

Abstract
Pyridoxamine‐5′‐phosphate oxidase (PNPO) deficiency is an autosomal recessive pyridoxal 5′‐phosphate (PLP)‐vitamin‐responsive epileptic encephalopathy. The emerging feature of PNPO deficiency is the occurrence of refractory seizures in the first year of life. Pre‐maturity and fetal distress, combined with neonatal seizures, are other associated key characteristics. The phenotype results from a dependency of PLP which regulates several enzymes in the body. We present the phenotypic and genotypic spectrum of (PNPO) deficiency based on a literature review (2002‐2020) of reports (n = 33) of patients with confirmed PNPO deficiency (n = 87). All patients who received PLP (n = 36) showed a clinical response, with a complete dramatic PLP response with seizure cessation observed in 61% of patients. In spite of effective seizure control with PLP, approximately 56% of patients affected with PLP‐dependent epilepsy suffer developmental delay/intellectual disability. There is no diagnostic biomarker, and molecular testing required for diagnosis. However, we noted that cerebrospinal fluid (CSF) PLP was low in 81%, CSF glycine was high in 80% and urinary vanillactic acid was high in 91% of the cases. We observed only a weak correlation between the severity of PNPO protein disruption and disease outcomes, indicating the importance of other factors, including seizure onset and time of therapy initiation. We found that pre‐maturity, the delay in initiation of PLP therapy and early onset of seizures correlate with a poor neurocognitive outcome. Given the amenability of PNPO to PLP therapy for seizure control, early diagnosis is essential.
Keywords: neonatal epileptic encephalopathy, PLP, PNPO, seizures, vitamin‐response epilepsy

1. INTRODUCTION
Pyridoxamine‐5′‐phosphate oxidase (PNPO) deficiency is a recently recognized autosomal recessive neonatal epileptic encephalopathy (MIM#610090). This deficiency manifests within hours of birth as a severe seizure disorder that does not respond to anticonvulsant drugs and can be fatal, if untreated. Seizures can cease with the administration of the active form of B6 pyridoxal 5′‐phosphate (PLP) but are not always responsive to pyridoxine (PN). 1
The first case was identified based on a clinical and biochemical profile that was consistent with PLP‐responsive neonatal epileptic encephalopathy. 2 Later, Mills et al 1 pinpointed that this condition is due to PNPO deficiency and the PNPO gene is the underlying genetic defect. Since the initial report, more than 30 studies have reported 87 cases.
PNPO uses flavin mononucleotide (FMN) as a cofactor to catalyze the oxidation of pyridoxine 5′‐phosphate (PNP) to PLP. Dysfunctional variants of PNPO result in an inability to catalyze the production of PLP. As a result, the body suffers from a scarcity of the active form of vitamin B6. PLP works as a cofactor for over 140 enzymes, representing every major class of enzymes except ligases 3 (Supplementary Table S1). PLP‐dependent enzymes have essential roles in a variety of biochemical processes, including amino acid metabolism, glycolysis, gluconeogenesis, glycogenolysis, transsulfuration, polyamine biosynthesis, and synthesis of sphingoid bases, and the heme precursor δ‐aminolevulinic acid. 4 , 5 Hence, PLP is one of the most central molecules for the general cellular metabolism.
Its metabolism in the liver requires many enzymes, including (1) pyridoxal kinase, which is responsible for vitamin B6 phosphorylation, an important step for the transfer of vitamin B6 to pyridoxal‐5‐phosphate; (2) pyridoxal phosphate phosphatase, the enzyme that is involved in the preferred degradation route of PLP by aldehyde oxidase to 4‐pyridoxic acid through PLP dephosphorylation; and (3) PNPO then catalyzes the last step in PLP synthesis 6 (Figure 1).
FIGURE 1.
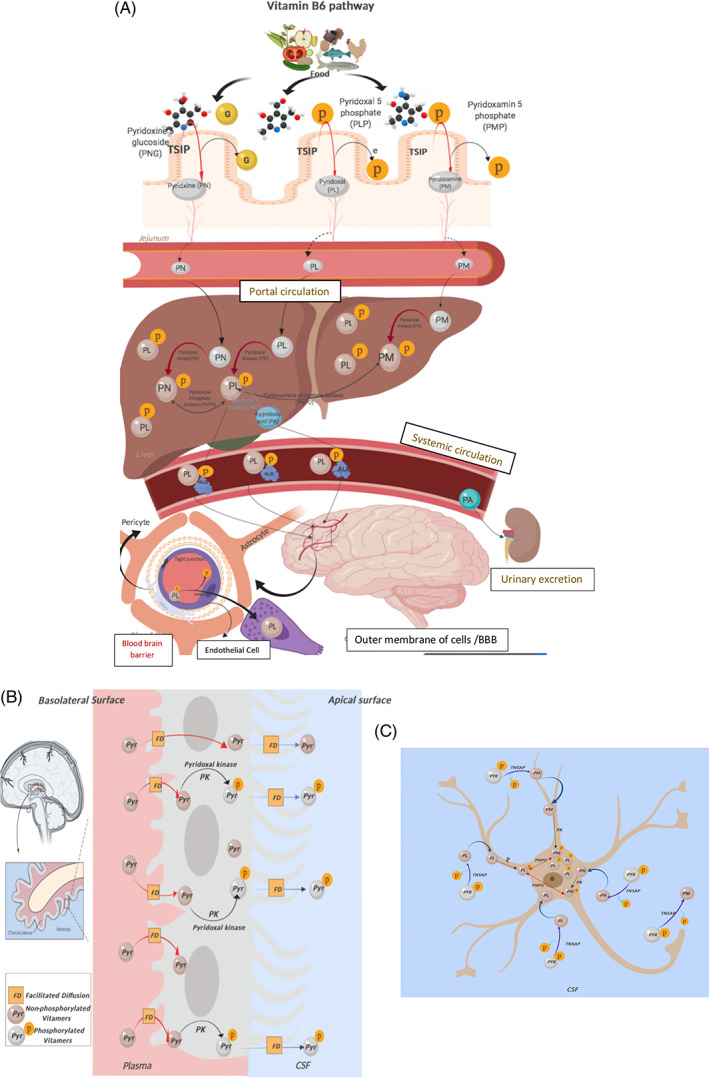
Vitamin B6 metabolism as it travels from the intestine to the portal circulation, crosses the blood‐brain barrier, and enters the brain cells. A, In the intestine, the dietary phosphorylated form is hydrolyzed to the free form by intestinal hydrolase (IH)/tissue‐specific intestinal phosphatase (TSIP) prior to absorption. This is followed by its uptake by intestinal cells, which is believed to occur through simple diffusion. Through portal circulation, the free B6 forms reach the liver, where metabolism in the liver is catalyzed by many enzymes. (1) Pyridoxal kinase (PK), (2) pyridoxal phosphate phosphatase, (3) pyridox(am)ine‐5′‐phosphate oxidase. B, The unphosphorylated forms of vitamin B6 are able to cross the blood‐brain barrier, probably by facilitated diffusion, mostly at the choroid plexus (CP). The CP traps PLP via pyridoxal kinase and can release PLP to a remarkable extent (and pyridoxal to a lesser extent). C, Excessive PLP in the CSF and extracellular space enters brain cells, and the B6 vitamers must be dephosphorylated so that they can enter brain cells and then metabolically trapped by being rephosphorylated by pyridoxal kinase. Pyridoxine phosphate and pyridoxamine phosphate are then oxidized by PNPO to form the active cofactor, PLP. PLP, pyridoxal 5′‐phosphate
Given the variety of PLP‐dependent enzymes, PLP deficiency might be expected to have diverse clinical presentations. However, the neurological phenotype is the predominant phenotype of PNPO deficiency, sometimes co‐occurring with non‐neurological manifestations such as impaired growth and hypochromic microcytic anemia that responds dramatically to treatment with PLP. Epileptiform convulsions in infants are a common presentation due to defective conversion of glutamic acid into γ‐aminobutyric acid (GABA). Other neurological manifestations including irritability and peripheral neuritis arise due to improper production of serotonin, epinephrine, norepinephrine, and GABA. Defects in the synthesis of sphingolipids lead to nerve demyelination, which is manifested as neuropathy. 4 , 5
This article reviews 87 cases of PNPO deficiency describing the spectrum of the neurological and non‐neurological phenotypes of PNPO deficiency as well as its diagnostic biochemical profile, genotypic basis, and therapeutic response to PLP and PN.
2. METHODS
For the literature review, we searched PubMed and Google scholar (http://www.ncbi.nlm.nih.gov/pubmed and https://scholar.google.com; 2002‐2020) using a combination of the following terms (restricted to humans): PNPO, pyridox(am)ine 5′ phosphate oxidase, PNPO, PLP‐dependent seizures, PLP‐responsive seizures. We selected all articles that met the following criteria: (1) published after the first case of PLP‐dependent seizure was reported in 2002; (2) published in English; and (3) reporting one or more PLP‐dependent epilepsy patients with confirmed PNPO deficiency, including a description of the clinical symptoms.
We extracted the clinical phenotypes, biochemical profiles, electroencephalography (EEG) results, and neuroimaging features from all the reviewed articles and all published data compiled them in one table (Suppl. Table S2). We studied the molecular effect of these variants on the PNPO structure and function, and correlated this information with the disease outcome. Other factors were included to predict outcomes, including pre‐maturity, fetal distress, seizure onset and time of initiation of PLP therapy.
3. RESULTS
3.1. Clinical aspect of PNPO Deficiency
The clinical data of 33 papers presented in the literature were collected and studied, for a total of 87 patients. Among the 87 cases, gender was identified in only 46 patients: 25 males and 21 females. Consanguinity was found in 25 patients and 24 cases had a family history of previously affected siblings or an early death with an undiagnosed condition manifested with refractory seizures (Supplementary Table S2). 22% of the patients (n = 19) died in the first year of life, one of them only after introduction of PLP. We have classified the clinical manifestations as neurological or extraneurological. The classical presentation is a neonatal epileptic encephalopathy, which is unresponsive to conventional anticonvulsant drugs and also to PN sometimes.
3.1.1. Neurological manifestations of PNPO deficiency
The described neurological changes varied widely among the literature. The most prominent neurological manifestation is a seizure within the first day of life, occurring in 59% of the reported cases. Abnormal fetal movement was noticed by the mother in the third trimester in 11% of the patients who have shown seizure within the first day of life (Figure 2).
FIGURE 2.
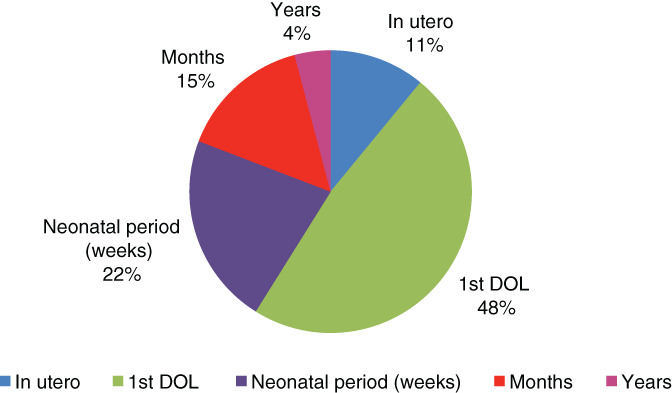
Seizure onset data for 87 reported cases with PNPO deficiency. PNPO, pyridoxamine‐5′‐phosphate oxidase
Different types of clinical seizures have been observed in patients with PLP‐dependent epilepsy. The classical seizure pattern consists of prolonged seizures and recurrent episodes of status epilepticus or breakthrough seizures controlled with PN and/or PLP. 80% of the cases showed clonic seizures (focal and multifocal), tonic‐clonic seizures, generalized seizures, abnormal movement, or a combination of two or more of those types in the same episode or in alternation between episodes; absence/staring episodes occurred in 7% of cases. 7 , 8 Furthermore, paroxysmal movement disorder was observed in 10% of cases. 9 Abnormal eye movement such as eye deviation was reported in 17% of cases. 1 , 2 , 10 , 11 , 12 , 13 Movement disorder including hyperkinetic movements was identified in one patient. 14 Approximately 56% of patients affected with PLP‐dependent epilepsy suffer developmental delay/intellectual disability.
Moreover, microcephaly was described in five cases. 15 , 16 , 17 , 18 Autistic spectrum disorders and attention‐deficit/hyperactivity disorder (ADHD) were observed in three patients. 7 , 13 , 19
3.1.2. PLP/PN clinical seizure responsiveness
PN was tried in most of the patients earlier considering pyridoxine‐dependent epilepsy (PDE) as one of the causes of refractory neonatal seizures. PN has tried 67 patients with variable clinical response; 44% (n = 29) showed remarkable clinical response and continued on PN alone, and 36% (n = 24) there was no response. 20% (n = 14) in whom PN was tried initially showed some sort of initial response then relapse of the seizure later and/or partial response with breakthrough seizures while on PN, which prompted a trial of PLP.
PLP trial was the second therapeutic option after PN in neonatal refectory seizure, used in 36 patients. Responses to PLP varied; 61% (n = 22) showed a marked immediate clinical response with seizures cessation including one patient required combined PN/PLP and riboflavin therapy to achieve seizure cessation 20 and another patient never experienced seizure as PLP was initiated prophylactically, 21 25% (n = 9) of the responsive patients developed an immediate adverse event either like apneia‐hypotonia prior to seizure cessation (n = 4) or developed status epileptics upon switching to PLP (n = 5) and 14% (n = 5) responded only after several days with gradual seizure control. We noted that all of the responsive patients became PLP‐dependent and dose adjustment was needed to manage sick days. Abnormal liver function was observed in four patients after long time of administration of PLP (two were on 50 mg/kg/day and two other patients were on100 mg/kg/day). 22 , 23 , 24 , 25 In spite of effective seizure control with PLP, approximately 56% of patients affected with PLP‐dependent epilepsy suffer developmental delay/intellectual disability.
3.1.3. Brain imaging
In the surveyed literature, neuroimaging findings were described in 55 patients (63%) with PNPO deficiency. There were no specific imaging features identified in the brains of the reviewed patients. Different radiological findings were described; however, 34 patients (61.8%) had normal brain imaging. 7 , 8 , 9 , 11 , 12 , 13 , 16 , 21 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 Di Salvo et al reported three patients with a c.347G > A (p. Arg116Gln) mutation in the PNPO gene who had normal brain imaging. These patients had residual enzyme activity accompanied by later onset and a milder seizure phenotype than patients with total loss of PNPO function. 7 One patient reported with homozygous for a c.283C > T; (p. R95C) mutation at PNPO gene who had a normal brain MRI initially at 1 month of age but his follow‐up imaging at 3 years showed severe diffuse atrophy. 11 The most common abnormality revealed on brain imaging was diffuse atrophy, which was observed in eight patients (14.5%), 8 , 9 , 16 , 20 followed by ischemic changes and encephalomalacia in five patients (5.5%), 1 , 13 , 16 , 17 and delayed myelination and atrophy in another three patients. 18 , 34 , 35 An additional two patients (2.5%) had diffuse or focal edema. 1 , 10 The remaining four patients were found to have venous sinus thrombosis, a small subdural hemorrhage, simplified pattern of gyrus and sulcus anatomy and nonspecific T2 signal in the periventricular white matter, which were probably not related to PNPO deficiency. 9 , 33 , 36
3.1.4. EEG features
The EEG features of patients with PNPO deficiency are variable. Burst‐suppression patterns are the most common feature, reported in approximately half of the cases, followed by multifocal spikes and sharp waves, and generalized spikes and waves discharge. Hypsarrhythmia has been reported in some cases. A minority have normal EEG. Table S3 shows details regarding the patients' EEG features.
3.2. Non‐neurological manifestations of PNPO deficiency
Most of the reported cases showed pre‐ and perinatal complications including pre‐maturity, fetal distress and intrauterine growth restriction (IUGR) with oligohydramnios.
Pre‐maturity was commonly observed in 50% of PNPO‐deficient patients and 58% of the pre‐mature cases suffered fetal distress. There were two detected cases of oligohydramnios 11 , 35 and two cases of IUGR. 11 , 17 Metabolic acidosis and/or lactic acidosis were reported in eight cases (Supplementary Table S2).
Hematological manifestations in the form of normocytic anemia, normochromic anemia or pancytopenia, ophthalmological changes if form of pigmentary retinopathy and gastrointestinal tract (GIT) manifestations including abdominal distension, constipation and feeding intolerance often occur in PNPO deficiency (Table 1).
TABLE 1.
Manifestations concomitant with PNPO deficiency seizures
| Concomitant non‐neurological disorders | No. of cases | References |
|---|---|---|
| Pre‐maturity | 44 | 1 , 2 , 8 , 9 , 10 , 11 , 13 , 14 , 15 , 16 , 17 , 18 , 20 , 21 , 25 , 26 , 27 , 30 , 34 , 35 , 36 , 43 |
| Oligohydraminous | 3 | 10 , 20 , 35 |
| IUGR | 2 | 10 , 17 |
| Lactic acidosis/metabolic acidosis | 8 | 1 , 2 , 15 , 17 , 20 , 26 |
| Ophthalmological. Pigmentary retinopathy | 2 | 9 , 14 |
| Anemia | 9 | 1 , 9 , 30 , 36 |
| Poor weight gain | 6 | 1 , 9 , 17 |
| GIT features | – | – |
| Abdominal distention | 2 | 1 |
| Dilated bowel loops | 1 | 25 |
| Constipation | 5 | 9 |
| Feeding intolerance | 1 | 8 |
| Hypoglycemia | 3 | 17 , 34 |
| Hypothyroidism | 1 | 9 |
| Cardiac disorders | 2 | 28 , 36 |
Abbreviations: IUGR, intrauterine growth restriction; PNPO, pyridoxamine‐5′‐phosphate oxidase.
3.3. Biochemical profiles in PNPO deficiency
The biochemical analysis protocols varied among the included studies. Notably, there was no available panel with specific biomarkers in the reviewed literature. The analytical testing in the majority of reports covered urine organic acids, with the main focus being on vanillactic acid (VLA), cerebrospinal fluid (CSF) neurotransmitters, and CSF and plasma amino acids.
Herein, we present the biochemical results for 59 cases provided by 22 reports (Supplementary Table S4). The biochemical tests used for diagnosis and their reference ranges vary among these studies. To allow a significant comparison, we only considered tests that were available for at least 10 patients. CSF 5‐HIAA and PLP were most frequently performed (>20 patients; Supplementary Figure S1). Elevation of urine VLA and decrease of CSF PLP were significant in the majority of patients, which make these measurements potential suggestive biomarkers for PNPO deficiency. Other tests are summarized in Table 2.
TABLE 2.
Summary of the most significant biochemical profiles in PNPO deficiency biofluids. Results were acquired based on the reference range provided by each study and patients were not on PLP or PN treatments
| Biofluid | Biomarker | Variables | Total number of patients | Rank | ||
|---|---|---|---|---|---|---|
| Low (%) | Normal % | High % | ||||
| Urine | VLA | 0 | 9 | 91 | 11 | 1 |
| CSF | PLP | 81 | 19 | 0 | 21 | 2 |
| CSF | Glycine | 6 | 12 | 80 | 16 | 3 |
| CSF | Threonine | 0 | 25 | 75 | 20 | 4 |
| CSF | HVA | 52 | 24 | 18 | 17 | 5 |
| CSF | 5‐HIAA | 40 | 36 | 24 | 25 | 6 |
| Plasma | Arginine | 39 | 56 | 6 | 18 | 7 |
| CSF | 3‐OMD | 0 | 63 | 37 | 19 | 8 |
Abbreviations: CSF, cerebrospinal fluid; HVA, homovanillic acid; 5HIAA, 5‐hydroxyindoleacetic acid; 3 OMD, 3‐ortho‐methyldopa; PLP, pyridoxal 5‐phosphate; PN, pyridoxine; PNPO, pyridoxamine‐5′‐phosphate oxidase; VLA, vanillactic acid.
3.4. Molecular aspect of PNPO deficiency
3.4.1. Structural analysis and predicted effect of variants
Based on their position in the protein structure (Protein Data Bank (PDB) accession number 1nrg 37 and type of mutation, PNPO variants can be separated into four categories. The largest category (here termed category I) contains mutations that directly affect the catalytic site and its capacity for ligand and cofactor binding (colored magenta in Figure 3 and Table 3). These mutations include R95C, R116Q, R141C, R161C, R225C/L/H, and R229W/Q. Strikingly, all these substitutions change a long and positively charged arginine into a polar or hydrophobic residue with different side chain dimensions. Accordingly, all of them impair the coordination of the negatively charged phosphate moieties of PLP or FMN, and sometimes have additional effects (eg, loss of π‐stacking interactions with PLP in the case of R225 mutations, or loss of inter‐chain ionic bonds for R116Q).
FIGURE 3.
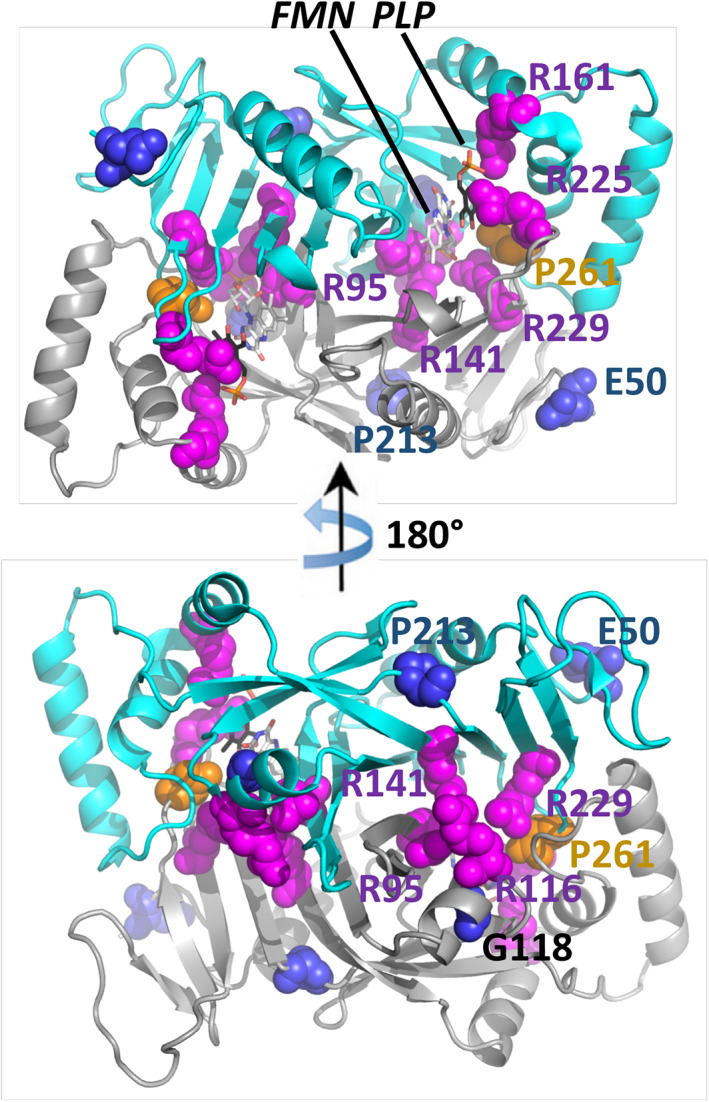
Overview of the reported point mutations. The crystal structure (PDB ID 1nrg) shows the post‐catalytic PLP (magenta) and FMN (dark lilac) as stick models. Point mutations are shown in sphere representation. Green coloration indicates that the mutation directly affects the binding and processing of the substrate and cofactor. Orange coloration indicates that the mutation decreases the protein's overall stability. FMN. flavin mononucleotide; PLP, pyridoxal 5′‐phosphate
TABLE 3.
Molecular analysis results shows the PLP and PN responsiveness as well as the clinical outcome of each variant
| Effect | Mutation | Clinical observations | Molecular basis | Reference |
|---|---|---|---|---|
| Normal | D33V | Normal neurodevelopmental outcome at 11 y | Non‐homologous substitution in disordered N‐terminal extension | 11 , 21 , 22 |
| D33V | Normal neurodevelopmental outcome at 11 mo | Non‐homologous substitution in disordered N‐terminal extension | 28 | |
| D33V | Normal neurodevelopmental outcome at 7 y | Non‐homologous substitution in disordered N‐terminal extension | 21 | |
| R116Q | Normal neurodevelopmental outcome at 3 y and 7 mo | Destabilizing FMN, loss of inter‐molecular ionic bond with E143 | 7 | |
| G118R | Normal neurodevelopmental outcome at 6 y | Severe destabilizing clashes with surrounding | 20 | |
| R141C | Normal IQ, recurrent breakthrough seizure and SE, seizure free at the age of 26 y for the last 4 y (cp hetero) | Loss of FMN phosphate coordination | 16 | |
| P213S | Normal neurodevelopmental outcome at 5 y | Loss of minor hydrophobic interactions | 21 | |
| R225H | Normal neurocognitive development at 3 y and 10 mo | Loss of PLP coordination | 16 | |
| R225H | Normal neuro‐cognitive outcome at 6 years and 10 months | Loss of PLP coordination | 16 | |
| R225C | Normal neurodevelopmental status, and improved axial tone | Loss of PLP coordination | 16 | |
| Mild to Moderate | R116Q | Moderate GDD at 12 y | Destabilizing FMN, loss of inter‐molecular ionic bond with E143 | 7 |
| R116Q | Mild GDD at 6 y | Destabilizing FMN, loss of inter‐molecular ionic bond with E143 | 7 | |
| G118R | GDD | Severe destabilizing clashes with surrounding | 9 | |
| G118R | GDD | Severe destabilizing clashes with surrounding | 9 | |
| G118R | Mild hypotonia and GDD at 28 mo | Severe destabilizing clashes with surrounding | 12 | |
| R141H | Normal growth and development patterns neurological at 4 mo | Loss of FMN phosphate coordination | 30 | |
| P150Rfs*27 | Normal neurodevelopmental outcome at the age of 12 mo | Non‐functional fragment | 27 | |
| R161C | Normal neurodevelopmental outcome at the age of 14 mo | Loss of PLP phosphate coordination | 29 | |
| R225H | ASD | Loss of PLP coordination | 13 | |
| R225H | Normal neurocognitive development and seizure free at 1 y and 8 mo. Unsteady gait | Loss of PLP coordination | 16 | |
| R225H | Normal neurocognitive development and seizure free at 1 y and 5 mo. Unsteady gait | Loss of PLP coordination | 16 | |
| R225H | GDD, seizure free at 9 y and 9 mo | Loss of PLP coordination | 16 | |
| R225H | GDD, recurrent breakthrough seizure | Loss of PLP coordination | 16 | |
| R225L | GDD | Loss of PLP coordination | 9 | |
| R225L | GDD | Loss of PLP coordination | 9 | |
| R225L | GDD | Loss of PLP coordination | 9 | |
| R225L | GDD | Loss of PLP coordination | 9 | |
| Severe | E50K | Severe DD, microcephaly, spasticity at 2 y | Loss of minor stabilizing interactions | 1 , 15 |
| E50K | Died at 6 wk | Loss of minor stabilizing interactions | 1 , 15 | |
| R95C | Died at 7 mo | Loss of FMN phosphate coordination | 11 | |
| R95C | Severe psychomotor delay at 2 y and 5 mo | Loss of FMN phosphate coordination | 11 | |
| R95C | Died | Loss of FMN phosphate coordination | 11 | |
| R95C | Died | Loss of FMN phosphate coordination | 11 | |
| R95C | Severe psychomotor delay | Loss of FMN phosphate coordination | 11 | |
| R95C | Death at 47 DOL | Loss of FMN phosphate coordination | 36 | |
| R95C | Died at 31 d | Loss of FMN phosphate coordination | 26 | |
| R95C | Psychomotor development was severely delayed and epileptic phenomena were almost constantly present at 3 y old | Loss of FMN phosphate coordination | 26 | |
| P150Rfs*27/R161G (cp heteroz) | Neurodevelopmental outcome was poor with severe developmental delay, cortical visual impairment and autistic features | Non‐functional fragment/loss of PLP phosphate coordination | 19 | |
| A174X | Died at 48 d of life (DOL) | Non‐functional fragment | 35 | |
| R225H | Died at 14 mo | Loss of PLP coordination | 18 | |
| R225H | Psychomotor retardation with autonomous gait at 2.5 y and a slight intellectual disability | Loss of PLP coordination | 30 | |
| R225H | Severe GDD, spastic tetraplegia, progressive microcephaly, recurrent breakthrough seizure at 8 y | Loss of PLP coordination | 16 | |
| R225H | Severe GDD, spastic tetraplegia, progressive microcephaly, recurrent breakthrough seizure while on PN | Loss of PLP coordination | 16 | |
| R225C | Global psychomotor delay at the age of 3 y | Loss of PLP coordination | 17 | |
| R229Q | Normal head growth, pigmentary retinopathy, diffuse hypotonia, and hyperkinetic movements | Loss of FMN phosphate coordination | 14 | |
| R229Q | Hemiparesis at 21 mo after middle cerebral artery ischemic stroke | Loss of FMN phosphate coordination | 13 | |
| R229W | Died at 21 DOL | Loss of FMN phosphate coordination | 1 , 2 | |
| R229W | Died on first DOL | Loss of FMN phosphate coordination | 1 , 2 | |
| R229W | Died at 6 mo | Loss of FMN phosphate coordination | 1 , 2 | |
| R229W | Died at 17 DOL | Loss of FMN phosphate coordination | 1 , 2 | |
| R229W | Died at 19 DOL | Loss of FMN phosphate coordination | 1 , 2 | |
| X262Q | Died at 23 DOL | Additional residues disrupt ligand binding and 3D‐fold | 1 | |
| X262Q | Died at 15 DOL | Additional residues disrupt ligand binding and 3D‐fold | 1 |
Abbreviations: DD, developmental delay; FMN. flavin mononucleotide; PLP, pyridoxal 5′‐phosphate; PN, pyridoxine.
The second category, category II, contains mutations that affect the fold and stability of the protein because of a non‐conservative side chain substitution (blue in Figure 3 and Table 3). Their effects range from a mild loss of stabilizing surface interactions (E50K) and loss of hydrophobic interactions (P213S) to severely destabilizing clashes (G118Q/R).
Category III is constituted by variants causing residue deletion (S93S_A94Ldel), pre‐mature stop codons (A174X), loss of stop codons (X262Q) and frame shifts combined with loss of subsequent protein sequences (L83W_fs*17, P150R_fs*27). In the compact structure of the PNPO dimer, a significant loss of residues would severely compromise the protein's structure and function. Hence, the truncating mutations are expected to lead to non‐functional proteins. PNPO has the particularity that its C‐terminus folds back inside the protein and terminates just before the substrate‐binding pocket. Accordingly, the loss of stop codon would disrupt both, ligand binding and protein fold (orange in Figure 3).
D33V is in a human‐specific N‐terminal extension and does not affect the 3D structure or catalytic function of the protein, placing it in category IV. 38 D33V might mildly affect the (unknown) function of the N‐terminal extension.
3.4.2. Correlation between molecular effects and clinical outcomes
Our overview reveals a striking lack of correlation between the reported clinical effects of PNPO variants and the molecular effects they have on the protein structure and function (Table 3). Some individuals showed apparently normal phenotypes, despite encoding for a clearly deleterious variant (eg, R116Q, G118R or P150R_fs*27), whereas others experienced most severe effects for a substitution that is expected to only mildly affect the protein's function (E50K). Moreover, the same variant may cause no or only mild effects in some individuals, but severely affect others (eg, R225H). However, the most severe complications appeared to be caused by category I and III variants. In particular, mutations of R95 and R229 were only found in the severe cases, and four out of five category III variants were ranked as “severe.” The unexpected severity of E50K (category II) appears not justified by its mildly destabilizing function and might indicate supplementary, unidentified human‐specific roles of this residue.
3.5. PNPO deficiency outcome
Finally, we assessed factors that impact the outcomes of PNPO deficiency based on cognitive function and/or developmental assessment. Pre‐maturity, fetal distress, seizure onset and initiation of PLP therapy have been documented and correlated with outcome variables including death, developmental delay and normal development (Supplementary Table S2).
To evaluate the impact of pre‐maturity, we studied all 87 cases based on gestational age at birth. Pre‐maturity of birth was observed in the majority of patients (n = 44); 24 of them had shown fetal distress during the neonatal period. Only seven patients developed normally given their pre‐maturity, while 37 patients either died or showed variable degrees of developmental delay. We have categorized patients based on the maturity and outcome (Figure 4A). Categories A, B and C represent the percentage of normal development within full term patients, pre‐mature patients and rest of patients, respectively. The apparent high correlation between pre‐maturity and unfavorable outcome could be caused by complications due to pre‐maturity itself and/or by the earlier onset of the disease.
FIGURE 4.
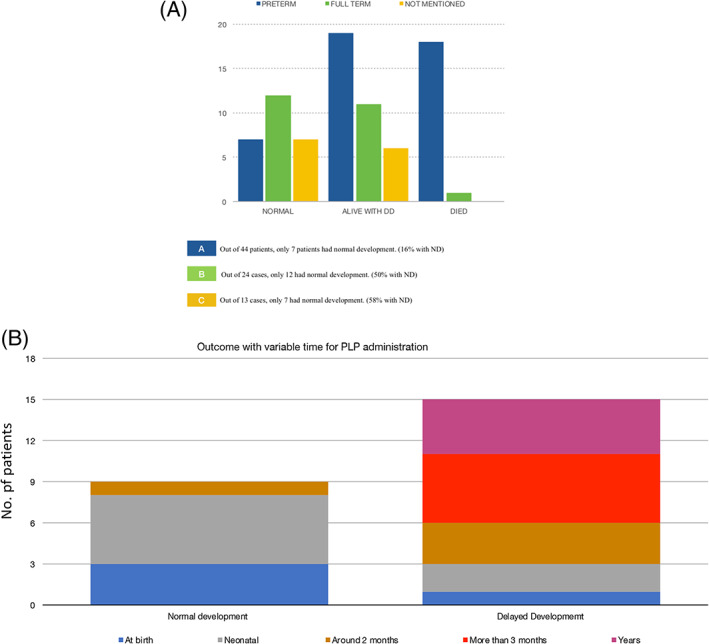
A, The impact of pre‐maturity on clinical outcomes, including death and developmental delay (DD). B, Variable outcomes with variable age of PLP administration. The earlier PLP was introduced as a therapy, the better the prognosis was reported. PLP, pyridoxal 5′‐phosphate
We explored seizure onset and time for diagnosis and treatment as factors for the outcome. Seizures generally manifest early after birth, commonly during the first days of life. Almost more than half of patients (59%) developed seizures within their first day of life; 50% of them showed variable developmental delay, 25% died and the others developed normally.
Various clinical courses and outcomes were reported for patient treatment with PN and/or PLP. Fourteen cases lacked information about the treatment strategy. Among the 87 patients who received PN treatment (n = 67) in this review, 36% of the patients showed a negative response to PN, and 64% of the patients showed variable degrees of response; all of them survived, but most of them had various degrees of developmental delay. Only 16 patients who responded to PN as a main treatment developed normally; six of them were full term. Of the 19 patients who died, all of them are pre‐mature newborns and they did not respond to PN, one only showed an initial response to PN, and neither of them received PLP treatment.
PLP trial was introduced for 36 patients where it showed a dramatic response in 61% (n = 22) of the patients including 36% (n = 8) of them had not responded to PN as their initial therapy and 4% (n = 1) of them started PLP since birth prophylactically. We have documented developmental outcome and the time of PLP initiation in the surveyed literatures for 24 patients. We noticed that the earlier PLP was introduced as a therapy, the better was the prognosis (Figure 4B). Normal development was found in nine cases where PLP therapy was started as early as possible. Four of them were full term. The remaining 15 cases survived but had variable degrees of developmental delay, and some patients suffered hypotonia, microcephaly or increased tone in all four limbs. In majority of those cases, PLP was introduced late (Figure 4B).
4. CONCLUSION
Herein we provided a comprehensive review of the clinical, biochemical and molecular observations reported for PNPO deficiency. Our survey shows that the phenotypic spectrum of PNPO deficiency is wide, including a multitude of neurological and systemic manifestations. Its characteristic clinical feature is refractory seizures during the first year of life. On a molecular level, most of the PNPO variants either affect substrate and cofactor binding and processing, or weaken/disrupt the 3‐dimensional structure of PNPO. The molecular effect generally correlates only in some cases with the severity of the symptoms. For example, mutations that disrupt the protein's catalytic function produce a more pronounced clinical outcome than mutations that are only mildly destabilizing. However, in many cases the clinical outcomes for the same PNPO variant can vary significantly between patients, highlighting the importance of the timing and nature of treatment administered.
Indeed, we noted that pre‐maturity, delayed therapeutic intervention and an earlier onset of clinical seizures correlated with a poorer neurocognitive outcome. Given the amenability of PNPO‐dependent epilepsy to treatment with PLP and PN therapy for optimal seizure control and favorable developmental outcomes, early diagnosis is essential. Therefore, emphasis should be placed on early testing and disease detection, especially in cases of a suspected family history indicating PNPO deficiency. Furthermore, prenatal supplementation of suspected cases should be considered in the presence of certain conditions, such as prenatal seizures or an index case in the family. Prenatal supplementation in PNPO deficiency has not been indicated yet, particularly as a therapy for cessation or prevention of prenatal seizures and/or fetal distress.
Although several biochemical changes indicate multiple enzyme defects (Table 2), there is currently no specific biomarker for PNPO deficiency. However, we noted that the combination of elevated urinary VLA, CSF glycine and decreasing CSF PLP might be a suggestive profile that could be used for infants presenting with unexplained refractory seizures and motivate a PNPO molecular analysis. Measuring PNPO enzyme activity through a rapid LC‐MS/MS‐based dried blood spot assay has also been proposed for rapid diagnosis of PNPO deficiency. 39
It is important to differentiate between PNPO deficiency and other B6‐dependent epilepsies, including alpha‐aminoadipic semialdehyde dehydrogenase (ALDH7A1) deficiency and pyridoxal phosphate‐binding protein (PLPBP) deficiency. Reliable biomarkers for ALDH7A1 disorder exist, and include an elevated urinary AASA/creatinine ratio, elevated plasma pipecolic acid, 6‐oxo‐pipecolate, 40 and a characteristic peak in the analysis of CSF monoamine metabolites by HPLC. 41 PLPBP deficiency, however, is another B6‐dependent epilepsy associated with low‐CSF‐PLP akin to PNPO deficiency. Hence, diagnosis of different B6‐dependent epilepsies is best established by the identification of biallelic pathogenic variants in the ALDH7A1, PNPO and PLPBP genes.
Four cases of PNPO deficiency were presented with liver cirrhosis or abnormal liver function test after receiving PLP treatment. Although abnormal liver function might expand the phenotypic spectrum of PNPO deficiency, the PLP administration is probably the cause of liver impairment. Schmitt et al. reported a 2 years and 6 months old boy with PNPO deficiency who developed an abnormal liver function test after escalating a PLP dose to 100 mg/kg/day and it had to be reduced to 53 mg/kg/day. 22 Sudarsanam et al have postulated that liver dysfunction in their patient was due to a high dose of administered PLP (100 mg/kg/day). 23 Although reduction of the PLP dose and frequency resulted in substantial reduction in the liver transaminases, episodes of uncontrolled seizures and encephalopathy required high doses of PLP, which negatively affected the liver function. 23 Porri et al has also reported a mild elevation in alanine aminotransferase and aspartate aminotransferase levels on a PNPO‐deficient patient treated with 50 mg/kg/day of PLP. 25 Later, Coman et al described a 4 years old boy with PNPO deficiency in whom liver cirrhosis has been showed while receiving a 50 mg/kg/day of PLP. 24 Last two cases have never received “high dose” of PLP, rather a dosage range of 30 to 50 mg/kg/day. 24 , 25 In all PNPO‐deficient patients, liver derangement occurred after long‐term administration of PLP with substantial improvement after dose adjustment which indicates that liver toxicity is probably related to PLP administration and should be carefully monitored. 22 , 23 , 24 , 25 In addition, a previously reported liver toxicity case secondary to high‐dose PLP for treating homocystinuria was documented by Yoshida et al supported further this hypothesis. 42 Collectively, these reports highlight the possibility of PLP‐related liver dysfunction in PNPO‐deficient patient, and hence surveillance for evidence of liver cirrhosis should be part of management of PNPO‐deficient patients receiving PLP.
We conclude that early detection of PNPO deficiency combined with early PLP treatment is key to optimizing the clinical outcome. While newborn screening is useful for the early detection of some diseases, it might not be feasible in PNPO deficiency due to the absence of sensitive biomarkers. However, we identified suggestive biochemical profiles in the literature that should motivate a definitive molecular diagnosis of PNPO gene variants, especially in cases of a suspected family history indicating PNPO deficiency.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.13843.
Supporting information
Figure S1 The most common biochemical tests requested for PNPO diagnosis (2002‐2020)
Table S1 The list of genes coding for PLP‐dependent enzymes.
Table S2 Patient information included patient age, gender, seizure onset, family history, parents' consanguinity, pregnancy pre‐maturity (gestational age), Apgar score, condition at birth, status of failure to thrive, neurological changes, electroencephalogram (EEG) changes, MRI tests, the results for molecular and biochemical analysis and responsiveness to antiepileptic therapy, pyridoxine therapy, and PLP trial.
Table S3 EEG changes in PNPO deficiency.
Table S4 comprehensive biochemical profile for each reported case.
ACKNOWLEDGEMENTS
This research was supported by a Dallah Health Grant. The research by Stefan T. Arold reported in this publication was supported by funding from King Abdullah University of Science and Technology (KAUST) Award No. FCC/1/1976‐25 from the Office of Sponsored Research (OSR).
Alghamdi M, Bashiri FA, Abdelhakim M, et al. Phenotypic and molecular spectrum of pyridoxamine‐5′‐phosphate oxidase deficiency: A scoping review of 87 cases of pyridoxamine‐5′‐phosphate oxidase deficiency. Clinical Genetics. 2021;99:99–110. 10.1111/cge.13843
Funding information College of Medicine Research Center, Deanship of Scientific Research, King Saud University, Riyadh, Saudi Arabia; Dallah healthcare grant, Grant/Award Number: CMRC‐DHG‐2/003; The research by STA reported in this publication was supported by funding from King Abdullah University of Science and Technology (KAUST), Office of Sponsored Research (OSR), Grant/Award Number: FCC/1/1976‐25
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article
REFERENCES
- 1. Mills PB, Surtees RA, Champion MP, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 50‐phosphate oxidase. Hum Mol Genet. 2005;14(8):1077‐1086. [DOI] [PubMed] [Google Scholar]
- 2. Bräutigam C, Hyland K, Wevers R, et al. Clinical and laboratory findings in twins with neonatal epileptic encephalopathy mimicking aromatic L‐amino acid decarboxylase deficiency. Neuropediatrics. 2002;33(3):113‐117. [DOI] [PubMed] [Google Scholar]
- 3. Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000;28(1):304‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stover PJ, Field MS. Vitamin B‐6. Adv Nutr. 2015;6(1):132‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson MP, Plecko B, Mills PB, Clayton PT. Disorders affecting vitamin B6 metabolism. J Inherit Metab Dis. 2019;42(4):629‐646. 10.1002/jimd.12060 Epub 2019 Mar 20. PMID: 30671974. [DOI] [PubMed] [Google Scholar]
- 6. Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez gene: gene‐centered information at NCBI. Nucleic Acids Res. 2005;33(Database issue:D54‐D58. 10.1093/nar/gki031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. di Salvo ML, Mastrangelo M, Nogués I, et al. Pyridoxine‐5′‐phosphate oxidase (Pnpo) deficiency: clinical and biochemical alterations associated with the C.347g > a (P.·Arg116gln) mutation. Mol Genet Metab. 2017;122(1–2):135‐142. [DOI] [PubMed] [Google Scholar]
- 8. Xue J, Chang X, Zhang Y, Yang Z. Novel phenotypes of pyridox(am)ine‐5′‐phosphate oxidase deficiency and high prevalence of c.445_448del mutation in Chinese patients. Metab Brain Dis. 2017;32(4):1081‐1087. [DOI] [PubMed] [Google Scholar]
- 9. Guerriero RM, Patel AA, Walsh B, et al. Systemic manifestations in pyridox(am)ine phosphate oxidase (PNPO) deficiency. Pediatr Neurol. 2017;76:47‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veeravigrom M, Damrongphol P, Ittiwut R, Ittiwut C, Suphapeetiporn K, Shotelersuk V. Pyridoxal 5′‐phosphate‐responsive epilepsy with novel mutations in the PNPO gene: a case report. Genet Mol Res. 2015;14(4):14130‐14135. [DOI] [PubMed] [Google Scholar]
- 11. Hoffmann GF, Schmitt B, Windfuhr M, et al. Pyridoxal 5′‐phosphate may be curative in early‐onset epileptic encephalopathy. J Inherit Metab Dis. 2007;30:96‐99. [DOI] [PubMed] [Google Scholar]
- 12. Pearl PL, Hyland K, Chiles J, McGavin CL, Yu Y, Taylor D. Partial pyridoxine responsiveness in PNPO deficiency. JIMD Rep. 2013;9:139‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ware TL, Earl J, Salomons GS, et al. Typical and atypical phenotypes of PNPO deficiency with elevated CSF and plasma pyridoxamine on treatment. Dev Med Child Neurol. 2013;56(5):498‐502. [DOI] [PubMed] [Google Scholar]
- 14. Olson HE, Kelly M, LaCoursiere CM, et al. Genetics and genotype‐phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann Neurol. 2017;81(3):419‐429. 10.1002/ana.24883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clayton P, Surtees R, DeVile C, Hyland K, Heales S. Neonatal epileptic encephalopathy. Lancet. 2003;361:1614. [DOI] [PubMed] [Google Scholar]
- 16. Plecko B1, Paul K, Mills P, et al. Pyridoxine responsiveness in novel mutations of the PNPO gene. Neurology. 2014;82(16):1425‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alghamdi M, Arold S, Hassan H, Bashiri F. Pyridox(am)ine 5′‐phosphate oxidase deficiency: severe prenatal presentation with hypoxic ischemic encephalopathy. J Pediatr Epilepsy. 2019;08:49‐55. 10.1055/s-0039-1697676. ISSN 2146‐457X. [DOI] [Google Scholar]
- 18. Levtova A, Camuzeaux S, Laberge AM, et al. Normal cerebrospinal fluid pyridoxal‐phosphate level in a PNPO‐deficient patient with neonatal‐onset epileptic encephalopathy. JIMD Rep. 2015;22:67‐75. 10.1007/8904_2015_413 Epub 2015 Mar 12. PMID: 25762494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fung CW, Kwong AK, Wong VC. Gene panel analysis for nonsyndromic cryptogenic neonatal/infantile epileptic encephalopathy. Epilepsia Open. 2017;2(2):236‐243. 10.1002/epi4.12055 PMID: 29588952; PMCID: PMC5719849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mohanlal S, Bindu PS, Sureshbabu S, Kumar S. Variable treatment response in a patient with pyridoxal N phosphate oxidase (PNPO) deficiency‐understanding the paradox. Epilepsy Behav Rep. 2020;14:100357 10.1016/j.ebr.2020.100357 eCollection 2020. PMID: 32395712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hatch J, Coman D, Clayton P, et al. Normal neurodevelopmental outcomes in PNPO deficiency: a case series and literature review. JIMD Rep. 2016;26:91‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmitt B, Baumgartner M, Mills PB, et al. Seizures and paroxysmal events: symptoms pointing to the diagnosis of pyridoxine‐dependent epilepsy and pyridoxine phosphate oxidase deficiency. Dev Med Child Neurol. 2010;52(7):e133‐e142. [DOI] [PubMed] [Google Scholar]
- 23. Sudarsanam A, Singh H, Wilcken B, et al. Cirrhosis associated with pyridoxal 5′‐phosphate treatment of pyridoxamine 5′‐phosphate oxidase deficiency. JIMD Rep. 2014;17:67‐70. 10.1007/8904_2014_338 Epub 2014 Sep 26. PMID: 25256445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coman D, Lewindon P, Clayton P, Riney K. PNPO deficiency and cirrhosis: expanding the clinical phenotype? JIMD Rep. 2016;25:71‐75. 10.1007/8904_2015_456 Epub 2015 Jun 25. PMID: 26108646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Porri S, Fluss J, Plecko B, Paschke E, Korff CM, Kern I. Positive outcome following early diagnosis and treatment of pyridoxal‐5′‐phosphate oxidase deficiency: a case report. Neuropediatrics. 2014;45(1):64‐68. [DOI] [PubMed] [Google Scholar]
- 26. Bagci S, Zschocke J, Hoffmann GF, et al. Pyridoxal phosphate‐dependent neonatal epileptic encephalopathy. Arch Dis Child Fetal Neonatal Ed. 2008;93:F151‐F152. PIMD: 17216302. [DOI] [PubMed] [Google Scholar]
- 27. Guerin A, Aziz AS, Mutch C, Lewis J, Go CY, Mercimek‐Mahmutoglu S. Pyridox(am)ine‐5‐phosphate oxidase deficiency treatable cause of neonatal epileptic encephalopathy with burst suppression: case report and review of the literature. J Child Neurol. 2015;30(9):1218‐1225. PMID: 25296925. [DOI] [PubMed] [Google Scholar]
- 28. Goyal M, Fequiere PR, McGrath TM, Hyland K. Seizures with decreased levels of pyridoxal phosphate in cerebrospinal fluid. Pediatr Neurol. 2013;48(3):227‐231. [DOI] [PubMed] [Google Scholar]
- 29. Jaeger B, Abeling NG, Salomons GS, Struys EA, Simas‐Mendes M, Geukers VG. Poll‐the BT. Pyridoxine responsive epilepsy caused by a novel homozygous PNPO mutation. Mol Genet Metab Rep. 2016;6:60‐63. 10.1016/j.ymgmr.2016.01.004 eCollection 2016 Mar. PMID: 27014579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lloreda‐Garcia JM, Fernandez‐Fructuoso JR, Martinez‐Ferrandez C, Fuentes‐ Gutierrez C. Severe fetal anemia and neonatal epileptic encephalopathy caused by a novel PNPO mutation. Rev Neurol. 2017;65(7):335‐336. PMID: 28929476. [PubMed] [Google Scholar]
- 31. Lugli L, Bariola MC, Ori L, Lucaccioni L, Berardi A, Ferrari F. Further delineation of pyridoxine‐responsive pyridoxine phosphate oxidase deficiency epilepsy: report of a new case and review of the literature with genotype‐phenotype correlation. J Child Neurol. 2019;34(14):937‐943. 10.1177/0883073819863992 Epub 2019 Aug 9 PMID: 31397616. [DOI] [PubMed] [Google Scholar]
- 32. Riikonen R, Mankinen K, Gaily E. Long‐term outcome in pyridoxine‐responsive infantile epilepsy. Eur J Paediatr Neurol. 2015;19(6):647‐651. 10.1016/j.ejpn.2015.08.001 Epub 2015 Aug 18 PMID: 2631086. [DOI] [PubMed] [Google Scholar]
- 33. Veerapandiyan A, Winchester SA, Gallentine WB, et al. Electroencephalographic and seizure manifestations of pyridoxal‐phosphate‐dependent epilepsy. Epilepsy Behav. 2011;20:494‐501. PMID: 21292558. [DOI] [PubMed] [Google Scholar]
- 34. Kuo MF, Wang HS. Pyridoxal phosphate‐responsive epilepsy with resistance to pyridoxine. Pediatr Neurol. 2002;26:146‐147. [DOI] [PubMed] [Google Scholar]
- 35. Ruiz A, García‐Villoria J, Ormazabal A, et al. A new fatal case of pyridox(am)ine 5′‐phosphate oxidase (PNPO) deficiency. Mol Genet Metab. 2008;93:216‐218. [DOI] [PubMed] [Google Scholar]
- 36. Khayat M, Korman SH, Frankel P, et al. PNPO deficiency: an under diagnosed inborn error of pyridoxine metabolism. Mol Genet Metab. 2008;94(4):431‐434. [DOI] [PubMed] [Google Scholar]
- 37. Musayev FN, Di Salvo ML, Ko TP, Schirch V, Safo MK. Structure and properties of recombinant human pyridoxine 5′‐phosphate oxidase. Protein Sci. 2003;12(7):1455‐1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Safo MK, Musayev FN, di Salvo ML, Schirch V. X‐ray structure of Escherichia coli pyridoxine 5′‐phosphate oxidase complexed with pyridoxal 5′‐phosphae at 2.0 a resolution. J Mol Biol. 2001;310(4):817–826. https://doi:10.1006/jmbi.2001.4734. [DOI] [PubMed] [Google Scholar]
- 39. Wilson MP, Footitt EJ, Papandreou A, et al. An LC‐MS/MS based method for the quantification of pyridox(am)ine 5′‐phosphate oxidase activity in dried blood spots from patients with epilepsy. Anal Chem. 2017;89(17):8892‐8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wempe MF, Kumar A, Kumar V, et al. Identification of a novel biomarker for pyridoxine dependent epilepsy: implications for newborn screening. J Inherit Metab Dis. 2019;42:565‐574. 10.1002/jimd.12059. [DOI] [PubMed] [Google Scholar]
- 41. Hyland K, Arnold LA. Value of lumbar puncture in the diagnosis of infantile epilepsy and folinic acid‐responsive seizures. J Child Neurol. 2002;17(suppl 3):3S48‐S56. [PubMed] [Google Scholar]
- 42. Yoshida I, Sakaguchi Y, Nakano M, Yamashita F, Hitoshi T. Pyridoxal phosphate‐induced liver injury in a patient with homocystinuria. J Inherit Metab Dis. 1985;8:91. [DOI] [PubMed] [Google Scholar]
- 43. Mills PB, Camuzeaux SS, Footitt EJ, et al. Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome. Brain. 2014;137(5):1350‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The most common biochemical tests requested for PNPO diagnosis (2002‐2020)
Table S1 The list of genes coding for PLP‐dependent enzymes.
Table S2 Patient information included patient age, gender, seizure onset, family history, parents' consanguinity, pregnancy pre‐maturity (gestational age), Apgar score, condition at birth, status of failure to thrive, neurological changes, electroencephalogram (EEG) changes, MRI tests, the results for molecular and biochemical analysis and responsiveness to antiepileptic therapy, pyridoxine therapy, and PLP trial.
Table S3 EEG changes in PNPO deficiency.
Table S4 comprehensive biochemical profile for each reported case.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article