

Key Points
Questions
What is the effect of early treatment with antispike neutralizing antibodies on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral load in outpatients with mild to moderate coronavirus disease 2019 (COVID-19)?
Findings
In the phase 2 portion of a randomized phase 2/3 clinical trial with 577 patients, there was no significant difference in change in viral load with 3 different doses of bamlanivimab monotherapy compared with placebo; treatment with a combination of bamlanivimab and etesevimab significantly decreased SARS-CoV-2 log viral load at day 11 compared with placebo (between-group difference, –0.57 [95% CI, –1.00 to –0.14], P = .01).
Meaning
Treatment with bamlanivimab and etesevimab combination therapy, but not bamlanivimab monotherapy, resulted in a reduction in SARS-CoV-2 log viral load at day 11 in patients with mild to moderate COVID-19.
Abstract
Importance
Coronavirus disease 2019 (COVID-19) continues to spread rapidly worldwide. Neutralizing antibodies are a potential treatment for COVID-19.
Objective
To determine the effect of bamlanivimab monotherapy and combination therapy with bamlanivimab and etesevimab on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral load in mild to moderate COVID-19.
Design, Setting, and Participants
The BLAZE-1 study is a randomized phase 2/3 trial at 49 US centers including ambulatory patients (N = 613) who tested positive for SARS-CoV-2 infection and had 1 or more mild to moderate symptoms. Patients who received bamlanivimab monotherapy or placebo were enrolled first (June 17-August 21, 2020) followed by patients who received bamlanivimab and etesevimab or placebo (August 22-September 3). These are the final analyses and represent findings through October 6, 2020.
Interventions
Patients were randomized to receive a single infusion of bamlanivimab (700 mg [n = 101], 2800 mg [n = 107], or 7000 mg [n = 101]), the combination treatment (2800 mg of bamlanivimab and 2800 mg of etesevimab [n = 112]), or placebo (n = 156).
Main Outcomes and Measures
The primary end point was change in SARS-CoV-2 log viral load at day 11 (±4 days). Nine prespecified secondary outcome measures were evaluated with comparisons between each treatment group and placebo, and included 3 other measures of viral load, 5 on symptoms, and 1 measure of clinical outcome (the proportion of patients with a COVID-19–related hospitalization, an emergency department [ED] visit, or death at day 29).
Results
Among the 577 patients who were randomized and received an infusion (mean age, 44.7 [SD, 15.7] years; 315 [54.6%] women), 533 (92.4%) completed the efficacy evaluation period (day 29). The change in log viral load from baseline at day 11 was –3.72 for 700 mg, –4.08 for 2800 mg, –3.49 for 7000 mg, –4.37 for combination treatment, and –3.80 for placebo. Compared with placebo, the differences in the change in log viral load at day 11 were 0.09 (95% CI, –0.35 to 0.52; P = .69) for 700 mg, –0.27 (95% CI, –0.71 to 0.16; P = .21) for 2800 mg, 0.31 (95% CI, –0.13 to 0.76; P = .16) for 7000 mg, and –0.57 (95% CI, –1.00 to –0.14; P = .01) for combination treatment. Among the secondary outcome measures, differences between each treatment group vs the placebo group were statistically significant for 10 of 84 end points. The proportion of patients with COVID-19–related hospitalizations or ED visits was 5.8% (9 events) for placebo, 1.0% (1 event) for 700 mg, 1.9% (2 events) for 2800 mg, 2.0% (2 events) for 7000 mg, and 0.9% (1 event) for combination treatment. Immediate hypersensitivity reactions were reported in 9 patients (6 bamlanivimab, 2 combination treatment, and 1 placebo). No deaths occurred during the study treatment.
Conclusions and Relevance
Among nonhospitalized patients with mild to moderate COVID-19 illness, treatment with bamlanivimab and etesevimab, compared with placebo, was associated with a statistically significant reduction in SARS-CoV-2 viral load at day 11; no significant difference in viral load reduction was observed for bamlanivimab monotherapy. Further ongoing clinical trials will focus on assessing the clinical benefit of antispike neutralizing antibodies in patients with COVID-19 as a primary end point.
Trial Registration
ClinicalTrials.gov Identifier: NCT04427501
This randomized clinical trial compares the effects of 3 doses of bamlanivimab monotherapy (700 vs 2800 vs 7000 mg) vs combination bamlanivimab and etesevimab vs placebo on change in day 11 severe acute respiratory syndrome coronavirus 2 viral load in patients with mild to moderate coronavirus disease 2019 (COVID-19).
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) continues to spread rapidly worldwide, fueling the coronavirus disease 2019 (COVID-19) global pandemic. Patients infected with the virus display a wide range of symptoms including cough, fever, malaise, myalgias, gastrointestinal symptoms, ageusia, and anosmia; some individuals progress to acute respiratory distress syndrome and death. Severe illness typically occurs approximately 1 week after the onset of symptoms and can rapidly progress from mild symptoms.1 The risk factors for severe COVID-19 include being male, older age, and having cardiovascular disease, lung disease, hypertension, diabetes, or obesity.2,3
Currently, only remdesivir (a viral RNA–dependent RNA polymerase inhibitor) has been approved by the US Food and Drug Administration for COVID-19 treatment, although steroids are now recommended by many professional societies, including the World Health Organization, as the primary treatment.4,5,6 However, convalescent plasma and neutralizing monoclonal antibodies, a class of therapeutics that have exhibited efficacy in other viral infections and show promise in the reduction of SARS-CoV-2 viral load, have been granted Emergency Use Authorization.7,8,9,10,11,12
Bamlanivimab (also known as LY3819253 or LY-CoV555) and etesevimab (LY3832479 or LY-CoV016) are potent antispike neutralizing monoclonal antibodies that were derived from 2 separate patients who recovered from COVID-19 in North America and China, respectively.13,14 In preclinical experiments, etesevimab was shown to bind a different epitope from bamlanivimab and to neutralize resistant variants with mutations in the epitope bound by bamlanivimab (eTable 1 in Supplement 1). Combining these 2 neutralizing monoclonal antibodies in clinical use may enhance viral load reduction and decrease treatment-emergent resistant variants.15
Interim results from the Blocking Viral Attachment and Cell Entry with SARS-CoV-2 Neutralizing Antibodies (BLAZE-1) trial with data for the 3 monotherapy doses of the neutralizing antibody bamlanivimab have been published.9 The current report presents the final data set for patients randomized to the 4 treatment groups and the placebo group in the initial portion of the trial, including findings for the bamlanivimab and etesevimab combination group, the 3 bamlanivimab monotherapy groups, and the placebo group.
Methods
Study Design
This clinical trial is an ongoing, multipart, phase 2/3, randomized, double-blind, placebo-controlled, single-infusion study including patients with recently diagnosed mild or moderate COVID-19 in the outpatient setting.9 The original and final protocol for the phase 2 trial, including the original and final statistical analysis plan, appear in Supplement 2. The trial complied with the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice, and applicable local regulations. The protocol was reviewed and approved by the ethics committees of all participating centers, and patients provided written informed consent before study entry.
Patients
All patients were aged 18 years or older, tested positive for SARS-CoV-2 infection, had 1 or more mild to moderate symptoms, and presented within 3 days of their first positive test result for SARS-CoV-2 (either direct antigen or reverse transcriptase–polymerase chain reaction). Mild to moderate COVID-19 was defined per US Food and Drug Administration guidance and included symptoms such as fever, cough, sore throat, malaise, headache, muscle pain, gastrointestinal symptoms, and shortness of breath with exertion. Investigators reviewed symptoms, risk factors, and other noninvasive inclusion and exclusion criteria prior to enrollment (the full list of inclusion and exclusion criteria appears in the clinical protocol in Supplement 2). Patient-reported race and ethnicity categories were collected as part of the demographic characteristics.
Randomization and Intervention
This study evaluated the effect of bamlanivimab monotherapy and combination therapy with bamlanivimab and etesevimab on change in viral load during treatment of mild to moderate COVID-19. All participants were centrally randomized to each study intervention using an interactive web response system (Figure 1). Before the study was initiated, the log-in information and directions for the interactive web response system was provided to each of the 49 US study sites.
Figure 1. Patient Enrollment and Treatment Assignment of the BLAZE-1 Trial of Bamlanivimab for Mild to Moderate COVID-19.
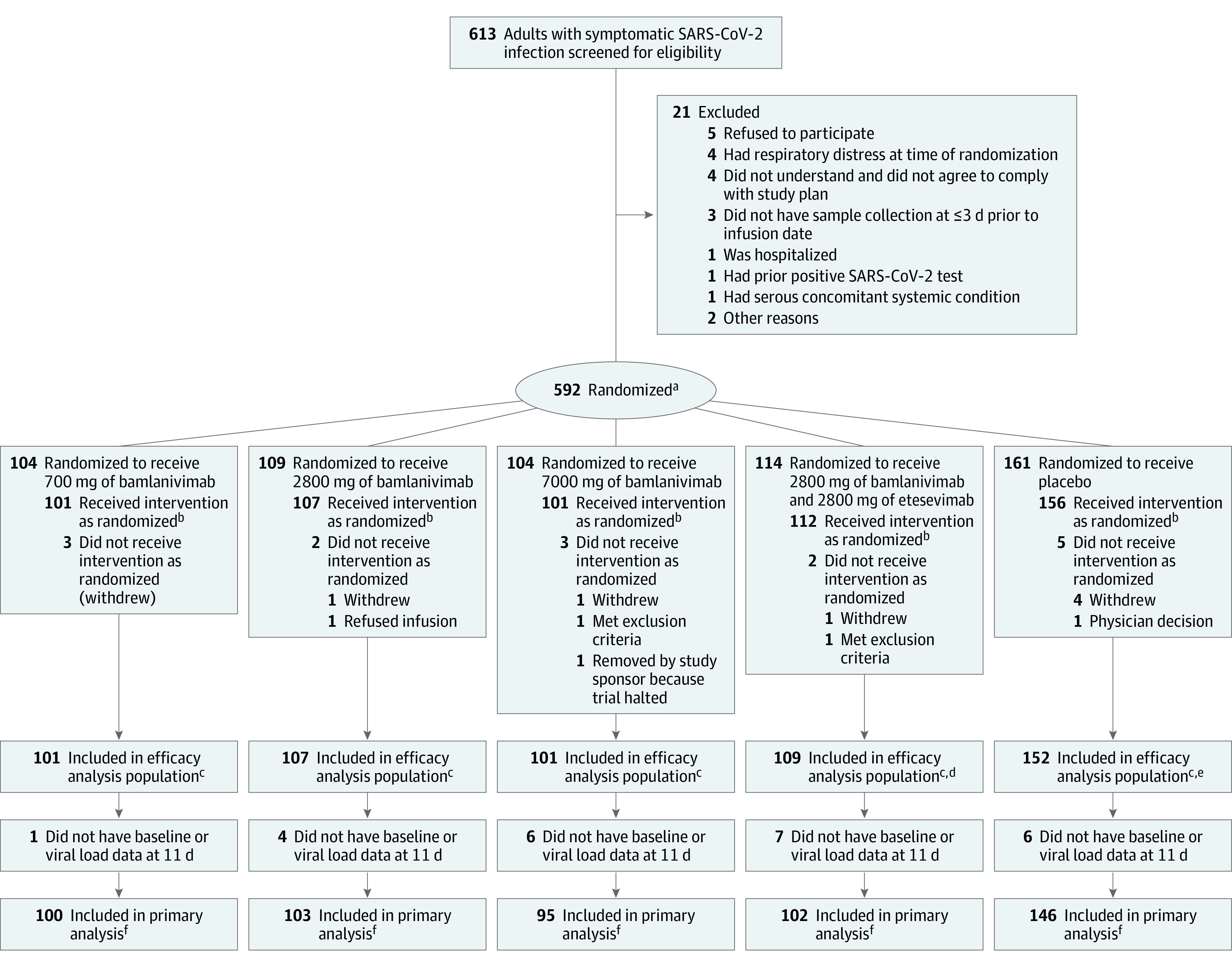
SARS-CoV-2 indicates severe acute respiratory syndrome coronavirus 2.
aStratified by duration since symptom onset to randomization (≤8 days vs >8 days).
bIncluded in the adverse event analysis.
cHad data on at least 1 postbaseline viral load.
dThree patients were excluded from the efficacy analysis because they did not have data on at least 1 postbaseline viral load. However, these patients were included in the safety analysis because they did receive the intervention as randomized.
eFour patients were excluded from the efficacy analysis because they did not have data on at least 1 postbaseline viral load. However, these patients were included in the safety analysis because they did receive the intervention as randomized.
fHad data on viral load for both baseline and at day 11.
Randomization was stratified by patients’ duration of symptoms (≤8 days vs >8 days) because symptom duration has an effect on prognosis.9 The treatment was administered within 3 days of the first positive SARS-CoV-2 test sample collection. Each patient in the trial received a single, 1-hour, intravenous infusion of placebo, bamlanivimab, or bamlanivimab and etesevimab. This final analysis includes results for the 5 treatment groups: placebo, 700 mg of bamlanivimab, 2800 mg of bamlanivimab, 7000 mg of bamlanivimab, and a combination treatment with 2800 mg of bamlanivimab and 2800 mg of etesevimab.
Patients who received bamlanivimab monotherapy or placebo were enrolled first (June 17-August 21, 2020) followed by patients who received bamlanivimab and etesevimab or placebo (August 22-September 3, 2020). The analysis was triggered on October 6, 2020, when the last patient randomized to treatment with bamlanivimab and etesevimab reached day 29 and includes all virological and symptom data available at that database lock. A previous report summarized earlier interim results of the 3 monotherapy doses of LY-CoV555 (bamlanivimab) vs placebo.9 The interim analysis was triggered on September 5, 2020.
Primary and Secondary Outcomes
The primary outcome characterized the effect of bamlanivimab monotherapy and combination therapy with bamlanivimab and etesevimab compared with placebo on SARS-CoV-2 log viral load from baseline to day 11 (±4 days). Viral load was measured by nasopharyngeal swab followed by quantitative reverse transcriptase–polymerase chain reaction at a central laboratory. Derivation of the viral load measure is described in §6.10 of the statistical analysis plan (Supplement 2).
A total of 9 prespecified secondary outcome measures were evaluated. Three focused on viral load (time to viral clearance; proportion of patients with viral clearance at days 7, 11, 15, and 22; and viral load area under the curve [AUC] at day 29), 5 focused on symptoms (change in symptom score at days 7, 11, 15, and 22; time to symptom improvement; time to symptom resolution; and the proportion of patients showing symptom improvement or resolution at days 7, 11, 15, and 22), and 1 focused on clinical outcomes (the proportion of patients with a COVID-19–related hospitalization, emergency department visit, or death) at day 29.
A questionnaire was used to assess symptom severity. The total symptom score (range, 0-24) was achieved by rating 8 symptom domains (cough, shortness of breath, feeling feverish, fatigue, body aches and pain, sore throat, chills, headache) from none or absent (score of 0) to severe (score of 3) and combining them to provide an overall score (excluding loss of appetite, taste, and smell).9
Adverse events or serious adverse events also were evaluated. The subgroup analyses for participants enrolled with shorter (≤8 days) and longer (>8 days) duration of symptoms prior to randomization were prespecified and performed, but because the subgroup with a symtom duration of longer than 8 days was only approximately 8% of the participants, the results of these analyses are not reported.
Exploratory Outcomes
The total symptom score AUC from day 0 to day 11 and from day 0 to day 29 were analyzed using a linear model, which contained treatment as a fixed effect and baseline severity as a covariate. To assess the prevalence of resistance variants, nasopharyngeal samples were obtained at study enrollment (baseline sample), and then subsequent sampling was done at days 3, 7, 11, 15, 22, and 29. A treatment-emergent variant was determined by comparing the sequencing results from each study participant’s baseline sample with the posttreatment samples. For instances in which a baseline next-generation sequencing result was not available (n = 37/448), the baseline status for these variants was imputed to the reference sequence of BetaCoV/Wuhan/IPBCAMS-WH-04/2019. Additional information about the methods used to detect resistance variants appears in the eMethods in Supplement 1.
Sample Size
A viral dynamic model was used to simulate viral loads over time for participants treated with bamlanivimab monotherapy, the bamlanivimab and etesevimab combination treatment, and placebo. This simulated population and Monte Carlo methods were used to estimate the statistical power associated with the comparison of change from baseline to day 11 (±4 days) in SARS-CoV-2 viral load between the treatment groups and the placebo group (additional details appear in §5.2 of the statistical analysis plan in Supplement 2).
Given these assumptions, a sample size of 100 participants per group was estimated to provide 91% power to test the superiority of bamlanivimab monotherapy or the bamlanivimab and etesevimab combination treatment vs placebo for the effect on viral load, as measured by change from baseline to day 11 (±4 days) at the 2-sided α level of .05.
Statistical Analyses
The SARS-CoV-2 viral load data were evaluated using a log base 10 scale. The treatment effects were compared using 2-sided tests with an α level of .05. Significance testing for the primary end point was performed using mixed-model repeated-measure analysis at the 2-sided .05 level. When the mixed-model repeated-measure analysis was used, it included: (1) treatment group, (2) stratification factor of duration since symptom onset to randomization (≤8 days vs >8 days), (3) baseline value in the model, (4) visit day (ie, 1, 3, 7, and 11), and (5) the treatment × visit interaction as fixed factors.
For the primary end point, the stratification factor of duration since symptom onset to randomization was not used in the model to avoid collinearity with baseline viral load. The Fisher exact test was used for the comparison of binary variables across treatment groups. Continuous outcome variables with a single time point were analyzed using analysis of covariance with (1) treatment group, (2) stratification factor of duration since symptom onset to randomization (≤8 days vs >8 days), and (3) baseline value in the model.
A post hoc analysis was performed evaluating COVID-19–related deterioration for patients aged 65 years or older or those with a body mass index (BMI; calculated as weight in kilograms divided by height in meters squared) of 35 or greater. COVID-19–related deterioration was defined as a patient experiencing a COVID-19–related hospitalization, an emergency department visit, or death.
Adjustments for multiple testing were not conducted for this study; therefore, the findings should be interpreted as exploratory. The full statistical analysis methods appear in §6.1 of the statistical analysis plan in Supplement 2. The statistical analyses were performed using Enterprise Guide 7.1 for SAS version 9.4 (SAS Institute Inc).
Results
Patient Demographics and Clinical Characteristics
At the time of the database lock (October 6, 2020), 577 patients had been randomized and had received an infusion of neutralizing monoclonal antibodies or placebo (Figure 1). There were 101 patients assigned to 700 mg of bamlanivimab, 107 patients assigned to 2800 mg of bamlanivimab, 101 patients assigned to 7000 mg of bamlanivimab, 112 patients assigned to combination therapy (2800 mg of bamlanivimab and 2800 mg of etesevimab), and 156 patients assigned to placebo. Patients in the bamlanivimab monotherapy groups, the bamlanivimab and etesevimab combination therapy group, and the placebo group were generally well balanced at the time of enrollment (Table 1).
Table 1. Patient Demographics and Baseline Clinical Characteristics.
| Characteristic | Bamlanivimab monotherapy | Combination therapy (2800 mg bamlanivimab and 2800 mg of etesevimab) (n = 112) |
Placebo (n = 156) |
||
|---|---|---|---|---|---|
| 700 mg (n = 101) |
2800 mg (n = 107) |
7000 mg (n = 101) |
|||
| Age | |||||
| Median (IQR), y | 39 (31-58) | 45 (31-56) | 46 (34-55) | 44 (30-60) | 46 (35-57) |
| ≥65 y, No. (%) | 11 (10.9) | 8 (7.5) | 14 (13.9) | 13 (11.6) | 23 (14.7) |
| Sex, No. (%) | |||||
| Female | 63 (62.4) | 51 (47.7) | 58 (57.4) | 58 (51.8) | 85 (54.5) |
| Male | 38 (37.6) | 56 (52.3) | 43 (42.6) | 54 (48.2) | 71 (45.5) |
| Self-reported race, No./total (%) | |||||
| White | 90/101 (89.1) | 90/104 (86.5) | 89/100 (89.0) | 105/111 (94.6) | 133/151 (88.1) |
| Black | 7/101 (6.9) | 7/104 (6.7) | 8/100 (8.0) | 4/111 (3.6) | 7/151 (4.6) |
| Asian | 1/101 (1.0) | 5/104 (4.8) | 3/100 (3.0) | 2/111 (1.8) | 8/151 (5.3) |
| American Indian or Alaska Native | 1/101 (1.0) | 0/104 | 0/100 | 0/111 | 2/151 (1.3) |
| Native Hawaiian or other Pacific Islander | 0/101 | 1/104 (1.0) | 0/100 | 0/111 | 0/151 |
| Multiple | 2/101 (2.0) | 1/104 (1.0) | 0/100 | 0/111 | 1/151 (0.7) |
| Self-reported ethnicity, No. (%) | |||||
| Hispanic | 49 (48.5) | 47 (43.9) | 39 (38.6) | 42 (37.5) | 68 (43.6) |
| Not Hispanic | 52 (51.5) | 60 (56.1) | 62 (61.4) | 70 (62.5) | 88 (56.4) |
| BMIa | |||||
| Median (IQR) | (n = 100) 28.8 (25.1-35.4) |
(n = 106) 30.4 (25.6-34.0) |
(n = 97) 27.8 (24.7-32.3) |
(n = 109) 27.2 (22.9-33.0) |
(n = 152) 29.2 (25.9-34.2) |
| ≥30 but <40, No./total (%) | 34/100 (34.0) | 50/106 (47.2) | 28/97 (28.9) | 33/109 (30.3) | 63/152 (41.4) |
| ≥40, No./total (%) | 11/100 (11.0) | 6/106 (5.7) | 7/97 (7.2) | 7/109 (6.4) | 9/152 (5.9) |
| Risk factors for severe COVID-19, No. (%)b | 74 (73.3) | 78 (72.9) | 63 (62.4) | 67 (59.8) | 105 (67.3) |
| COVID-19 severity, No. (%)c | |||||
| Mild | 83 (82.2) | 79 (73.8) | 70 (69.3) | 92 (82.1) | 125 (80.1) |
| Moderate | 18 (17.8) | 28 (26.2) | 31 (30.7) | 20 (17.9) | 31 (19.9) |
| Duration of symptoms, median (IQR), dd | 5 (3-6) | 4 (3-6) | 4 (2-7) | 4 (3-5) | 4 (3-6) |
| SARS-CoV-2 cycle threshold, mean (SD)e | 23.8 (6.5) | 24.5 (7.6) | 23.4 (6.8) | 22.7 (8.0) | 23.8 (7.8) |
Abbreviations: BMI, body mass index; COVID-19, coronavirus disease 2019; IQR, interquartile range; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Calculated as weight in kilograms divided by height in meters squared.
Aged 55 years or older; BMI of 30 or greater; medical history of diabetes, chronic kidney disease, cardiovascular disease, chronic respiratory disease, or immunosuppressive disease; or receiving immunosuppressive treatment.
Based on 8 symptom domains (cough, shortness of breath, feeling feverish, fatigue, body aches and pain, sore throat, chills, headache) that were rated from none or absent (score of 0) to severe (score of 3), which were combined to provide an overall score (range, 0-24; symptom score excluded loss of appetite, taste, and smell).
Calculated based on the patient-reported start date of symptom onset and compared with the date of treatment infusion.
The cycle threshold is the number of polymerase chain reaction cycles required for a viral sample to be detected. Lower numbers suggest more infecting organisms and an increased burden of disease. Values range between 0 and 45; the (log base 10) viral load was calculated from the cycle threshold value (45 − cycle threshold)/log210, or (45 − cycle threshold)/3.321928.
The mean age of patients was 44.7 years (SD, 15.7 years). A total of 315 patients (54.6%) were female, 245 patients (42.5%) identified as Hispanic, and 387 patients (67.1%) had at least 1 risk factor for severe COVID-19 (aged ≥55 years, BMI ≥30, or ≥1 relevant comorbidity such as hypertension). Patients were randomized and received study infusions within a median of 4 days of symptom onset. At the time of randomization, 449 patients (77.8%) had mild symptoms. On the day of the infusion, the observed mean polymerase chain reaction cycle threshold value (a measure of viral load) was 23.7 (SD, 7.4), demonstrating a high viral burden in the population. There were 533 patients (92.4%) who completed the efficacy evaluation period (day 29).
Primary Outcome
The change in log viral load from baseline to day 11 was –3.72 for the 700 mg group, –4.08 for the 2800 mg group, –3.49 for the 7000 mg group, –4.37 for the combination therapy group, and –3.80 for the placebo group. Compared with the placebo group, the change from baseline to day 11 in log viral load was not significantly different for any of the monotherapy groups (0.09 [95% CI, –0.35 to 0.52], P = .69 for the 700 mg group; –0.27 [95% CI, –0.71 to 0.16], P = .21 for the 2800 mg group; and 0.31 [95% CI, –0.13 to 0.76], P = .16 for the 7000 mg group), but the change was statistically significantly different for the combination therapy group (–0.57 [95% CI, –1.00 to –0.14], P = .01; Figure 2 and Table 2).
Figure 2. Change in Log Viral Load and in Viral Load Cycle Threshold Over Time With Bamlanivimab Monotherapy and Bamlanivimab and Etesevimab Combination Therapy.

Randomization and infusion occurred on day 1. A, The middle line represents the median change from baseline for log viral load; the boxes represent the interquartile range; the squares inside each box represent the mean; the whiskers extend to the highest and lowest values within 1.5 x the interquartile range of the nearer quartile; and the dots represent observed values outside that range. B, The cycle threshold is defined as the number of cycles required for the fluorescent signal of the polymerase chain reaction assay to cross the threshold (ie, exceeds background level). Cycle threshold levels are inversely proportional to the number of copies of the virus and thus serve to estimate viral load. Virus is presumed to be undetectable beyond approximately 40 cycle thresholds. SARS-CoV-2 indicates severe acute respiratory syndrome coronavirus 2.
Table 2. Outcomes for Primary and Prespecified Secondary End Points.
| Outcome | Bamlanivimab monotherapy | Combination therapy (2800 mg of bamlanivimab and 2800 mg of etesevimab) (n = 109) |
Placebo (n = 152) |
||
|---|---|---|---|---|---|
| 700 mg (n = 101) |
2800 mg (n = 107) |
7000 mg (n = 101) |
|||
| Primary outcome | |||||
| No. of patients for SARS-CoV-2 viral load at day 11 | 100 | 103 | 95 | 102 | 146 |
| Viral load, mean (SD)a | 2.64 (1.80) | 2.21 (1.73) | 2.85 (1.76) | 2.16 (1.82) | 2.43 (1.78) |
| Change from baseline to day 11 vs placebo, mean (95% CI)b | 0.09 (–0.35 to 0.52) | –0.27 (–0.71 to 0.16) | 0.31 (–0.13 to 0.76) | –0.57 (–1.00 to –0.14) | |
| P value | .69 | .21 | .16 | .01 | |
| Secondary outcomesc | |||||
| No. of patients for SARS-CoV-2 viral load area under the curve (AUC) at day 29 | 85 | 91 | 84 | 72 | 118 |
| Viral load AUC, mean (SD) | 70.17 (29.68) | 63.74 (28.97) | 71.53 (30.15) | 61.69 (28.39) | 74.45 (35.30) |
| Change from baseline to day 29 vs placebo, mean (95% CI)d | –6.25 (–13.21 to 0.71) | –9.50 (–16.32 to –2.68) | –5.38 (–12.36 to 1.61) | –17.91 (–25.25 to –10.58) | |
| P value | .08 | .006 | .13 | <.001 | |
| No. of patients for SARS-CoV-2 viral clearance at day 7e | 99 | 101 | 99 | 100 | 145 |
| Viral clearance, No. (%) | 10 (9.9) | 12 (11.2) | 8 (7.9) | 14 (12.8) | 16 (10.5) |
| Change from baseline to day 7 vs placebo, % (95% CI) | –0.6 (–8.2 to 7.0) | 0.7 (–7.0 to 8.4) | –2.6 (–9.8 to 4.6) | 2.3 (–5.6 to 10.3) | |
| P value | >.99 | >.99 | .52 | .56 | |
| No. of patients for SARS-CoV-2 viral clearance at day 11e | 92 | 100 | 94 | 104 | 137 |
| Viral clearance, No. (%) | 13 (12.9) | 21 (19.6) | 14 (13.9) | 30 (27.5) | 27 (17.8) |
| Change from baseline to day 11 vs placebo, % (95% CI) | –4.9 (–13.8 to 4.0) | 1.9 (–7.8 to 11.5) | –3.9 (–13.0 to 5.2) | 9.8 (–0.6 to 20.1) | |
| P value | .38 | .75 | .49 | .07 | |
| No. of patients for SARS-CoV-2 viral clearance at day 15e | 91 | 97 | 94 | 98 | 132 |
| Viral clearance, No. (%) | 25 (24.8) | 30 (28.0) | 25 (24.8) | 36 (33.0) | 34 (22.4) |
| Change from baseline to day 15 vs placebo, % (95% CI) | 2.4 (–8.3 to 13.1) | 5.7 (–5.1 to 16.5) | 2.4 (–8.3 to 13.1) | 10.7 (–0.4 to 21.7) | |
| P value | .76 | .31 | .76 | .07 | |
| No. of patients for SARS-CoV-2 viral clearance at day 22e | 85 | 93 | 86 | 82 | 122 |
| Viral clearance, No. (%) | 41 (40.6) | 43 (40.2) | 37 (36.6) | 40 (36.7) | 56 (36.8) |
| Change from baseline to day 22 vs placebo, % (95% CI) | 3.8 (–8.5 to 16.0) | 3.3 (–8.7 to 15.4) | –0.2 (–12.3 to 11.9) | –0.1 (–12.0 to 11.7) | |
| P value | .60 | .61 | >.99 | >.99 | |
| No. of patients for COVID-19–related hospitalization or emergency department visit at day 29f | 101 | 107 | 101 | 112 | 156 |
| Had hospitalization or emergency department visit, No. (%) | 1 (1.0) | 2 (1.9) | 2 (2.0) | 1 (0.9) | 9 (5.8) |
| Change from baseline to day 29 vs placebo, % (95% CI) | –4.8 (–8.9 to –0.6) | –3.9 (–8.4 to 0.6) | –3.8 (–8.3 to 0.8) | –4.9 (–8.9 to –0.8) | |
| P value | .09 | .21 | .21 | .049 | |
| No. of patients for total symptom score at day 7g | 98 | 98 | 97 | 103 | 143 |
| Total symptom score, mean (SD) | 1.90 (2.49) | 2.07 (2.93) | 2.22 (2.97) | 2.14 (2.98) | 2.43 (2.67) |
| Change from baseline to day 7 vs placebo, mean (95% CI)h | –0.48 (–1.17 to 0.21) | –0.33 (–1.01 to 0.35) | –0.39 (–1.08 to 0.30) | –0.31 (–0.98 to 0.37) | |
| P value | .17 | .34 | .27 | .37 | |
| No. of patients for total symptom score at day 11g | 94 | 92 | 93 | 95 | 134 |
| Total symptom score, mean (SD) | 1.06 (1.58) | 1.59 (2.24) | 1.56 (2.61) | 1.28 (2.48) | 1.88 (2.50) |
| Change from baseline to day 11 vs placebo, mean (95% CI)h | –0.78 (–1.37 to –0.20) | –0.32 (–0.91 to 0.26) | –0.45 (–1.04 to 0.13) | –0.60 (–1.18 to –0.03) | |
| P value | .009 | .27 | .13 | .04 | |
| No. of patients for total symptom score at day 15g | 86 | 96 | 93 | 94 | 133 |
| Total symptom score, mean (SD) | 1.00 (2.25) | 1.20 (2.03) | 1.00 (2.07) | 1.04 (2.43) | 1.24 (2.05) |
| Change from baseline to day 15 vs placebo, mean (95% CI)h | –0.16 (–0.71 to 0.38) | –0.07 (–0.60 to 0.46) | –0.39 (–0.93 to 0.15) | –0.25 (–0.78 to 0.28) | |
| P value | .56 | .80 | .16 | .35 | |
| No. of patients for total symptom score at day 22g | 86 | 90 | 84 | 96 | 129 |
| Total symptom score, mean (SD) | 0.46 (1.16) | 0.74 (1.67) | 0.71 (1.54) | 0.76 (2.00) | 0.77 (1.67) |
| Change from baseline to day 22 vs placebo, mean (95% CI)h | –0.17 (–0.60 to 0.25) | –0.03 (–0.45 to 0.38) | –0.22 (–0.64 to 0.21) | 0.03 (–0.38 to 0.44) | |
| P value | .42 | .88 | .32 | .89 | |
| No. of patients for COVID-19 symptom improvement at day 7i | 99 | 98 | 98 | 103 | 143 |
| Had symptom improvement, No. (%) | 47 (46.5) | 37 (34.6) | 46 (45.5) | 50 (45.9) | 62 (40.8) |
| Change from baseline to day 7 vs placebo, % (95% CI) | 5.7 (–6.7 to 18.2) | –6.2 (–18.1 to 5.7) | 4.8 (–7.7 to 17.2) | 5.1 (–7.1 to 17.3) | |
| P value | .44 | .36 | .52 | .45 | |
| No. of patients for COVID-19 symptom improvement at day 11i | 95 | 92 | 94 | 95 | 134 |
| Had symptom improvement, No. (%) | 60 (59.4) | 48 (44.9) | 59 (58.4) | 58 (53.2) | 66 (43.4) |
| Change from baseline to day 11 vs placebo, % (95% CI) | 16.0 (3.6 to 28.4) | 1.4 (–10.8 to 13.7) | 15.0 (2.6 to 27.4) | 9.8 (–2.5 to 22.0) | |
| P value | .02 | .90 | .02 | .13 | |
| No. of patients for COVID-19 symptom improvement at day 15i | 87 | 96 | 94 | 94 | 133 |
| Had symptom improvement, No. (%) | 63 (62.4) | 63 (58.9) | 69 (68.3) | 69 (63.3) | 83 (54.6) |
| Change from baseline to day 15 vs placebo, % (95% CI) | 7.8 (–4.6 to 20.1) | 4.3 (–8.0 to 16.5) | 13.7 (1.7 to 25.8) | 8.7 (–3.3 to 20.7) | |
| P value | .24 | .53 | .04 | .17 | |
| No. of patients for COVID-19 symptom improvement at day 22i | 87 | 90 | 85 | 96 | 129 |
| Had symptom improvement, No. (%) | 70 (69.3) | 69 (64.5) | 71 (70.3) | 78 (71.6) | 96 (63.2) |
| Change from baseline to day 22 vs placebo, % (95% CI) | 6.1 (–5.7 to 18.0) | 1.3 (–10.5 to 13.2) | 7.1 (–4.6 to 18.9) | 8.4 (–3.0 to 19.8) | |
| P value | .35 | .90 | .28 | .18 | |
| No. of patients for COVID-19 symptom resolution at day 7j | 99 | 98 | 98 | 103 | 143 |
| Had symptom resolution, No. (%) | 37 (36.6) | 33 (30.8) | 34 (33.7) | 38 (34.9) | 48 (31.6) |
| Change from baseline to day 7 vs placebo, % (95% CI) | 5.1 (–6.9 to 17.0) | –0.7 (–12.2 to 10.7) | 2.1 (–9.7 to 13.9) | 3.3 (–8.3 to 14.9) | |
| P value | .42 | >.99 | .78 | .60 | |
| No. of patients for COVID-19 symptom resolution at day 11j | 95 | 92 | 94 | 95 | 134 |
| Had symptom resolution, No. (%) | 51 (50.5) | 43 (40.2) | 44 (43.6) | 50 (45.9) | 56 (36.8) |
| Change from baseline to day 11 vs placebo, % (95% CI) | 13.7 (1.2 to 26.1) | 3.3 (–8.7 to 15.4) | 6.7 (–5.6 to 19.1) | 9.0 (–3.1 to 21.1) | |
| P value | .04 | .61 | .30 | .16 | |
| No. of patients for COVID-19 symptom resolution at day 15j | 87 | 96 | 94 | 94 | 133 |
| Had symptom resolution, No. (%) | 56 (55.4) | 59 (55.1) | 60 (59.4) | 63 (57.8) | 70 (46.1) |
| Change from baseline to day 15 vs placebo, % (95% CI) | 9.4 (–3.1 to 21.9) | 9.1 (–3.2 to 21.4) | 13.4 (0.9 to 25.8) | 11.7 (–0.5 to 23.9) | |
| P value | .16 | .17 | .04 | .08 | |
| No. of patients for COVID-19 symptom resolution at day 22j | 87 | 90 | 85 | 96 | 129 |
| Had symptom resolution, No. (%) | 68 (67.3) | 63 (58.9) | 62 (61.4) | 75 (68.8) | 88 (57.9) |
| Change from baseline to day 22 vs placebo, % (95% CI) | 9.4 (–2.6 to 21.5) | 1.0 (–11.2 to 13.2) | 3.5 (–8.8 to 15.8) | 10.9 (–0.8 to 22.6) | |
| P value | .15 | .90 | .60 | .09 | |
Abbreviations: COVID-19, coronavirus disease 2019; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
The (log base 10) viral load was calculated from the cycle threshold value (45 − cycle threshold)/log210, or (45 − cycle threshold)/3.321928. The cycle threshold is the number of polymerase chain reaction cycles required for a viral sample to be detected. If the SARS-CoV-2 viral load for day 11 was missing, the earliest measurement closest to the day 11 visit within 4 days (ie, days 7-15) was used for the day 11 value.
Baseline was defined as the last nonmissing assessment recorded on or prior to the date of first study drug injection. The mixed-model repeated-measure analysis included log base 10–transformed baseline as a covariate and treatment, day, and treatment × day interaction as fixed effects. The stratification factor of duration since symptom onset to randomization was not used in the model to avoid collinearity with baseline viral load.
Time from baseline to day of SARS-CoV-2 viral clearance, COVID-19 symptom improvement, and COVID-19 symptom resolution were ran as Kaplan-Meier product limit curves (eFigures 1-3 in Supplement 1).
This analysis was conducted using a linear model with treatment as a fixed effect and log base 10–transformed baseline viral load as a covariate. No imputations of missing data were conducted. No AUC values from baseline to day 29 were calculated when the day 1 predose or the day 29 value was missing or if there were more than 3 values missing in the profile.
Earliest date of the 2 consecutive negative polymerase chain reaction test results for SARS-CoV-2. Treatment and symptom onset strata were used as factors in the logistic regression analysis (with a Firth penalized likelihood).
All randomized patients were included in this analysis. Treatment and symptom onset strata were used as factors in the logistic regression analysis (with a Firth penalized likelihood). Of the 15 hospitalizations or emergency department visits, 12 were hospitalizations.
The total symptom score has a range from 0 to 24 points based on 8 symptom domains (cough, shortness of breath, feeling feverish, fatigue, body aches and pain, sore throat, chills, headache) that were rated from none or absent (score of 0) to severe (score of 3) and were combined to provide an overall score (excluding loss of appetite, taste, and smell).
Baseline was defined as the last nonmissing assessment recorded on or prior to the date of first study drug injection. The mixed-model repeated-measure analysis included log base 10–transformed baseline as a covariate and treatment, day, and treatment × day interaction as fixed effects.
Defined by (1) all symptoms on the symptom questionnaire scored as moderate or severe at baseline were subsequently scored as mild or absent and (2) all symptoms on the symptom questionnaire scored as mild or absent at baseline were subsequently scored as absent. Treatment and symptom onset strata were used as factors in the logistic regression analysis (with a Firth penalized likelihood).
All symptoms (excluding the loss of appetite and changes in taste and smell symptoms) on the symptom questionnaire were scored as absent. Treatment and symptom onset strata were used as factors in the logistic regression analysis (with a Firth penalized likelihood).
Secondary Outcomes
Among the secondary outcome measures, differences between each treatment group vs the placebo group were statistically significant for 10 of 84 end points. The change from baseline to day 29 in viral load AUC was not significantly different for the 700 mg (difference, –6.25 [95% CI, –13.21 to 0.71]; P = .08) and 7000 mg monotherapy dose groups (difference, –5.38 [95% CI, –12.36 to 1.61]; P = .13), but the change was statistically significantly different for the 2800 mg dose group (difference, –9.50 [95% CI, –16.32 to –2.68]; P = .006) and for the combination treatment group (difference, –17.91 [95% CI, –25.25 to –10.58]; P < .001). However, viral clearance (defined as 2 consecutive negative test results for SARS-CoV-2) did not differ among any of the treatment groups at any time point (Table 2).
Compared with the placebo group, the change in mean total symptom score from baseline to day 11 was statistically significantly different for the 700 mg monotherapy group (mean difference, –0.78 [95% CI, –1.37 to –0.20]; P = .009) and for the combination group (mean difference, –0.60 [95% CI, –1.18 to –0.03]; P = .04), but the change was not significantly different for the 2800 mg monotherapy group (mean difference, –0.32 [95% CI, –0.91 to 0.26]; P = .27) or for the 7000 mg group (mean difference, –0.45 [95% CI, –1.04 to 0.13]; P = .13).
Compared with the placebo group, the change in symptom improvement from baseline to day 11 was statistically significantly different for the 700 mg group (difference, 16.0% [95% CI, 3.6% to 28.4%]; P = .02) and the 7000 mg group (difference, 15.0% [95% CI, 2.6% to 27.4%]; P = .02), but the change was not significant for the 2800 mg group (difference, 1.4% [95% CI, –10.8% to 13.7%]; P = .90) and the combination treatment group (difference, 9.8% [95% CI, –2.5% to 22.0%]; P = .13). Compared with the placebo group, the change in symptom resolution from baseline to day 11 was statistically significantly different for the 700 mg group (difference, 13.7% [95% CI, 1.2% to 26.1%]; P = .04), but the change was not significant for the 2800 mg group (difference, 3.3% [95% CI, –8.7% to 15.4%]; P = .61), the 7000 mg group (difference, 6.7% [95% CI, –5.6% to 19.1%]; P = .30), or the combination group (difference, 9.0% [95% CI, –3.1% to 21.1%]; P = .16).
The proportion of patients with COVID-19–related hospitalizations or emergency department visits at day 29 was 1.0% (1 event/101 patients) in the 700 mg group, 1.9% (2 events/107 patients) in the 2800 mg group, 2.0% (2 events/101 patients) in the 7000 mg group, 0.9% (1 event/112 patients) in the combination therapy group, and 5.8% (9 events/156 patients) in the placebo group. The difference vs placebo was –4.8% (95% CI, –8.9% to –0.6%; P = .09) for the 700 mg group, –3.9% (95% CI, –8.4% to 0.6%; P = .21) for the 2800 mg group, –3.8% (95% CI, –8.3% to –0.8%; P = .21) for the 7000 mg group, and –4.9% (95% CI, –8.9% to –0.8%; P = .049) for the combination group (Table 2).
The results from additional secondary end points (including time to viral clearance, symptom resolution, and symptom improvement) appear in eFigures 1, 2, and 3 in Supplement 1.
Post Hoc Analyses
Among patients aged 65 years or older or with a BMI of 35 or greater, those who received bamlanivimab monotherapy had a lower hospitalization rate (2.7% [1/37 patients] in the 700 mg group and a difference of –10.8% [95% CI, –21.4% to –0.1%]; 3.3% [1/30 patients] in the 2800 mg group and a difference of –10.1% [95% CI, –21.4% to 1.2%]; and 5.9% [2/34 patients] in the 7000 mg group and a difference of –7.6% [95% CI, –19.8% to 4.6%]) as well as those who received combination therapy (0% [0/31 patients] in the bamlanivimab and etesevimab group and a difference of –13.5% [95% CI, –22.7% to –4.2%]; P = .04) compared with those who received placebo (13.5% [7/52 patients]; eTable 2 in Supplement 1). Only 1 patient in the study (in the placebo group) was admitted to the intensive care unit. Additional post hoc analyses appear in the eResults and eFigure 4 in Supplement 1.
Exploratory Outcomes
Total symptom score AUC from baseline to day 11 was assessed in an exploratory analysis. Compared with placebo, the difference in mean change in total symptom score AUC from baseline to day 11 was –8.28 (95% CI, –14.04 to –2.53; P = .005) for the 700 mg group, –6.59 (95% CI, –12.46 to –0.72; P = .03) for the 2800 mg group, –8.09 (95% CI, –14.05 to –2.13; P = .008) for the 7000 mg group, and –8.63 (95% CI, –14.39 to –2.88; P = .003) for the combination therapy group (eTable 2 in Supplement 1).
In an exploratory analysis to assess the ability of bamlanivimab and etesevimab to reduce the levels of treatment-emergent bamlanivimab-resistant variants, the frequency of these variants in baseline samples across cohorts in the study population was low (0.4% [2/523 patients]) and is similar to the global prevalence of these variants.
Putative treatment-emergent bamlanivimab-resistant variants were detected in 7.1% of patients (7/98) in the 700 mg group, in 9.8% of patients (10/102) in the 2800 mg group, in 11.3% of patients (11/97) in the 7000 mg group, in 1% of patients (1/102) in the bamlanivimab and etesevimab combination group, and in 4.8% of patients (7/145) in the placebo group (eTable 2 in Supplement 1). The patient with a treatment-emergent bamlanivimab-resistant variant in the combination group had a single sample with an S494P spike variant on day 11 at an allele fraction of 0.198 and a viral load of 3.64 (N1 cycle threshold of approximately 32). This variant was transient in nature and was not detected in subsequent samples through study day 25. The bamlanivimab monotherapy groups had a higher frequency of patients who had a variant detected at more than 1 time point during the viral time course (4.1% for the 700 mg group, 5.9% for the 2800 mg group, and 7.2% for the 7000 mg group) than the placebo group or the bamlanivimab and etesevimab combination group (both 0%).
Adverse Events
Serious adverse events unrelated to SARS-CoV-2 infection or considered related to the study drug by the investigator occurred in 0% (0/309) of patients in the bamlanivimab monotherapy groups, in 0.9% (1/112) of patients in the bamlanivimab and etesevimab combination group, and in 0.6% (1/156) of patients in the placebo group (Table 3). The serious adverse event observed in the combination group was a urinary tract infection that was deemed unrelated to the study drug. The serious adverse event observed in the placebo group was upper abdominal pain and was deemed unrelated to the study drug.
Table 3. Adverse Events.
| Adverse events, No. (%)a | |||||
|---|---|---|---|---|---|
| Bamlanivimab monotherapy | Combination therapy (2800 mg of bamlanivimab and 2800 mg of etesevimab) (n = 112) |
Placebo (n = 156) |
|||
| 700 mg (n = 101) |
2800 mg (n = 107) |
7000 mg (n = 101) |
|||
| Patients with ≥1 treatment-emergent adverse eventb | 27 (26.7) | 26 (24.3) | 22 (21.8) | 19 (17.0) | 42 (26.9) |
| Severity of treatment-emergent adverse eventb,c | |||||
| Mild | 17 (16.8) | 18 (16.8) | 10 (9.9) | 15 (13.4) | 21 (13.5) |
| Moderate | 7 (6.9) | 5 (4.7) | 7 (6.9) | 3 (2.7) | 18 (11.5) |
| Severe | 2 (2.0) | 3 (2.8) | 5 (5.0) | 1 (0.9) | 3 (1.9) |
| Most common treatment-emergent adverse events (occurring in ≥4 patients)b | |||||
| Chest discomfort | 0 | 2 (1.9) | 1 (1.0) | 0 | 1 (0.6) |
| Chills | 0 | 1 (0.9) | 3 (3.0) | 0 | 0 |
| Diarrhea | 1 (1.0) | 2 (1.9) | 6 (5.9) | 1 (0.9) | 7 (4.5) |
| Dizziness | 3 (3.0) | 3 (2.8) | 3 (3.0) | 1 (0.9) | 3 (1.9) |
| Headache | 3 (3.0) | 2 (1.9) | 0 | 0 | 3 (1.9) |
| Nasal congestion | 2 (2.0) | 1 (0.9) | 0 | 0 | 1 (0.6) |
| Nausea | 3 (3.0) | 4 (3.7) | 5 (5.0) | 4 (3.6) | 6 (3.8) |
| Pruritus | 2 (2.0) | 3 (2.8) | 0 | 2 (1.8) | 1 (0.6) |
| Pyrexia | 1 (1.0) | 2 (1.9) | 1 (1.0) | 1 (0.9) | 0 |
| Rash | 1 (1.0) | 0 | 1 (1.0) | 1 (0.9) | 1 (0.6) |
| Syncope | 0 | 1 (0.9) | 1 (1.0) | 0 | 2 (1.3) |
| Vomiting | 1 (1.0) | 3 (2.8) | 1 (1.0) | 1 (0.9) | 4 (2.6) |
| Serious adverse eventd | 0 | 0 | 0 | 1 (0.9) | 1 (0.6) |
Includes full randomized population that received at least 1 infusion.
A treatment-emergent adverse event was defined as an event that first occurred or worsened in severity after baseline. Adverse events were reported by the participant, or, when appropriate, by a caregiver, surrogate, or the participant’s legally authorized representative.
Patients with multiple occurrences of these categories were counted once for each category. Patients with multiple occurrences of the same event were included in the count for the severe category. The investigator assessed the intensity for each adverse event reported during the study and assigned it to one of the following categories, which together with serious criteria (life-threatening or death) were aligned with the Division of AIDS table for grading the severity of adult and pediatric adverse events (trial protocol in Supplement 2; §10.3.3, version 2.1, July 2017).
Defined as any untoward medical occurrence that at any dose resulted in death, was life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent disability or incapacity, or caused a congenital anomaly (trial protocol in Supplement 2; §10.3.2 with exceptions listed in §10.3.1). No deaths occurred during study treatment.
The most frequently reported adverse events were nausea (3.0% for the 700 mg group, 3.7% for the 2800 mg group, 5.0% for the 7000 mg group, 3.6% for the combination therapy group, and 3.8% for the placebo group) and diarrhea (1.0%, 1.9%, 5.9%, 0.9%, and 4.5%, respectively). Immediate hypersensitivity reactions that could have been infusion related were reported in 9 patients (6 in the bamlanivimab monotherapy groups, 2 in the bamlanivimab and etesevimab group, and 1 in the placebo group). Most reactions occurred during infusion and were reported as mild in severity and not dose related. There were no changes in vital signs and symptoms included pruritus, flushing, rash, and facial swelling. The infusions were completed in all instances.
Discussion
In this phase 2/3 clinical trial that evaluated the efficacy and adverse effects of bamlanivimab monotherapy and bamlanivimab and etesevimab combination therapy in outpatients with recently diagnosed mild to moderate COVID-19, the primary end point, mean change from baseline in log viral load at day 11, was not significantly different for the bamlanivimab monotherapy groups compared with the placebo group, but was significantly different for the bamlanivimab and etesevimab combination therapy group compared with the placebo group.
Among the secondary outcomes, there were no consistent differences between the monotherapy groups or the combination therapy group vs placebo for the other measures of viral load or clinical symptom scores. The proportion of patients with COVID-19–related hospitalizations or emergency department visits was numerically lower for the monotherapy groups and the combination therapy group compared with the placebo group, but the difference was only significant for the combination group. Additional study is needed to understand whether the greater reduction of viral load shown by combination therapy would eventually translate to clinical benefit compared with monotherapy.
Consistent with the literature,16,17,18,19 the post hoc analyses indicated that hospitalization rates were higher in placebo-treated patients with the comorbidities of advanced age (≥65 years) or morbid obesity (BMI ≥35) (13.5%), although no hospitalizations were observed in this high-risk subgroup in the combination therapy group. These preliminary data are hypothesis generating and suggest the need for further study to determine whether patients with these risk factors should be prioritized for this particular treatment.
In the exploratory analysis of ongoing viral sequencing, putative bamlanivimab-resistant variants were observed in all treatment groups, including placebo. Even though the combination group had the largest reductions in viral load, the monotherapy groups all performed comparably with the combination group on several clinical end points (eg, mean total symptom score and hospitalization rate). Therefore, the clinical significance of the resistant variants remains unclear.
Currently, only remdesivir is approved by the US Food and Drug Administration for the treatment of patients with COVID-19 who are seriously ill, although corticosteroids are generally considered the treatment of choice in this population and baricitinib recently received Emergency Use Authorization. COVID-19 convalescent plasma is available for use in hospitalized patients through Emergency Use Authorization; although, efficacy has not been established definitively and it is still considered investigational.7,20 Recently, the 700 mg dose of bamlanivimab has been authorized for emergency use in the US and Canada for the treatment of outpatients with mild to moderate COVID-19. Additional studies, including the ongoing subsequent portions of this trial in high-risk patients, are needed to fully elucidate the clinical benefit of therapeutic monoclonal antibodies for COVID-19.
Limitations
This study had several limitations. First, the trial was originally designed as a safety and biomarker study.
Second, the patient population was small, which made detecting clinically meaningful differences between treatment groups more difficult.
Third, only 1 combination dose was chosen for this study. Because the antiviral activity of etesevimab monotherapy or different combination doses was not investigated, it is difficult to determine whether the greater reduction in viral load observed in the combination group was due to additive or synergistic effects vs differential efficacy of etesevimab.
Fourth, the primary end point at day 11 may have been too late in the immune response to optimally detect treatment effects. All patients, including those who received placebo, demonstrated substantial viral reduction by day 11. An earlier time point like day 3 or day 7 could possibly have been more appropriate to measure viral load.
Fifth, the full genotypic and phenotypic analysis of the trial is still ongoing, and the resistance data presented here are limited to the sample sequences that were available at the time of this analysis.
Conclusions
Among nonhospitalized patients with mild to moderate COVID-19 illness, treatment with bamlanivimab and etesevimab, compared with placebo, was associated with a statistically significant reduction in SARS-CoV-2 viral load at day 11; no significant difference in viral load reduction was observed for bamlanivimab monotherapy. Further ongoing clinical trials will focus on assessing the clinical benefit of antispike neutralizing antibodies in patients with COVID-19 as a primary end point.
eTable 1. Pseudovirus neutralization of spike variants in the presence of bamlanivimab and etesevimab
eTable 2. Outcomes for post hoc and exploratory analyses
eMethods
eResults
eFigure 1. Time to SARS-CoV-2 clearance
eFigure 2. Time to symptom improvement
eFigure 3. Time to symptom resolution
eFigure 4. Viral load over time for all patients
BLAZE-1 Investigators
Support staff
Trial protocol and statistical analysis plan
Data sharing statement
References
- 1.Berlin DA, Gulick RM, Martinez FJ. Severe Covid-19. N Engl J Med. 2020;383(25):2451-2460. doi: 10.1056/NEJMcp2009575 [DOI] [PubMed] [Google Scholar]
- 2.Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934-943. doi: 10.1001/jamainternmed.2020.0994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williamson EJ, Walker AJ, Bhaskaran K, et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 2020;584(7821):430-436. doi: 10.1038/s41586-020-2521-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569-1578. doi: 10.1016/S0140-6736(20)31022-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beigel JH, Tomashek KM, Dodd LE, et al. ; ACTT-1 Study Group Members . Remdesivir for the treatment of Covid-19—final report. N Engl J Med. 2020;383(19):1813-1826. doi: 10.1056/NEJMoa2007764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prescott HC, Rice TW. Corticosteroids in COVID-19 ARDS: evidence and hope during the pandemic. JAMA. 2020;324(13):1292-1295. doi: 10.1001/jama.2020.16747 [DOI] [PubMed] [Google Scholar]
- 7.Li L, Zhang W, Hu Y, et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. JAMA. 2020;324(5):460-470. Published correction appears in JAMA. 2020;324(5):519. doi: 10.1001/jama.2020.10044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renn A, Fu Y, Hu X, Hall MD, Simeonov A. Fruitful neutralizing antibody pipeline brings hope to defeat SARS-Cov-2. Trends Pharmacol Sci. 2020;41(11):815-829. doi: 10.1016/j.tips.2020.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen P, Nirula A, Heller B, et al. ; for the BLAZE-1 Investigators . SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with Covid-19. N Engl J Med. Published online October 28, 2020. doi: 10.1056/NEJMoa2029849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.US Food and Drug Administration FDA issues Emergency Use Authorization for convalescent plasma as potential promising COVID–19 treatment, another achievement in administration’s fight against pandemic. Published August 23, 2020. Accessed December 22, 2020. https://www.fda.gov/news-events/press-announcements/fda-issues-emergency-use-authorization-convalescent-plasma-potential-promising-covid-19-treatment
- 11.US Food and Drug Administration Coronavirus (COVID-19) update: FDA authorizes monoclonal antibody for treatment of COVID-19. Published November 9, 2020. Accessed December 22, 2020. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibody-treatment-covid-19
- 12.US Food and Drug Administration Coronavirus (COVID-19) update: FDA authorizes monoclonal antibodies for treatment of COVID-19. Published November 21, 2020. Accessed December 22, 2020. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19
- 13.Jones BE, Brown-Augsburger PL, Corbett KS, et al. LY-CoV555, a rapidly isolated potent neutralizing antibody, provides protection in a non-human primate model of SARS-CoV-2 infection. bioRxiv. Published online October 9, 2020. doi: 10.1101/2020.09.30.318972 [DOI]
- 14.Shi R, Shan C, Duan X, et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584(7819):120-124. doi: 10.1038/s41586-020-2381-y [DOI] [PubMed] [Google Scholar]
- 15.Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369(6506):1014-1018. doi: 10.1126/science.abd0831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497-506. doi: 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garg S, Kim L, Whitaker M, et al. Hospitalization rates and characteristics of patients hospitalized with laboratory-confirmed coronavirus disease 2019—COVID-NET, 14 States, March 1-30, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:458-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrilli CM, Jones SA, Yang J, et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ. 2020;369:m1966. doi: 10.1136/bmj.m1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko JY, Danielson ML, Town M, et al. ; COVID-NET Surveillance Team . Risk factors for COVID-19-associated hospitalization: COVID-19-associated hospitalization surveillance network and behavioral risk factor surveillance system. Clin Infect Dis. Published online September 18, 2020. doi: 10.1093/cid/ciaa1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joyner MJ, Wright RS, Fairweather D, et al. Early safety indicators of COVID-19 convalescent plasma in 5000 patients. J Clin Invest. 2020;130(9):4791-4797. doi: 10.1172/JCI140200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Pseudovirus neutralization of spike variants in the presence of bamlanivimab and etesevimab
eTable 2. Outcomes for post hoc and exploratory analyses
eMethods
eResults
eFigure 1. Time to SARS-CoV-2 clearance
eFigure 2. Time to symptom improvement
eFigure 3. Time to symptom resolution
eFigure 4. Viral load over time for all patients
BLAZE-1 Investigators
Support staff
Trial protocol and statistical analysis plan
Data sharing statement