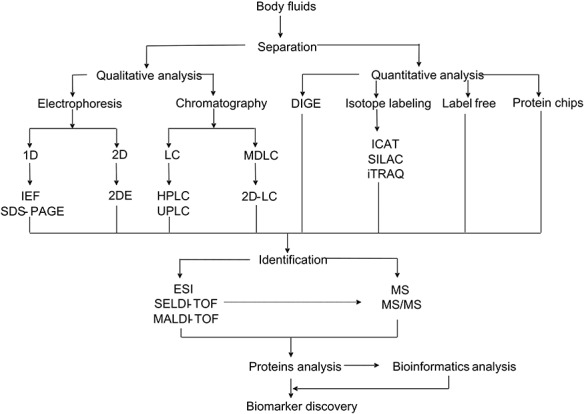
Overview of different strategies used for human body fluids analysis aiming biomarker discovery.
The qualitative separation was mainly through 1-DE, 2-DE and chromatography. Although 2-DE is low-cost, reproducible and visual, questions remain concerning its ability of handling protein co-migration [31] and limitations in protein analysis for high- or low-molecular weight proteins as well as those of proteins with extreme isoelectric point (pI) values [32]. In contrast, multiple liquid chromatography (LC) techniques and their continuous improvements in separation components are providing further advances and enabling increasingly effective large-scale proteomics [33].
A number of isotope-labeling approaches are available for quantitative proteomic analysis [34], including 2D difference gel electrophoresis [35], isotope-coded affinity tag [36], stable isotope labeling by amino acids in cell culture [37], isobaric tags for relative and absolute quantification [38]. Although in general isotopic labeling technology is deemed successful, it has some technical limitations due to the high costs of the labeling reagents, computational difficulties and the error-prone nature [39]. The ion intensity-based label-free quantitative approach has gradually gained more popularity and provides an alternative powerful tool to resolve and identify thousands of proteins from a complex biological sample [40]. It is rapid and sensitive and can increase the protein dynamic range by 3- to 4-fold compared with 2-DE [41]. Similarly, protein chip has also been employed as a simple-to-use technology that offers the capability of differentiating proteins and quantifying the abundance [42,43].
MS has become an indispensable analytical tool in quantitative protein analysis. Particularly, both matrix-assisted laser desorption ionization–time of flight (MALDI–TOF) MS and tandem MS (MS/MS) can provide excellent mass accuracy, high resolution, high sensitivity and direct analysis from complex mixtures [44].
Proteomic analysis on 16 types of human body fluids
In this section, we review proteomic research on 16 major types of body fluids since 2001 and summarize the major discovery of body-fluid proteins on Figure 3 shows the distribution of the 16 types of body fluids in human body.
Figure 3.
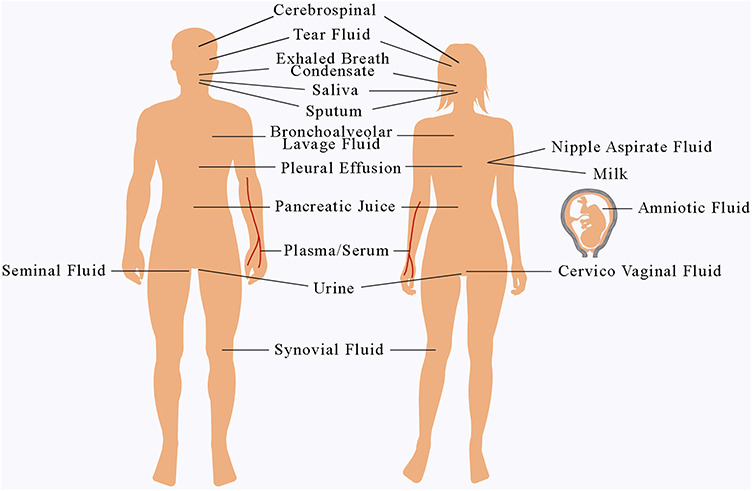
The distribution of 16 types of body fluids in human body.
Plasma/serum
Blood plasma is believed to have the most complex human-derived proteome [45] and has attracted high volume of research attentions [45–111]. Owing to the importance of plasma proteins, several large-scale proteomic efforts have been carried out on human plasma proteins [112]. To date, over 12 000 different plasma proteins have been identified with high confidence, which provides the largest set of circulating proteins as the most commonly-used pool for finding potential biomarkers for clinical diagnosis. In the meantime, great challenges remain because of the complex modification of proteins in blood.
Saliva
Saliva mainly comes from parotid, submandibular, sublingual and several minor glands, and is a dilute aqueous solution consisting of electrolytes, minerals, buffers and proteins [113]. The collection of saliva is simple, noninvasive and cheap and can be easily repeated [28]. The saliva proteome research has led to the identification of more than 4000 different protein species [4,105,107,113–142]. In the context of clinical proteomics, it has gained increasing potential for disease diagnosis using saliva proteins, especially in oral cancers [105, 130,137] and periodontal diseases [129].
Urine
Urine is a complex fluid comprised of proteins from the different sources, including the filtration of the blood within the glomerulus and secretion form the kidneys and the urogenital tract [143]. Urine has the advantage to be obtained in large volume. In 1979, the Anderson’s group published the first studies by 2-DE on normal urine [144], in which they identified only the major components. Up to today, more than 8000 proteins have been identified in human urine [17,18,107,143, 145–165]. Early success has made toward the development of candidate biomarker in urine for various urogenital diseases, including acute kidney injury, bladder cancer and diabetic nephropathy [164].
Cerebrospinal fluid
Cerebrospinal fluid (CSF) is in continuum with the extra-cellular fluid of the central nervous system (CNS) and is produced by the choroid plexus that surrounds the brain [14]. Several proteomic studies were conducted to identify the proteome of human cerebrospinal fluid, and over 6000 proteins have been identified [107,150,166–179]. CSF is a promising source for studying protein biomarkers of diseases in the CNS [170] and provides an accessible liquid pool in the brain [173].
Seminal fluid
Seminal fluid is the liquid component of sperm [180]. In the case of studying human seminal plasma, the main aim would be the discovery of new biomarkers for prostate and testis cancers [181]. In addition, it also sheds new light into the fundamental aspects of the human sperm and points to new potential proteins involved in male infertility [182].
Amniotic fluid
Amniotic fluid (AF) contains cells of fetal origin and a wide range of fetal proteins, and is formed from fetal urine and secretions [183]. Proteomic profiles of amniotic fluid have been generated by several groups using different methods since 1997 [184]. As an important source of biomarkers for fetal pathologies, amniotic fluid has been widely studied for diagnosis of many pregnancy-related pathologies and genetic diseases [15,185–189], including fetal abnormalities [15], gestational age-dependent changes [189] and so on.
Tear fluid
Tear fluid (TF) is a complex mixture of secretions produced by the lacrimal gland, goblet cells, cornea and vascular sources [190]. Many methods have been used to map tear protein profiles, including different MS technologies [191], such as MALDI–TOF [192] and LC/MS [193]. TF is becoming an increasingly important source for finding biomarkers for eye-related diseases, such as Graves’ ophthalmopathy [194].
Bronchoalveolar lavage fluid
Bronchoalveolar lavage fluid (BALF) is a clinical body fluid used in sampling of the soluble protein contents of the airway lumen [195]. One of the earliest attempts to map the protein components of normal human BALF has identified 49 proteins [196]. Since then, more than 1000 proteins have been identified [195–205]. BALF also has the great advantage of easy collection and lung-disease indication therefore has been widely studied in ventilator-associated pneumonia [201], lung cancer [198,204], lung adenocarcinoma [197] and chronic obstructive pulmonary disease (COPD) [206].
Milk
Human milk contains many bioactive proteins that serve as the first source of nutrition for mammalian infants [207]. Over 1700 proteins have been identified in human milk [208]. Among them, milk fat globule membrane [209] and human colostrum [210] have become important targets for proteomics research. In an effort to explore the benefits that human milk can provide, numerous proteomic studies investigated the proteins in milk whey [211,212], which comprises 40.0% of the total milk proteins and has strong implication in growth/maintenance and immunity support.
Synovial fluid
Synovial fluid is a serum filtrate located in the joints that contains proteins from surrounding tissues, articular cartilage, synovial membrane and bone [213]. Many research on synovial-fluid proteome focused on the rheumatoid arthritis [213,214] and osteoarthritis [2, 214–220]. To date, only less than 1000 proteins can be identified in synovial fluid.
Nipple aspirate fluid
Nipple aspirate fluid (NAF) is a fluid secreted by the epithelial cells of the mammary ductal and lobular system, and it contains a set of specific breast tissue proteins [221, 222]. Therefore, NAF proteome is a valuable source of breast cancer biomarkers. For decades, the literature on NAF and breast secretions has expanded considerably and more than 2000 proteins have been identified [221–228].
Cervical-vaginal fluid
Cervical-vaginal fluid (CVF) consists of water, electrolytes, low-molecular-weight organic compounds, cells and a wide range of proteins and proteolytic enzymes [229]. Up to today, about 600 proteins were identified in CVF by seven research groups [189,229–234]. CVF could play a critical role in spontaneous preterm birth by detecting biomarkers and potential molecular networks.
Pleural effusion
Pleural effusion (PE) is the excess fluid in the pleural space, which exists in lung cancer patients and also forms due to many benign ailments [235]. To date, about 1300 proteins have been detected in PE [236–240] and a number of potential biomarkers were evaluated, such as lung surfactant protein A, cystatin-C, vascular endothelial growth factor and so on.
Sputum
Sputum is a readily accessible biological fluid, and its composition may change by different disease [241]. Sputum contains biomarkers of inflammation in common chronic airway diseases, such as asthma and COPD [242].
Exhaled breath condensate
Exhaled breath condensate (EBC) is a biological fluid consisting of aerosol droplets and water vapor, and can be obtained by freezing exhaled air under conditions of spontaneous breathing [243]. EBC composition reflects the physiological state of the lung and consequently, and, in principle, can be used to identify and monitor several pathologies, including asthma, COPD, bronchiectasis, cystic fibrosis, acute respiratory distress syndrome, infectious and neoplastic lung diseases [243]. Approximately 220 proteins were identified in EBC, which is considerably lower than those identified in other body fluids [243–249].
Pancreatic juice
Pancreatic juice is often used for pancreatic cancer detection [250]. Only a few studies have been published on the identification of pancreatic juice proteins. Over 740 unique proteins were identified including known pancreatic cancer tumor markers and proteins over expressed in pancreatic cancers [250–253].
Clearly, apart from the applausive progresses made in the field of human body-fluid proteomics, there are significant discrepancies between different proteomic discoveries, which is mainly caused by biased sample selection and preparation, technical difference of proteomic profiling, and distinct rules toward result interpretation. Nevertheless, the accumulation of publically-available proteomics data has shown great potential in facilitating various quantitative analysis in a broad array of biomedical applications.
Computational predictions on body-fluid proteome
In the last decade, the large-scale proteomics studies have encounter challenges in large dynamic range of the protein abundance [22] and high experimental costs (both in material and time) [254]. As alternative strategies, several computational methods for protein prediction based on statistics and machine learning have been developed and demonstrated promising performance [20–26].
Overview of learning-based prediction models
Intuitively, the discovery of proteins in different body fluids can be formulated into a classification problem, where published experimental data can be used for training a classifier to infer undiscovered instances. In fact, different learning-based approaches have been documented in the literature, including the following: (i) SVM-based classification: In 2008, Cui et al. [20] firstly proposed a computational method for prediction if a secreted protein was likely to enter into bloodstream based on a SVM classifier. Since then, similar other works include a classifier that used physiochemical properties and amino acid composition features to infer whether a protein can be excreted into urine [22,23], and a computational model for identification of origins of detected proteins in urine; classifiers for identifying human salivary proteins and applications in head and neck cancer biomarker discovery [24,25]; (ii) ranking-based prediction: Liu et al. [21] presented a computational framework for blood-secretory protein prediction using manifold ranking algorithm, which ranks all the candidate proteins according to the possibility of being blood-secreted. (iii) Network-based prediction: Hu et al. [26] has developed a novel approach that employed protein–protein interaction (PPI) network to predict human secreted proteins related to different body fluids.
In general, all these data-driven predictions require the collection of known body-fluid proteins for training and validation of the model [20], as well as molecular features as instance descriptors, as shown in Figure 4. Each approach introduces a unique set of analytical or computational challenges. In the next section, we will focus on several key issues on each topic.
Figure 4.
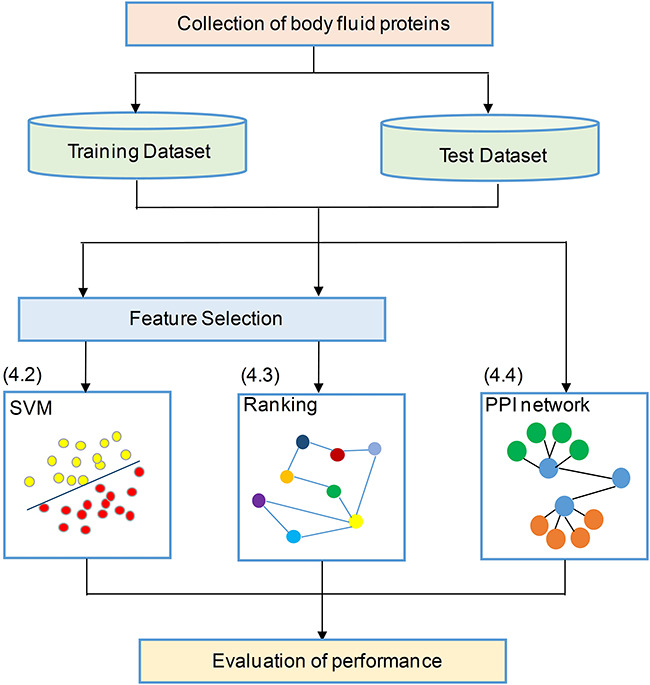
Summary workflow of the statistical and machine learning process for prediction of body-fluid proteomes.
SVM-based secreted protein prediction
Among all the learning-based methods, SVM has become the most popular and powerful classifier for body-fluid proteins because of its easy use and its compelling performance. Note that SVM emphasizes the idea of maximizing the margin or degree of separation in the two-class or multiple-class classification in the training process [23]. To ensure a successful application in body-fluid proteomic study, the following steps are key to train a reliable SVM classifier.
Data collection
It is essential to collect human body-fluid proteins that are experimentally detected by multiple proteomic studies from the public databases or literatures. For examples, Sys-BodyFluid database [13] contains over 10 000 proteins from 11 kinds of body fluids. Plasma proteome database contains information on 10 546 proteins detected in serum/plasma [112]. In addition, some body-fluid protein datasets can be collected from published literature. In [20], the authors collected a total of 1620 human proteins that are annotated as secretory proteins from the Swissprot and SPD database [255]. Approximately 305 of those proteins match at least two peptides and hence are considered as secreted proteins into blood—a common practice for protein identification based on MS data. To ensure the good quality, this study only used 305 proteins that has met two criteria (both secreted and serum/plasma detected), as the positive dataset and did not include proteins that leak into the blood as a result of cell damage (e.g. cardiac myoglobin released into plasma after a heart attack).
For binary classification through SVM, a negative dataset of non-body-fluid proteins is always required. Since such data are often not well-defined, it often requires a reliable way to generate the negative datasets, e.g. through a random selection of representative from all non-secreted related protein families, as defined in Pfam protein families [23]. Specifically, the negative data generation includes the following steps: (i) obtaining all human proteins and Pfam families from the UniProt database, (ii) mapping the known fluid proteins (positive data) to Pfam family, (iii) excluding the families which include known fluid proteins and (iv) randomly selecting representatives from each remaining family to construct the negative data with comparable size. Cui et al. used a large test set containing 98 secretory proteins and 6601 non-secretory proteins of human along with other additional data to evaluate the models [20].
Feature selection
Numerous protein features have been used to train the classification model that can predict human body-fluid proteins, which can be categorized into four types of property [24]: (i) sequence properties, (ii) structural properties, (iii) domains and motifs properties and (iv) physicochemical properties, as shown in Table 1.
Table 1.
The major type of features and the number of selected contributing features in each referenced study
| Feature category | Feature description | Feature dimentionality | Sources | Number of selected contributing features in each referenced study |
|---|---|---|---|---|
| Sequence properties | Sequence length | 1 | Uniprot [256], Profeat [257] | – |
| Amino acid composition | 20 | Hong et al. (2010):13 [22]; Wang et al. (2016):4 [23]; Wang et al. (2013):5 [25]; Sun et al. (2015):3 [24] | ||
| Di-peptides composition | 400 | Hong et al. (2010):12 [22]; Wang et al. (2016):33 [23]; Wang et al. (2013):25 [25]; Sun et al. (2015):20 [24] | ||
| Normalized Moreau–Broto autocorrelation | 240 | Wang et al. (2016):8 [23]; Wang et al. (2013):4 [25]; Sun et al. (2015):5 [24] | ||
| Moran autocorrelation | 240 | Wang et al. (2016):8[23]; Wang et al. (2013):5[25]; Sun et al. (2015):7 [24] | ||
| Geary autocorrelation | 240 | Wang et al. (2016):8 [23]; Wang et al. (2013):6 [25]; Sun et al. (2015):6 [24] | ||
| Sequence order | 160 | Wang et al. (2016):10 [23]; Sun et al. (2015):6 [24] | ||
| Pseudo amino acid composition | 50 | Hong et al. (2010):11 [22]; Wang et al. (2016):3 [23]; Sun et al. (2015):3 [24] | ||
| Physicochemical properties | Hydrophobicity | 21 | Profeat [257], Fldbin [258], ExPASy Tools [259] | Cui et al. (2008):8 [20]; Hong et al. (2010):4 [22]; Wang et al. (2013):2 [25]; Sun et al. (2015):5 [24] |
| Normalized Van der Waals volume | 21 | Cui et al. (2008):11 [20]; Hong et al. (2010):5 [22]; Wang et al. (2016):2 [23] | ||
| Polarity | 21 | Cui et al. (2008):12 [20]; Hong et al. (2010):4 [22]; Wang et al. (2013):2 [25]; Sun et al. (2015):1 [24] | ||
| Polarizability | 21 | Cui et al. (2008):8 [20]; Hong et al. (2010):4 [22]; Wang et al. (2016):1 [23]; Wang et al. (2013):1 [25]; Sun et al. (2015):1 [24] | ||
| Charge | 21 | Cui et al. (2008):11 [20]; Hong et al. (2010):5 [22]; Wang et al. (2016):1 [23]; Wang et al. (2013):1 [25]; Sun et al. (2015):1 [24] | ||
| Secondary structure | 21 | Cui et al. (2008):13 [20]; Hong et al. (2010):4 [22]; Wang et al. (2016):1 [23]; Wang et al. (2013):2 [25]; Sun et al. (2015):2 [24] | ||
| Solvent accessibility | 21 | Cui et al. (2008):6 [20]; Hong et al. (2010):3 [22]; Wang et al. (2016):1 [23]; Sun et al. (2015):2 [24] | ||
| Unfoldability | 1 | Cui et al. (2008):1 [20]; Hong et al. (2010):1 [22] | ||
| Fldbin charge | 1 | |||
| Hydrophobicity | 1 | Cui et al. (2008):1 [20]; Wang et al. (2013):1 [25] | ||
| Longest disordered regions | 1 | Cui et al. (2008):1 [20]; Wang et al. (2016):1 [23] | ||
| Isoelectric point | 1 | Hong et al. (2010):1 [22]; Wang et al. (2016):1 [23]; Sun et al. (2015):1 [24] | ||
| Charge | 1 | Cui et al. (2008):1 [20]; Hong et al. (2010):1 [22]; Wang et al. (2016):1 [23] | ||
| Molecular weight | 1 | |||
| Percentage of disordered region | 1 | Hong et al. (2010):1 [22] | ||
| Percentage of disordered residues | 1 | |||
| Relative surface accessibility | 3 | Cui et al. (2008):3 [20] | ||
| Domains/motifs properties | Beta-barrel transmembrane (BBTM) score | 1 | SingalP [260], TMB-Hunt [261], TatP [262], Phobius [263], NetOglyc [264], NetNGlyc [265] | Cui et al. (2008):1 [20] |
| Log P BBTM/Non-BBTM protein ratio | 1 | Cui et al. (2008):1 [20] | ||
| Twin-arginine signal peptide | 1 | Cui et al. (2008):1 [20] | ||
| Transmembrane domains | 1 | Cui et al. (2008):1 [20]; Wang et al. (2016):1 [23]; Sun et al. (2015):1 [24]; Hong et al. (2010):1 [22] | ||
| Signal peptide | 1 | Cui et al. (2008):1 [20]; Hong et al. (2010):1 [22]; Sun et al. (2015):1 [24] | ||
| Glycosylation number and presence | 4 | Cui et al. (2008):1 [20]; Hong et al. (2010):2 [22] | ||
| C-mannosylated | 1 | |||
| Phosphorylation sites | 1 | |||
| Cleavage site | 2 | Wang et al. (2016):1 [23] | ||
| Subcellular location | 4 | |||
| Percentage of coil-content | 1 | Hong et al. (2010):1 [22] | ||
| Structural properties | Secondary structural content | 4 | SSCP [266], Radius of Gyration | Cui et al. (2008):1 [20]; Wang et al. (2016):1 [23]; Sun et al. (2015):2 [24] |
| Radius gyration | 1 | Cui et al. (2008):1 [20] | ||
| Radius | 1 | Wang et al. (2016):1 [23]; Wang et al. (2013):1 [25]; Sun et al. (2015):1 [24] |
Since not all the initial features are related to a specific application, it is often useful to remove features that are noisy or irrelevant when predicting a specific group of body-fluid proteins [267]. A simple t-test is often used to determine the significance of a feature in terms of distinguishing two classes. Based on the derived P-value, a q-value is calculated to control the false discovery rate [268], where 0.005 is used as the threshold for removing non-contributing features. Furthermore, a classic feature-selection method known as recursive feature elimination [269] based on SVM is employed to remove features with weak classification power.
Table 1 listed the selected features in each of the published classifier, which are mostly related to transmembrane domains, signal peptide, sequence order, amino acid composition, Moran autocorrelation and so on. These selected features can better predict body-fluid proteins and improve the performance of classifier compared to using the whole original set of features.
Model learning
In SVM, the hyperplane of a high-dimensional space, called feature space, is constructed to separate two classes [23], where one class represents body-fluid proteins and the other represents non-body fluid proteins. The SVM makes prediction based on the function [270]:
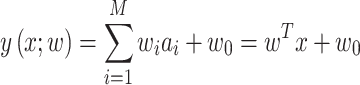 |
(1) |
where 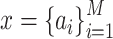 represents one input vector. Each
represents one input vector. Each 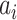 represents one aforementioned feature vector for each protein in the training set.
represents one aforementioned feature vector for each protein in the training set. 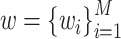 and
and 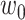 represent the unknown weights to compute. The output is 1 or − 1 representing if the input protein is movable to human body fluid or not. Among all available kernels in SVM (e.g. linear, polynomial and Gaussian), the Gaussian kernel [271] has been most extensively used in protein studies using SVM [20,22–24].
represent the unknown weights to compute. The output is 1 or − 1 representing if the input protein is movable to human body fluid or not. Among all available kernels in SVM (e.g. linear, polynomial and Gaussian), the Gaussian kernel [271] has been most extensively used in protein studies using SVM [20,22–24].
Model evaluation and selection
The classification performance is often evaluated by the sensitivity, specificity, precision, accuracy and Matthews correlation coefficient (MCC) value [25]. The formulas are shown in Equations (2–6).
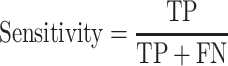 |
(2) |
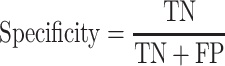 |
(3) |
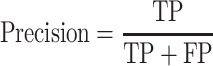 |
(4) |
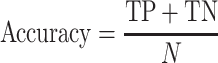 |
(5) |
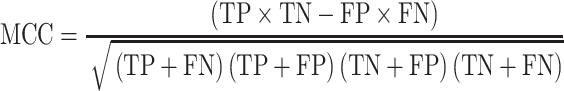 |
(6) |
where TP, FP, TN and FN mean the number of true positives, false positives, true negatives and false negatives, respectively. N is the total number of proteins for prediction in a given test set [23]. For instance, the sensitivity relates to the classifier’s ability to correctly identify the positive examples, that is, body-fluids proteins, while the specificity relates to the ability to correctly identify the negative examples, that is, non-body-fluids proteins. Note that MCC [272] is generally used as a balance measure of the quality of two-class classifications when the classes are of very different sizes. Additionally, the area under curve (AUC) is the average value of sensitivity for all possible values of specificity [273]. Last, the performance can be assessed using k-fold cross-validation [274] to identify the optimized model, e.g. the one achieves the highest AUC of the recall-precision curve precision. For example, in the study of blood-secreted protein [20], the SVM classifier achieved ∼90% sensitivity and ∼98% specificity on the test set containing 98 secretory proteins and 6601 non-secretory proteins of human with AUC as 0.96. Several additional datasets were used to further assess the performance in that study [20].
Ranking-based models
Different from SVM-based binary classifier that often requires a clean negative dataset of non-body-fluid for training, ranking-based algorithms can be employed to rank all the candidate proteins according to the possibility of being in body fluids [21]. For example, the manifold ranking algorithm [275], initially proposed to rank data points along their underlying manifold by analyzing their relationship in Euclidean space [276], has been used for to identify proteins in blood [21]. Specifically, a manifold ranking algorithm uses two datasets, a true sample set (as positive set) and an unknown sample set (as background set). According to the relevance of the unknown sample set with the true samples, the individual members of the unknown sample set can be ranked [21]. An intuitive description of this algorithm is as follows: a weighted graph is first formed, where each node represents one sample and an edge with weight score represents the similarity between the two nodes in the feature space; all the nodes then propagate their scores to the nearby points via the weighted graph; the propagation process is repeated until a global stable state is reached (which means convergence), and all the nodes except the true sample will have their own scores according to which they will be ranked.
Specifically, Liu et al. performed the analysis following the steps shown in Figure 5 [21]. A total of 11 394 proteins was used to training the model, where 253 high-confidence secreted proteins were used as positive data the rest are background data. As a result, 3681 proteins were identified as human plasma proteins. Novel blood proteins were ranked based on their relevance to the core set of experimentally validated blood proteins. The higher the ranked proteins, the more likely to be body-fluid proteins. The AUC to evaluate the prediction performance is 66.3% in Liu’s study [21]. Although ranking method provides an alternative solution for single class classification, it is not always advantageous over binary SVM when one can generate pseudo negative examples.
Figure 5.
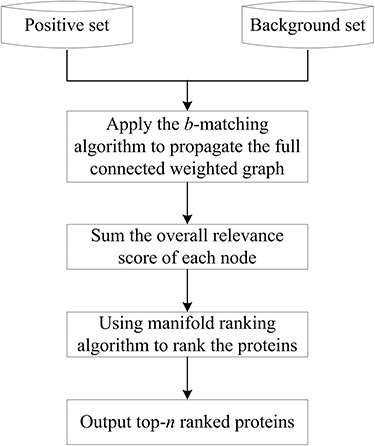
The workflow of the ranking-based models.
Network-based models
Considering interacted proteins may be secreted into the same body fluid to perform their functions [26], the PPI information was used in the prediction. The PPI community has been characterized by a wide and open distribution of proteomic data through the collection of PPI and pathway information [277]. For example, the human PPI networks were retrieved from STRING, a database dedicated to both physical and functional interactions of human proteins [26].
PPI networks can be intuitively modeled as a static graph 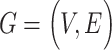 , where
, where 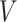 is the set of nodes (proteins), and
is the set of nodes (proteins), and 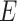 is the set of edges (PPI) [278]. The weight of undirected edge between each pair of nodes represents the interaction confidence score in the PPI network.
is the set of edges (PPI) [278]. The weight of undirected edge between each pair of nodes represents the interaction confidence score in the PPI network.
This network method for body-fluid proteome prediction requires only a true sample set and the rest of the procedure is follows:
Define the relationship
 between the protein set (the true sample set) and the body fluid.
between the protein set (the true sample set) and the body fluid.  means this protein can be secreted into the certain body fluid, otherwise
means this protein can be secreted into the certain body fluid, otherwise 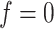 ;
;Denote the interaction confidence score
 between the query proteins with the protein set in the PPI network (
between the query proteins with the protein set in the PPI network ( means no interaction);
means no interaction);Formulate the likelihood score
 as the sum of the interaction confidence scores of the query protein with its interacting proteins that can be secreted into a certain body fluid
as the sum of the interaction confidence scores of the query protein with its interacting proteins that can be secreted into a certain body fluid 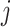 ;
;The most likely body fluid
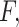 where the protein is secreted should be the one with the maximum score.
where the protein is secreted should be the one with the maximum score.
Jackknife test cross-validation methods are used to examine a predictor for its effectiveness in practical application. For the 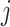 th order prediction, the accuracy
th order prediction, the accuracy 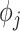 obtained by the jackknife test can be formulated as
obtained by the jackknife test can be formulated as
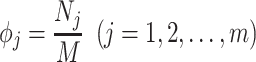 |
(7) |
where 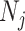 represents the number of the secreted proteins, whose
represents the number of the secreted proteins, whose 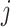 th order predicted body fluid is one of the true body fluids, and
th order predicted body fluid is one of the true body fluids, and 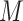 represents the total number of proteins in the PPI network.
represents the total number of proteins in the PPI network.
Given a query protein, the higher the likelihood score, the more likely they are to be secreted into a certain body fluid [26]. In [26], a breakdown of the 529 human secreted proteins from 11 different types of body fluids based on the literature search were used in the training dataset according to the, and 57 blood-secreted proteins were used to test this method. The model achieved 96% accuracy based on validation.
As shown in Table 2, all those methods have shown promising prediction power in the identification of body-fluid proteins. Particularly, the average performance (AUC or accuracy) of independent test across all these computational methodologies is 87.0%, while the average accuracy of SVM-based methods is 90.0%.
Table 2.
An overview of the protein prediction and application of disease biomarker. N/A = not application (no application discussion in this article)
| Study | Body fluid | Algorithm | Size of training data set | # of selected features | Performance | Application outcome | Ref |
|---|---|---|---|---|---|---|---|
| Cui et al. (2008) | Bloodstream | SVM | 6696 (151/6545) | 85 | 0.94 (AUC) | 13 biomarkers in gastric cancer; 26 biomarkers in lung cancer | [20] |
| Hong et al. (2011) | Urine | 3940 (1313/2627) | 74 | 0.90 (AUC) | Six biomarkers in gastric cancer | [22] | |
| Wang et al. (2013) | Saliva | 7077 (261/6816) | 55 | 0.81 (AUC) | 37 biomarkers in breast cancer | [25] | |
| Sun et al. (2015) | Saliva | 2757 (556/2201) | 68 | 0.90 (Accuracy) | 29 biomarkers in head and neck cancer | [24] | |
| Wang et al. (2016) | Urine | 2000 (1000/1000) | 87 | 0.93 (AUC) | 29 biomarkers in lung cancer | [23] | |
| Liu et al. (2010) | Bloodstream | Ranking | 11 394 (253/11 141) | 85 | 0.66 (AUC) | N/A | [21] |
| Hu et al. (2011) | Body fluids | PPI | 529 | N/A | 0.96 (Accuracy) | N/A | [26] |
Discussions and future perspective
As mentioned earlier, a useful repertoire of proteomics technologies is currently available for disease diagnosis and clinical-related applications. Our article reviews a large collection of different approaches involved in the proteomic data analysis of human body fluids, both experimentally and computationally. Current successes of the wet experimental technologies for protein characterization have been obvious. So far, there are over 15 000 different proteins discovered in major human body fluids. As discussed earlier, the largest sample dataset includes over 12 000 plasma and serum proteins; on the contrary, the smallest set is on EBC, including approximate 220 proteins. Further development of those technologies, especially with MS, will likely reduce sample requirement, increase the throughput and more effectively uncover various types of protein alterations such as post-translational modifications [279].
In the meantime, a great variety of computational tools has been developed to assist the analysis of body-fluid proteome and has shown promising performance, especially in novel protein discovery. Since the first predictor was proposed in 2008 to annotate the body fluids where human protein can be secreted into blood stream [20], it is anticipated that such methods will benefit the relevant experimental researches and stimulate a series of follow-up investigations into this emerging and challenging area. As reviewed previously, machine learning-based prediction through, e.g. SVM, has proved to be highly effective in terms of identifying novel secreted proteins and disease biomarkers. Similarly, both ranking and PPI network methods have made promising progress in body-fluid proteomics research. Although SVM-based prediction has achieved decent performance, it still has room for improvement through possibly increasing the size and quality of the positive training set and including more relevant features. This, however, may raise concern of computation complexity in the ranking algorithm. In general, a few key aspects to ensure a good performance of those computational predictions include a proper data collection of high quality experimentally-detected proteins to train the model, a comprehensive collection of molecular features underlying possible mechanisms of the secretion and effective techniques for feature and model selection.
Note that in general when learning larger dataset with high-dimensional features, new challenges arise. Conventional machine-learning techniques were somewhat limited in processing high-dimensional data [280]. New approach based on deep learning will likely lead to more successes in the near future because it requires very little engineering by hand and can easily take advantage of the increasingly-accumulated data available in the field [281]. As an example, the deep neural network-based model introduced in [282] can be another promising method that facilitates the understanding of body-fluid proteome and accelerate biomarker discovery in human disease.
A reliable prediction of human body-fluid proteins allows for effective targeted search for biomarker in body fluids. When further combined with other information such as disease-associated transcriptomic data, as reviewed above, such framework provides an upstream tool that is highly useful for finding candidate biomarkers associated with human diseases or physiological phenotypes. Often a combination of several proteins can form a signature panel for non-invasive test in clinical practice for diseases diagnosis. Ongoing effort in identifying and designing such effective panels for disease detection represents other major research topics in this field, which is beyond the scope of this review. All in all, a highly innovative and integrative approach leveraging the strength of both experimental profiling and computational prediction should be further pursued along the current research line to accelerate the process toward successful clinical applications.
Key Points
A tremendous progress in disclosing the body-fluid proteomes through high-throughput technologies has led to a collection of over 15 000 different proteins detected in major human body fluids. However, common challenges remain with current proteomics technologies about how to effectively handle the large variety of protein modifications in those fluids.
Major computational studies focused on the prediction of body-fluid-related secreted proteins have been reviewed in this article along with discussion on various bioinformatics techniques and tools.
Machine-learning models have been successfully applied in the prediction of various types of pf human secretome, as well as disease biomarker discovery.
Circulating proteins play important roles as disease markers for diagnosis and prognosis application.
Further research focused on data-driven discovery of disease protein markers through reliable modeling and computational prediction are emerging. New insights about future applications are presented.
Conflict of interest
No.
Acknowledgements
We thank all members of Key laboratory of Symbol Computation and Knowledge Engineering or their helpful discussion and comments. This research was funded by the National Natural Science Foundation of China (Nos. 61572227, 61772227 and 61702214), the Development Project of Jilin Province of China (Nos. 20180414012GH, 20190201273JC and 20190201293JC). This work was also supported by Jilin Provincial Key Laboratory of Big Date Intelligent Computing (No. 20180622002JC).
Lan Huang is a professor at the College of Computer Science and Technology in the Jilin University. Her research primarily focuses on bioinformatics, data mining and machine learning.
Dan Shao is a PhD candidate in the College of Computer Science and Technology in Jilin University and an assistant professor at the College of Computer Science and Technology in Changchun University. Her research focuses on proteomics and biomarker discovery.
Yan Wang is a professor at the College of Computer Science and Technology in the Jilin University. His research primarily focuses on bioinformatics, data mining and machine learning.
Xueteng Cui is a research assistant at the College of Computer Science and Technology in the Changchun University.
Yufei Li is a research assistant at the College of Computer Science and Technology in the Changchun University.
Qian Chen is a PhD candidate at the College of Computer Science and Technology in the Jilin University. Her research primarily focuses on bioinformatics.
Juan Cui is an assistant professor at the Department of Computer Science and Engineering in the University of Nebraska-Lincoln and an associated member of Nebraska Center for the Prevention of Obesity Diseases. Her primary research interests are biomedical informatics, systems biology and data mining.
References
- 1. Wu CC, Duan JC, Liu T, et al. Contributions of immunoaffinity chromatography to deep proteome profiling of human biofluids. J Chromatogr B Anal Technol Biomed Life Sci 2016;1021:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peffers MJ, Mcdermott B, Clegg PD, et al. Comprehensive protein profiling of synovial fluid in osteoarthritis following protein equalization. Osteoarthr Cartil 2015;23:1204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanaka Y, Akiyama H, Kuroda T, et al. A novel approach and protocol for discovering extremely low-abundance proteins in serum. Proteomics 2006;6:4845–55. [DOI] [PubMed] [Google Scholar]
- 4. Hu S, Wang JH, Meijer J, et al. Salivary proteomic and genomic biomarkers for primary sjögren’s syndrome. Arthritis Rheum 2007;56:3588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tiselius A. Electrophoresis of serum globulin: electrophoretic analysis of normal and immune sera. Biochem J 1937;31:313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Margolis J, Kenrick KG. Two-dimensional resolution of plasma proteins by combination of polyacrylamide disc and gradient gel electrophoresis. Nature 1969;221:1056–7. [DOI] [PubMed] [Google Scholar]
- 7. Freeman T, Smith J. Human serum protein fractionation by gel filtration. Biochem J 1970;118:869–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rabilloud T, Chevallet M, Luche S, et al. Two-dimensional gel electrophoresis in proteomics: past, present and future. J Proteome 2010;73:2064–77. [DOI] [PubMed] [Google Scholar]
- 9. Thomson JJ. Rays of positive electricity and their application to chemical analyses. Nature 1914;92:549–50. [Google Scholar]
- 10. Burlingame AL, Boyd RK, Gaskell SJ. Mass spectrometry. Anal Chem 1976;60:268–303. [Google Scholar]
- 11. Roepstorff P. Mass spectrometry in protein studies from genome to function. Curr Opin Biotechnol 1997;8:6–13. [DOI] [PubMed] [Google Scholar]
- 12. Omenn GS. The human proteome organization plasma proteome project pilot phase: reference specimens, technology platform comparisons, and standardized data submissions and analyses. Proteomics 2004;4:1235–40. [DOI] [PubMed] [Google Scholar]
- 13. Li SJ, Peng M, Li H, et al. Sys-BodyFluid: a systematical database for human body fluid proteome research. Nucleic Acids Res 2009;37:D907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogata Y, Charlesworth MC, Muddiman DC. Evaluation of protein depletion methods for the analysis of total-, phospho- and glycoproteins in lumbar cerebrospinal fluid. J Proteome Res 2005;4:837–45. [DOI] [PubMed] [Google Scholar]
- 15. Cho CK, Shan SJ, Winsor EJ, et al. Proteomics analysis of human amniotic fluid. Mol Cell Proteomics 2007;6:1406–15. [DOI] [PubMed] [Google Scholar]
- 16. Zeng Z, Hincapie M, Pitteri SJ, et al. A proteomics platform combining depletion, multi-lectin affinity chromatography (M-LAC), and isoelectric focusing to study the breast cancer proteome. Anal Chem 2011;83:4845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marimuthu A, O'Meally RN, Chaerkady R, et al. A comprehensive map of the human urinary proteome. J Proteome Res 2011;10:2734–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hogan MC, Johnson KL, Zenka RM, et al. Subfractionation, characterization, and in-depth proteomic analysis of glomerular membrane vesicles in human urine. Kidney Int 2014;85:1225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature 2003;422:198–207. [DOI] [PubMed] [Google Scholar]
- 20. Cui J, Liu Q, Puett D, et al. Computational prediction of human proteins that can be secreted into the bloodstream. Bioinformatics 2008;24:2370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu Q, Cui J, Yang Q, et al. In-silico prediction of blood-secretory human proteins using a ranking algorithm. BMC Bioinformatics 2010;11:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hong CS, Cui J, Ni ZH, et al. A computational method for prediction of excretory proteins and application to identification of gastric cancer markers in urine. PLoS One 2011;6:e16875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Y, Du W, Liang YC, et al. PUEPro: A Computational Pipeline for Prediction of Urine Excretory Proteins. Advanced Data Mining and Applications (ADMA). QLD, Australia: Gold Coast, 2016. [Google Scholar]
- 24. Sun Y, Du W, Zhou C, et al. A computational method for prediction of saliva-secretory proteins and its application to identification of head and neck cancer biomarkers for salivary diagnosis. IEEE Trans Nanobiosci 2015;14:167–74. [DOI] [PubMed] [Google Scholar]
- 25. Wang JX, Liang YC, Wang Y, et al. Computational prediction of human salivary proteins from blood circulation and application to diagnostic biomarker identification. PLoS One 2013;8:e80211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu LL, Huang T, Cai YD, et al. Prediction of body fluids where proteins are secreted into based on protein interaction network. PLoS One 2011;6:e22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schulze WX, Usadel B. Quantitation in mass-spectrometry-based proteomics. Annu Rev Plant Biol 2010;61:491–516. [DOI] [PubMed] [Google Scholar]
- 28. De Bock M, de Seny D, Meuwis MA, et al. Challenges for biomarker discovery in body fluids using SELDI-TOF-MS. J Biomed Biotechnol 2010;2010:906082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vitorino R, Guedes S, Manadas B, et al. Toward a standardized saliva proteome analysis methodology. J Proteome 2012;75:5140–65. [DOI] [PubMed] [Google Scholar]
- 30. Tolstikov VV, Lommen A, Nakanishi K, et al. Monolithic silica-based capillary reversed-phase liquid chromatography/electrospray mass spectrometry for plant metabolomics. Anal Chem 2003;75:6737–40. [DOI] [PubMed] [Google Scholar]
- 31. Gygi SP, Corthals GL, Zhang Y, et al. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc Natl Acad Sci U S A 2000;97:9390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang J, Gao MX, Deng CH, et al. Recent development of multi-dimensional chromatography strategies in proteome research. J Chromatogr B Anal Technol Biomed Life Sci 2008;866:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao YY, Lin RC. UPLC–MSE application in disease biomarker discovery: the discoveries in proteomics to metabolomics. Chem Biol Interact 2014;215:7–16. [DOI] [PubMed] [Google Scholar]
- 34. Zhu WH, Smith JW, Huang CM. Mass spectrometry-based label-free quantitative proteomics. J Biomed Biotechnol 2010;2010:840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Unlü M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis 1997;18:2071–7. [DOI] [PubMed] [Google Scholar]
- 36. Gygi SP, Rist B, Gerber SA, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 1999;17:994–9. [DOI] [PubMed] [Google Scholar]
- 37. Hoedt E, Zhang GA, Neubert TA. Stable isotope labeling by amino acids in cell culture (SILAC) for quantitative proteomics. Adv Mass Spectrom Biomed Res 2014;93–106. [DOI] [PubMed] [Google Scholar]
- 38. Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 2004;3:1154–69. [DOI] [PubMed] [Google Scholar]
- 39. Neilson KA, Ali NA, Muralidharan S, et al. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 2011;11:535–53. [DOI] [PubMed] [Google Scholar]
- 40. Bantscheff M, Schirle M, Sweetman G, et al. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem 2007;389:1017–31. [DOI] [PubMed] [Google Scholar]
- 41. Wang M, You JS, Bemis KG, et al. Label-Free Mass Spectrometry-Based Protein Quantification Technologies in Protein Biomarker Discovery. New Jersey, USA: Humana Press, 2008. [DOI] [PubMed] [Google Scholar]
- 42. Fung ET, Enderwick C. ProteinChip® clinical proteomics: computational challenges and solutions. Biotechniques 2002. (Suppl:34–38;40–1). [PubMed] [Google Scholar]
- 43. Media M. SELDI ProteinChip® array in oncoproteomic research. Technol Cancer Res Treat 2002;1:273–9. [DOI] [PubMed] [Google Scholar]
- 44. Salzano AM, Crescenzi M. Mass spectrometry for protein identification and the study of post translational modifications. Ann Ist Super Sanita 2005;41:443–50. [PubMed] [Google Scholar]
- 45. Jin WH, Jie D, Li S, et al. Human plasma proteome analysis by multidimensional chromatography prefractionation and linear ion trap mass spectrometry identification. J Proteome Res 2004;4:613–9. [DOI] [PubMed] [Google Scholar]
- 46. Acosta-Martin AE, Panchaud A, Chwastyniak M, et al. Quantitative mass spectrometry analysis using PAcIFIC for the identification of plasma diagnostic biomarkers for abdominal aortic aneurysm. PLoS One 2011;6:e28698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adkins JN, Varnum SM, Auberry KJ, et al. Toward a human blood serum proteome. Mol Cell Proteomics 2002;1:947–55. [DOI] [PubMed] [Google Scholar]
- 48. Ahn Y, Kang UB, Kim J, et al. Mining of serum glycoproteins by an indirect approach using cell line secretome. Mol Cell 2010;29:123–30. [DOI] [PubMed] [Google Scholar]
- 49. Al-Daghri NM, Al-Attas OS, Johnston HE, et al. Whole serum 3D LC-nESI-FTMS quantitative proteomics reveals sexual dimorphism in the milieu Intérieur of overweight and obese adults. J Proteome Res 2014;13:5094–6015. [DOI] [PubMed] [Google Scholar]
- 50. Anderson NL, Polanski M, Pieper R, et al. The human plasma proteome: a nonredundant list developed by combination of four separate sources. Mol Cell Proteomics 2004;3:311–26. [DOI] [PubMed] [Google Scholar]
- 51. Beer LA, Tang H, Sriswasdi S, et al. Systematic discovery of ectopic pregnancy serum biomarkers using 3-D protein profiling coupled with label-free quantitation. J Proteome Res 2011;10:1126–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bell LN, Jlvuppalanchi T. Serum proteomics and biomarker discovery across the spectrum of nonalcoholic fatty liver disease. Hepatology 2010;51:111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bell LN, Vuppalanchi R, Watkins PB, et al. Serum proteomic profiling in patients with drug-induced liver injury. Aliment Pharmacol Ther 2012;35:600–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bjelosevic S, Pascovici D, Ping H, et al. Quantitative age-specific variability of plasma proteins in healthy neonates, children and adults. Mol Cell Proteomics 2017;16:924–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boccardi C, Rocchiccioli S, Antonella C, et al. An automated plasma protein fractionation design: high-throughput perspectives for proteomic analysis. BMC Res Notes 2012;5:612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boichenko AP, Govorukhina N, Klip HG, et al. A panel of regulated proteins in serum from patients with cervical intraepithelial neoplasia and cervical cancer. J Proteome Res 2014;13:4995–5007. [DOI] [PubMed] [Google Scholar]
- 57. Bortner JD, Richie JP, Das A, et al. Proteomic profiling of human plasma by iTRAQ reveals down-regulation of ITI-HC3 and VDBP by cigarette smoking. J Proteome Res 2011;10:1151–9. [DOI] [PubMed] [Google Scholar]
- 58. Chen LZ, Gu H, Li J, et al. Comprehensive maternal serum proteomics identifies the cytoskeletal proteins as non-invasive biomarkers in prenatal diagnosis of congenital heart defects. Sci Rep 2016;6:19248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheon DH, Nam EJ, Park KH, et al. Comprehensive analysis of low-molecular-weight human plasma proteome using top-down mass spectrometry. J Proteome Res 2016;15:229–44. [DOI] [PubMed] [Google Scholar]
- 60. Cole RN, Ruczinski I, Schulze K, et al. The plasma proteome identifies expected and novel proteins correlated with micronutrient status in undernourished Nepalese children. J Nutr 2013;143:1540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Jesus JR, da Silva FR, de Souza PG, et al. Depleting high-abundant and enriching low-abundant proteins in human serum: an evaluation of sample preparation methods using magnetic nanoparticle, chemical depletion and immunoaffinity techniques. Talanta 2017;170:199–209. [DOI] [PubMed] [Google Scholar]
- 62. Domanski D, Percy AJ, Yang J, et al. MRM-based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma. Proteomics 2012;12:1222–43. [DOI] [PubMed] [Google Scholar]
- 63. Farrah T, Deutsch EW, Omenn GS, et al. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol Cell Proteomics 2011;10:M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gautam P, Nair SC, Ramamoorthy K, et al. Analysis of human blood plasma proteome from ten healthy volunteers from Indian population. PLoS One 2013;8:e72584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Glorieux G, Mullen W, Duranton F, et al. New insights in molecular mechanisms involved in chronic kidney disease using high-resolution plasma proteome analysis. Nephrol Dial Transplant : Off Publ Eur Dial Transplant Assoc - Eur Renal Assoc 2015;30:1842–52. [DOI] [PubMed] [Google Scholar]
- 66. gnjatovic V, Lai C, Summerhayes R, et al. Age-related differences in plasma proteins: how plasma proteins change from neonates to adults. PLoS One 2011;6:e17213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Haqqani AS, Hutchison JS, Ward R, et al. Protein biomarkers in serum of pediatric patients with severe traumatic brain injury identified by ICAT-LC-MS/MS. J Neurotrauma 2007;24:54–74. [DOI] [PubMed] [Google Scholar]
- 68. Harel M, Oren-Giladi P, Kaidar-Person O, et al. Proteomics of microparticles with SILAC quantification (PROMIS-Quan): a novel proteomic method for plasma biomarker quantification. Mol Cell Proteomics 2015;14:1127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. He T, Hu JY, Han J, et al. Identification of differentially expressed serum proteins in infectious purpura fulminans. Dis Markers 2014;2014:698383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Juhasz P, Lynch M, Sethuraman M, et al. Semi-targeted plasma proteomics discovery workflow utilizing two-stage protein depletion and off-line LC-MALDI MS/MS. J Proteome Res 2011;10:34–45. [DOI] [PubMed] [Google Scholar]
- 71. Keshishian H, Burgess MW, Gillette MA, et al. Multiplexed, quantitative workflow for sensitive biomarker discovery in plasma yields novel candidates for early myocardial injury. Mol Cell Proteomics 2015;14:2375–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim YJ, Sertamo K, Pierrard MA, et al. Verification of the biomarker candidates for non-small-cell lung cancer using a targeted proteomics approach. J Proteome Res 2015;14:1412–9. [DOI] [PubMed] [Google Scholar]
- 73. Kramer G, Woolerton Y, Straalen JP, et al. Accuracy and reproducibility in quantification of plasma protein concentrations by mass spectrometry without the use of isotopic standards. PLoS One 2015;10:e0140097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kuzyk MA, Smith D, Yang JC, et al. Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol Cell Proteomics 2009;8:1860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lai X, Liangpunsakul S, Crabb D, et al. A proteomic workflow for discovery of serum carrier protein-bound biomarker candidates of alcohol abuse using LC-MS/MS. Electrophoresis 2009;30:2207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lee MY, Eun YK, Kim SH, et al. Discovery of serum protein biomarkers in drug-free patients with major depressive disorder. Prog Neuro-Psychopharmacol Biol Psychiatry 2016;69:60–8. [DOI] [PubMed] [Google Scholar]
- 77. Lee SE, West KP, Cole RN, et al. Plasma proteome biomarkers of inflammation in school aged children in Nepal. PLoS One 2015;10:e0144279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li L, Xu Y, Yu CX. Proteomic analysis of serum of women with elevated Ca-125 to differentiate malignant from benign ovarian tumors. Asian Pac J Cancer Prev 2012;13:3265–70. [DOI] [PubMed] [Google Scholar]
- 79. Li SL, Liu X, Wei L, et al. Plasma biomarker screening for liver fibrosis with the N-terminal isotope tagging strategy. Sci China Life Sci 2011;54:393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Limonier F, Van Steendam K, Waeterloos G, et al. An application of mass spectrometry for quality control of biologicals: highly sensitive profiling of plasma residuals in human plasma-derived immunoglobulin. J Proteome 2017;152:312–320. [DOI] [PubMed] [Google Scholar]
- 81. Liu X, Valentine SJ, Plasencia MD, et al. Mapping the human plasma proteome by SCX-LC-IMS-MS. J Am Soc Mass Spectrom 2007;18:1249–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu Z, Fan S, Liu H, et al. Enhanced detection of low-abundance human plasma proteins by integrating polyethylene glycol fractionation and immunoaffinity depletion. PLoS One 2016;11:e0166306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Miike K, Aoki M, Yamashita R, et al. Proteome profiling reveals gender differences in the composition of human serum. Proteomics 2010;10:2678–91. [DOI] [PubMed] [Google Scholar]
- 84. Oller Moreno S, Cominetti O, Núñez Galindo A, et al. The differential plasma proteome of obese and overweight individuals undergoing a nutritional weight loss and maintenance intervention. Proteomics Clin Appl 2017;12:1600150. [DOI] [PubMed] [Google Scholar]
- 85. Omenn GS, States DJ, Adamski M, et al. Overview of the HUPO plasma proteome project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics 2005;5:3226–45. [DOI] [PubMed] [Google Scholar]
- 86. Pan S, Chen R, Crispin DA, et al. Protein alterations associated with pancreatic cancer and chronic pancreatitis found in human plasma using global quantitative proteomics profiling. J Proteome Res 2011;10:2359–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Percy AJ, Chambers AG, Yang J, et al. Advances in multiplexed MRM-based protein biomarker quantitation toward clinical utility. Biochim Biophys Acta 1844;2014:917–26. [DOI] [PubMed] [Google Scholar]
- 88. Pietzner M, Engelmann B, Kacprowski T, et al. Plasma proteome and metabolome characterization of an experimental human thyrotoxicosis model. BMC Med 2017;15:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Qian WJ, Monroe ME, Liu T, et al. Quantitative proteome analysis of human plasma following in vivo lipopolysaccharide administration using 16O/18O labeling and the accurate mass and time tag approach. Mol Cell Proteom MCP 2005;4:700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Riley CP, Zhang X, Nakshatri H, et al. A large, consistent plasma proteomics data set from prospectively collected breast cancer patient and healthy volunteer samples. J Transl Med 2011;9:80 9, 1(2011-05-27). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Schenk S, Schoenhals GJ, Souza GD, et al. A high confidence, manually validated human blood plasma protein reference set. BMC Med Genet 2008;1:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sennels L, Salek M, Lomas L, et al. Proteomic analysis of human blood serum using peptide library beads. J Proteome Res 2007;6:4055–62. [DOI] [PubMed] [Google Scholar]
- 93. Sheng S, Chen D, Van Eyk JE. Multidimensional liquid chromatography separation of intact proteins by chromatographic focusing and reversed phase of the human serum proteome: optimization and protein database. Mol Cell Proteom MCP 2006;5:26–34. [DOI] [PubMed] [Google Scholar]
- 94. Sinclair J, Timms JF. Quantitative profiling of serum samples using TMT protein labelling, fractionation and LC–MS/MS. Methods 2011;54:361–9. [DOI] [PubMed] [Google Scholar]
- 95. Song F, Poljak A, Kochan NA, et al. Plasma protein profiling of mild cognitive impairment and Alzheimer’s disease using iTRAQ quantitative proteomics. Proteome Sci 2014;12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Suh EJ, Kabir MH, Kang UB, et al. Comparative profiling of plasma proteome from breast cancer patients reveals thrombospondin-1 and BRWD3 as serological biomarkers. Exp Mol Med 2012;44:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Surinova S, Choi M, Tao S, et al. Prediction of colorectal cancer diagnosis based on circulating plasma proteins. EMBO Mol Med 2015;7:1166–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tirumalai RS, Chan KC, Prieto DA, et al. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics 2003;2:1096–103. [DOI] [PubMed] [Google Scholar]
- 99. Tu CJ, Dai J, Li SJ, et al. High-sensitivity analysis of human plasma proteome by immobilized isoelectric focusing fractionation coupled to mass spectrometry identification. J Proteome Res 2005;4:1265–73. [DOI] [PubMed] [Google Scholar]
- 100. Valentine SJ, Plasencia MD, Liu X, et al. Toward plasma proteome profiling with ion mobility-mass spectrometry. J Proteome Res 2006;5:2977–84. [DOI] [PubMed] [Google Scholar]
- 101. Wang D, Liem DA, Lau E, et al. Characterization of human plasma proteome dynamics using deuterium oxide. Proteomics - Clin Appl 2015;8:610–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wei-Jun Q, Petritis BO, Amit K, et al. Plasma proteome response to severe burn injury revealed by 18O-labeled "universal" reference-based quantitative proteomics. J Proteome Res 2010;9:4779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wu DJ, Zhu D, Xu M, et al. Analysis of transcriptional factors and regulation networks in patients with acute renal allograft rejection. J Proteome Res 2011;10:175–81. [DOI] [PubMed] [Google Scholar]
- 104. Yadav AK, Bhardwaj G, Basak T, et al. A systematic analysis of eluted fraction of plasma post immunoaffinity depletion: implications in biomarker discovery. PLoS One 2011;6:e24442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yan W, Apweiler R, Balgley BM, et al. Systematic comparison of the human saliva and plasma proteomes. Proteomics - Clin Appl 2010;3:116–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zeng Z, Hincapie M, Pitteri SJ, et al. A proteomics platform combining depletion, multi-lectin affinity chromatography(M-LAC), and isoelectric focusing to study the breast cancer proteome. Anal Chem 2011;83:4845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhao M, Yang Y, Guo Z, et al. A comparative proteomics analysis of five body fluids: plasma, urine, cerebrospinal fluid, amniotic fluid and saliva. Proteomics Clin Appl 2018;e1800008. [DOI] [PubMed] [Google Scholar]
- 108. Zhao Y, Chang C, Qin P, et al. Mining the human plasma proteome with three-dimensional strategies by high-resolution Quadrupole Orbitrap mass spectrometry. Anal Chim Acta 2016;904:65–75. [DOI] [PubMed] [Google Scholar]
- 109. Zhi W, Sharma A, Purohit S, et al. Discovery and validation of serum protein changes in type 1 diabetes patients using high throughput two dimensional liquid chromatography-mass spectrometry and immunoassays. Mol Cell Proteom MCP 2011;10:M111.012203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhou C, Simpson KL, Lancashire LJ, et al. Statistical considerations of optimal study design for human plasma proteomics and biomarker discovery. J Proteome Res 2012;11:2103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhou M, Prieto DA, Lucas DA, et al. Identification of the SELDI ProteinChip human serum retentate by microcapillary liquid chromatography-tandem mass spectrometry. J Proteome Res 2006;5:2207–16. [DOI] [PubMed] [Google Scholar]
- 112. Nanjappa V, Thomas JK, Marimuthu A, et al. Plasma proteome database as a resource for proteomics research: 2014 update. Nucleic Acids Res 2014;42:D959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wilmarth PA, Riviere MA, Rustvold DL, et al. Two-dimensional liquid chromatography study of the human whole saliva proteome. J Proteome Res 2004;3:1017–23. [DOI] [PubMed] [Google Scholar]
- 114. Amado FM, Ferreira RP, Vitorino R. One decade of salivary proteomics: current approaches and outstanding challenges. Clin Biochem 2013;46:506–17. [DOI] [PubMed] [Google Scholar]
- 115. Aboodi GM, Sima C, Moffa EB, et al. Salivary cytoprotective proteins in inflammation and resolution during experimental gingivitis—a pilot study. Front Cell Infect Microbiol 2016;5:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ambatipudi KS, Swatkoski S, Moresco JJ, et al. Quantitative proteomics of parotid saliva in primary Sjögren's syndrome. Proteomics 2012;12:3113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Aqrawi LA, Galtung HK, Vestad B, et al. Identification of potential saliva and tear biomarkers in primary Sjögren’s syndrome, utilising the extraction of extracellular vesicles and proteomics analysis. Arthritis Res Ther 2017;19:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bandhakavi S, Stone MD, Onsongo G, et al. A dynamic range compression and three-dimensional peptide fractionation analysis platform expands proteome coverage and the diagnostic potential of whole saliva. J Proteome Res 2009;8:5590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cho HR, Kim HS, Park JS, et al. Construction and characterization of the Korean whole saliva proteome to determine ethnic differences in human saliva proteome. PLoS One 2017;12:e0181765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jong EP, Xie HW, Onsongo G, et al. Quantitative proteomics reveals myosin and actin as promising saliva biomarkers for distinguishing pre-malignant and malignant oral lesions. PLoS One 2010;5:e11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Denny P, Hagen FK, Hardt M, et al. The proteomes of human parotid and submandibular/sublingual gland salivas collected as the ductal secretions. J Proteome Res 2008;7:1994–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Devic I, Shi M, Schubert MM, et al. Proteomic analysis of saliva from patients with oral chronic graft-versus-host disease. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant 2014;20:1048–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dominy SS, Brown JN, Ryder MI, et al. Proteomic analysis of saliva in HIV-positive heroin addicts reveals proteins correlated with cognition. PLoS One 2014;9:e89366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Fleissig Y, Deutsch O, Reichenberg E, et al. Different proteomic protein patterns in saliva of Sjögren's syndrome patients. Oral Dis 2009;15:61–8. [DOI] [PubMed] [Google Scholar]
- 125. Gonzalezbegne M, Lu BW, Liao LJ, et al. Characterization of the human submandibular/sublingual saliva glycoproteome using lectin affinity chromatography coupled to multidimensional protein identification technology. J Proteome Res 2011;10:5031–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Guo T, Rudnick PA, Wang WJ, et al. Characterization of the human salivary proteome by capillary isoelectric focusing/nanoreversed-phase liquid chromatography coupled with ESI-tandem MS. J Proteome Res 2006;5:1469–78. [DOI] [PubMed] [Google Scholar]
- 127. Hardt M, Thomas LR, Dixon SE, et al. Toward defining the human parotid gland salivary proteome and peptidome: identification and characterization using 2D SDS-PAGE, ultrafiltration, HPLC, and mass spectrometry. Biochemistry 2005;44:2885–99. [DOI] [PubMed] [Google Scholar]
- 128. Hu S, Arellano M, Boontheung P, et al. Salivary proteomics for oral cancer biomarker discovery. Clin Cancer Res 2008;14:6246–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hu S, Xie Y, Ramachandran P, et al. Large-scale identification of proteins in human salivary proteome by liquid chromatography/mass spectrometry and two-dimensional gel electrophoresis-mass spectrometry. Proteomics 2010;5:1714–28. [DOI] [PubMed] [Google Scholar]
- 130. Hu S, Xie YM, Ramachandra P, et al. Large-scale identification of proteins in human salivary proteome by liquid chromatography/mass spectrometry and two-dimensional gel electrophoresis-mass spectrometry. Proteomics 2005;5:1714–28. [DOI] [PubMed] [Google Scholar]
- 131. Huang CM. Comparative proteomic analysis of human whole saliva. Arch Oral Biol 2004;49:951–62. [DOI] [PubMed] [Google Scholar]
- 132. Jagtap P, McGowan T, Bandhakavi S, et al. Deep metaproteomic analysis of human salivary supernatant. Proteomics 2012;12:992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Marvin RK, Saepoo MB, Ye S, et al. Salivary protein changes in response to acute stress in medical residents performing advanced clinical simulations: a pilot proteomics study. Biomarkers 2017;22:372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ramachandran P, Boontheung P, Xie YM, et al. Identification of N-linked glycoproteins in human saliva by glycoprotein capture and mass spectrometry. J Proteome Res 2006;5:1493–503. [DOI] [PubMed] [Google Scholar]
- 135. Salih E, Siqueira WL, Helmerhorst EJ, et al. Large-scale phosphoproteome of human whole saliva using disulfide–thiol interchange covalent chromatography and mass spectrometry. Anal Biochem 2010;407:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Siqueira WL, Salih E, Wan DL, et al. Proteome of human minor salivary gland secretion. J Dent Res 2008;87:445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sivadasan P, Kumar Gupta M, Sathe GJ, et al. Data from human salivary proteome – a resource of potential biomarkers for oral cancer. J Proteome 2015;4:374–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sondej M, Denny PA, Xie Y, et al. Glycoprofiling of the human salivary proteome. Clin Proteomics 2009;5:52–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Thumbigere-Math V, Michalowicz BS, de Jong EP, et al. Salivary proteomics in bisphosphonate-related osteonecrosis of the jaw. Oral Dis 2015;21:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ventura TMDS, Ribeiro NR, Dionizio AS, et al. Standardization of a protocol for shotgun proteomic analysis of saliva. J Appl Oral Sci Revista Fob 2018;26:e20170561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Winck FV, Ribeiro ACP, Domingues RR, et al. Insights into immune responses in oral cancer through proteomic analysis of saliva and salivary extracellular vesicles. Sci Rep 2015;5:16305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Xie H, Rhodus NL, Griffin RJ, et al. A catalogue of human saliva proteins identified by free flow electrophoresis-based peptide separation and tandem mass spectrometry. Mol Cell Proteomics 2005;4:1826–30. [DOI] [PubMed] [Google Scholar]
- 143. Spahr CS, Davis MT, Mcginley MD, et al. Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry. I: Profiling an unfractionated tryptic digest. Proteomics 2001;1:93–107. [DOI] [PubMed] [Google Scholar]
- 144. Anderson NG, Anderson NL, Tollaksen SL. Concentration and analysis by two-dimensional electrophoresis. Clin Chem 1979;25:1199–210. [PubMed] [Google Scholar]
- 145. Adachi J, Kumar C, Zhang YL, et al. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol 2006;7:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Alamgir K, Packer NH. Simple urinary sample preparation for proteomic analysis. J Proteome Res 2006;5:2824–38. [DOI] [PubMed] [Google Scholar]
- 147. Castagna A, Cecconi D, Sennels L, et al. Exploring the hidden human urinary proteome via ligand library beads. J Proteome Res 2005;4:1917–30. [DOI] [PubMed] [Google Scholar]
- 148. Gonzales PA, Pisitkun T, Hoffert JD, et al. Large-scale proteomics and phosphoproteomics of urinary exosomes. J Am Soc Nephrol 2009;20:363–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Guo Z, Wang Z, Lu C, et al. Analysis of the differential urinary protein profile in IgA nephropathy patients of Uygur ethnicity. BMC Nephrol 2018;19:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Guo ZG, Zhang Y, Zou LL, et al. A proteomic analysis of individual and gender variations in normal human urine and cerebrospinal fluid using iTRAQ quantification. PLoS One 2015;10:e0133270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Li QR, Fan KX, Li RX, et al. A comprehensive and non-prefractionation on the protein level approach for the human urinary proteome: touching phosphorylation in urine. Rapid Commun Mass Spectr RCM 2010;24:823–32. [DOI] [PubMed] [Google Scholar]
- 152. Lin L, Yu Q, Zheng JX, et al. Fast quantitative urinary proteomic profiling workflow for biomarker discovery in kidney cancer. Clin Proteomics 2018;15:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Liu XJ, Shao C, Wei LL, et al. An individual urinary proteome analysis in normal human beings to define the minimal sample number to represent the normal urinary proteome. Proteome Sci 2012;10:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Nielsen HH, Beck HC, Kristensen LP, et al. The urine proteome profile is different in neuromyelitis optica compared to multiple sclerosis: a clinical proteome study. PLoS One 2015;10:e0139659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Oh J, Pyo JH, Jo EH, et al. Establishment of a near-standard two-dimensional human urine proteomic map. Proteomics 2004;4:3485–97. [DOI] [PubMed] [Google Scholar]
- 156. Onile OS, Calder B, Soares NC, et al. Quantitative label-free proteomic analysis of human urine to identify novel candidate protein biomarkers for schistosomiasis. PLoS Negl Trop Dis 2017;11:e0006045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Pieper R, Gatlin CL, Mcgrath AM, et al. Characterization of the human urinary proteome: a method for high-resolution display of urinary proteins on two-dimensional electrophoresis gels with a yield of nearly 1400 distinct protein spots. Proteomics 2010;4:1159–74. [DOI] [PubMed] [Google Scholar]
- 158. Ru QC, Katenhusen RA, Zhu LA, et al. Proteomic profiling of human urine using multi-dimensional protein identification technology. J Chromatogr A 2006;1111:166–74. [DOI] [PubMed] [Google Scholar]
- 159. Santucci L, Candiano G, Petretto A, et al. From hundreds to thousands: widening the normal human Urinome. Data Brief 2014;1:25–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Simona P, Yunee K, Simona F, et al. Identification of prostate-enriched proteins by in-depth proteomic analyses of expressed prostatic secretions in urine. J Proteome Res 2012;11:2386–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang Z, Hill S, Luther JM, et al. Proteomic analysis of urine exosomes by multidimensional protein identification technology (MudPIT). Proteomics 2012;12:329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zerefos PG, Aivaliotis M, Baumann M, et al. Analysis of the urine proteome via a combination of multi-dimensional approaches. Proteomics 2012;12:391–400. [DOI] [PubMed] [Google Scholar]
- 163. Zerefos PG, Vougas K, Dimitraki P, et al. Characterization of the human urine proteome by preparative electrophoresis in combination with 2-DE. Proteomics 2006;6:4346–55. [DOI] [PubMed] [Google Scholar]
- 164. Zhao MD, Li ML, Yang YH, et al. A comprehensive analysis and annotation of human normal urinary proteome. Sci Rep 2017;7:3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Zheng JH, Liu LG, Wang J, et al. Urinary proteomic and non-prefractionation quantitative phosphoproteomic analysis during pregnancy and non-pregnancy. BMC Genomics 2013;14:777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Pan S, Wang Y, Quinn JF, et al. Identification of glycoproteins in human cerebrospinal fluid with a complementary proteomic approach. J Proteome Res 2006;5:2769–79. [DOI] [PubMed] [Google Scholar]
- 167. Bora A, Anderson C, Bachani M, et al. Robust two-dimensional separation of intact proteins for bottom-up tandem mass spectrometry of the human CSF proteome. J Proteome Res 2012;11:3143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Borg J, Campos A, Diema C, et al. Spectral counting assessment of protein dynamic range in cerebrospinal fluid following depletion with plasma-designed immunoaffinity columns. Clin Proteomics 2011;8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Collins MA, An J, Hood BL, et al. Label-free LC-MS/MS proteomic analysis of cerebrospinal fluid identifies protein/pathway alterations and candidate biomarkers for amyotrophic lateral sclerosis. J Proteome Res 2015;14:4486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Guldbrandsen A, Vethe H, Farag Y, et al. In-depth characterization of the cerebrospinal fluid (CSF) proteome displayed through the CSF proteome resource (CSF-PR). Mol Cell Proteom MCP 2014;13:3152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hu ZY, Zhang HY, Zhang Y, et al. Nanoparticle size matters in the formation of plasma protein coronas on Fe3O4 nanoparticles. Colloids Surf B: Biointerfaces 2014;121:354–61. [DOI] [PubMed] [Google Scholar]
- 172. Hyung SW, Piehowski PD, Moore RJ, et al. Microscale depletion of high abundance proteins in human biofluids using IgY14 immunoaffinity resin: analysis of human plasma and cerebrospinal fluid. Anal Bioanal Chem 2014;406:7117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Mouton-Barbosa E, Roux-Dalvai F, Bouyssié D, et al. In-depth exploration of cerebrospinal fluid by combining peptide ligand library treatment and label-free protein quantification. Mol Cell Proteomics 2010;9:1006–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Ogata Y, Charlesworth MC, Higgins L, et al. Differential protein expression in male and female human lumbar cerebrospinal fluid using iTRAQ reagents after abundant protein depletion. Proteomics 2010;7:3726–34. [DOI] [PubMed] [Google Scholar]
- 175. Pan S, Zhu D, Quinn JF, et al. A combined dataset of human cerebrospinal fluid proteins identified by multi-dimensional chromatography and tandem mass spectrometry. Proteomics 2007;7:469–73. [DOI] [PubMed] [Google Scholar]
- 176. Perrin RJ, Payton JE, Malone JP, et al. Quantitative label-free proteomics for discovery of biomarkers in cerebrospinal fluid: assessment of technical and inter-individual variation. PLoS One 2013;9:e64314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Schutzer SE, Angel TE, Liu T, et al. Distinct cerebrospinal fluid proteomes differentiate post-treatment Lyme disease from chronic fatigue syndrome. PLoS One 2011;6:e17287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Schutzer SE, Liu T, Natelson BH, et al. Establishing the proteome of normal human cerebrospinal fluid. PLoS One 2010;5:e10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Zougman A, Pilch B, Podtelejnikov A, et al. Integrated analysis of the cerebrospinal fluid peptidome and proteome. J Proteome Res 2008;7:386–99. [DOI] [PubMed] [Google Scholar]
- 180. Pilch B, Mann M. Large-scale and high-confidence proteomic analysis of human seminal plasma. Genome Biol 2006;7:R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Amaral A, Castillo J, Ramalhosantos J, et al. The combined human sperm proteome: cellular pathways and implications for basic and clinical science. Hum Reprod Update 2014;20:40–62. [DOI] [PubMed] [Google Scholar]
- 182. de Mateo S, Martínez Heredia J, Estanyol JM, et al. Marked correlations in protein expression identified by proteomic analysis of human spermatozoa. Proteomics 2010;7:4264–77. [DOI] [PubMed] [Google Scholar]
- 183. Nilsson S, M. R, Palmblad M, et al. Explorative study of the protein composition of amniotic fluid by liquid chromatography electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. J Proteome Res 2004;3:884–9. [DOI] [PubMed] [Google Scholar]
- 184. Liberatori S, Bini L, de Felice C, et al. A two-dimensional protein map of human amniotic fluid at 17 weeks' gestation. Electrophoresis 1997;18:2816–22. [DOI] [PubMed] [Google Scholar]
- 185. Liu X, Song YJ, Guo ZG, et al. A comprehensive profile and inter-individual variations analysis of the human normal amniotic fluid proteome. J Proteome 2019;192:1–9. [DOI] [PubMed] [Google Scholar]
- 186. Chen CP, Lai TC, Chern SR, et al. Proteome differences between male and female fetal cells in amniotic fluid. Omics-J Integr Biol 2013;17:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Cho CK, Smith CR, Diamandis EP. Amniotic fluid proteome analysis from down syndrome pregnancies for biomarker discovery. J Proteome Res 2010;9:3574–82. [DOI] [PubMed] [Google Scholar]
- 188. Gianazza E, Wait R, Begum S, et al. Mapping the 5-50-kDa fraction of human amniotic fluid proteins by 2-DE and ESI-MS. Proteomics Clin Appl 2007;1:167–75. [DOI] [PubMed] [Google Scholar]
- 189. Michaels JE, Dasari S, Pereira L, et al. Comprehensive proteomic analysis of the human amniotic fluid proteome: gestational age-dependent changes. J Proteome Res 2007;6:1277–85. [DOI] [PubMed] [Google Scholar]
- 190. Huang Z, Du CX, Pan XD. The use of in-strip digestion for fast proteomic analysis on tear fluid from dry eye patients. PLoS One 2018;13:e0200702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. de Souza GA, de Godoy LM, Mann M. Identification of 491 proteins in the tear fluid proteome reveals a large number of proteases and protease inhibitors. Genome Biol 2006;7:R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Li N, Wang N, Zheng J, et al. Characterization of human tear proteome using multiple proteomic analysis techniques. J Proteome Res 2005;4:2052–61. [DOI] [PubMed] [Google Scholar]
- 193. Zhou L, Zhao SZ, Koh SK, et al. In-depth analysis of the human tear proteome. J Proteome 2012;75:3877–85. [DOI] [PubMed] [Google Scholar]
- 194. Aass C, Norheim I, Eriksen EF, et al. Single unit filter-aided method for fast proteomic analysis of tear fluid. Anal Biochem 2015;480:1–5. [DOI] [PubMed] [Google Scholar]
- 195. Plymoth A, Yang ZP, Löfdahl CG, et al. Rapid proteome analysis of bronchoalveolar lavage samples of lifelong smokers and never-smokers by micro-scale liquid chromatography and mass spectrometry. Clin Chem 2006;52:671–9. [DOI] [PubMed] [Google Scholar]
- 196. Sabounchi-Schütt F, Aström J, Eklund A, et al. Detection and identification of human bronchoalveolar lavage proteins using narrow-range immobilized pH gradient DryStrip and the paper bridge sample application method. Electrophoresis 2001;22:1851–60. [DOI] [PubMed] [Google Scholar]
- 197. Almatroodi SA, Mcdonald CF, Collins AL, et al. Quantitative proteomics of bronchoalveolar lavage fluid in lung adenocarcinoma. Cancer Genomics Proteomics 2015;12:39–48. [PubMed] [Google Scholar]
- 198. Carvalho AS, Cuco CM, Lavareda C, et al. Bronchoalveolar lavage proteomics in patients with suspected lung cancer. Sci Rep 2017;7:42190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Chen JZ, Ryu S, Gharib SA, et al. Exploration of the normal human bronchoalveolar lavage fluid proteome. Proteomics Clin Appl 2008;2:585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Foster MW, Thompson JW, Que LG, et al. Proteomic analysis of human bronchoalveolar lavage fluid after subsgemental exposure. J Proteome Res 2013;12:2194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Nguyen EV, Gharib SA, Palazzo SJ, et al. Proteomic profiling of bronchoalveolar lavage fluid in critically ill patients with ventilator-associated pneumonia. PLoS One 2013;8:e58782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Ortea I, Rodríguez-Ariza A, Chicano-Gálvez E, et al. Discovery of potential protein biomarkers of lung adenocarcinoma in bronchoalveolar lavage fluid by SWATH MS data-independent acquisition and targeted data extraction. J Proteome 2016;138:106–14. [DOI] [PubMed] [Google Scholar]
- 203. Tu CJ, Mammen MJ, Li J, et al. Large-scale, ion-current-based proteomics investigation of bronchoalveolar lavage fluid in chronic obstructive pulmonary disease patients. J Proteome Res 2014;13:627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Uribarri M, Hormaeche I, Zalacain R, et al. A new biomarker panel in bronchoalveolar lavage for an improved lung cancer diagnosis. J Thorac Oncol Off Public Int Assoc Stud Lung Cancer 2014;9:1504–12. [DOI] [PubMed] [Google Scholar]
- 205. Wu J, M. K, Sousa EA, et al. Differential proteomic analysis of bronchoalveolar lavage fluid in asthmatics following segmental antigen challenge. Mol Cell Proteomics 2005;4:1251–64. [DOI] [PubMed] [Google Scholar]
- 206. Pastor MD, Nogal A, Molina-Pinelo S, et al. Identification of proteomic signatures associated with lung cancer and COPD. J Proteome 2013;89:227–37. [DOI] [PubMed] [Google Scholar]
- 207. Liao YL, Alvarado R, Phinney B, et al. Proteomic characterization of specific minor proteins in the human milk casein fraction. J Proteome Res 2011;10:5409–15. [DOI] [PubMed] [Google Scholar]
- 208. Liao Y, Weber D, Xu W, et al. Absolute quantification of human milk caseins and the whey/casein ratio during the first year of lactation. J Proteome Res 2017;16:4113–21. [DOI] [PubMed] [Google Scholar]
- 209. Fortunato D, Giuffrida MG, Cavaletto M, et al. Structural proteome of human colostral fat globule membrane proteins. Proteomics 2003;3:897–905. [DOI] [PubMed] [Google Scholar]
- 210. Palmer DJ, Kelly VC, Smit A-M, et al. Human colostrum: identification of minor proteins in the aqueous phase by proteomics. Proteomics 2006;6:2208–16. [DOI] [PubMed] [Google Scholar]
- 211. Liao Y, Alvarado R, Phinney B, et al. Proteomic characterization of human milk whey proteins during a twelve-month lactation period. J Proteome Res 2011;10:1746–54. [DOI] [PubMed] [Google Scholar]
- 212. Zhang Q, Cundiff JK, Maria SD, et al. Quantitative analysis of the human milk whey proteome reveals developing milk and mammary-gland functions across the first year of lactation. Proteomes 2013;1:128–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Mateos J, Lourido L, Fernández-Puente P, et al. Differential protein profiling of synovial fluid from rheumatoid arthritis and osteoarthritis patients using LC–MALDI TOF/TOF. J Proteome 2012;75:2869–78. [DOI] [PubMed] [Google Scholar]
- 214. Balakrishnan L, Bhattacharjee M, Ahmad S, et al. Differential proteomic analysis of synovial fluid from rheumatoid arthritis and osteoarthritis patients. Clin Proteomics 2014;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Balakrishnan L, Nirujogi RS, Ahmad S, et al. Proteomic analysis of human osteoarthritis synovial fluid. Clin Proteomics 2014;11:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Chen CP, Hsu CC, Yeh WL, et al. Optimizing human synovial fluid preparation for two-dimensional gel electrophoresis. Proteome Sci 2011;9:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Gobezie R, Kho A, Krastins B, et al. High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res Ther 2007;9:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Liao W, Li Z, Li T, et al. Proteomic analysis of synovial fluid in osteoarthritis using SWATH-mass spectrometry. Mol Med Rep 2018;17:2827–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Ritter SY, Subbaiah R, Bebek G, et al. Proteomic analysis of synovial fluid from the osteoarthritic knee: comparison with transcriptome analyses of joint tissues. Arthritis Rheum 2013;65:981–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Sohn DH, Sokolove J, Sharpe O, et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via toll-like receptor 4. Arthritis Res Ther 2012;14:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Shaheed SU, Tait C, Kyriacou K, et al. Nipple aspirate fluid-a liquid biopsy for diagnosing breast health. Proteomics - Clin Appl 2017;11:1700015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Pavlou MP, Kulasingam V, Sauter ER, et al. Nipple aspirate fluid proteome of healthy females and patients with breast cancer. Clin Chem 2010;56:848–55. [DOI] [PubMed] [Google Scholar]
- 223. Giusti L, Iacconi P, Ciregia F, et al. Proteomic analysis of human thyroid fine needle aspiration fluid. J Endocrinol Investig 2007;30:865–9. [DOI] [PubMed] [Google Scholar]
- 224. He J, Gornbein J, Shen D, et al. Detection of breast cancer biomarkers in nipple aspirate fluid by SELDI-TOF and their identification by combined liquid chromatography-tandem mass spectrometry. Int J Oncol 2007;30:145–54. [PubMed] [Google Scholar]
- 225. Varnum SM, Covington CC, Woodbury RL, et al. Proteomic characterization of nipple aspirate fluid: identification of potential biomarkers of breast cancer. Breast Cancer Res Treat 2003;80:87–97. [DOI] [PubMed] [Google Scholar]
- 226. Alexander H, Stegner AL, Wagnermann C, et al. Proteomic analysis to identify breast cancer biomarkers in nipple aspirate fluid. Clin Cancer Res Off J Am Assoc Cancer Res 2004;10:7500–10. [DOI] [PubMed] [Google Scholar]
- 227. Brunoro GV, Carvalho PC, Ferreira AT, et al. Proteomic profiling of nipple aspirate fluid (NAF): exploring the complementarity of different peptide fractionation strategies. J Proteome 2015;117:86–94. [DOI] [PubMed] [Google Scholar]
- 228. Kurono S, Kaneko Y, Matsuura N, et al. Identification of potential breast cancer markers in nipple discharge by protein profile analysis using two-dimensional nano-liquid chromatography/nanoelectrospray ionization-mass spectrometry. Proteomics Clin Appl 2016;10:605–13. [DOI] [PubMed] [Google Scholar]
- 229. Dasari S, Pereira L, Reddy AP, et al. Comprehensive proteomic analysis of human cervical-vaginal fluid. J Proteome Res 2007;6:1258–68. [DOI] [PubMed] [Google Scholar]
- 230. Pereira L, Reddy AP, Jacob T, et al. Identification of novel protein biomarkers of preterm birth in human cervical-vaginal fluid. J Proteome Res 2007;6:1269–76. [DOI] [PubMed] [Google Scholar]
- 231. Shaw JL, Smith CR, Diamandis EP. Proteomic analysis of human cervico-vaginal fluid. J Proteome Res 2007;6:2859–65. [DOI] [PubMed] [Google Scholar]
- 232. Tang LJ, De Seta F, Odreman F, et al. Proteomic analysis of human cervical-vaginal fluids. J Proteome Res 2007;6:2874–83. [DOI] [PubMed] [Google Scholar]
- 233. Venkataraman N, Cole AL, Svoboda P, et al. Cationic polypeptides are required for anti-HIV-1 activity of human vaginal fluid. J Immunol 2005;175:7560–7. [DOI] [PubMed] [Google Scholar]
- 234. Zegels G, Van Raemdonck GA, Coen EP, et al. Comprehensive proteomic analysis of human cervical-vaginal fluid using colposcopy samples. Proteome Sci 2009;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Domanski D, Perzanowska A, Kistowski M, et al. A multiplexed cytokeratin analysis using targeted mass spectrometry reveals specific profiles in cancer-related pleural effusions. Neoplasia 2016;18:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Yu CJ, Wang CL, Wang CI, et al. Comprehensive proteome analysis of malignant pleural effusion for lung cancer biomarker discovery by using multidimensional protein identification technology. J Proteome Res 2011;10:4671–82. [DOI] [PubMed] [Google Scholar]
- 237. Liu PJ, Chen CD, Wang CL, et al. In-depth proteomic analysis of six types of exudative pleural effusions for nonsmall cell lung cancer biomarker discovery. Mol Cell Proteom MCP 2015;14:917–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Domanski D, Perzanowska A, Kistowski M, et al. A multiplexed cytokeratin analysis using targeted mass spectrometry reveals specific profiles in cancer-related pleural effusions. Neoplasia 2016;18:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Mundt F, Johansson HJ, Forshed J, et al. Proteome screening of pleural effusions identifies galectin 1 as a diagnostic biomarker and highlights several prognostic biomarkers for malignant mesothelioma. Mol Cell Proteom MCP 2014;13:701–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Tyan YC, Wu HY, Lai WW, et al. Proteomic profiling of human pleural effusion using two-dimensional nano liquid chromatography tandem mass spectrometry. J Proteome Res 2005;4:1274–86. [DOI] [PubMed] [Google Scholar]
- 241. Nicholas B, Skipp P, Mould R, et al. Shotgun proteomic analysis of human-induced sputum. Proteomics 2006;6:4390–401. [DOI] [PubMed] [Google Scholar]
- 242. Burg D, Schofield JPR, Brandsma J, et al. Large-scale label-free quantitative mapping of the sputum proteome. J Proteome Res 2018;17:2072–91. [DOI] [PubMed] [Google Scholar]
- 243. Muccilli V, Saletti R, Cunsolo V, et al. Protein profile of exhaled breath condensate determined by high resolution mass spectrometry. J Pharm Biomed Anal 2015;105:134–49. [DOI] [PubMed] [Google Scholar]
- 244. Cheng ZJ, Chan AK, Lewis CR, et al. Analysis of exhaled breath condensate in lung cancer patients. J Cancer Ther 2011;2:1–8. [Google Scholar]
- 245. Fumagalli M, Dolcini L, Sala A, et al. Proteomic analysis of exhaled breath condensate from single patients with pulmonary emphysema associated to alpha1-antitrypsin deficiency. J Proteome 2008;71:211–21. [DOI] [PubMed] [Google Scholar]
- 246. Fumagalli M, Ferrari F, Luisetti M, et al. Profiling the proteome of exhaled breath condensate in healthy smokers and COPD patients by LC-MS/MS. Int J Mol Sci 2012;13:13894–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Hayes SA, Haefliger S, Harris B, et al. Exhaled breath condensate for lung cancer protein analysis: a review of methods and biomarkers. J Breath Res 2016;10:034001. [DOI] [PubMed] [Google Scholar]
- 248. Kononikhin AS, Chagovets VV, Starodubtseva NL, et al. Determination of proteomic and metabolic composition of exhaled breath condensate of newborns. Mol Biol 2016;50:470–3. [DOI] [PubMed] [Google Scholar]
- 249. Kurova VS, Anaev EC, Kononikhin AS, et al. Proteomics of exhaled breath: methodological nuances and pitfalls. Clin Chem Lab Med 2009;47:706–12. [DOI] [PubMed] [Google Scholar]
- 250. Grønborg M, Bunkenborg J, Kristiansen TZ, et al. Comprehensive proteomic analysis of human pancreatic juice. J Proteome Res 2004;3:1042–55. [DOI] [PubMed] [Google Scholar]
- 251. Doyle CJ, Yancey K, Pitt HA, et al. The proteome of normal pancreatic juice. Pancreas 2012;41:186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Marchegiani G, Paulo JA, Sahora K, et al. The proteome of postsurgical pancreatic juice. Pancreas 2015;44:574–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Paulo JA, Kadiyala V, Gaun A, et al. Analysis of endoscopic pancreatic function test (ePFT)-collected pancreatic fluid proteins precipitated via ultracentrifugation. J Pancreas 2013;14:176–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Roy P, Truntzer C, Maucortboulch D, et al. Protein mass spectra data analysis for clinical biomarker discovery: a global review. Brief Bioinform 2011;12:176–86. [DOI] [PubMed] [Google Scholar]
- 255. Chen Y, Zhang Y, Yin Y, et al. SPD: a web-based secreted protein database. Nucleic Acids Res 2005;33:D169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Bateman A, Martin MJ, O'Donovan C, et al. UniProt: a hub for protein information. Nucleic Acids Res 2015;43:D204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Zhang P, Tao L, Zeng X, et al. PROFEAT update: a protein features web-server with added facility to compute network descriptors for studying omics-derived networks. J Mol Biol 2016;429:416–25. [DOI] [PubMed] [Google Scholar]
- 258. Prilusky J, Felder CE, Zeev-Ben-Mordehai T, et al. FoldIndex©: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005;21:3435–8. [DOI] [PubMed] [Google Scholar]
- 259. Wilkins MR, Gasteiger E, Bairoch A, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 1999;112:531–52. [DOI] [PubMed] [Google Scholar]
- 260. Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 2019;37:420–3. [DOI] [PubMed] [Google Scholar]
- 261. Garrow AG, Alison A, TMB-Hunt WDR. A web server to screen sequence sets for transmembrane beta-barrel proteins. Nucleic Acids Res 2005;33:W188–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Bendtsen JD, Nielsen H, Widdick D, et al. Prediction of twin-arginine signal peptides. BMC Bioinformatics 2005;6:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Käll L, Krogh A, Sonnhammer EL. Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res 2007;35:W429–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Steentoft C, Vakhrushev SY, Joshi HJ, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J 2013;32:1478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Gupta R, Jung E, Brunak S. Prediction of N-glycosylation sites in human. Proteins 2004. http://www.cbs.dtu.dk/services/NetNGlyc/. [Google Scholar]
- 266. Eisenhaber F, Imperiale F, Argos P, et al. Prediction of secondary structural content of proteins from their amino acid composition alone. I. New analytic vector decomposition methods. Proteins-structure function. Bioinformatics 2015;25:157–68. [DOI] [PubMed] [Google Scholar]
- 267. Zhang J, Chai HT, Guo S, et al. High-throughput identification of mammalian secreted proteins using species-specific scheme and application to human proteome. Molecules 2018;23:1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Hu S, Loo JA, Wong DT. Human saliva proteome analysis and disease biomarker discovery. Expert Rev Proteomics 2007;4:531–8. [DOI] [PubMed] [Google Scholar]
- 269. Du W, Sun Y, Wang Y, et al. A novel multi-stage feature selection method for microarray expression data analysis. Int J Data Mining Bioinform 2013;7:58–77. [DOI] [PubMed] [Google Scholar]
- 270. Khan NM, Ksantini R, Ahmad IS, et al. A novel SVM+NDA model for classification with an application to face recognition. Pattern Recogn 2012;45:66–79. [Google Scholar]
- 271. Schölkopf B, Burges CJ, Smola AJ. Advances in Kernel Methods: Support Vector Machine. Annual Neural Information Processing Systems (NIPS) Conference. Colorado: Breckenridge, 1999. [Google Scholar]
- 272. Klee EW, Sosa CP. Computational classification of classically secreted proteins. Drug Discov Today 2007;12:234–40. [DOI] [PubMed] [Google Scholar]
- 273. Ma H, Bandos AI, Gur D. On the use of partial area under the ROC curve for comparison of two diagnostic tests. Biom J 2015;57:304–20. [DOI] [PubMed] [Google Scholar]
- 274. Tang ZQ, Han LY, Lin HH, et al. Derivation of stable microarray cancer-differentiating signatures using consensus scoring of multiple random sampling and gene-ranking consistency evaluation. Cancer Res 2007;67:9996–10003. [DOI] [PubMed] [Google Scholar]
- 275. Xiong T, Cherkassky VA. Combined SVM and LDA Approach for Classification. IEEE International Joint Conference on Neural Networks. Canada, Montreal, Quebec: IEEE, 2005. [Google Scholar]
- 276. He JR, Li MJ, Zhang HJ, et al. Generalized manifold-ranking-based image retrieval. New York, NY, USA: 12th ACM International Conference on Multimedia, 2004. [Google Scholar]
- 277. Klingström T, Plewczynski D. Protein–protein interaction and pathway databases, a graphical review. Brief Bioinform 2011;12:702–13. [DOI] [PubMed] [Google Scholar]
- 278. Wang X, Wang Z, Ye JHKC. An algorithm to predict protein complexes in protein-protein interaction networks. Biomed Res Int 2015;2011:480294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Mcdermaid A, Monier B, Zhao J, et al. Interpretation of differential gene expression results of RNA-seq data: review and integration. Brief Bioinform 2018;bby067 10.1093/bib/bby067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 2017;18:851–69. [DOI] [PubMed] [Google Scholar]
- 281. Lv Z, Ao C, Zou Q. Protein function prediction: from traditional classifier to deep learning. Proteomics 2019;19:e1900119. [DOI] [PubMed] [Google Scholar]
- 282. Lecun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521:436–44. [DOI] [PubMed] [Google Scholar]