

Abstract
As a novel rylene type dye a diimine ligand with a fully rigid and extended π‐system in its backbone was prepared by directly fusing a 1,10‐phenanthroline building block with 1,8‐naphthalimide. The corresponding heteroleptic ruthenium photosensitizer bearing one biipo and two tbbpy ligands was synthesized and extensively analyzed by a combination of NMR, single crystal X‐ray diffraction, steady‐state absorption and emission, time‐resolved spectroscopy and different electrochemical measurements supported by time‐dependent density functional theory calculations. The cyclic and differential pulse voltammograms revealed, that the naphthaloylenebenzene moiety enables an additional second reduction of the ligand. Moreover, this ligand possesses a very broad absorption in the visible region. In the RuII complex this causes an overlap of ligand‐centered and metal‐to‐ligand charge transfer transitions. The emission of the complex is clearly redshifted compared to the ligand emission with very long‐lived excited states lifetimes of 1.7 and 24.7 μs in oxygen‐free acetonitrile solution. This behavior is accompanied by a surprisingly high oxygen sensitivity. Finally, this photosensitizer was successfully applied for the effective evolution of singlet oxygen challenging some of the common RuII prototype complexes.
Keywords: electron storage, excited state properties, extended π-systems, ruthenium photosensitizers, singlet oxygen evolution
The constant evolution of phenanthroline leads to a new type of a polycyclic, fully conjugated, and rigid diimine ligand with a naphthaloylenebenzene moiety in the back. The so‐called biipo ligand enabled a RuII photosensitizer, which has an excited state lifetime of up to 24.7 μs and is active for the light‐driven singlet oxygen production.
Introduction
The capture, storage and usage of solar light for light‐driven reactions in organic synthesis[ 1 , 2 , 3 , 4 ] and for the production of solar fuels[ 5 , 6 , 7 , 8 ] or singlet oxygen (1O2) is of strongly increasing importance.[ 9 , 10 , 11 , 12 ] As 1O2 is one of the most relevant reactive oxygen species (ROS) for photodynamic therapy (PDT), there is a great need for effective singlet oxygen generating chromophores.[ 13 , 14 , 15 , 16 ] This requires the design of efficient and stable photosensitizers with suitable redox potentials and excited state lifetimes. Moreover, for some applications (e.g., the reduction of protons to hydrogen) photosensitizers are desired that are able to store multiple charges.[ 17 , 18 , 19 , 20 , 21 , 22 ] In this context especially RuII polypyridine complexes containing an additional electron storage moiety at the diimine ligand have been extensively studied.[ 17 , 19 , 20 , 23 , 24 ] For instance, different types and number of methyl viologen, anthraquinone, naphthalimide or pyrene derivatives had been introduced as an additional substituent to 2,2’‐bipyridine (bpy) or 1,10‐phenanthroline (phen).[ 23 , 24 , 25 , 26 , 27 , 28 ]
The integration of donor–acceptor units like naphthalene dicarboxylic acid monoimide (NMI) or other rylene derivatives enabled easy synthetic access to photosensitizers with high thermal and (photo)chemical stabilities and to adapt the photophysical as well as electrochemical properties of the chromophores in a wide range. In addition, rylene dyes often provide high fluorescence quantum yields, large extinction coefficients over a broad spectral range and the option to store multiple electrons.[ 29 , 30 , 31 , 32 , 33 , 34 , 35 ] The usage of related dyes like naphthalene tetracarboxylic acid bisbenzimidazole (perinone) allows for low energy gaps and light absorption over a wide range.[ 36 , 37 , 38 ] However, due to their low solubility, these ligand systems received only limited attention.[ 36 , 37 , 38 ]
In contrast to the extension of the π‐system of the diimine ligand by different substituents, the main aim of this study was the direct integration of a naphthaloylenebenzene unit in the backbone of the 1,10‐phenanthroline ligand (Figure 1) to obtain a fully conjugated and rigid ligand. It is expected that this modification not only affects the redox behavior, but also the charge transfer processes and efficiency. To this end, 16H‐benzo[4′,5′]isoquinolino[2′,1′:1,2]imidazo[4,5‐f]‐[1,10]‐phenanthrolin‐16‐one (biipo) and its heteroleptic RuII complex [(tbbpy)2Ru(biipo)]2+ (Rubiipo, with tbbpy=4,4′‐tert‐butyl‐2,2′‐bipyridine) were prepared and analyzed (including single crystal X‐ray diffraction (XRD)) for the first time (Figure 1). Cyclic and differential pulse voltammetry were used to evaluate the effect of the internal electron storage on the redox chemistry. A combination of steady‐state and time‐resolved emission spectroscopy along with transient absorption spectroscopy and (time‐dependent) density functional theory (TD‐)DFT calculations were then used to explore the photophysical properties and charge‐transfer processes. Moreover, Rubiipo was successfully used for the evolution of 1O2 competing some of the parent standard complexes like [Ru(bpy)3]2+ (Rubpy) or [(tbbpy)2Ru(phen)]2+ (Ruphen).
Figure 1.
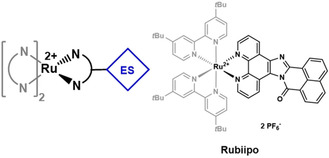
Schematic representation of a RuII photosensitizer containing an additional electron storage (ES) moiety (left) and the structure of Rubiipo investigated in this study (right). The biipo ligand is depicted in black.
Results and Discussion
Synthesis and structural characterization
The synthesis of the biipo ligand was performed via a four‐step procedure (Scheme S2.1 in the Supporting Information) starting from 1,10‐phenanthroline. In the final step 5,6‐diamino‐1,10‐phenanthroline and a slight excess of 1,8‐naphthalic anhydride were condensed to yield biipo as a yellow solid in 94 % (Figure 2). Subsequently, due to its low solubility biipo was coordinated to the [(tbbpy)2RuCl2] precursor using hot ethylene glycol as solvent. This procedure is also applied for similar poorly soluble diimine ligands and is known in literature.[ 39 , 40 ] Rubiipo was then purified via silica gel column chromatography (for further details see Supporting Information) and counter ion exchange to obtain the complex in a total yield of 71 %.
Figure 2.
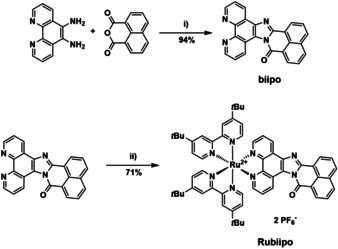
Preparation of the biipo ligand and its corresponding RuII complex Rubiipo. Conditions: i) acetic acid, Ar, 140 °C, 24 h and ii) [(tbbpy)2RuCl2], ethylene glycol, 120 °C, 20 h.
Rubiipo was fully characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, high‐resolution mass spectrometry (HRMS, electron spray ionization), IR spectroscopy, elemental analysis, and single crystal XRD (see all spectra in the Supporting Information). Rubiipo and biipo both exhibit a characteristic carbonyl stretch vibration at around 1700 cm−1 in their IR spectra (Figures S3.15 and S3.16) originating from the biipo ligand framework. The high‐resolution mass spectra of both the ligand and the complex match very well with theoretical data and simulated isotopic patterns (see Figures S3.9 and S3.10). In the 1H NMR spectra the chemical shifts of all aromatic protons appear as expected.[ 41 , 42 ] The proton signals of the two bipyridines, the phenanthroline and the naphthaloylenebenzene moiety suggest an asymmetric octahedral RuII complex. Moreover, the aliphatic protons of the tertiary butyl (tBu) groups show a small splitting (i.e., four separated singlet signals with 9 protons each), which is caused by the asymmetric structure of the biipo ligand. Caused by this asymmetry the four pyridines and tBu groups of the two tbbpy ligands are not equivalent within the Rubiipo complex and possess a different chemical environment. Further, 1H NMR spectra with different concentrations of Rubiipo (in the range from 1.28 mm to 32.1 mm, Figure S3.6) exhibit considerable shifts of the proton signals of up to 0.5 ppm. According to literature protocols, a dimerization constant of 27 m −1 was determined for Rubiipo (see Supporting Information).[ 43 , 44 , 45 , 46 ] This clearly suggests intermolecular interactions between different Rubiipo species in solution, which are caused by the quite large and planar biipo ligand. This observation is in line with other RuII complexes bearing polycyclic diimine ligands such as dipyridophenazine, tetrapyridophenazine (dimerization constant=289±17 m −1), di(pyridin2‐yl)tetraazaphenazine (83±17 m −1) or tetraazatetra‐pyridopentacene, which reveal even stronger interactions between the extended π‐systems of these ligands.[ 26 , 27 , 29 , 45 , 46 ] This might be important, because in case of high intermolecular association behavior self‐quenching of excited states can get more favorable.[ 29 , 47 , 48 , 49 ]
Single crystals of Rubiipo suitable for X‐ray diffraction analysis were obtained by slow evaporation from a saturated acetonitrile/water mixture. Rubiipo crystallizes in the monoclinic space group C2/c (Figure 3). Due to the asymmetric structure of biipo, two enantiomers of Rubiipo are possible causing a geometrical disorder (see Figure S3.11) of the respective solid‐state structure. Please note, that Rubiipo does not have different diastereomers, but exists as a mixture of enantiomers (Figure S3.13). [50] As clearly evident from the X‐ray structural data, there are the expected enantiomers of the corresponding regioisomers of Rubiipo (Figure S3.13). In addition, the biipo ligand facilitates a certain degree of π–π interactions between the individual metal complex cations (Figure S3.12) similar to other extended rigid polyaromatic diimine ligands.[ 47 , 51 ] The obtained unit cell contains 8 Ru complexes along with 16 PF6 − anions and 30 acetonitrile molecules. This resulted in a challenging crystal structure, where the second hexafluorophosphate anion is overlaid by several acetonitrile molecules. Interestingly, this obstacle and the need for several restraints was found in multiple measurements, but prevents a detailed discussion of exact bond lengths and angles. Nevertheless, with a final R‐value of 6.4 % the solid‐state structure is still well suited to show the general properties, for example, the conventional distorted octahedral type coordination environment around the ruthenium center [52] and the planar structure of biipo.
Figure 3.
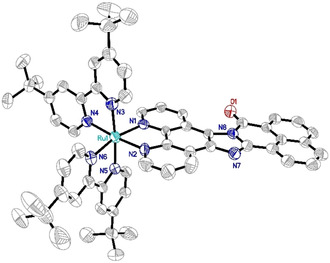
Solid‐state structure (ORTEP representation) of Rubiipo with thermal ellipsoids at a probability level of 50 %. Hydrogen atoms, PF6 − counterions and solvent molecules are omitted for clarity. Deposition number 1993565 (Rubiipo) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Electrochemical properties
The redox properties of Rubiipo and biipo were studied by cyclic voltammetry and differential pulse voltammetry (Figure 4, Table 1) in acetonitrile solution containing 0.1 m [Bu4N][PF6]. The reversible RuII/RuIII oxidation couple is located at +0.80 V vs. Fc/Fc+, which is in accordance to related RuII complexes, for example, Ruphen (+0.78 V) or Rubpy (+0.89 V).[ 53 , 54 ]
Figure 4.
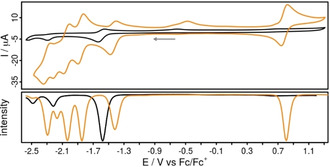
Cyclic voltammograms (top) and differential pulse voltammograms (DPV, bottom) of biipo (black, concentration <1 mm due to low solubility in acetonitrile) and Rubiipo (orange, 1 mm) in acetonitrile solution referenced vs. the ferrocene/ferricenium (Fc/Fc+) couple. The current values of the DPV are normalized. Conditions: scan rate of 100 mV s−1, [Bu4N][PF6] (0.1 m) as supporting electrolyte.
Table 1.
Summary of the photophysical and electrochemical properties of biipo, Rubiipo, and of selected reference compounds in acetonitrile solution at room temperature. The electrochemical data were obtained from deaerated acetonitrile solutions at room temperature and are referenced versus the ferrocene/ferricenium (Fc/Fc+) couple.
|
Compound |
λ abs,max [nm] (ϵ [103 m −1 cm−1]) |
λ em [nm] inert |
ϕem inert |
ϕem O2 |
τ em [ns] inert |
τ em [ns] O2 |
E 1/2ox [V] |
E 1/2red [V] |
|---|---|---|---|---|---|---|---|---|
|
biipo |
409 (6.2) |
540 |
0.258 |
0.428 |
<10[b] |
<10[b] |
– |
−1.59, −2.29[c] |
|
phen [a] |
265 (32.0) |
365 |
<0.01 |
|
|
|
|
|
|
Rubiipo |
411 (26.3), 460 (24.7) |
623 |
0.022 |
0.006 |
1668, 24 717 |
222 |
+0.80 |
−1.44, −1.86 −2.04, −2.21[c], −2.31 |
|
Ruphen [a] |
426 (16), 455 (19) |
602 |
0.03 |
– |
272 |
– |
+0.79 |
−1.79, −1.99, −2.26 |
[a] Taken from reference [57]; [b] the lifetime was below the detection limit; [c] irreversible redox event.
The reversible reduction waves at −1.44, −1.86, −2.04 and −2.31 V are characteristic for [(bpy)2Ru(L)]2+ type complexes, where L represents a phen derivative with an extended π‐system.[ 41 , 47 ] These potentials can be assigned to the stepwise reduction of (i) the naphthaloylenebenzene moiety of biipo, (ii)/(iii) the two tbbpy ligands and (iv) the phen part of the biipo ligand, respectively.[ 55 , 56 ] The first reduction event of Rubiipo (−1.44 V) is 150 mV shifted to more positive potentials compared to biipo (−1.59 V). This reduction is reversible for both compounds as also evidenced by scan‐rate dependent CV measurements (Figure S5.2). This process can be attributed to the naphthaloylenebenzene moiety of biipo, which is supported by the DFT calculations of the reduced ligand, showing that the spin density is located at the backbone of the biipo ligand (Figure S8.2). However, the irreversible reduction process at −2.21 V of Rubiipo seems to be related to the imidazole unit as known from the literature. [41]
Absorption and emission properties
The biipo ligand absorbs in acetonitrile in a broad visible range up to 500 nm (Figure 5). This is a significant redshift compared to the phen ligand, which possesses an absorption maximum at around 365 nm. [57] The energetically lowest lying band could be assigned by TD‐DFT calculations to a ligand centered (LC) π–π* transition from the phenanthroline/imidazole part to the naphthaloylenebenzene moiety of biipo (480 nm, transition 1, Figure 5 and Supporting Information). At higher energies (i.e., the band at 350 nm) the situation is different and a transition from the naphthaloylenebenzene part to the phenanthroline unit occurs. These results indicate that although in the biipo ligand the naphthaloylenebenzene moiety is directly fused to the phenanthroline part, this ligand is still split into two separated halves concerning its orbitals in the ground and excited states. This results in a very broad absorption of the pure ligand.
Figure 5.
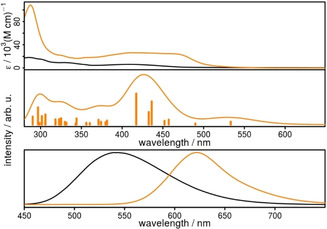
Spectra of biipo (black) and Rubiipo (orange). Top) UV/Vis absorption spectrum in acetonitrile. Middle) Calculated absorption spectrum of Rubiipo resulting from the convolution (std. deviation 40) of the transitions above 290 nm (vertical bars) with a Gaussian function. The transition energies are calculated at full TD‐DFT level of theory (B3LYP‐D3(BJ)/def2‐TZVP. Bottom) Emission spectra in acetonitrile under oxygen‐free conditions.
In comparison to Ruphen, [57] having an absorption maximum at 444 nm in the metal‐to‐ligand charge transfer (MLCT) transition, Rubiipo possesses a broader MLCT band with two maxima located at 411 and 460 nm, respectively. This latter prominent feature can be explained by MLCT transitions from the ruthenium center to the two tbbpy ligands and to the phen part of biipo (Table S9.2) Additionally, the MLCT transition with the lowest energy ends also directly at the naphthaloylenebenzene moiety (c.f. inset of Figure 6, Supporting Information) as supported by TDDFT calculations. However, in the calculations the LC π–π* transitions of Rubiipo are redshifted from 320 nm to 370 nm compared to the transitions of the uncoordinated biipo ligand. Contrary, the π–π* transition to the naphthaloylenebenzene moiety is clearly blueshifted from 480 nm (biipo) to 417 nm (Rubiipo, transition 12, Table S9.2). For these reasons, the absorption maximum at around 411 nm is very intense causing the high attenuation coefficient of Rubiipo in this region of the absorption spectrum. Nevertheless, above 420 nm the MLCT transitions in Rubiipo are not overlaid by the LC transitions, which is commonly observed for related systems containing diimine ligands with an extended π‐system.[ 41 , 49 , 58 , 59 , 60 ] There, the MLCT transitions are often superimposed by the LC transitions.[ 41 , 49 ]
Figure 6.
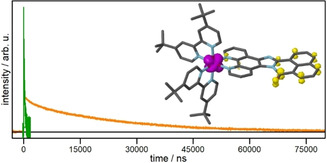
Emission lifetime of Rubiipo in acetonitrile under ambient (green) and inert conditions (orange). The respective fits and the residuals are depicted in the Supporting Information. The inset (differential density plot of Rubiipo) shows one of the occurring MLCT transitions from the ruthenium d orbital to the π* orbital located at the naphthaloylenebenzene moiety.
The emission properties of biipo and Rubiipo differ significantly from each other. The ligand emits at about 540 nm and is almost unaffected by the presence of oxygen in the acetonitrile solution (Figure 5, bottom). In strong contrast, Rubiipo is almost non‐emissive under aerated conditions, but exhibits a clear emission at 623 nm in the absence of oxygen with a quantum yield of about 2.2 % (Table 1). This quantum yield is in the same order of magnitude as some related RuII complexes, [52] but lower than a heteroleptic RuII complex with an azabenz‐annulated perylene bisimide (11 %). [28] Remarkably, the emission intensity is drastically reduced already by small traces of oxygen. Thus, Rubiipo is highly sensitive vs. oxygen and a good oxygen sensor due to the strong quenching of emission. [61]
In accordance with the steady state emission measurements the emission lifetimes of Rubiipo were also determined in acetonitrile (Figure 6). Under aerated conditions a lifetime of 222 ns was obtained (Figure S6.1), which is comparable to the related complex [(bpy)2Ru(PNI‐phen)]2+ (with PNI=4‐piperidinyl‐1,8‐naphthalimide), where the π‐extension is not directly fused to the phenanthroline part. [25] Under inert conditions the emissive excited state lives remarkably longer (Figure 6). Analysis of the kinetics revealed a biexponential decay with two time constants of τ1=1.7 μs and τ2=24.7 μs, which belong to LC or MLCT transitions. Thus, Rubiipo emits about 100 times longer when changing from ambient to oxygen free conditions. This is again comparable to [(bpy)2Ru(PNI‐phen)]2+ with a lifetime of 27.6 μs under inert conditions. Nevertheless, to the best of our knowledge this is a large increase in excited state lifetime for such a RuII complex containing a diimine ligand with a fully conjugated and extended π‐system. Interestingly, the second time constant is highly oxygen sensitive. As soon as Rubiipo gets in contact with O2, τ2 considerably decreases to only sub microseconds. This again underlines the strong oxygen sensitivity of Rubiipo.
Transient absorption spectroscopy
To verify the obtained excited state lifetimes by a different method time‐resolved transient absorption spectroscopy was performed. Under inert conditions and excited at 355 nm, a very broad excited state absorption from 480 to 700 nm was obtained for Rubiipo in acetonitrile solution (Figure 7). This is a typical observation, if ligand‐centred transitions are detected by transient absorption spectroscopy.[ 60 , 62 ] Due to this feature, the expected ground state bleach was only extended up to 440 nm and not up to 500 nm, as estimated from the steady state absorption spectra (Figure 5 top). The kinetics (Figure 7) were analyzed globally over the whole range from 370 to 700 nm by a biexponentially fit function. The received lifetimes τ1=2.5 and τ2=24.9 μs are comparable to the ones obtained for the emission lifetime under oxygen‐free conditions. It should be noted that especially the longer lifetime is very oxygen sensitive. As soon as there are traces of oxygen present, this time constant decreases significantly. Thus, it is very important to work under fully inert conditions for the determination of the excited state lifetimes of Rubiipo. Under aerobe conditions the monoexponential fit of the data results in one time constant, that is, τ=261 ns, which is also comparable to the obtained emission lifetimes in the presence of oxygen.
Figure 7.
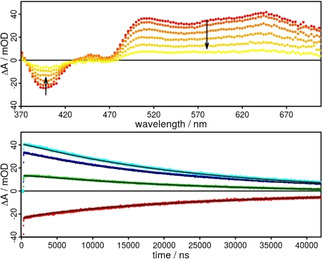
Transient absorption spectra (top) from 500 (red) to 40 000 ns (yellow) and respective kinetics (bottom) of Rubiipo in acetonitrile excited at 355 nm under inert conditions. Kinetics at 390 (red), 480 (green), 500 (blue), and 650 nm (light blue), the black line belongs to the fits with two time constants of 2.5 and 24.9 μs.
Oxygen sensitivity and productivity
Motivated by the strong oxygen sensitivity Rubiipo was tested for its ability to produce singlet oxygen upon light irradiation. 1O2 is the leading actor in Type II pathway of Photodynamic Therapy (PDT) actions. An efficient production of reactive singlet oxygen under oxygen containing conditions is still a crucial point for this type of application.[ 5 , 63 ] To verify the 1O2 production by Rubiipo, a solution of ABDA (9,10‐anthracenediyl‐bis(methylene)dimalonic acid) in PBS (phosphate buffered solution) was added to distinct quantities (c=1 μm) of Rubiipo, Rubpy and Ruphen (ABDA:Ru=50:1), respectively. ABDA is a commonly applied singlet oxygen detection agent, due to its very selective reaction with this reactive oxygen species (ROS). 1O2 and ABDA build up an endoperoxide (Figure S4.5), which loses the characteristic anthracene absorption bands at around 350 to 410 nm.[ 64 , 65 , 66 ] Upon irradiation with blue light (LED, 470 nm, 50 mW cm−2) under constant cooling Rubiipo (Figure 8, top) shows very fast depletion of the ABDA absorbance at around 360, 380, and 400 nm. In order to evaluate the singlet oxygen productivity, the decrease of this absorption band was plotted against time until complete consumption of ABDA (Figure 8, bottom). The observed decay was not linear, but seemed to be an exponential decrease, which is much faster compared to Ruphen (Figure 8, bottom). Whereas also Rubpy evolves 1O2 much slower compared to Rubiipo (Figures S4.6–S4.10). Nevertheless, a better energy transfer rate from Rubiipo to oxygen can neither be assessed nor excluded based on the current data and further investigations concerning this issue are needed.
Figure 8.
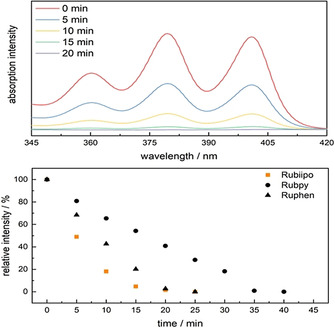
Top) Singlet oxygen evolution experiment using Rubiipo (1 equiv.) and ABDA (50 equiv.) in PBS after irradiation with blue light (50 mWcm−2, 470 nm) in the range of ABDA absorbance (see also Figure S4.5). Bottom) Comparison of the relative ABDA absorbance as function of time during irradiation of Rubiipo (squares), Rubpy (circles) and Ruphen (triangles) with blue light (50 mWcm−2, 470 nm). It becomes clear, that Rubiipo degrades ABDA the fastest. See for the specific ABDA test in the Supporting Information the section 4.
Conclusions
In summary, starting from 1,10‐phenanthroline a new type of a polycyclic, fully conjugated, and rigid diimine ligand with a naphthaloylenebenzene moiety in the back was prepared for the first time. Based on the special ligand structure, a RuII photosensitizer (Rubiipo), also verified by single crystal X‐ray diffraction analysis, with some unique properties was formed. For instance, Rubiipo shows a reversible redox chemistry (i.e., four reductions and one oxidation) as well as a strong and broad absorption in the visible with two maxima at 411 and 460 nm, respectively. More importantly, Rubiipo possesses a clear emission at 623 nm with very long‐lived excited states of 1.7 and 24.7 μs in acetonitrile in the absence of oxygen. Interestingly, the emission intensity and lifetimes are drastically decreased (by a factor of >100) by O2, which indicates a high oxygen sensitivity of this complex. In the future, temperature‐dependent emission, emission lifetime, and spectroelectrochemical measurements are planned, to clarify the nature of the excited states further. In addition, a detailed investigation of the oxygen sensing ability of Rubiipo will be assessed by Stern–Volmer quenching experiments.
Most importantly, beside potential applications as an oxygen sensor Rubiipo is able to produce singlet oxygen under visible light irradiation with very fast kinetics. Therefore, the study of further applications of the Rubiipo photosensitizer as well as of other transition metal complexes using the new biipo ligand are currently ongoing.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Y.Y., M.K., and S.T. gratefully acknowledge the German Science Foundation (DFG, Priority Program SPP 2102 “Light‐controlled reactivity of metal complexes” (KA 4671/2‐1 and TS 330/4‐1) and the DFG project TS 330/3‐1) for funding. S.R. is thankful for financial support from the DFG within the collaborative research center TRR 234 “Catalight”. J.B. gratefully acknowledges the Fonds der Chemischen Industrie (FCI) for a Kekulé‐Stipendium. The authors acknowledge support by the state of Baden‐Württemberg through bwHPC and by the DFG (INST 40/467‐1 FUGG). The authors also thank Bernd Maier, Martin Rentschler, and Julian Bösking for their synthetic work and Dr. Dieter Sorsche for providing X‐ray data. Open access funding enabled and organized by Projekt DEAL.
Y. Yang, J. Brückmann, W. Frey, S. Rau, M. Karnahl, S. Tschierlei, Chem. Eur. J. 2020, 26, 17027.
Contributor Information
Dr. Michael Karnahl, Email: michael.karnahl@oc.uni-stuttgart.de.
Prof. Dr. Stefanie Tschierlei, Email: s.tschierlei@tu-bs.de.
References
- 1. Shaw M. H., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Malzkuhn S., Wenger O. S., Coord. Chem. Rev. 2018, 35952–35956. [Google Scholar]
- 3. Kainz Q. M., Matier C. D., Bartoszewicz A., Zultanski S. L., Peters J. C., Fu G. C., Science 2016, 351, 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Twilton J., Le C. C., Zhang P., Shaw M. H., Evans R. W., MacMillan D. W. C., Nat. Rev. Chem. 2017, 1, 0052. [Google Scholar]
- 5. Eisenberg R., Science 2009, 324, 44. [DOI] [PubMed] [Google Scholar]
- 6. Frischmann P. D., Mahata K., Würthner F., Chem. Soc. Rev. 2013, 42, 1847–1870. [DOI] [PubMed] [Google Scholar]
- 7. Berardi S., Drouet S., Francàs L., Gimbert-Suriñach C., Guttentag M., Richmond C., Stoll T., Llobet A., Chem. Soc. Rev. 2014, 43, 7501–7519. [DOI] [PubMed] [Google Scholar]
- 8. Yuan Y.-J., Yu Z.-T., Chen D.-Q., Zou Z.-G., Chem. Soc. Rev. 2017, 46, 603–631. [DOI] [PubMed] [Google Scholar]
- 9. Dougherty T. J., Gomer C. J., Henderson B. W., Jori G., Kessel D., Korbelik M., Moan J., Peng Q., J. Natl. Cancer Inst. 1998, 90, 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ogilby P. R., Chem. Soc. Rev. 2010, 39, 3181–3209. [DOI] [PubMed] [Google Scholar]
- 11. Van Straten D., Mashayekhi V., De Bruijn S. H., Oliveira S., Robinson J. D., Cancers 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Monro S., Colón K. L., Yin H., Roque J., Konda P., Gujar S., Thummel R. P., Lilge L., Cameron C. G., McFarland S. A., Chem. Rev. 2019, 119, 797–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeRosa M. C., Crutchley R. J., Coord. Chem. Rev. 2002, 233–234, 351–234371. [Google Scholar]
- 14. Mari C., Pierroz V., Ferrari S., Gasser G., Chem. Sci. 2015, 6, 2660–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi G., Monro S., Hennigar R., Colpitts J., Fong J., Kasimova K., Yin H., DeCoste R., Spencer C., Chamberlain L., Mandel A., Lilge L., McFarland S. A., Coord. Chem. Rev. 2015, 282–283, 127–138. [Google Scholar]
- 16. Heinemann F., Karges J., Gasser G., Acc. Chem. Res. 2017, 50, 2727–2736. [DOI] [PubMed] [Google Scholar]
- 17. Konduri R., de Tacconi N. R., Rajeshwar K., MacDonnell F. M., J. Am. Chem. Soc. 2004, 126, 11621–11629. [DOI] [PubMed] [Google Scholar]
- 18. Singh S., de Tacconi N. R., Diaz N. R. G., Lezna R. O., Muñoz Zuñiga J., Abayan K., MacDonnell F. M., Inorg. Chem. 2011, 50, 9318–9328. [DOI] [PubMed] [Google Scholar]
- 19. Karlsson S., Boixel J., Pellegrin Y., Blart E., Becker H.-C., Odobel F., Hammarström L., Faraday Discuss. 2012, 155, 233–252. [DOI] [PubMed] [Google Scholar]
- 20. Aslan J. M., Boston D. J., MacDonnell F. M., Chem. Eur. J. 2015, 21, 17314–17323. [DOI] [PubMed] [Google Scholar]
- 21. Hammarström L., Acc. Chem. Res. 2015, 48, 840–850. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y., Traber P., Zedler L., Kupfer S., Gräfe S., Schulz M., Frey W., Karnahl M., Dietzek B., Phys. Chem. Chem. Phys. 2018, 20, 24843–24857. [DOI] [PubMed] [Google Scholar]
- 23. Yonemoto E. H., Riley R. L., Kim Y. I., Atherton S. J., Schmehl R. H., Mallouk T. E., J. Am. Chem. Soc. 1992, 114, 8081–8087. [Google Scholar]
- 24. Lavie-Cambot A., Lincheneau C., Cantuel M., Leydet Y., McClenaghan N. D., Chem. Soc. Rev. 2010, 39, 506–515. [DOI] [PubMed] [Google Scholar]
- 25. Tyson D. S., Luman C. R., Zhou X., Castellano F. N., Inorg. Chem. 2001, 40, 4063–4071. [DOI] [PubMed] [Google Scholar]
- 26. Polyansky D. E., Danilov E. O., Castellano F. N., Inorg. Chem. 2006, 45, 2370–2372. [DOI] [PubMed] [Google Scholar]
- 27. Hankache J., Wenger O. S., Phys. Chem. Chem. Phys. 2012, 14, 2685–2692. [DOI] [PubMed] [Google Scholar]
- 28. Schulze M., Steffen A., Würthner F., Angew. Chem. Int. Ed. 2015, 54, 1570–1573; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1590–1593. [Google Scholar]
- 29. Langhals H., Heterocycles 1995, 40, 477–500. [Google Scholar]
- 30. Herrmann A., Müllen K., Chem. Lett. 2006, 35, 978–985. [Google Scholar]
- 31. Weil T., Vosch T., Hofkens J., Peneva K., Müllen K., Angew. Chem. Int. Ed. 2010, 49, 9068–9093; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9252–9278. [Google Scholar]
- 32. Pan M., Lin X.-M., Li G.-B., Su C.-Y., Coord. Chem. Rev. 2011, 255, 1921–1936. [Google Scholar]
- 33. Castellano F. N., Dalton Trans. 2012, 41, 8493–8501. [DOI] [PubMed] [Google Scholar]
- 34. Suraru S.-L., Würthner F., Angew. Chem. Int. Ed. 2014, 53, 7428–7448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7558–7578. [Google Scholar]
- 35. Würthner F., Saha-Möller C. R., Fimmel B., Ogi S., Leowanawat P., Schmidt D., Chem. Rev. 2016, 116, 962–1052. [DOI] [PubMed] [Google Scholar]
- 36. Langhals H., Jaschke H., Chem. Eur. J. 2006, 12, 2815–2824. [DOI] [PubMed] [Google Scholar]
- 37. Yuan Z., Xiao Y., Li Z., Qian X., Org. Lett. 2009, 11, 2808–2811. [DOI] [PubMed] [Google Scholar]
- 38. Mamada M., Pérez-Bolívar C., Kumaki D., Esipenko N. A., Tokito S., P. Anzenbacher, Jr. , Chem. Eur. J. 2014, 20, 11835–11846. [DOI] [PubMed] [Google Scholar]
- 39. Karnahl M., Tschierlei S., Kuhnt C., Dietzek B., Schmitt M., Popp J., Schwalbe M., Krieck S., Görls H., Heinemann F. W., Rau S., Dalton Trans. 2010, 39, 2359–2370. [DOI] [PubMed] [Google Scholar]
- 40. Yin H., Stephenson M., Gibson J., Sampson E., Shi G., Sainuddin T., Monro S., McFarland S. A., Inorg. Chem. 2014, 53, 4548–4559. [DOI] [PubMed] [Google Scholar]
- 41. Isakov D., Giereth R., Nauroozi D., Tschierlei S., Rau S., Inorg. Chem. 2019, 58, 12646–12653. [DOI] [PubMed] [Google Scholar]
- 42. Skaisgirski M., Guo X., Wenger O. S., Inorg. Chem. 2017, 56, 2432–2439. [DOI] [PubMed] [Google Scholar]
- 43. Steullet V., Dixon D. W., J. Chem. Soc. Perkin Trans. 2 1999, 1547–1558. [Google Scholar]
- 44. de Tacconi N. R., Chitakunye R., MacDonnell F. M., Lezna R. O., J. Phys. Chem. A 2008, 112, 497–507. [DOI] [PubMed] [Google Scholar]
- 45. Pfeffer M. G., Pehlken C., Staehle R., Sorsche D., Streb C., Rau S., Dalton Trans. 2014, 43, 13307–13315. [DOI] [PubMed] [Google Scholar]
- 46. Huber F., Nauroozi D., Mengele A. K., Rau S., Eur. J. Inorg. Chem. 2017, 4020–4027. [Google Scholar]
- 47. Bolger J., Gourdon A., Ishow E., Launay J.-P., Inorg. Chem. 1996, 35, 2937–2944. [Google Scholar]
- 48. Bergman S. D., Kol M., Inorg. Chem. 2005, 44, 1647–1654. [DOI] [PubMed] [Google Scholar]
- 49. Troian-Gautier L., Moucheron C., Molecules 2014, 19, 5028–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gong L., Wenzel M., Meggers E., Acc. Chem. Res. 2013, 46, 2635–2644. [DOI] [PubMed] [Google Scholar]
- 51. Karnahl M., Kuhnt C., Heinemann F. W., Schmitt M., Rau S., Popp J., Dietzek B., Chem. Phys. 2012, 393, 65–73. [Google Scholar]
- 52. Juris A., Balzani V., Barigelletti F., Campagna S., Belser P., von Zelewsky A., Coord. Chem. Rev. 1988, 8485–8277. [Google Scholar]
- 53. Rau S., Fischer R., Jäger M., Schäfer B., Meyer S., Kreisel G., Görls H., Rudolf M., Henry W., Vos J., Eur. J. Inorg. Chem. 2004, 2001–2003. [Google Scholar]
- 54. Büldt L. A., Prescimone A., Neuburger M., Wenger O. S., Eur. J. Inorg. Chem. 2015, 4666–4677. [Google Scholar]
- 55. Wintergerst P., Mengele A. K., Nauroozi D., Tschierlei S., Rau S., Eur. J. Inorg. Chem. 2019, 1988–1992. [Google Scholar]
- 56. Fan S.-H., Zhang A.-G., Ju C.-C., Gao L.-H., Wang K.-Z., Inorg. Chem. 2010, 49, 3752–3763. [DOI] [PubMed] [Google Scholar]
- 57. Karnahl M., Krieck S., Görls H., Tschierlei S., Schmitt M., Popp J., Chartrand D., Hanan G. S., Groarke R., Vos J. G., Rau S., Eur. J. Inorg. Chem. 2009, 4962–4971. [Google Scholar]
- 58. Giereth R., Reim I., Frey W., Junge H., Tschierlei S., Karnahl M., Sustainable Energy Fuels 2019, 3, 692–700. [Google Scholar]
- 59. Sun Y., Joyce L. E., Dickson N. M., Turro C., Chem. Commun. 2010, 46, 2426–2428. [DOI] [PubMed] [Google Scholar]
- 60. Schindler J., Zhang Y., Traber P., Lefebvre J.-F., Kupfer S., Demeunynck M., Gräfe S., Chavarot-Kerlidou M., Dietzek B., J. Phys. Chem. C 2018, 122, 83–95. [DOI] [PubMed] [Google Scholar]
- 61. Demas J. N., DeGraff B. A., Coleman P. B., Anal. Chem. 1999, 71, 793A-800A. [DOI] [PubMed] [Google Scholar]
- 62. Tyson D. S., Henbest K. B., Bialecki J., Castellano F. N., J. Phys. Chem. A 2001, 105, 8154–8161. [Google Scholar]
- 63. Plaetzer K., Krammer B., Berlanda J., Berr F., Kiesslich T., Lasers Med. Sci. 2009, 24, 259–268. [DOI] [PubMed] [Google Scholar]
- 64. Wang T., Zabarska N., Wu Y., Lamla M., Fischer S., Monczak K., Ng D. Y. W., Rau S., Weil T., Chem. Commun. 2015, 51, 12552–12555. [DOI] [PubMed] [Google Scholar]
- 65. Planas O., Macia N., Agut M., Nonell S., Heyne B., J. Am. Chem. Soc. 2016, 138, 2762–2768. [DOI] [PubMed] [Google Scholar]
- 66. Chakrabortty S., Agrawalla B. K., Stumper A., Vegi N. M., Fischer S., Reichardt C., Kögler M., Dietzek B., Feuring-Buske M., Buske C., Rau S., Weil T., J. Am. Chem. Soc. 2017, 139, 2512–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary