

Abstract
Amino acids (AAs) are key structural motifs with widespread applications in organic synthesis, biochemistry, and material sciences. Recently, with the development of milder and more versatile radical‐based procedures, the use of strategies relying on radical chemistry for the synthesis and modification of AAs has gained increased attention, as they allow rapid access to libraries of novel unnatural AAs containing a wide range of structural motifs. In this Minireview, we provide a broad overview of the advancements made in this field during the last decade, focusing on methods for the de novo synthesis of α‐, β‐, and γ‐AAs, as well as for the selective derivatisation of canonical and non‐canonical α‐AAs.
Keywords: amino acids, peptides, photochemistry, radical reactions, synthetic methods
This Minireview provides a broad overview of the advancements made in the synthesis and modification of amino acid derivatives using radical chemistry during the last decade. The overview is divided in two sections: methods for the de novo synthesis of α‐, β‐, and γ‐amino acids, and methods for the selective derivatisation of canonical and non‐canonical α‐amino acids.
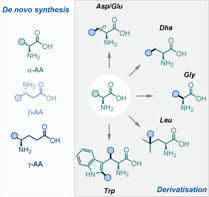
1. Introduction
Amino acids (AAs) are widely used in applications across several scientific fields; for example, they can be used as ligands in transition‐metal catalysis, [1] as key components in polymers and materials, [2] or as synthons for the preparation of biologically active molecules and peptidomimetic drugs. [3] To fully exploit the versatility offered by AAs, it is important to have efficient and straightforward methods to access structural motifs beyond those exhibited by proteomic AAs. To this end, chemists have developed a myriad of efficient and straightforward strategies for the synthesis of unnatural amino acids (UAAs) using enzymatic or transition‐metal‐catalysed processes. [4] A less explored approach, however, is the use of methods exploiting one‐electron pathways. [5]
Radical chemistry offers exciting and highly attractive approaches to access new chemical space in a rapid fashion. This is due, in part, to the plethora of synthetic precursors available to generate open‐shell species. [6] During the last decade, mild and versatile radical‐based methods have been developed to access a wide range of structural motifs in organic synthesis. An application that is becoming ever more prominent is the use of radical strategies to gain rapid access to libraries of novel UAAs or for the site‐selective modification of peptides and proteins. [7]
The aim of this Minireview is to provide a broad overview of the advances made in this field during the last decade. For clarity, it is organised in two sections: de novo synthesis, which deals with strategies to access the main three classes of AAs, namely α‐, β‐, and γ‐AAs, and modification, which describes strategies for the derivatisation of canonical and non‐canonical AAs. Furthermore, to avoid significant overlap with recent reviews, [8] special attention is paid to non‐photoredox‐mediated strategies.
2. De Novo Synthesis
Arguably, α‐, β‐, and γ‐AAs constitute the most common types of AAs. In this section, strategies to access these key structural motifs, either in their racemic or enantioenriched forms, are discussed.
2.1. α‐Amino Acids
The synthesis of α‐AAs using radical reactions can be broadly classified in three categories: addition of an open‐shell species to an imine, which can afford racemic or enantioenriched α‐AAs depending on the N‐substituents of the imine, C(sp3)−H aminations, or use of CO2 as a C1 building block.
2.1.1. Addition to Imines
In 2015, Inoue and co‐workers reported a BEt3‐mediated procedure for the synthesis of α,β‐diamino acid derivatives starting from α‐aminoacyl tellurides 1, which can be readily prepared from the corresponding α‐AA and diphenyl telluride, as well as ethyl glyoxylate oxime 2 (Scheme 1 A). [9] The key step in this reaction is the homolytic cleavage of the C−Te bond in 1, which leads to the formation of an acyl radical intermediate that undergoes facile decarbonylation to deliver a highly stabilised α‐amino radical species. [10] Subsequent addition to 2, followed by protonation furnishes the desired α,β‐diamino acid. This strategy can also be applied to the synthesis of γ‐AAs if acrylates are used as radical acceptors.
Scheme 1.
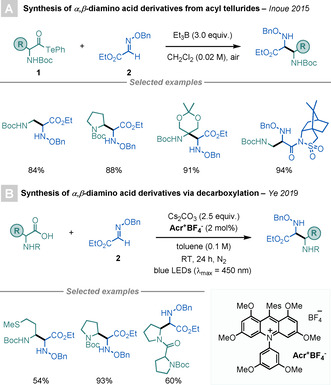
Syntheses of α,β‐diamino esters.
A more direct method for the synthesis of α,β‐diamino esters using α‐AAs and 2 was reported by Ye and co‐workers (Scheme 1 B). [11] This light‐mediated procedure employs an acridinium‐based photocatalyst (PC) to access the key α‐amino radical intermediate directly from α‐AAs.
Mariano, Wang, and co‐workers reported in 2019 a light‐mediated, decarboxylative procedure for the synthesis of C‐glyco‐α‐AAs (Scheme 2). [12] This catalyst‐free method proceeds by the addition of C‐centred glycosyl radicals, generated from the corresponding N‐hydroxyphthalimide (NHP) derived redox‐active esters (RAEs) 3, to α‐imino ester derivatives 4. The transformation shows a broad scope in terms of both the saccharide and imine motifs. Although the reaction is completely stereoselective regarding the α‐oxo stereocentre, only modest levels of diastereoselectivity were observed for the α‐amino stereocentre. The key step of the reaction is a photoinduced electron transfer (PET) between the excited Hantzsch ester (*HE) and the NHP‐derived RAE to afford glycosyl radical intermediate 5, which subsequently adds to 4 to deliver the targeted C‐glyco‐α‐AA after a hydrogen atom transfer (HAT) step.
Scheme 2.
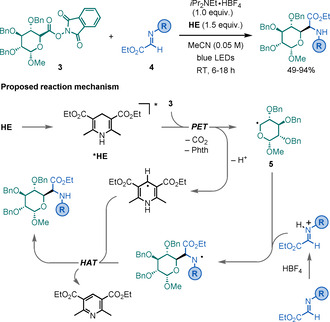
Synthesis of C‐glyco‐α‐AAs.
Recently, Nishikata, Yazaki, Ohshima, and co‐workers developed a Cu‐catalysed method for the synthesis of quaternary α‐AA derivatives from tertiary alkyl halides and AA‐derived Schiff bases (Scheme 3 A). [13] The transformation proceeds through the radical–radical coupling of a tertiary and an azaallyl radical, both generated by the Cu catalyst. This mild method shows a broad scope and functional group (FG) tolerance.
Scheme 3.
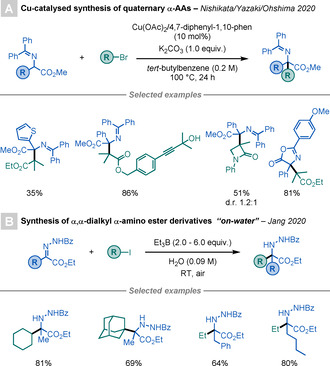
Syntheses of quaternary α‐amino esters.
An alternative metal‐free method for the synthesis of quaternary α‐amino esters was reported by Nam and Jang (Scheme 3 B). [14] This procedure employs BEt3 as a radical initiator to generate open‐shell species from alkyl iodides. The generated radical intermediates undergo radical addition to hydrazine derivatives of α‐keto esters to produce the desired products. Notably, this transformation works better when using H2O as the solvent and under air. The authors rationalised this by the hydrophobic effect and hydrogen bonding between the interface of the water and the substrate, which would accelerate the reaction.
Since it is possible to form imines in situ from a combination of amines and carbonyl motifs, multicomponent reactions for the synthesis of α‐AAs have also been designed.
In 2018, Lu, Gong, and co‐workers presented a three‐component, one‐pot reaction for the formation of masked α‐amino aldehydes (Scheme 4 A). [15] The reaction proceeds through an AIBN‐initiated radical chain mechanism, and affords the desired α‐amino aldehydes after the addition of an α‐oxo radical, generated from 1,3‐dioxolane, to an in situ generated imine.
Scheme 4.
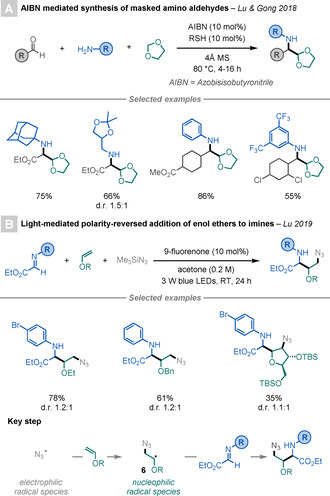
Three‐component syntheses of α‐AAs.
Later, the same group developed a light‐mediated, redox‐neutral strategy to access γ‐azide‐α‐AAs (Scheme 4 B). [16] This three‐component, one‐pot reaction proceeds through the addition of an electrophilic azide radical (generated from Me3SiN3) to an enol ether to afford nucleophilic radical intermediate 6, which can subsequently react with the imine to deliver the targeted product.
The use of chiral auxiliaries on the N‐substituent of the imine allows for the formation of enantiopure α‐AAs through diastereoselective radical additions. The most common auxiliary employed in these processes is the mesitylsulfinyl group, as its large steric demand allows for high levels of diastereocontrol during the key radical‐addition step.
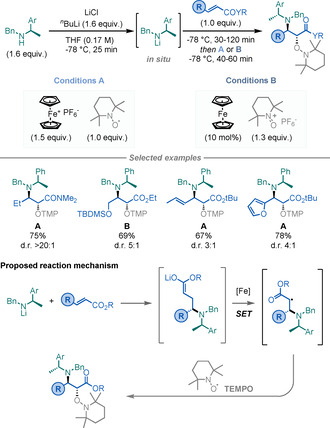
In 2018, Baran and co‐workers reported a Ni‐catalysed method for the diastereoselective synthesis of α‐AAs by using NHP‐derived RAEs as radical precursors (Scheme 5 A). [17] These RAEs undergo facile single‐electron transfer (SET) with NiI species to give alkyl radicals, which add diastereoselectively to chiral imine 7 to afford the targeted α‐AAs. The utility of this mild and scalable method was highlighted by its impressive scope, with broad FG tolerance, including the derivatisation of natural products and pharmaceuticals.
Scheme 5.
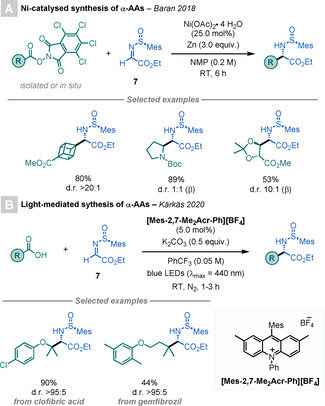
Enantiopure α‐AAs by radical decarboxylations.
Recently, Kärkäs and co‐workers expanded this strategy by using photoredox catalysis (PRC; Scheme 5 B). [18] This procedure complements Baran's method by using simpler radical precursors (carboxylic acids vs. NHP‐derived RAEs) and employing an organic‐based photocatalyst.
In 2018, Shenvi and co‐workers reported the use of unactivated olefins as alkyl‐radical precursors for the synthesis of α‐AAs (Scheme 6). [19] By using a combination of Fe(acac)3 and a silane it is possible to promote a metal‐hydride HAT (MHAT) with unactivated alkenes to afford alkyl radicals that can subsequently add to chiral sulfinimine 8. The method displays a broad scope and delivers the α‐AAs in reasonable yields and good to excellent diastereoselectivities.
Scheme 6.
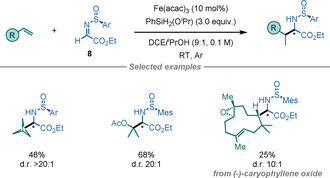
Synthesis of α‐AAs by MHAT.
2.1.2. C(sp3)−H Amination
Zhang and co‐workers reported a Co‐catalysed approach for the synthesis of α‐AAs by intramolecular radical C(sp3)−H aminations (Scheme 7). [20] In this work, a CoII‐porphyrin complex was used to generate an N‐centred radical, which, through a 1,6‐HAT process, generates a C‐centred radical. Subsequent intramolecular α‐amination affords the targeted cyclic α‐AA. The selectivity towards the less hydridic C−H bond is achieved via the formation of a nucleophilic CoIII‐nitrene radical intermediate. The use of enantiopure starting materials allows for high levels of diastereocontrol over the reaction.
Scheme 7.
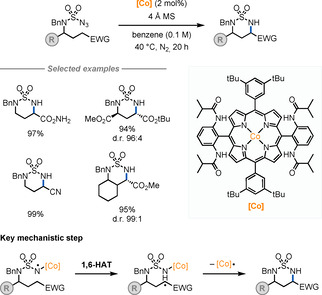
α‐AA synthesis by C(sp3)−H amination.
2.1.3. CO2 as a C1 Building Block
Recently, the use of CO2 as a C1 source to access α‐AAs has attracted increasing attention. [21] Studies by Prikhod'ko, Walter, Py et al., [22] as well as Radosevich and co‐workers [23] and Mita and Sato, [24] focused on the use of (super)stoichiometric metal reductants (SmI2, Mg, or Mn) to generate α‐amino radicals that can react with CO2 to access α‐AAs. A similar strategy was reported by the groups of Yu [25] and Walsh, [26] through the use of PRC. In this case, the key α‐amino radicals, generated after single‐electron reduction of imines, are further reduced to the corresponding carbanion, which is subsequently trapped by CO2 to yield α‐AAs (Scheme 8 A).
Scheme 8.
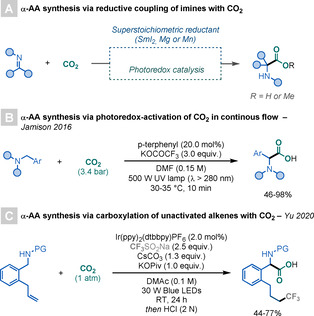
CO2 as a C1 building block to access α‐AAs.
Finally, the groups of Jamison [27] and Yu [28] have reported the synthesis of α‐AAs by α‐C−H functionalisation. Whereas Jamison's strategy proceeds by a radical–radical coupling of α‐amino radicals with CO2 .−, Yu's proceeds by an intramolecular 1,5‐HAT, which generates the key α‐amino radical that can be subsequently reduced to the corresponding carbanion and trapped by CO2 (Scheme 8 B,C).
2.2. β‐Amino Acids
The synthesis of β‐AAs by radical methods remains highly rare.
In 2018, Jahn and co‐workers reported a diastereoselective strategy for the asymmetric synthesis of anti‐β‐amino‐α‐(aminoxy) esters and amides (Scheme 9). [29] The reaction proceeds through a polar, asymmetric aza‐Michael addition of lithium amides onto α,β‐unsaturated carboxylic acid derivatives, followed by a diastereoselective radical recombination with the persistent free radical TEMPO, which acts as an oxygen source. The resulting oxygen protecting group (TMP) is stable towards acidic, basic, hydridic, and hydrogenolytic conditions, but can be readily deprotected using zinc and acetic acid. Remarkably, these conditions are compatible with tBuMe2Si (TBDMS) or Boc protecting groups without causing epimerization.
Scheme 9.
Asymmetric synthesis of β‐amino‐α‐(aminoxy) esters and amides.
Recently, Zard reported the synthesis of β‐AAs using xanthates as radical precursors (Scheme 10).[ 30 , 31 ] This lauroyl peroxide promoted transformation proceeds by the radical addition of β‐phthalimido‐α‐xanthylpropionic acid derivatives to vinylic olefins or heteroaromatic molecules. When using the free acid, spontaneous decarboxylation of the intermediate occurs in some cases, thereby leading to the formation of β‐heteroarylethylamines.
Scheme 10.
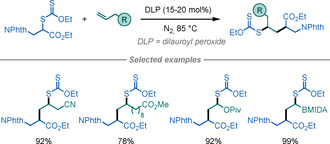
β‐AA synthesis using xanthates.
2.3. γ‐Amino Acids
The main strategy for the synthesis of γ‐AAs is the addition of α‐amino radicals to acrylate derivatives. The key nucleophilic α‐amino radical species can be accessed by several approaches, such as decarboxylation of α‐AAs, [32] proton‐coupled electron‐transfer (PCET) reduction of imines, [33] or HAT on primary [34] or benzylic amines. [35]
In 2014, MacMillan and co‐workers reported their seminal work on the photoredox‐mediated Giese reaction (Scheme 11 A). [32a] By employing an Ir‐based PC, it is possible to induce a single‐electron oxidation of α‐amino carboxylates to generate acyloxy radicals, which undergo rapid decarboxylation to afford α‐amino radical intermediates. These intermediates subsequently add to acrylates to deliver the targeted γ‐AAs. Variations of this method were later reported by the groups of Rüping [32b] and König, [32c] with the latter using NHP‐derived RAEs as radical precursors (Scheme 11 B,C). In addition, MacMillan and co‐workers have exploited this approach to accomplish peptide macrocyclisations [32e] and site‐selective bioconjugation of proteins. [32f]
Scheme 11.
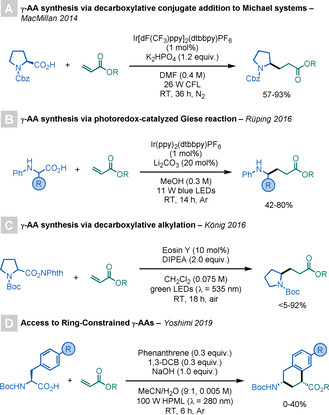
Syntheses of γ‐AAs by radical decarboxylations.
Recently, Yoshimi and co‐workers expanded MacMillan's procedure for the synthesis of ring‐constrained γ‐AAs by a radical addition/intramolecular radical cyclisation sequence (Scheme 11 D). [32d]
Dixon and co‐workers reported an elegant strategy for the synthesis of primary α‐tertiary amines by the α‐C−H functionalisation of α‐secondary amines (Scheme 12 A). [33] This bio‐inspired method proceeds through the reaction of quinone with primary amines to generate ketimine intermediates in situ, which can readily react with C‐centred nucleophiles. The versatility of this transformation was demonstrated by its broad scope. In addition, it is possible to expand this concept to the synthesis of γ‐AAs. It was shown that the in situ generated ketimine intermediate can be reduced by HE and an excited PC through a PCET process to produce a nucleophilic α‐amino radical, which can then undergo addition to acrylates to yield γ‐AAs.
Scheme 12.
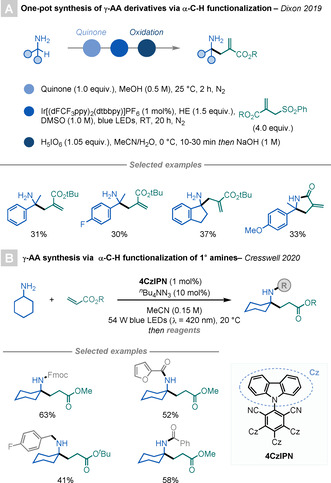
Syntheses of γ‐AAs by ketimine umpolung or HAT.
Recently, Cresswell and co‐workers reported a new method based on PRC for the synthesis of α‐tertiary amines by the α‐C−H alkylation of unmasked primary amines with different Michael acceptors (Scheme 12 B). [34] The reaction is promoted by an organophotocatalyst and nBu4NN3 as the HAT catalyst. The authors demonstrated the potential of this transformation with an impressive scope and showing its applicability in continuous‐flow syntheses.
In 2017, Xie and Zhu reported a diastereoselective strategy for the synthesis of five‐membered heterocyclic γ‐AAs (Scheme 13). [35] The key step in this light‐mediated method is a HAT reaction between a thiyl radical and the benzylic C−H bond of a benzyl‐protected amine, which results in the formation of a benzylic α‐amino radical intermediate. A subsequent 5‐exo‐trig radical cyclisation onto an α,β‐unsaturated carbonyl moiety delivers the targeted γ‐AA. A variety of heterocyclic γ‐AAs were synthesised in moderate to good yields and diastereoselectivities.
Scheme 13.
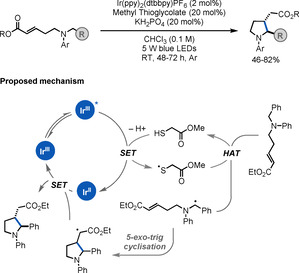
Synthesis of heterocyclic γ‐AAs.
Following the seminal work by Phipps and co‐workers on asymmetric Minisci‐type reactions, [36] Zheng and Studer reported an elegant approach for the synthesis of enantioenriched γ‐AAs (Scheme 14). [37] This light‐mediated, three‐component radical cascade reaction proceeds in a highly chemo‐, regio‐, and enantioselective fashion. The key for the reaction to succeed is the philicity of the open‐shell intermediates: SET reduction of bromoacetate derivative 9 generates electrophilic radical intermediate 10, which subsequently adds to the electron‐rich N‐vinylacetamide. This affords a nucleophilic α‐amino radical species 11, which adds to the activated electron‐poor heteroarene. The is the enantiodetermining step of the reaction and it is controlled by the use of a chiral Brønsted acid catalyst.
Scheme 14.
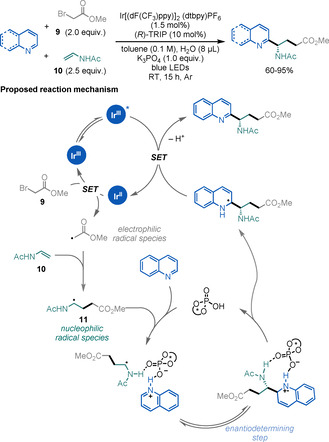
Asymmetric synthesis of γ‐AAs.
3. Modifications
This section highlights radical‐based modifications of canonical and non‐canonical AAs. Cysteine derivatisations, which have recently been reviewed, [8b] as well as deaminative lysine modifications, which are highly rare, are not included. [38]
3.1. Aspartic (Asp) and Glutamic Acid (Glu)
The primary carboxylate motif present at the β‐ or γ‐position of Asp and Glu, respectively, can be exploited through radical decarboxylative processes to provide a wide range of UAAs. These transformations can be mediated by light or transition metals.
3.1.1. Light‐Mediated Procedures
Building upon the radical spirocyclisation procedure developed by Guindeuil and Zard, [39] as well as the use of NHP‐derived RAEs as radical precursors by Schnermann and Overman, [40] Reiser and co‐workers developed in 2013 a procedure for the synthesis of (spiro)anellated furans using PRC. [41] To demonstrate the utility of this strategy, the authors carried out the synthesis of (S)‐(+)‐lycoperdic acid in seven steps, using commercially available 5‐bromofurfural and (S)‐Asp dimethyl ester as starting materials (Scheme 15 A). Interestingly, in the proposed reaction mechanism, Ir(ppy)2(dtbbpy)PF6 interacts with the phthalimide moiety of the substrate through an energy‐transfer event, rather than the more common SET process.
Scheme 15.
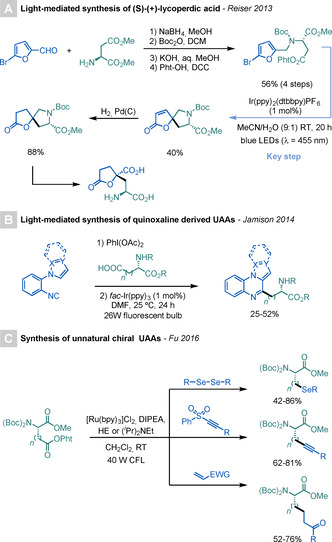
Syntheses of UAAs using RAEs.
In 2014, Jamison and co‐workers developed a light‐mediated method for the synthesis of polycyclic quinoxaline derivatives using PhI(O2CR)2 reagents, which are readily accessed from the corresponding carboxylic acids and PhI(OAc)2, as RAEs. [42] To further highlight the versatility of this procedure, the authors conceived the synthesis of UAAs by reacting Asp and Glu derivatives with the quinoxaline core under photoredox conditions, which provided the target products in moderate yields (Scheme 15 B).
Finally, Fu and co‐workers reported a photoredox‐mediated synthesis of chiral UAAs using NHP‐derived Glu and Asp RAEs as radical precursors (Scheme 15 C). [43] The key step of the method is the quenching of the excited Ru‐based PC by either HE or (iPr)2NEt to generate a highly reducing RuI species, which can engage in SET with the RAE to deliver the required alkyl radical. This can then react with Michael acceptors, [43a] alkynyl sulfones, [43a] or diselenides [43b] to give the targeted UAA.
3.1.2. Transition‐Metal‐Mediated Procedures
The groups of Weix [44] and Baran [45] independently reported in 2016 that NHP‐derived RAEs can readily react with NiI species by SET, thereby inducing the formation of alkyl radicals, which can be subsequently engaged in cross‐coupling reactions. [46] Since then, Baran and co‐workers have demonstrated that Fe‐ and Cu‐based catalysts are also competent in these processes.[ 47 , 48 ]
This highly versatile and powerful strategy has been applied to the modification of Asp and Glu derivatives through arylation,[ 44 , 47 , 49 ] alkylation, [45] alkenylation, [50] alkynylation, [51] borylations,[ 48 , 52 ] Giese reactions or Barton decarboxylation, [53] and even C‐isotope exchanges. [54] These reactions afford moderate to good yields of the desired UAA, and, in many cases, proceed without racemization of the α‐amino stereocentre (Scheme 16). This strategy can also be exploited for the selective modification of Asp and Glu residues on peptides.
Scheme 16.
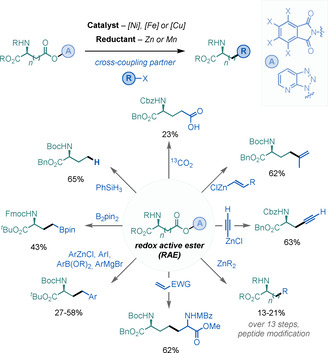
Asp/Glu derivatisations by reductive cross‐coupling.
Baran and co‐workers reported the use of CITU as a coupling reagent to generate RAEs. [55] This commercially available reagent opened the door for the synthesis and modification of peptidic RAEs, both in the solid phase and in solution, to afford a straightforward procedure for the introduction of non‐natural side chains in peptides (Scheme 17 A).
Scheme 17.
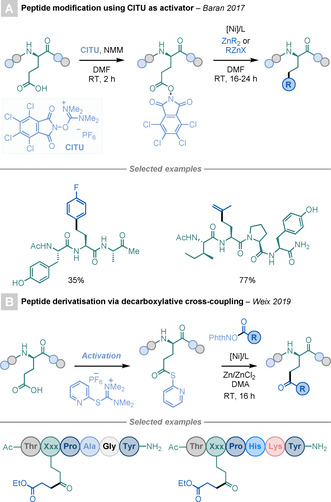
Modification of Asp/Glu residues on peptides.
Recently, Weix and co‐workers reported a sophisticated Ni‐catalysed decarboxylative cross‐coupling method for the synthesis of ketones, starting from two different types of ester derivatives, such as NHP‐derived RAEs and S‐2‐pyridyl thioesters (Scheme 17 B). [56] Both esters can be readily accessed from carboxylic acids, which broadens the scope of the method. The key to the reaction is that one of the esters acts as a radical precursor—RAE—while the other acts as an acyl surrogate—thioester. The high versatility and FG tolerance of the method was showcased by solid‐phase modification of several Asp and Glu residues on peptides.
3.2. Dehydroalanine (Dha)
Dha derivatives are highly versatile synthons that usually behave as Michael acceptors. [57] Dha modifications by radical pathways can be broadly classified depending on whether the reaction affords a racemic or enantioenriched α‐AA.
3.2.1. Racemic Procedures
Several methods have been developed for Giese‐type reactions of Dha using RAEs, [58] carboxylic acids, [59] imines, [60] ketals, [61] trifluoroborates, [62] alkyl halides, [63] or thioesters [64] as radical precursors.
In 2018, Baran and co‐workers reported a modification of his Ni‐catalysed Giese reaction, [53] which allowed the use of NHP‐derived RAEs as radical precursors for the synthesis of DNA‐encoded libraries.[ 58 , 65 ] The procedure must be carried out under highly diluted conditions (1 mM) and on a small scale (0.01 mmol) to account for the poor solubility of DNA in water. The reaction showed broad scope, with examples using a Dha derivatives as a radical acceptor affording a wide range of UAAs in high yields (Scheme 18 A).
Scheme 18.
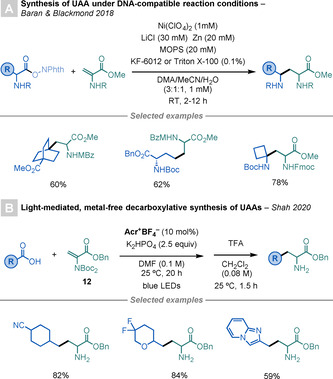
Modifications of Dha by decarboxylative strategies.
Recently, Shah et al. reported a light‐mediated, metal‐free, decarboxylative method for the synthesis of UAAs (Scheme 18 B). [59] This one‐pot, two‐step procedure uses readily available 1°, 2°, or 3° carboxylic acids as radical precursors, and an N‐Boc2‐Dha derivative (12) as radical acceptor, to deliver a wide range of unprotected UAAs.
Organoborate species are versatile radical precursors in PRC, which have been employed for the modification of Dha derivatives. [62]
de Bruijn and Roelfes reported a light‐mediated alkylation of Dha residues in antimicrobial peptides using an Ir‐based PC and trifluoroborate derivatives (Scheme 19 A). [62a]
Scheme 19.
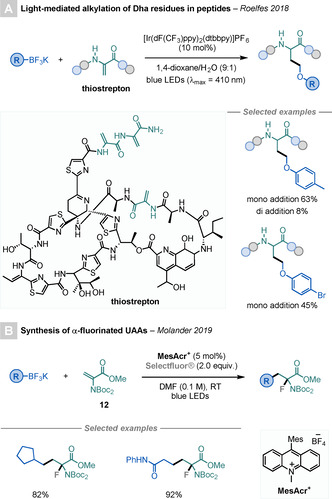
Derivatisations of Dha using organoborates.
Molander and co‐workers reported a photoredox‐mediated three‐component reaction for the synthesis of α‐fluorinated UAAs using alkyltrifluoroborates, 12, and Selectfluor® (Scheme 19 B). [62b] The key for this procedure to proceed is the electron‐withdrawing character of the N‐protecting group in 12, as it is essential to favour the attack of the nucleophilic alkyl radical on 12, rather than its engagement in a fluorine‐transfer reaction with Selectfluor®.
Dixon and co‐workers have recently reported a couple of light‐mediated methods for the modification of Dha derivatives using imines [60] and ketals [61] as radical precursors. The first is a three‐component procedure where in situ generated imines are reduced by an Ir‐based PC to generate α‐amino radicals that add to 12, thereby affording UAAs bearing 1,3‐diamine motifs (Scheme 20 A). [60] Although the products are generated in good to excellent yields, the method displays low to moderate diastereoselectivities.
Scheme 20.
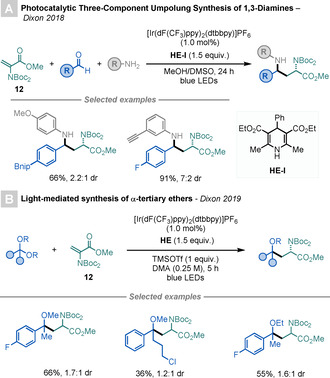
Dha modifications by reduction of in situ generated intermediates.
Lewis acids can be used to generate oxocarbenium species from ketals. Dixon and co‐workers have cleverly exploited this reactivity to develop a procedure for the synthesis of α‐tertiary ethers (Scheme 20 B). [61] The key step in this process is the reaction of Me3SiOTf with a ketal to in situ generate an oxocarbenium species, which is subsequently reduced by an Ir‐based PC to afford α‐alkoxy radicals. These add to 12 to afford UAAs. As in the previous transformation, low to moderate diastereoselectivities are observed.
Recently, several methods have been reported for the modification of Dha using alkyl halides. [63] In 2016, Davis and co‐workers designed a bio‐orthogonal strategy for the side‐chain modification of proteins (Scheme 21 A). [63b] The procedure proceeds through the selective addition of alkyl radicals, generated from a combination of alkyl bromides/iodides and NaBH4 in aqueous solution, to Dha residues.
Scheme 21.
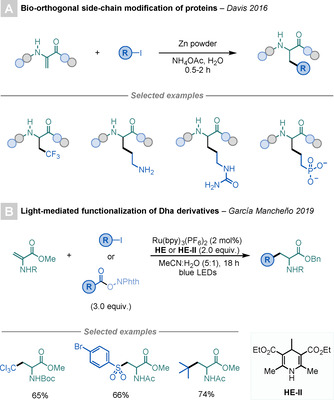
Modifications of Dha using alkyl halides.
In 2019, Brandhofer and García Mancheño reported a straightforward light‐mediated method for the derivatisation of Dha derivatives (Scheme 21 B). [63a] Although the majority of the reactions were carried out using fluorinated alkyl halides, arylsulfonyl chlorides or NHP‐derived RAEs are also suitable precursors in this Ru‐catalysed process. To highlight the versatility of the method, peptide modifications containing Dha residues were carried out.
Finally, Scanlan and co‐workers reported various chemoselective strategies for the synthesis of cysteinyl peptide thioesters through the addition of thioesters to Dha residues. [64] This transformation can be efficiently accomplished by either ionic or radical‐mediated pathways, and can be carried out under aqueous buffered conditions at neutral pH. The radical approach uses UVA light and 4′‐methoxyacetophenone as a photosensitiser to promote the generation of the key thiyl radical intermediate.
3.2.2. Diastereoselective Procedures
The Beckwith‐Karady alkene 13 is a chiral Dha derivative developed in the early 1990s. [66] Over the years, 13 has been employed to establish highly diastereoselective routes for the synthesis of UAAs. With the advent of PRC, the use of 13 in radical processes has experienced a resurgence. As a result, methods using alcohols, [67] heteroaryl halides, [68] amines, [69] C(sp3)−H bonds, [70] or α‐keto and carboxylic acids as radical precursors have been developed. [71]
In 2017, Jui and co‐workers reported a practical and scalable light‐mediated synthesis of heteroaryl α‐AAs (Scheme 22 A). [68] The key step in this photoredox‐mediated procedure is the single‐electron reduction of heteroaryl halides by a highly reducing IrII species, generated from the excited‐state quenching of an IrIII‐based PC with HE. The method displays a broad FG tolerance and scope, and it is readily amenable to large‐scale synthesis. Examples using both 12 and 13 as radical acceptors were shown.
Scheme 22.
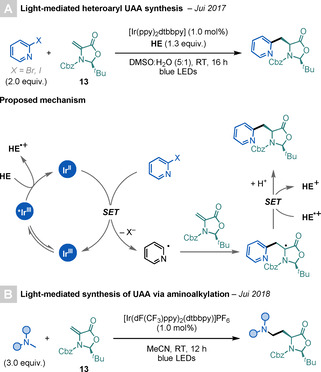
Diastereoselective syntheses of UAAs.
A year later, the same group reported the synthesis of UAAs and peptides by a light‐mediated aminoalkylation method (Scheme 22 B). [69a] The key step of the transformation is the generation of an amine radical cation, by the oxidation of tertiary amines by an excited Ir‐based photocatalyst, which, after deprotonation, affords α‐amino radicals that can engage in radical additions to 13. This method can be applied for the late‐stage modification of complex molecules, as well as, for the modification of Dha residues in peptides.
In 2018, Gaunt and co‐workers disclosed an elegant light‐mediated strategy for the synthesis tertiary alkylamines. [69b] This three‐component method combines dialkyl amines with carbonyl compounds (either aldehydes or ketones) to give iminium ions in situ. These ions are subsequently reduced by a PC to afford α‐amino radical intermediates that can engage in Giese‐type reactions with a wide range of Michael acceptors. The procedure presents a broad scope and FG tolerance, with several examples of using 13 as a radical acceptor, and affords UAA derivatives in moderate to good yields and excellent diastereoselectivities (Scheme 23). The versatility of this method was later highlighted by the rapid synthesis of several natural products. [69c]
Scheme 23.
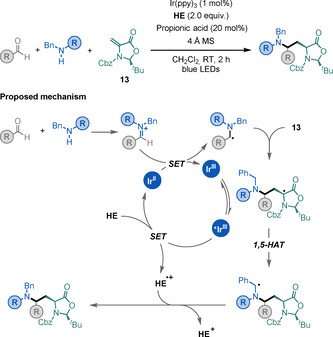
Three‐component synthesis of UAAs.
Recently, the groups of Gómez‐Suárez, [71b] Wang, [71a] and Shubert [71c] independently reported a photoredox‐mediated decarboxylative synthesis of UAAs using 13 as a radical acceptor (Scheme 24). Whereas the procedure developed by Gómez‐Suárez and co‐workers presents a broad scope with a variety of aromatic and aliphatic α‐keto acids, as well as aliphatic carboxylic acids as radical precursors, [71b] the approach developed by Wang and co‐workers allows the synthesis of α‐deuterated UAAs, [71a] and the method developed by the Shubert group facilitates the modification of peptides under metal‐free conditions. [71c] All three methods allow the straightforward synthesis of a wide range of UAAs in good to excellent yields and high diastereoselectivities.
Scheme 24.
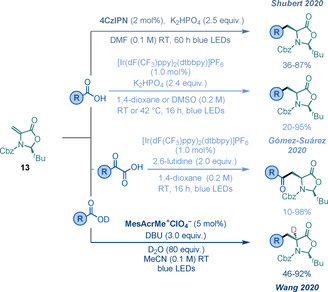
Decarboxylative functionalisations of 13.
3.3. Glycine (Gly)
The direct C−H functionalisation of Gly has been a greatly sought‐after route for the facile synthesis of UAAs. In 2015, You and co‐workers reported a Ni‐catalysed strategy for the C−H benzylation of Gly (Scheme 25). [72] This transformation proceeds by coordination‐activation of a 2‐picolinamido α‐amino ester with a high‐valent NiIII species. The system avoids formation of an imine intermediate through direct trapping of a benzylic radical species by the amine. A wide range of β‐aromatic α‐AAs can be accessed by using a variety of (hetero)arylmethanes, arylmethylenes, or arylmethines.
Scheme 25.
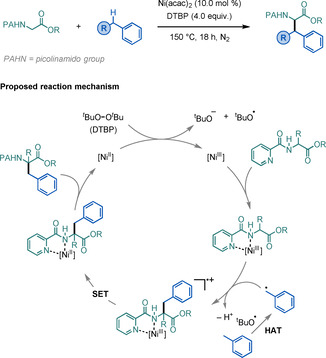
Ni‐catalysed C−H benzylation of Gly.
Very recently, Xu and co‐workers reported a remarkable light‐mediated, catalyst‐free C−H alkylation of Gly using alkylpyridinium salts as alkyl radical precursors (Scheme 26). [73] The key step in the reaction is the formation of an electron‐donor‐acceptor (EDA) complex between Gly and the pyridinium salt, which upon irradiation undergoes an intermolecular charge transfer to generate an alkyl radical—after homolytic cleavage of the N−C bond—and a Gly‐derived α‐amino radical. These two species can undergo a radical–radical coupling, thereby allowing the straightforward derivatisation of Gly under very mild conditions. This was highlighted by the selective derivatisation of Gly residues on several peptides.
Scheme 26.
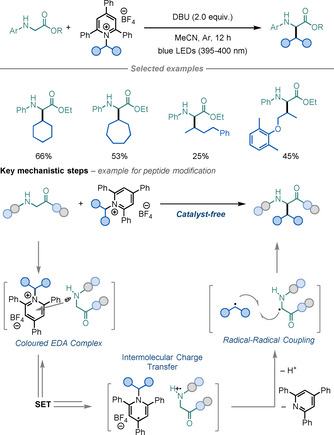
Deaminative Gly modification.
A different strategy for the modification of Gly involves its derivatisation in situ to generate imines that can be further functionalised. [74]
In 2015, Wu and co‐workers described a dual photoredox/Co‐catalysed Gly modification that takes advantage of this approach (Scheme 27 A). [74a] In this procedure, the in situ generated imine is further derivatised by a nucleophilic attack from a β‐keto ester/indole derivative to access highly functionalised UAAs.
Scheme 27.
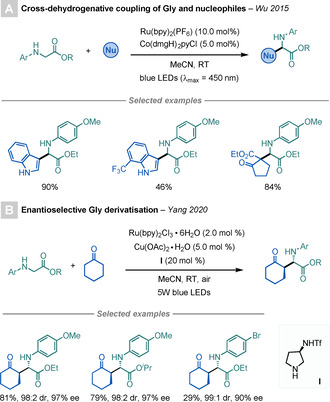
Gly derivatisations by in situ generated imines.
Recently, Yang et al. have also exploited this strategy to develop an enantioselective Gly derivatisation procedure (Scheme 27 B). [74b] This light‐mediated method uses a dual photoredox/Cu‐catalysed strategy to generate the key imine intermediate, which is subsequently involved in a proline‐catalysed asymmetric Mannich‐type reaction to produce a wide range of enantioenriched UAAs.
3.4. Leucine (Leu)
Leu modifications by C−H functionalisation of the isopropyl motif through HAT reactions have been targeted.
In 2015, Di Rocco, Britton, and co‐workers reported a light‐mediated procedure for the preparation of a fluorinated Leu derivative employed in the synthesis of a cathepsin K inhibitor used in the treatment of osteoporosis (Scheme 28 A). [75] The reaction involves photoexcitation of a decatungstate catalyst, which produces an excited‐state intermediate that abstracts a hydrogen atom from the γ‐position of Leu. The newly formed C‐centred radical undergoes selective fluorine atom transfer with N‐fluorobenzenesulfonimide (NFSI).
Scheme 28.
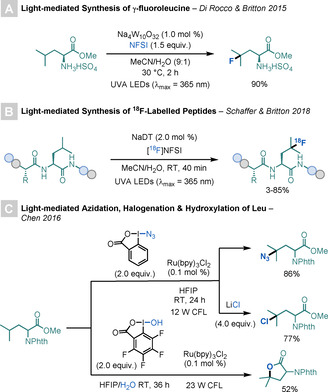
C−H functionalisations of Leu.
Later, the groups of Britton and Schaffer partnered to expand this strategy to the synthesis of 18F‐labeled peptides, by the C−H fluorination of Leu residues in unprotected, unmodified peptides (Scheme 28 B). [76] The selectivity of this reaction for the γ‐position of Leu, combined with the tolerance of charged, polar, and hydrophobic AAs is particularly notable.
In 2016, Chen and co‐workers developed a system for the functionalisation of tertiary aliphatic C−H bonds of several substrates, including Leu (Scheme 28 C). [77] By using Zhdankin's reagent, Ru(bpy)3Cl2, and visible‐light irradiation, the reaction allows azidation of Leu under mild conditions. By adding LiCl or nBu4NBr to the reaction, halogenated Leu derivatives can also be prepared.
Later, the Chen group expanded this method to the hydroxylation and amidation of tertiary and benzylic C−H bonds, including Leu, thereby suggesting a possible efficient method for the late‐stage modification of biomolecules. [78]
3.5. Phenylalanine (Phe)
Phe presents two main vectors for derivatisation by radical‐based methods: the benzylic C−H bond and the aromatic ring.
Several reports have dealt with the fluorination of the benzylic C−H bond in Phe using Selectfluor® as the fluorinating source. [79] Among them, the most relevant is the light‐mediated fluorination of Phe residues on peptides reported by Lectka and co‐workers in 2016 (Scheme 29). [80] Although the method afforded poor diastereoselectivities, the products could be successfully separated using flash chromatography. In peptides presenting a wide range of C−H bonds susceptible to HAT reactions, the reported transformations showcased a remarkable selectivity towards the fluorination of benzylic C−H bonds in Phe residues.
Scheme 29.
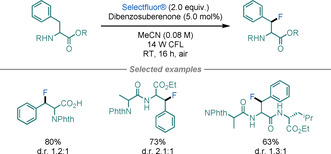
C−H fluorination of Phe.
Another strategy to modify Phe is the functionalisation of aromatic C−H bonds through amination reactions. Ritter and co‐workers reported a method for para‐selective C−H amination by charge‐transfer‐directed radical substitution of aromatic systems in a one‐pot, two‐step reaction (Scheme 30 A). [81] The method showed a broad substrate scope and was used for the selective para‐amination of phthalimide‐protected Phe in reasonable yield and with complete stereoretention.
Scheme 30.
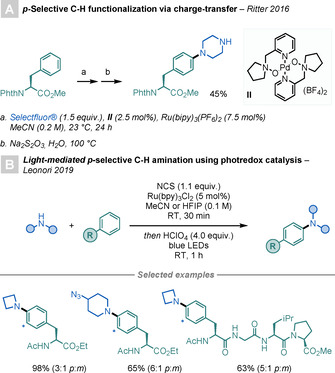
para‐Selective C−H aminations of Phe.
Recently, Leonori and co‐workers expanded the scope of para‐selective C−H aminations using PRC (Scheme 30 B). [82] The key step of the process is the generation of a highly electrophilic aminyl radical, which selectively adds at the para position of the arene. The method can be applied for the selected derivatisation of Phe residues in tetrapeptides.
3.6. Tryptophan (Trp)
The most prevalent modifications of Trp are by fluoroalkylation/trifluoromethylation reactions at the C2‐position of the indole residue. [83]
A notable example comes from a recent report by Guerrero and Correa on the radical C−H trifluoromethylation of Trp‐containing peptides using abundant, non‐precious materials under mild conditions (Scheme 31). [83c] This Cu‐catalysed system incorporates CF3 groups at the C2‐position of the indole, thereby resulting in trifluoromethylated Trp‐containing peptides with excellent selectivity.
Scheme 31.
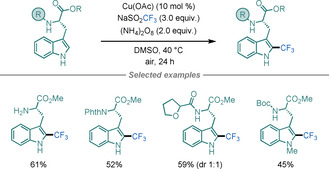
Cu‐catalysed trifluoromethylation of Trp residues.
Shi and co‐workers reported a chemoselective peptide modification at the β‐C(sp3)−H bond of Trp residues by using methyl acrylate and a PC (Scheme 32). [84] The reaction can be carried out with other Michael acceptors, and represents an innovative process for activation of the β‐C(sp3)−H bond of Trp for residue‐specific peptide modifications.
Scheme 32.
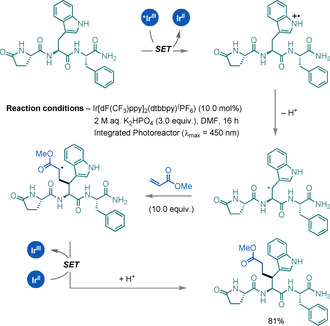
β‐C(sp3)−H alkylation of Trp.
3.7. Vinyl and (Homo)allylglycine
Despite its use as a starting material for several reactions, the application of vinyl‐Gly as a substrate in radical reactions has scarcely been investigated, possibly because of the presence of a labile, tertiary, and allylic hydrogen atom. To overcome this limitation, Zard and co‐workers investigated the intermolecular radical addition of xanthates to protected vinyl‐Gly. [85] This afforded UAAs in respectable yields and with complete stereoretention at the α‐amino position (Scheme 33).
Scheme 33.
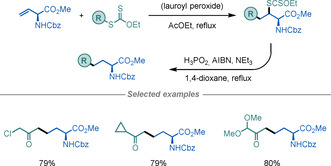
Modification of vinyl‐Gly using xanthates.
In 2019, Jackson and co‐workers reported a radical hydrofluorination of unsaturated AAs, including allyl‐Gly derivatives (Scheme 34). [86] This study expanded the scope of the radical method reported by Barker and Boger for the addition of HF to unactivated alkenes. [87] The combination of FeIII/NaBH4, and Selectfluor® or NaN3 resulted in enantiomerically pure protected fluorinated or azidated AAs in reasonable yields, without the need to remove protecting groups or use additional toxic reagents.
Scheme 34.
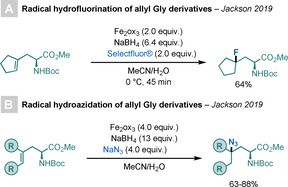
Fluorination or azidation of allyl‐Gly derivatives.
In addition, the groups of Shenvi [88] as well as Ruffoni and Leonori [89] have reported several methods where allylglycine is part of the reaction scope. Its modification usually proceeds smoothly and in moderate to good yields.
Finally, Deming and co‐workers have shown that it is possible to functionalise alkene side chains on poly(homoallylglycine) through radical thio‐ene reactions. [90] The incorporation of homoallyl Gly residues into polypeptides allowed alteration/control of the peptide‐chain conformations.
4. Conclusions and Outlook
During the last decade, significant advances have been made in the synthesis and modification of AAs by using radical‐based strategies. This progress has been driven by the development of milder and more general catalytic strategies for the generation of open‐shell species. As a result, straightforward procedures for the synthesis of α‐, β‐, and γ‐AAs, both in racemic and enantiopure forms, as well as methods for the chemoselective modification of AA residues in peptides, have been reported. Despite this progress, there is still room for improvement. For example, the main use of α‐AAs in syntheses involving radical chemistry is as readily available precursors of α‐amino radicals by decarboxylative transformations, [8c] which obliterates their stereochemical information. Moreover, although substantial developments have been made for site‐selective peptide modification using radical chemistry, bioorthogonal methods remain scarce. At the current pace of innovation, it is expected that methods capable of addressing these challenges will emerge in the near future. The radical‐based synthesis and derivatisation of AAs is an exciting field of research with a healthy present and a brilliant future, full of opportunities for innovation.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Francisco José Aguilar Troyano graduated in 2016 at the University of Jaén. For his M.Sc. he moved to the University of Granada, where he carried out his thesis under the supervision of Prof. Luisa Carlota López. In 2018, he joined Janssen Pharmaceutical Companies of Johnson & Johnson in Toledo (Spain), as a junior research scientist. In 2019, he moved to Wuppertal (Germany) and joined the Gómez‐Suárez group as a PhD student. His current research focuses on the development of deoxyfunctionalisation methods using radical chemistry.

Biographical Information
Kay Merkens received his M.Sc. from the Heinrich‐Heine University Düsseldorf in 2017. During his studies he carried out a five‐month internship with Prof. Mikael Bergdahl at San Diego State University and wrote his Master's thesis with Prof. Tobias Ritter at the Max‐Planck‐Institute for Coal Research. He then joined Prof. Hans‐Günther Schmalz at the University of Cologne. In 2019, he joined the Gómez‐Suárez group as a PhD student. His current research focuses on the synthesis and modification of amino acids using radical chemistry.

Biographical Information
Khadijah Anwar graduated from the University of Strathclyde, Scotland, with an M.Chem. in 2019. During her studies, she carried out a year‐long internship working with adhesives and additives at Sika Technology in Zürich, Switzerland. She carried out her thesis under the supervision of Prof. Eva Hevia, working on the synthesis of novel organometallic species with an emphasis on the influence of the binding ligand. In 2020, she moved to Wuppertal to join the Gómez‐Suárez group as a PhD student.

Biographical Information
Adrián Gómez‐Suárez graduated from the University of Santiago de Compostela in 2009. In 2014 he was awarded his PhD from the University of St. Andrews for his studies on the chemistry of dinuclear gold complexes under the supervision of Prof. Steven P. Nolan. For postdoctoral research he moved to the WWU Münster to work with Prof. Frank Glorius. In 2018 he started his habilitation at the Bergische Universität Wuppertal, under the mentorship of Prof. Stefan F. Kirsch. His research interests include radical chemistry, catalysis, and photochemistry.

Acknowledgements
This work was funded by the Fonds der Chemischen Industrie (a Liebig scholarship to A.G.S. and a PhD scholarship to F.J.A.T.) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project no. 443074366, and supported by the Bergische Universität Wuppertal (BUW). Prof. Stefan Kirsch (BUW) is greatly acknowledged for his continuous support. Open access funding enabled and organized by Projekt DEAL.
F. J. Aguilar Troyano, K. Merkens, K. Anwar, A. Gómez-Suárez, Angew. Chem. Int. Ed. 2021, 60, 1098.
References
- 1. Shao Q., Wu K., Zhuang Z., Qian S., Yu J. Q., Acc. Chem. Res. 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Pinazo A., Pons R., Pérez L., Infante M. R., Ind. Eng. Chem. Res. 2011, 50, 4805–4817; [Google Scholar]
- 2b. Rowland M. J., Appel E. A., Coulston R. J., Scherman O. A., J. Mater. Chem. B 2013, 1, 2904–2910. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Reetz M. T., Angew. Chem. Int. Ed. Engl. 1991, 30, 1531–1546; [Google Scholar]; Angew. Chem. 1991, 103, 1559–1573; [Google Scholar]
- 3b. Singh P., Samanta K., Das S. K., Panda G., Org. Biomol. Chem. 2014, 12, 6297–6339; [DOI] [PubMed] [Google Scholar]
- 3c. Walsh C. T., O'Brien R. V., Khosla C., Angew. Chem. Int. Ed. 2013, 52, 7098–7124; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7238–7265; [Google Scholar]
- 3d. Blaskovich M. A. T., J. Med. Chem. 2016, 59, 10807–10836. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Gröger H., Chem. Rev. 2003, 103, 2795–2828; [DOI] [PubMed] [Google Scholar]
- 4b. Nájera C., Sansano J. M., Chem. Rev. 2007, 107, 4584–4671; [DOI] [PubMed] [Google Scholar]
- 4c. Perdih A., Dolenc M. S., Curr. Org. Chem. 2007, 11, 801–832; [Google Scholar]
- 4d. Wang J., Liu X., Feng X., Chem. Rev. 2011, 111, 6947–6983; [DOI] [PubMed] [Google Scholar]
- 4e. Saladino R., Botta G., Crucianelli M., Mini-Rev. Med. Chem. 2012, 12, 277–300; [DOI] [PubMed] [Google Scholar]
- 4f. Ordóñez M., Cativiela C., Romero-Estudillo I., Tetrahedron: Asymmetry 2016, 27, 999–1055; [Google Scholar]
- 4g. Hedges J. B., Ryan K. S., Chem. Rev. 2020, 120, 3161–3209; [DOI] [PubMed] [Google Scholar]
- 4h. Noda H., Shibasaki M., Eur. J. Org. Chem. 2020, 2350–2361. [Google Scholar]
- 5.
- 5a. Easton C. J., Chem. Rev. 1997, 97, 53–82; [DOI] [PubMed] [Google Scholar]
- 5b. Hansen S. G., Skrydstrup T., in Radicals in Synthesis II (Ed.: Gansäuer A.), Springer, Berlin, Heidelberg, 2006, pp. 135–162; [Google Scholar]
- 5c. Deska J., in Amino Acids, Peptides and Proteins in Organic Chemistry: Building Blocks, Catalysis and Coupling Chemistry, Volume 3,(Ed.: B. Hughes A.), Wiley-VCH-Verlag & Co, Weinheim, 2011, pp. 115–141. [Google Scholar]
- 6.
- 6a. Zard S. Z., Org. Lett. 2017, 19, 1257–1269;28301943 [Google Scholar]
- 6b. Yan M., Lo J. C., Edwards J. T., Baran P. S., J. Am. Chem. Soc. 2016, 138, 12692–12714; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Studer A., Curran D. P., Angew. Chem. Int. Ed. 2016, 55, 58–102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 58–106. [Google Scholar]
- 7.
- 7a. Malins L. R., Peptide Sci. 2018, 110, e24049; [Google Scholar]
- 7b. Malins L. R., Curr. Opin. Chem. Biol. 2018, 46, 25–32; [DOI] [PubMed] [Google Scholar]
- 7c. Guerrero I., Correa A., Asian J. Org. Chem. 2020, 9, 898–909. [Google Scholar]
- 8.
- 8a. Brandhofer T., Mancheno O. G., Eur. J. Org. Chem. 2018, 6050–6067; [Google Scholar]
- 8b. Bottecchia C., Noel T., Chem. Eur. J. 2019, 25, 26–42; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Liu J.-Q., Shatskiy A., Matsuura B. S., Kärkäs M. D., Synthesis 2019, 51, 2759–2791. [Google Scholar]
- 9. Nagatomo M., Nishiyama H., Fujino H., Inoue M., Angew. Chem. Int. Ed. 2015, 54, 1537–1541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1557–1561. [Google Scholar]
- 10. Chatgilialoglu C., Crich D., Komatsu M., Ryu I., Chem. Rev. 1999, 99, 1991–2070. [DOI] [PubMed] [Google Scholar]
- 11. Wu G., Wang J., Liu C., Sun M., Zhang L., Ma Y., Cheng R., Ye J., Org. Chem. Front. 2019, 6, 2245–2249. [Google Scholar]
- 12. Ji P., Zhang Y., Wei Y., Huang H., Hu W., Mariano P. A., Wang W., Org. Lett. 2019, 21, 3086–3092. [DOI] [PubMed] [Google Scholar]
- 13. Matsumoto Y., Sawamura J., Murata Y., Nishikata T., Yazaki R., Ohshima T., J. Am. Chem. Soc. 2020, 142, 8498–8505. [DOI] [PubMed] [Google Scholar]
- 14. Nam T. K., Jang D. O., Tetrahedron Lett. 2020, 61, 151411. [Google Scholar]
- 15. Zeng H., Yang S., Li H., Lu D., Gong Y., Zhu J. T., J. Org. Chem. 2018, 83, 5256–5266. [DOI] [PubMed] [Google Scholar]
- 16. Yang S., Zhu S., Lu D., Gong Y., Org. Lett. 2019, 21, 8464–8468. [DOI] [PubMed] [Google Scholar]
- 17. Ni S., Garrido-Castro A. F., Merchant R. R., de Gruyter J. N., Schmitt D. C., Mousseau J. J., Gallego G. M., Yang S., Collins M. R., Qiao J. X., Yeung K. S., Langley D. R., Poss M. A., Scola P. M., Qin T., Baran P. S., Angew. Chem. Int. Ed. 2018, 57, 14560–14565; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14768–14773. [Google Scholar]
- 18.A. Shatskiy, A. Axelsson, B. Blomkvist, J.-Q. Liu, P. Dinér, M. D. Kärkäs, Chemrxiv 2020, 10.26434/chemrxiv.12084672.v1. [DOI]
- 19. Matos J. L. M., Vasquez-Cespedes S., Gu J., Oguma T., Shenvi R. A., J. Am. Chem. Soc. 2018, 140, 16976–16981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu H., Hu Y., Jiang H., Wojtas L., Zhang X. P., Org. Lett. 2012, 14, 5158–5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mita T., Sato Y., Chem. Asian J. 2019, 14, 2038–2047. [DOI] [PubMed] [Google Scholar]
- 22. Prikhod′ko A., Walter O., Zevaco T. A., Garcia-Rodriguez J., Mouhtady O., Py S., Eur. J. Org. Chem. 2012, 3742–3746. [Google Scholar]
- 23. Sathe A. A., Hartline D. R., Radosevich A. T., Chem. Commun. 2013, 49, 5040–5042. [DOI] [PubMed] [Google Scholar]
- 24. Mita T., Chen J., Sato Y., Org. Lett. 2014, 16, 2200–2203. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Ju T., Fu Q., Ye J.-H., Zhang Z., Liao L.-L., Yan S.-S., Tian X.-Y., Luo S.-P., Li J., Yu D.-G., Angew. Chem. Int. Ed. 2018, 57, 13897–13901; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14093–14097; [Google Scholar]
- 25b. Fu Q., Bo Z.-Y., Ye J.-H., Ju T., Huang H., Liao L.-L., Yu D.-G., Nat. Commun. 2019, 10, 3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fan X., Gong X., Ma M., Wang R., Walsh P. J., Nat. Commun. 2018, 9, 4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seo H., Katcher M. H., Jamison T. F., Nat. Chem. 2017, 9, 453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song L., Fu D. M., Chen L., Jiang Y. X., Ye J. H., Zhu L., Lan Y., Fu Q., Yu D. G., Angew. Chem. Int. Ed. 2020, 10.1002/anie.202008630; Angew. Chem. 2020, . [DOI] [Google Scholar]
- 29. Hidasová D., Janák M., Jahn E., Císařová I., Jones P. G., Jahn U., Eur. J. Org. Chem. 2018, 5222–5230. [Google Scholar]
- 30. Zard S. Z., CHIMIA 2020, 74, 9–17. [DOI] [PubMed] [Google Scholar]
- 31. Chen X., Zard S. Z., Org. Lett. 2020, 22, 3628–3632. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Chu L., Ohta C., Zuo Z., MacMillan D. W., J. Am. Chem. Soc. 2014, 136, 10886–10889; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32b. Millet A., Lefebvre Q., Rueping M., Chem. Eur. J. 2016, 22, 13464–13468; [DOI] [PubMed] [Google Scholar]
- 32c. Schwarz J., König B., Green Chem. 2016, 18, 4743–4749; [Google Scholar]
- 32d. Osaka K., Usami A., Iwasaki T., Yamawaki M., Morita T., Yoshimi Y., J. Org. Chem. 2019, 84, 9480–9488; [DOI] [PubMed] [Google Scholar]
- 32e. McCarver S. J., Qiao J. X., Carpenter J., Borzilleri R. M., Poss M. A., Eastgate M. D., Miller M. M., MacMillan D. W., Angew. Chem. Int. Ed. 2017, 56, 728–732; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 746–750; [Google Scholar]
- 32f. Bloom S., Liu C., Kolmel D. K., Qiao J. X., Zhang Y., Poss M. A., Ewing W. R., MacMillan D. W. C., Nat. Chem. 2018, 10, 205–211; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32g. Ma J., Lin J., Zhao L., Harms K., Marsch M., Xie X., Meggers E., Angew. Chem. Int. Ed. 2018, 57, 11193–11197; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11363–11367. [Google Scholar]
- 33. Vasu D., Fuentes de Arriba A. L., Leitch J. A., de Gombert A., Dixon D. J., Chem. Sci. 2019, 10, 3401–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ryder A. S. H., Cunningham W. B., Ballantyne G., Mules T., Kinsella A. G., Turner-Dore J., Alder C. M., Edwards L. J., McKay B. S. J., Grayson M. N., Cresswell A. J., Angew. Chem. Int. Ed. 2020, 59, 14986–14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu J., Xie J., Zhu C., Org. Chem. Front. 2017, 4, 2433–2436. [Google Scholar]
- 36.
- 36a. Proctor R. S. J., Davis H. J., Phipps R. J., Science 2018, 360, 419–422; [DOI] [PubMed] [Google Scholar]
- 36b. Reid J. P., Proctor R. S. J., Sigman M. S., Phipps R. J., J. Am. Chem. Soc. 2019, 141, 19178–19185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng D., Studer A., Angew. Chem. Int. Ed. 2019, 58, 15803–15807; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15950–15954. [Google Scholar]
- 38.
- 38a. Basch C. H., Liao J., Xu J., Piane J. J., Watson M. P., J. Am. Chem. Soc. 2017, 139, 5313–5316; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38b. Wu J., He L., Noble A., Aggarwal V. K., J. Am. Chem. Soc. 2018, 140, 10700–10704. [DOI] [PubMed] [Google Scholar]
- 39. Guindeuil S., Zard S. Z., Chem. Commun. 2006, 665–667. [DOI] [PubMed] [Google Scholar]
- 40. Schnermann M. J., Overman L. E., Angew. Chem. Int. Ed. 2012, 51, 9576–9580; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9714–9718. [Google Scholar]
- 41. Kachkovskyi G., Faderl C., Reiser O., Adv. Synth. Catal. 2013, 355, 2240–2248. [Google Scholar]
- 42. He Z., Bae M., Wu J., Jamison T. F., Angew. Chem. Int. Ed. 2014, 53, 14451–14455; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14679–14683. [Google Scholar]
- 43.
- 43a. Jiang M., Jin Y., Yang H., Fu H., Sci. Rep. 2016, 6, 26161; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43b. Jiang M., Yang H., Fu H., Org. Lett. 2016, 18, 1968–1971; [DOI] [PubMed] [Google Scholar]
- 43c. Jin Y., Yang H., Fu H., Chem. Commun. 2016, 52, 12909–12912. [DOI] [PubMed] [Google Scholar]
- 44. Huihui K. M., Caputo J. A., Melchor Z., Olivares A. M., Spiewak A. M., Johnson K. A., DiBenedetto T. A., Kim S., Ackerman L. K., Weix D. J., J. Am. Chem. Soc. 2016, 138, 5016–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qin T., Cornella J., Li C., Malins L. R., Edwards J. T., Kawamura S., Maxwell B. D., Eastgate M. D., Baran P. S., Science 2016, 352, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Murarka S., Adv. Synth. Catal. 2018, 360, 1735–1753. [Google Scholar]
- 47. Toriyama F., Cornella J., Wimmer L., Chen T. G., Dixon D. D., Creech G., Baran P. S., J. Am. Chem. Soc. 2016, 138, 11132–11135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang J., Shang M., Lundberg H., Feu K. S., Hecker S. J., Qin T., Blackmond D. G., Baran P. S., ACS Catal. 2018, 8, 9537–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang J., Qin T., Chen T. G., Wimmer L., Edwards J. T., Cornella J., Vokits B., Shaw S. A., Baran P. S., Angew. Chem. Int. Ed. 2016, 55, 9676–9679; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9828–9831. [Google Scholar]
- 50. Edwards J. T., Merchant R. R., McClymont K. S., Knouse K. W., Qin T., Malins L. R., Vokits B., Shaw S. A., Bao D. H., Wei F. L., Zhou T., Eastgate M. D., Baran P. S., Nature 2017, 545, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.
- 51a. Huang L., Olivares A. M., Weix D. J., Angew. Chem. Int. Ed. 2017, 56, 11901–11905; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12063–12067; [Google Scholar]
- 51b. Smith J. M., Qin T., Merchant R. R., Edwards J. T., Malins L. R., Liu Z., Che G., Shen Z., Shaw S. A., Eastgate M. D., Baran P. S., Angew. Chem. Int. Ed. 2017, 56, 11906–11910; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12068–12072. [Google Scholar]
- 52. Li C., Wang J., Barton L. M., Yu S., Tian M., Peters D. S., Kumar M., Yu A. W., Johnson K. A., Chatterjee A. K., Yan M., Baran P. S., Science 2017, 356, eaam7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qin T., Malins L. R., Edwards J. T., Merchant R. R., Novak A. J., Zhong J. Z., Mills R. B., Yan M., Yuan C., Eastgate M. D., Baran P. S., Angew. Chem. Int. Ed. 2017, 56, 260–265; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 266–271. [Google Scholar]
- 54. Kingston C., Wallace M. A., Allentoff A. J., deGruyter J. N., Chen J. S., Gong S. X., S. Bonacorsi, Jr. , Baran P. S., J. Am. Chem. Soc. 2019, 141, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. deGruyter J. N., Malins L. R., Wimmer L., Clay K. J., Lopez-Ogalla J., Qin T., Cornella J., Liu Z., Che G., Bao D., Stevens J. M., Qiao J. X., Allen M. P., Poss M. A., Baran P. S., Org. Lett. 2017, 19, 6196–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang J., Cary B. P., Beyer P. D., Gellman S. H., Weix D. J., Angew. Chem. Int. Ed. 2019, 58, 12081–12085; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12209–12213. [Google Scholar]
- 57. Bogart J. W., Bowers A. A., Org. Biomol. Chem. 2019, 17, 3653–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang J., Lundberg H., Asai S., Martin-Acosta P., Chen J. S., Brown S., Farrell W., Dushin R. G., O'Donnell C. J., Ratnayake A. S., Richardson P., Liu Z., Qin T., Blackmond D. G., Baran P. S., Proc. Natl. Acad. Sci. USA 2018, 115, E6404–E6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shah A. A., M. J. Kelly, 3rd , Perkins J. J., Org. Lett. 2020, 22, 2196–2200. [DOI] [PubMed] [Google Scholar]
- 60. Rossolini T., Leitch J. A., Grainger R., Dixon D. J., Org. Lett. 2018, 20, 6794–6798. [DOI] [PubMed] [Google Scholar]
- 61. Rossolini T., Ferko B., Dixon D. J., Org. Lett. 2019, 21, 6668–6673. [DOI] [PubMed] [Google Scholar]
- 62.
- 62a. de Bruijn A. D., Roelfes G., Chem. Eur. J. 2018, 24, 11314–11318; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62b. Sim J., Campbell M. W., Molander G. A., ACS Catal. 2019, 9, 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.
- 63a. Brandhofer T., Mancheno O. G., ChemCatChem 2019, 11, 3797–3801; [Google Scholar]
- 63b. Wright T. H., Bower B. J., Chalker J. M., Bernardes G. J., Wiewiora R., Ng W. L., Raj R., Faulkner S., Vallee M. R., Phanumartwiwath A., Coleman O. D., Thezenas M. L., Khan M., Galan S. R., Lercher L., Schombs M. W., Gerstberger S., Palm-Espling M. E., Baldwin A. J., Kessler B. M., Claridge T. D., Mohammed S., Davis B. G., Science 2016, 354, aag1465. [DOI] [PubMed] [Google Scholar]
- 64. Petracca R., Bowen K. A., McSweeney L., O'Flaherty S., Genna V., Twamley B., Devocelle M., Scanlan E. M., Org. Lett. 2019, 21, 3281–3285. [DOI] [PubMed] [Google Scholar]
- 65. Flood D. T., Kingston C., Vantourout J. C., Dawson P. E., Baran P. S., Isr. J. Chem. 2020, 60, 268–280. [Google Scholar]
- 66.
- 66a. Beckwith A. L. J., Chai C. L. L., J. Chem. Soc. Chem. Commun. 1990, 1087–1088; [Google Scholar]
- 66b. Axon J. R., Beckwith A. L. J., J. Chem. Soc. Chem. Commun. 1995, 549–550. [Google Scholar]
- 67. Nawrat C. C., Jamison C. R., Slutskyy Y., MacMillan D. W. C., Overman L. E., J. Am. Chem. Soc. 2015, 137, 11270–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aycock R. A., Vogt D. B., Jui N. T., Chem. Sci. 2017, 8, 7998–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.
- 69a. Aycock R. A., Pratt C. J., Jui N. T., ACS Catal. 2018, 8, 9115–9119; [Google Scholar]
- 69b. Trowbridge A., Reich D., Gaunt M. J., Nature 2018, 561, 522–527; [DOI] [PubMed] [Google Scholar]
- 69c. Reich D., Trowbridge A., Gaunt M. J., Angew. Chem. Int. Ed. 2020, 59, 2256–2261; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 2276–2281. [Google Scholar]
- 70. Kanegusuku A. L. G., Castanheiro T., Ayer S. K., Roizen J. L., Org. Lett. 2019, 21, 6089–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.
- 71a. Ji P., Zhang Y., Dong Y., Huang H., Wei Y., Wang W., Org. Lett. 2020, 22, 1557–1562; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71b. Merkens K., Aguilar Troyano F. J., Djossou J., Gómez-Suárez A., Adv. Synth. Catal. 2020, 362, 2354–2359; [Google Scholar]
- 71c. Zhang O., Schubert J. W., J. Org. Chem. 2020, 85, 6225–6232. [DOI] [PubMed] [Google Scholar]
- 72. Li K., Wu Q., Lan J., You J., Nat. Commun. 2015, 6, 8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang C., Qi R., Xue H., Shen Y., Chang M., Chen Y., Wang R., Xu Z., Angew. Chem. Int. Ed. 2020, 59, 7461–7466; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7531–7536. [Google Scholar]
- 74.
- 74a. Gao X. W., Meng Q. Y., Li J. X., Zhong J. J., Lei T., Li X. B., Tung C. H., Wu L. Z., ACS Catal. 2015, 5, 2391–2396; [Google Scholar]
- 74b. Yang X. R., Xie Z. X., Li Y., Zhang Y., Chem. Sci. 2020, 11, 4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Halperin S. D., Kwon D., Holmes M., Regalado E. L., Campeau L. C., DiRocco D. A., Britton R., Org. Lett. 2015, 17, 5200–5203. [DOI] [PubMed] [Google Scholar]
- 76. Yuan Z., Nodwell M. B., Yang H., Malik N., Merkens H., Benard F., Martin R. E., Schaffer P., Britton R., Angew. Chem. Int. Ed. 2018, 57, 12733–12736; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12915–12918. [Google Scholar]
- 77. Wang Y., Li G. X., Yang G., He G., Chen G., Chem. Sci. 2016, 7, 2679–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li G. X., Morales-Rivera C. A., Gao F., Wang Y., He G., Liu P., Chen G., Chem. Sci. 2017, 8, 7180–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Amaoka Y., Nagatomo M., Inoue M., Org. Lett. 2013, 15, 2160–2163. [DOI] [PubMed] [Google Scholar]
- 80. Bume D. D., Pitts C. R., Jokhai R. T., Lectka T., Tetrahedron 2016, 72, 6031–6036. [Google Scholar]
- 81. Boursalian G. B., Ham W. S., Mazzotti A. R., Ritter T., Nat. Chem. 2016, 8, 810–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ruffoni A., Julia F., Svejstrup T. D., McMillan A. J., Douglas J. J., Leonori D., Nat. Chem. 2019, 11, 426–433. [DOI] [PubMed] [Google Scholar]
- 83.
- 83a. Wang Y., Wang J., Li G. X., He G., Chen G., Org. Lett. 2017, 19, 1442–1445; [DOI] [PubMed] [Google Scholar]
- 83b. Rahimidashaghoul K., Klimankova I., Hubalek M., Korecky M., Chvojka M., Pokorny D., Matousek V., Fojtik L., Kavan D., Kukacka Z., Novak P., Beier P., Chem. Eur. J. 2019, 25, 15779–15785; [DOI] [PubMed] [Google Scholar]
- 83c. Guerrero I., Correa A., Org. Lett. 2020, 22, 1754–1759. [DOI] [PubMed] [Google Scholar]
- 84. Yu Y., Zhang L. K., Buevich A. V., Li G., Tang H., Vachal P., Colletti S. L., Shi Z. C., J. Am. Chem. Soc. 2018, 140, 6797–6800. [DOI] [PubMed] [Google Scholar]
- 85. Li S. G., Portela-Cubillo F., Zard S. Z., Org. Lett. 2016, 18, 1888–1891. [DOI] [PubMed] [Google Scholar]
- 86. Reeve P. A. P., Grabowska U., Oden L. S., Wiktelius D., Wangsell F., Jackson R. F. W., ACS Omega 2019, 4, 10854–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Barker T. J., Boger D. L., J. Am. Chem. Soc. 2012, 134, 13588–13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.
- 88a. Green S. A., Vasquez-Cespedes S., Shenvi R. A., J. Am. Chem. Soc. 2018, 140, 11317–11324; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88b. Green S. A., Huffman T. R., McCourt R. O., van der Puyl V., Shenvi R. A., J. Am. Chem. Soc. 2019, 141, 7709–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Govaerts S., Angelini L., Hampton C., Malet-Sanz L., Ruffoni A., Leonori D., Angew. Chem. Int. Ed. 2020, 59, 15021–15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Perlin P., Gharakhanian E. G., Deming T. J., Chem. Commun. 2018, 54, 6196–6199. [DOI] [PubMed] [Google Scholar]