

Abstract
An orally administered, fixed‐dose coformulation of sodium phenylbutyrate‐taurursodiol (PB‐TURSO) significantly slowed functional decline in a randomized, placebo‐controlled, phase 2 trial in ALS (CENTAUR). Herein we report results of a long‐term survival analysis of participants in CENTAUR. In CENTAUR, adults with ALS were randomized 2:1 to PB‐TURSO or placebo. Participants completing the 6‐month (24‐week) randomized phase were eligible to receive PB‐TURSO in the open‐label extension. An all‐cause mortality analysis (35‐month maximum follow‐up post‐randomization) incorporated all randomized participants. Participants and site investigators were blinded to treatment assignments through the duration of follow‐up of this analysis. Vital status was obtained for 135 of 137 participants originally randomized in CENTAUR. Median overall survival was 25.0 months among participants originally randomized to PB‐TURSO and 18.5 months among those originally randomized to placebo (hazard ratio, 0.56; 95% confidence interval, 0.34‐0.92; P = .023). Initiation of PB‐TURSO treatment at baseline resulted in a 6.5‐month longer median survival as compared with placebo. Combined with results from CENTAUR, these results suggest that PB‐TURSO has both functional and survival benefits in ALS.
Keywords: amyotrophic lateral sclerosis, CENTAUR, motor neuron disease, sodium phenylbutyrate‐taurursodiol, survival analysis
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder associated with motor neuron degeneration in the motor cortex and spinal cord, leading to muscle weakness and atrophy. 1 Riluzole and edaravone are the only medications approved by the United States Food and Drug Administration for disease‐modifying treatment of ALS. 2 Riluzole has been shown to prolong survival in ALS. 3 , 4 , 5 The effect of edaravone on survival is unknown. 6 , 7
An orally administered, fixed‐dose coformulation of two compounds, sodium phenylbutyrate (PB) and taurursodiol (TURSO, also known as tauroursodeoxycholic acid), was designed to reduce neuronal death by mitigating both endoplasmic reticulum (ER) stress and mitochondrial dysfunction. Joint dysfunction of these two organelles within motor neurons has been recognized as a potential pathogenic factor in ALS, motivating development of this coformulation. 8 , 9 , 10 Preclinical data support a mitigating effect of PB and TURSO both alone 11 , 12 , 13 , 14 , 15 , 16 and in combination 17 on neuronal death and other disease‐specific features in models of neurodegenerative diseases and mitochondrial dysfunction. In addition, pilot clinical studies confirmed the individual safety of both compounds in individuals with ALS. 18 , 19
The recently completed randomized, double‐blind, placebo‐controlled CENTAUR trial evaluated efficacy and safety of sodium phenylbutyrate‐taurursodiol (PB‐TURSO) in individuals with ALS. 20 The trial met its primary efficacy endpoint—slowing of decline in Amyotrophic Lateral Sclerosis Functional Rating Scale—Revised (ALSFRS‐R) total score in participants treated with PB‐TURSO vs placebo over 6 months (24 weeks). Few participants died during the 6‐month duration of CENTAUR; a joint rank analysis of function and survival confirmed an effect of PB‐TURSO in slowing ALSFRS‐R progression when accounting for potentially informative loss to follow‐up from mortality. Participants who completed CENTAUR were eligible for enrollment in an open‐label extension (OLE) aimed at assessing long‐term safety and efficacy of PB‐TURSO in ALS. In this article we report the results of a nearly 3‐year survival analysis incorporating all participants who enrolled in CENTAUR, regardless of whether they continued long‐term treatment with PB‐TURSO in the OLE.
2. METHODS
2.1. Trial design and oversight
Both the randomized, controlled trial and the OLE were conducted in accordance with Good Clinical Practice guidelines of the International Conference on Harmonization and ethical principles of the Declaration of Helsinki. Protocol approval was obtained for all trial sites by a central institutional review board, the Partners Human Research Committee. 21 Participants provided written informed consent before entering each phase of the study.
2.2. Randomized, controlled trial
Detailed methods of the randomized study have been published elsewhere. 20 In brief, CENTAUR (NCT03127514) was a randomized, double‐blind, placebo‐controlled trial conducted at 25 Northeast ALS Consortium (NEALS) centers in the United States between June 2017 and September 2019. The trial enrolled participants, 18 to 80 years of age, with a diagnosis of definite ALS by revised El Escorial criteria (ie, clinical evidence of both upper and lower motor neuron signs in at least 3 body regions 22 ) and had symptom onset within the previous 18 months. Additional inclusion criteria were slow vital capacity of more than 60% of predicted value for sex, height, and age, 23 and either no use of riluzole at study entry or a stable dose of riluzole for at least 30 days before screening. The study protocol was amended after edaravone became available in August 2017 to allow for unrestricted use of the drug.
In the randomized trial, participants were randomized 2:1 to PB‐TURSO (3 g PB and 1 g TURSO per sachet) or matching placebo administered twice a day by mouth or feeding tube for a planned duration of 6 months (24 weeks).
2.3. Open‐label extension
Eligible participants in CENTAUR were given the option to enroll in an OLE (NCT03488524) and receive active drug for up to 30 months (132 weeks). Enrollment in the OLE occurred from March 2018 through September 2019. Participants who completed all visits in the randomized trial were eligible for OLE enrollment provided they enrolled within 28 days of their last visit in the randomized phase and did not prematurely discontinue study drug for reasons other than tracheostomy or initiation of permanent assisted ventilation (PAV). Four participants received a protocol waiver and enrolled in the OLE more than 28 days after completion of CENTAUR, as the PB‐TURSO drug supply for the OLE did not become available until approximately 2 months after the first participants had completed CENTAUR. Participants who received riluzole or edaravone during the randomized trial were permitted to continue these medications, and, as in the randomized phase, edaravone could be initiated during the OLE.
In the OLE, participants originally assigned to PB‐TURSO or placebo were administered PB‐TURSO by mouth or feeding tube, continuing the same dose of study drug they had been receiving at the end of the randomized phase of the trial (either 2 sachets daily [1 sachet dissolved in water, both in the morning and evening] or 1 sachet daily for 1 participant who had previously reduced dosage because of intermittent constipation). This regimen was implemented so that participants and investigators would remain blinded to original treatment assignment in the randomized phase during the OLE study.
2.4. Statistical analysis
The prespecified survival analysis compared time to death (all‐cause mortality) between participants originally randomized to active treatment and those originally randomized to placebo. A cutoff date of July 20, 2020 (longest follow‐up, 35 months after randomization) was used to incorporate the most mature data to date for the purposes of providing the most complete analysis. The vital status and date of death for all participants randomized into the CENTAUR trial (intent‐to‐treat population) were investigated by OmniTrace (a firm specializing in locating participants and ascertaining vital status via search of public records, obituary databases, property records, and social media). This vital status search was approved by the Partners Human Research Committee, which served as the central investigational review board. The censoring date was designated as 30 days before the date the survival check was performed for each participant, allowing for the maximum time it may have taken for public record of death to be updated. Participants for whom vital status could not be obtained were censored at the date of their last follow‐up.
The hazard ratio (HR) of death comparing the group originally randomized to active treatment vs the group originally randomized to placebo was estimated using a Cox proportional hazards model with covariates of age at randomization, pre‐baseline ALSFRS‐R slope, and baseline ALSFRS‐R total score, variables shown to be predictors of disease progression in historical data. 24 , 25 , 26 Pre‐baseline ALSFRS‐R slope, defined as the rate of decline in ALSFRS‐R total score between symptom onset and study baseline, was calculated during the baseline visit using the formula [(48 − baseline ALSFRS‐R) / (number of months between symptom onset and study baseline)]. 24 All covariates were prespecified before completing the vital status check. Median duration of survival and the associated 95% confidence interval (CI) were estimated from Kaplan‐Meier curves, and tests were declared significant if P ≤ .05 (two‐tailed). The duration of exposure to PB‐TURSO in each group was summarized using descriptive statistics.
Number and time‐to‐death‐equivalent events (defined as tracheostomy or PAV at least 22 hours/day for at least 7 days, with date of PAV onset being the first of the 7 days) were summarized for the two groups as potential confounders of survival outcomes; unlike the occurrence of death, which could be ascertained for all randomized participants, occurrence of death‐equivalent events was determined prospectively in the course of participant monitoring as part of the double‐blind period and the OLE, but could not be ascertained for those who discontinued from study or were lost to follow‐up. Sensitivity analyses were performed to assess the potential impact of use of concomitant medications at baseline on the overall survival analysis. Three sensitivity models were tested: adding baseline use of riluzole, edaravone, or both as covariates, with baseline ALSFRS‐R; age at randomization; and pre‐baseline ALSFRS‐R slope as additional covariates.
3. RESULTS
3.1. Participants
The disposition of participants in CENTAUR and the subsequent OLE is shown in Figure 1. A total of 137 participants were randomized in CENTAUR, of whom 89 were randomized to PB‐TURSO and 48 were randomized to placebo. Vital status was obtainable for all but two of the participants randomized in CENTAUR; according to the statistical analysis plan, these participants were censored at the date of last contact with the clinical site. Ninety‐eight participants who completed CENTAUR were eligible for the OLE; 97 participants completed the study on drug, and 1 participant had a brief (~1‐week) drug disruption at the very end of the study and was permitted to enter the OLE. Of these, 90 (92%) continued into the OLE, 34 of whom had been originally randomized to placebo and 56 to active drug. The longest follow‐up was 35 months after randomization in CENTAUR, with 18 participants originally randomized to active treatment and 9 participants originally randomized to placebo remaining in follow‐up.
FIGURE 1.
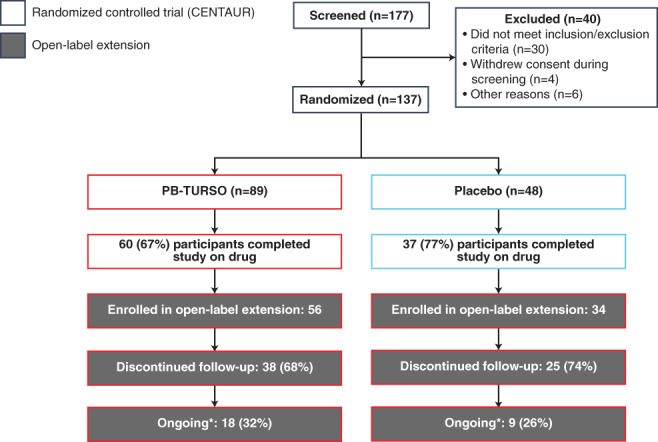
Participant disposition. Randomized, controlled trial and open‐label extension (through July 2020). Percentages may not add up precisely due to rounding (asterisk: as of July 2020. Vital status for participants who discontinued study participation was ascertained by OmniTrace, as noted earlier. PB‐TURSO, sodium phenylbutyrate‐taurursodiol
Baseline demographic and disease characteristics for participants at the start of the randomized phase were generally similar for the two treatment groups (see Table S1 online). Most (77%) participants were receiving riluzole or edaravone at or before study entry. Higher proportions of participants originally randomized to placebo were receiving edaravone and both edaravone and riluzole at or before study entry compared with the group originally randomized to PB‐TURSO.
3.2. Survival analysis
In the overall survival analysis encompassing all participants randomized in CENTAUR, the risk of death was 44% lower among those originally randomized to active treatment compared with those originally randomized to placebo (HR, 0.56; 95% CI, 0.34‐0.92; P = .023; Figure 2). Median survival duration was 25.0 months (95% CI, 19.0‐33.6 months) in the group originally randomized to active treatment and 18.5 months (95% CI, 13.5‐23.2 months) in the group originally randomized to placebo, a 6.5‐month difference. The estimated probability of survival at 12 months among participants originally randomized to PB‐TURSO and placebo was 80.9% (95% CI, 71.1%‐87.7%) and 72.9% (95% CI, 58.0%‐83.3%), respectively. At 24 months, the estimates were 51.6% (95% CI, 38.9%‐62.9%) and 33.9% (95% CI, 19.4%‐49.1%), respectively. The median time to censoring was 21.3 months. The duration of PB‐TURSO exposure is summarized for each randomized participant by originally randomized groups in Figure S1 online. Inclusive of all randomized participants, including those who did not enroll in the OLE, those originally randomized to active treatment had a median (range; first and third quartiles) PB‐TURSO exposure duration of 8.8 (0.1‐33.0; 3.7 and 15.8) months, and those originally randomized to placebo had a median PB‐TURSO exposure of 1.9 (0‐22.5; 0 and 9.1) months (all in the OLE). Mean PB‐TURSO exposure duration was 10.6 months in the group originally randomized to PB‐TURSO and 4.7 months in the group originally randomized to placebo.
FIGURE 2.
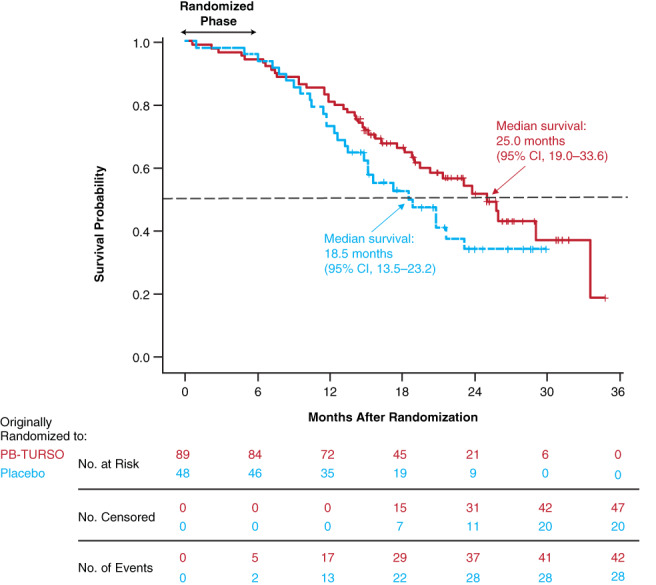
Overall survival in the entire randomized population. Starting at the conclusion of the randomized phase at month 6 (24 weeks), eligible participants could enroll in the OLE. Of 98 eligible participants, 90 (92%) continued on in the OLE, 34 of whom were originally randomized to placebo and 56 to active drug. The survival analysis encompassed all participants randomized at time 0, including those who did not enter the OLE, discontinued study participation, or were lost to follow‐up. The hazard ratio for death in the group originally randomized to active treatment was 0.56 (P = .023 vs group originally randomized to placebo). OLE, open‐label extension; PB‐TURSO, sodium phenylbutyrate‐taurursodiol
The two groups had similar rates of death‐equivalent events. Six (6.7%) participants originally randomized to PB‐TURSO and 4 (8.3%) participants originally randomized to placebo experienced death‐equivalent events.
Results of the three sensitivity analyses accounting for use of concomitant riluzole, edaravone, or both at baseline retained the statistically significant benefit of PB‐TURSO over placebo (Table 1), consistent with the survival analysis. These results suggest that the benefit of PB‐TURSO was independent of baseline concomitant medication use.
TABLE 1.
Comparative results of sensitivity analyses accounting for baseline concomitant medication use
| Model a | Hazard ratio | 95% CI | P value b |
|---|---|---|---|
| Primary | 0.56 | 0.34‐0.92 | .023 |
| Riluzole use at baseline | 0.54 | 0.33‐0.89 | .018 |
| Edaravone use at baseline | 0.53 | 0.32‐0.90 | .019 |
| Riluzole and edaravone use at baseline | 0.53 | 0.32‐0.88 | .016 |
Abbreviations: ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale—Revised; PB‐TURSO, sodium phenylbutyrate‐taurursodiol.
Cox proportional hazards models, including baseline ALSFRS‐R, age at randomization, and pre‐baseline ALSFRS‐R slope in addition to treatment (PB‐TURSO) and concomitant medication use at baseline.
Values obtained using the likelihood ratio test.
4. DISCUSSION
In this analysis of survival encompassing all participants randomized in CENTAUR, the group originally randomized to PB‐TURSO had a median overall survival that was 6.5 months longer than the group originally randomized to placebo. Median PB‐TURSO exposure duration differed by approximately 7 months between the groups. Notably, most of the participants originally randomized to placebo received some exposure to PB‐TURSO in the OLE, possibly diluting the survival benefit attributable to PB‐TURSO in this analysis; however, it is not known whether those who were originally randomized to placebo but later received PB‐TURSO in the OLE derived any survival benefit, as the OLE had no concurrent placebo control group.
Initiation of PB‐TURSO at baseline was associated with longer survival, a finding that suggests a neuroprotective effect of PB‐TURSO in ALS. The concept of neuroprotection involves early therapeutic intervention after an acute injury or in chronic neurodegenerative conditions with the aim of averting irreversible neuronal death or degeneration. Neuroprotection may be achieved by interrupting either upstream disease‐specific processes that lead to neurodegeneration or downstream processes that secondarily promote neuronal death. 27 The coformulated compounds in PB‐TURSO have shown mitigating effects on ER stress (PB) 28 , 29 and mitochondrial dysfunction (TURSO), 30 both mechanisms thought to underlie neurodegeneration in ALS. 10
Our analysis was limited by a relatively small sample size but was strengthened by inclusion of the entire randomized sample from CENTAUR and assessment of vital status up to 35 months after randomization. Confirmation of vital status was facilitated by the use of a participant locating service, a novel approach that differentiated our survival analysis from those in other ALS studies. Data on vital status that are incomplete because of discontinuation or loss to follow‐up may bias the results of survival analyses. 31 , 32 Participant tracing has been used in clinical trials to improve the accuracy of results by tracking long‐term outcomes for as many trial participants as possible. 33 , 34 , 35 This method may be particularly useful in trials with large dropout rates, such as those in ALS, 36 by accounting for potentially differing outcomes between participants who drop out and those who continue on in the study. Our confirmation of vital status for all but two randomized participants, regardless of duration of follow‐up, provided the basis for statistically robust and comprehensive overall survival analyses.
In ALS, decisions to implement life‐sustaining measures, such as tracheostomy and PAV, may be affected by patient choice and local practices; they have the potential to confound survival analyses and are thus considered to be death‐equivalent events. 37 In this study, identification of all death‐equivalent events may not have occurred, as these events were provided by the study sites and are not consistently searchable in public databases or records. Nevertheless, the low rates and similar proportions of participants affected by such events in both groups suggest survival was not likely to have been confounded by tracheostomy or initiation of PAV in either group. Results of sensitivity analyses suggest that the PB‐TURSO treatment effect was independent of baseline use of riluzole, edaravone, or both together.
Despite currently available therapies, ALS remains rapidly debilitating and fatal. PB‐TURSO has previously demonstrated functional benefit in a randomized, controlled trial. 20 The results presented here demonstrate a long‐term survival benefit from early initiation of PB‐TURSO treatment in participants with ALS, adding to the previously reported functional benefit.
CONFLICT OF INTEREST
S. Paganoni reports grants from Amylyx Pharmaceuticals, Inc, The ALS Association, and ALS Finding A Cure during the conduct of the study, and grants from Revalesio, Ra Pharma, Biohaven, Clene Nanomedicine, and Prilenia, outside the submitted work. S. Hendrix reports other personal fees from Pentara Corporation, outside the submitted work. S.P. Dickson reports other personal fees from Pentara outside the submitted work. E.A. Macklin reports grants from Amylyx, The ALS Association, and ALS Finding A Cure Foundation during the conduct of the study. He is also a data and safety monitoring board member (DSMB) and reports grants from Acorda Therapeutics; steering committee membership for Biogen; consultant work for Cerevance; grants from GlaxoSmithKline; consultant work for Inventram, Lavin Consulting, and Myolex; grants from Mitsubishi Tanabe Pharmaceuticals; and DSMB membership for Novartis Pharmaceuticals and Shire Human Genetic Therapies, outside the submitted work. J.D. Berry reports grants from ALS Finding A Cure, The ALS Association, and Amylyx during the conduct of the study; personal fees from Biogen and Clene Nanomedicine; and grants from Alexion, Biogen, MT Pharma of America, Anelixis Therapeutics, Brainstorm Cell Therapeutics, Genentech, nQ Medical, National Institute of Neurological Disorders and Stroke, and the Muscular Dystrophy Association, outside the submitted work. M.A. Elliott has received personal fees from Amylyx and personal fees from Biogen. C. Karam reports grants and personal fees from Akcea, Alnylam, and Genzyme, and personal fees from Acceleron, Biogen, Alexion, Argenx, Cytokinetics, and CSL Behring, outside the submitted work. J.B. Caress reports grants from Amylyx during the conduct of the study, and grants from Orion Pharmaceuticals, MTB Pharma, and Cytokinetics, outside the submitted work. J. Wymer reports grants from Amylyx during the conduct of the study. S.A. Goutman reports grants from The ALS Association during the conduct of the study; grants from the National Institutes of Health/National Institutes of Environmental Health Sciences, The ALS Association, and Target ALS, outside the submitted work; consultant work for Biogen and ITF Pharma, outside the submitted work; and personal fees from Biogen, ITF Pharma, Watermark Research Partners, and for expert testimony, outside the submitted work. T.D. Heiman‐Patterson reports grants from Mitsubishi Tanabe Pharma America, Amylyx Pharmaceuticals, The ALS Association, and Orion Pharma, and personal fees from Cytokinetics, ITF, and Biohaven, outside the submitted work. C.E. Jackson reports grants from Amylyx during the conduct of the study; grants and personal fees from Cytokinetics, personal fees from CSL Behring, grants and personal fees from Mitsubishi Tanabe Pharma America, DSMB membership, and personal fees from Brainstorm and Mallinckrodt during the conduct of this study; and personal fees from ITF Pharma, outside the submitted work. C. Quinn received personal fees for serving on an advisory board of Amylyx, outside the submitted work. J.D. Rothstein reports licensing agreement and nonfinancial support from Ionis Pharmaceuticals; nonfinancial support from Calico, Biogen, and IBM Watson; research grant support from the National Institute of Neurological Disorders and Stroke, National Institute on Aging, Department of Defense, the Chan Zuckerberg Initiative, Microsoft, The ALS Association, the Muscular Dystrophy Association, Target ALS, F Prime, ALS Finding A Cure, Answer ALS, Robert Packard Center for ALS Research, GlaxoSmithKline, Travelers Insurance, American Airlines, Caterpillar, and the National Football League; and personal consulting fees from Expansion Therapeutics and Team Gleason. He also reports that his institution was a trial site and thus had a contract with Amylyx to participate in the study. J. Katz reports personal fees from MT Pharma America, Denali Pharmaceuticals, Genentech, and Calico, outside the submitted work. S. Ladha reports grants from Amylyx, Biogen, and MT Pharma, and personal fees from Amylyx and Biogen. T.M. Miller reports licensing agreement and nonfinancial support from Ionis Pharmaceuticals, a licensing agreement with C2N, grants and personal fees from Biogen, and personal fees from Cytokinetics and Disarm Therapeutics, outside the submitted work. S.N. Scelsa reports grants from Amylyx during the conduct of the study, and grants from Orion Pharma, outside the submitted work. T.H. Vu reports personal fees from the speakers bureau of Mitsubishi Tanabe Pharmaceuticals, and has participated in clinical trials sponsored by Amylyx, Orion, Biogen, Mallinckrodt, and Cytokinetics during the conduct of the study. J.D. Glass reports that his institution was a trial site and thus had a contract from Amylyx to participate in the study. A. Swenson reports research support from Amylyx, The ALS Association, Massachusetts General Hospital, the National Institutes of Health/National Institute of Neurological Disorders and Stroke, and serving on an independent data monitoring committee for Alexion. P.L. Andres reports personal fees from Amylyx for consulting during the conduct of the study and has an isometric strength testing apparatus (US Patent 7493812B2) held by the Hospital Corporation. S. Babu reports research support from the American Academy of Neurology, the AANEM Foundation, The ALS Association, the Muscular Dystrophy Association, Biogen, Orion, Voyager Therapeutics, and Novartis. M. Chase reports grants to the Massachusetts General Hospital (MGH) from The ALS Association, grants to the MGH from ALS Finding A Cure, and fee for service from Amylyx during the conduct of the study. M. Hall reports grants for funding for clinical trial monitoring and outcomes training support from The ALS Association and Amylyx during the conduct of the study. G. Kittle reports grants from The ALS Association and Amylyx during the conduct of the study. J.M. Shefner reports grants and personal fees from Amylyx during the conduct of the study; personal fees for consulting work from Cytokinetics and Brainstorm; grants and personal fees for outcomes training and study design from Mitsubishi Pharma America, outside the submitted work; personal fees for consulting from Neurosense and Otsuka, outside the submitted work; and grants for outcomes training from Alexion, Medicinova, and Biogen, outside the submitted work. He is also Neuromuscular section editor for UpToDate. J. Wittes and Z.‐F. Yu report payments from Amylyx to their employer during the conduct of the study. J. Cohen and J. Klee report a relationship with Amylyx during the conduct of the study, and they serve as co‐CEOs of Amylyx, outside the submitted work, with multiple patents issued to Amylyx. K. Leslie reports being full‐time employee of Amylyx during the conduct of the study, and personal fees from Amylyx, outside the submitted work. R.E. Tanzi reports personal fees from Amylyx outside the submitted work; has helped with inception and design of the clinical trial but was not involved with running the trial and had no contact with the trial subjects; and owns founding equity in Amylyx and serves as head of the company's scientific advisory board. W. Gilbert was director of Amylyx during the conduct of the study and a company shareholder. P.D. Yeramian reports full‐time employment at Amylyx during the conduct of the study. D. Schoenfeld reports grants from The ALS Association, during the conduct of the study, and personal fees from Immunitypharma and Alexion, outside the submitted work. M.E. Cudkowicz reports grants from Massachusetts General Hospital during the conduct of the study; grants from Clene Nanomedicine, Ra Pharma, Biohaven, and Prilenia, outside the submitted work; and personal fees from Takeda, Biogen, Sunovian, Cytokinetics, and Immunity Pharma, outside the submitted work. The remaining authors declare no potential conflicts of interest.
ETHICAL PUBLICATION STATEMENT
The authors confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- ALSFRS‐R
Amyotrophic Lateral Sclerosis Functional Scale—Revised
- ER
endoplasmic reticulum
- HR
hazard ratio
- ITT
intent‐to‐treat
- NEALS
Northeast ALS Consortium
- OLE
open‐label extension
- PAV
permanent assisted ventilation
- PB
sodium phenylbutyrate
- PB‐TURSO
sodium phenylbutyrate‐taurursodiol
- SVC
slow vital capacity
- TURSO
taurursodiol
Supporting information
APPENDIX S1. Supporting information
FIGURE S1 Duration of PB‐TURSO exposure for each randomized participant. Each bar represents an individual participant, with grouping of bars by originally randomized treatment (PB‐TURSO or placebo). PB‐TURSO, sodium phenylbutyrate‐taurursodiol.
TABLE S1 Participants’ demographics and clinical characteristicsa
ACKNOWLEDGMENTS
The authors thank the individuals who participated in the CENTAUR and open‐label extension trials as well as their caregivers and families. We also thank Steve Kolb and the late Stephen Winthrop, who brought the voices of caregivers for persons with ALS and persons with ALS, respectively, to this trial as members of the Steering Committee; the Partners Human Research Committee, for serving as the single institutional review board of record for the study; the medical monitor, Dr Anne‐Marie Wills; data and safety monitoring board members, Dr Lorne Zinman and Dr Myles Keroack; and the CENTAUR coordination center and trial site staff (see Appendix online). This analysis was funded by Amylyx Pharmaceuticals, Inc, the ALS Finding A Cure Foundation, and The ALS Association. Lara Primak, MD, and Dipanwita Ghose, MS, PhD, of PRECISIONScientia provided medical writing assistance with the development and revision of the manuscript under the direction of the authors, with financial support from Amylyx and in compliance with international Good Publication Practice guidelines.
Paganoni S, Hendrix S, Dickson SP, et al. Long‐term survival of participants in the CENTAUR trial of sodium phenylbutyrate‐taurursodiol in amyotrophic lateral sclerosis . Muscle & Nerve. 2021;63:31–39. 10.1002/mus.27091
Portions of this manuscript were presented at the 2020 Annual Northeast Amyotrophic Lateral Sclerosis Consortium Meeting.
Funding information ALS Finding A Cure Foundation; Amylyx Pharmaceuticals, Inc.; The ALS Association
REFERENCES
- 1. Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:162‐172. [DOI] [PubMed] [Google Scholar]
- 2. Wong W. The role of managed care professionals in improving care for patients with ALS. Am J Manag Care. 2020;26(suppl 9):S198‐S205. [DOI] [PubMed] [Google Scholar]
- 3. Bensimon G, Lacomblez L, Meininger V, ALS/Riluzole Study Group . A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330:585‐591. [DOI] [PubMed] [Google Scholar]
- 4. Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. ALS/Riluzole study group‐II. Dose‐ranging study of riluzole in amyotrophic lateral sclerosis. Lancet. 1996;347:1425‐1431. [DOI] [PubMed] [Google Scholar]
- 5. Andrews JA, Jackson CE, Heiman‐Patterson TD, Bettica P, Brooks BR, Pioro EP. Real‐world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020;1‐10. 10.1080/21678421.2020.1771734. [DOI] [PubMed] [Google Scholar]
- 6. Abe K, Itoyama Y, Sobue G, et al. Confirmatory double‐blind, parallel‐group, placebo‐controlled study of efficacy and safety of edaravone (MCI‐186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:610‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Writing Group . Edaravone (MCI‐186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2017;16:505‐512. [DOI] [PubMed] [Google Scholar]
- 8. Bernard‐Marissal N, Chrast R, Schneider BL. Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: a broken relationship? Cell Death Dis. 2018;9:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lau DHW, Hartopp N, Welsh NJ, et al. Disruption of ER‐mitochondria signalling in fronto‐temporal dementia and related amyotrophic lateral sclerosis. Cell Death Dis. 2018;9:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Manfredi G, Kawamata H. Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol Dis. 2016;90:35‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cho JA, Zhang X, Miller GM, Lencer WI, Nery FC. 4‐Phenylbutyrate attenuates the ER stress response and cyclic AMP accumulation in DYT1 dystonia cell models. PLoS One. 2014;9:e110086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dionísio PA, Amaral JD, Ribeiro MF, Lo AC, D'Hooge R, Rodrigues CM. Amyloid‐β pathology is attenuated by tauroursodeoxycholic acid treatment in APP/PS1 mice after disease onset. Neurobiol Aging. 2015;36:228‐240. [DOI] [PubMed] [Google Scholar]
- 13. Kusaczuk M. Tauroursodeoxycholate—bile acid with chaperoning activity: molecular and cellular effects and therapeutic perspectives. Cells. 2019;8:1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ricobaraza A, Cuadrado‐Tejedor M, Pérez‐Mediavilla A, Frechilla D, Del Río J, García‐Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721‐1732. [DOI] [PubMed] [Google Scholar]
- 15. Ryu H, Smith K, Camelo SI, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti‐apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087‐1098. [DOI] [PubMed] [Google Scholar]
- 16. Wiley JC, Pettan‐Brewer C, Ladiges WC. Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell. 2011;10:418‐428. [DOI] [PubMed] [Google Scholar]
- 17. Leslie K, Cohen J, Klee J, Valsecchi F, Manfredi G. AMX0035, a novel combination therapeutic candidate for treatment of primary mitochondrial diseases. Poster presented at: 2017 MDA Scientific Conference, March 19‐22, 2017, Arlington, VA. 2017.
- 18. Cudkowicz ME, Andres PL, Macdonald SA, et al. Northeast ALS and National VA ALS Research Consortiums. Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph Lateral Scler. 2009;10:99‐106. [DOI] [PubMed] [Google Scholar]
- 19. Elia AE, Lalli S, Monsurrò MR, et al. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur J Neurol. 2016;23:45‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate‐taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383:919‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. NEALS Central Institutional Review Board . Northeast Amyotrophic Lateral Sclerosis Consortium. https://www.neals.org/for-als-researchers/central-irb-master-contracts. Accessed September 3, 2020.
- 22. Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on motor neuron diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293‐299. [DOI] [PubMed] [Google Scholar]
- 23. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow‐volume curve with growth and aging. Am Rev Respir Dis. 1983;127:725‐734. [DOI] [PubMed] [Google Scholar]
- 24. Labra J, Menon P, Byth K, Morrison S, Vucic S. Rate of disease progression: a prognostic biomarker in ALS. J Neurol Neurosurg Psychiatry. 2016;87:628‐632. [DOI] [PubMed] [Google Scholar]
- 25. Daghlas I, Lever TE, Leary E. A retrospective investigation of the relationship between baseline covariates and rate of ALSFRS‐R decline in ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:206‐211. [DOI] [PubMed] [Google Scholar]
- 26. Taylor AA, Fournier C, Polak M, et al. The Pooled Resource Open‐Access ALS Clinical Trials Consortium predicting disease progression in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2016;3:866‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cummings J. Disease modification and neuroprotection in neurodegenerative disorders. Transl Neurodegener. 2017;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaur B, Bhat A, Chakraborty R, et al. Proteomic profile of 4‐PBA treated human neuronal cells during ER stress. Mol Omics. 2018;14:53‐63. [DOI] [PubMed] [Google Scholar]
- 29. Suaud L, Miller K, Panichelli AE, Randell RL, Marando CM, Rubenstein RC. 4‐Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT‐3 in cystic fibrosis epithelial cells. J Biol Chem. 2011;286:45083‐45092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodrigues CM, Steer CJ. The therapeutic effects of ursodeoxycholic acid as an anti‐apoptotic agent. Expert Opin Investig Drugs. 2001;10:1243‐1253. [DOI] [PubMed] [Google Scholar]
- 31. Dettori JR. Loss to follow‐up. Evid Based Spine Care J. 2011;2:7‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shih W. Problems in dealing with missing data and informative censoring in clinical trials. Curr Control Trials Cardiovasc Med. 2002;3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bronner LE, Podewils LJ, Peters A, et al. Impact of community tracer teams on treatment outcomes among tuberculosis patients in South Africa. BMC Public Health. 2012;12:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McMahon JH, Elliott JH, Hong SY, Bertagnolio S, Jordan MR. Effects of physical tracing on estimates of loss to follow‐up, mortality and retention in low and middle income country antiretroviral therapy programs: a systematic review. PLoS One. 2013;8:e56047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Semeere A, Freeman E, Wenger M, et al. Updating vital status by tracking in the community among patients with epidemic Kaposi sarcoma who are lost to follow‐up in sub‐Saharan Africa. BMC Cancer. 2017;17:611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Atassi N, Yerramilli‐Rao P, Szymonifka J, et al. Analysis of start‐up, retention, and adherence in ALS clinical trials. Neurology. 2013;81:1350‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paganoni S, Cudkowicz M, Berry JD. Outcome measures in amyotrophic lateral sclerosis clinical trials. Clin Investig. 2014;4:605‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. Supporting information
FIGURE S1 Duration of PB‐TURSO exposure for each randomized participant. Each bar represents an individual participant, with grouping of bars by originally randomized treatment (PB‐TURSO or placebo). PB‐TURSO, sodium phenylbutyrate‐taurursodiol.
TABLE S1 Participants’ demographics and clinical characteristicsa