

Abstract
Objective:
We tested the hypothesis that sigma receptor-1 (σ1) modulates endothelial barrier function due to its influence on endothelial bioenergetics.
Methods:
Cultured human umbilical vein endothelial cell (HUVEC) monolayers were used to model the endothelial barrier. Electric cell-substrate impedance sensing (ECIS), Transwell assays, and immunofluorescence labeling of junctional proteins were used to evaluate endothelial barrier function. Endothelial cell bioenergetics were determined using extracellular flux analysis and direct ATP level measurements. The endothelial-specific contribution of σ1 was tested using the σ1-selective agonist, PRE-084, and with targeted knockdown of σ1 expression using siRNA.
Results:
Activation of σ1 with PRE-084 significantly enhanced endothelial barrier function and decreased permeability to albumin and dextran. Knockdown of σ1 with siRNA reduced barrier function and abolished PRE-084-induced endothelial barrier enhancement. PRE-084 upregulated endothelial glycolysis and glycolytic ATP production, but this response was abolished by siRNA-mediated knockdown of σ1 expression. PRE-084 also reduced the degree of endothelial barrier dysfunction caused by the mitochondrial oxidative phosphorylation uncoupler CCCP.
Conclusion:
Activation of σ1 enhances endothelial barrier function and modulates the ratio of glycolytic versus mitochondrial ATP production. These novel findings suggest that endothelial σ1 may prove beneficial as a novel therapeutic target for reducing microvascular hyperpermeability and counteracting mitochondrial dysfunction.
Keywords: Sigma-1 receptor, endothelial permeability, endothelial bioenergetics
INTRODUCTION
The sigma-1 receptor (σ1) is a single 25-kDa transmembrane protein residing primarily in the ER that acts as a chaperone protein. Its interaction with mitochondria at the mitochondria-associated ER membrane domain is well documented [1]. Following activation, σ1 has been reported to bind to the IP3R, and modulate intracellular calcium homeostasis [2]. Also, the activated σ1 modulates plasma membrane receptors and ion channel functions, and may regulate cellular excitability [3–6]. Further, σ1 affects trafficking of lipids and proteins essential for neurotransmission, cell growth and motility [7,8].
Activation of σ1 provides neuroprotection and cardio-protection in various models. Examples of neuroprotection include but not limited to its therapeutic use in neuro-psychiatric disorders, cognitive disorders, neurodegenerative diseases and stroke [9–13]. In regards to cardio-protection, Bhuiyan et al have shown that cardiac σ1 receptors have a role in ameliorating cardiac hypertrophy in pressure overload induced heart failure model [14]. In addition to neuro- and cardio-protection, there are reports of actions of σ1 in endothelium such as enhancement of nitric oxide production or modulation of calcium homeostasis [15,16]. The recent finding that σ1 activation lessens blood-brain barrier dysfunction in a vascular dementia mouse model suggests that σ1 may have a role in maintaining endothelial junctional integrity, which has not previously been studied in detail [17]. Another finding showed that σ1 knockout mice had impaired cellular energetics represented by a decrease in oxygen consumption rate in the heart [18]. Whether these properties extend directly to the endothelium, and if the barrier integrity and modulation of cellular energetics are linked, remains unknown.
Endothelial cells have a critical role in controlling blood-tissue gas and solute exchange [19]. The barrier function of the endothelium depends on the integrity of endothelial structure, which can be actively altered by the responses of endothelial cells to various inflammatory mediators, growth factors, and physical forces [20–25]. Endothelial junctional proteins and cytoskeleton dynamics have a well characterized role in endothelial health [23,26,27]. In addition, multiple reports utilizing shock models have shown that mitochondrial dysfunction contributes to increased microvascular permeability [28–30], and we recently reported that disruption of mitochondrial complex III with antimycin-A can severely impair endothelial barrier function [29].
As σ1 receptors reside in the ER-mitochondrial interface [1], their ability to impact the endothelial barrier may be related to mitochondrial functions such as cellular bioenergetics. Endothelial bioenergetics has been studied largely in the context of endothelial-dependent vasorelaxation [31,32], but its role has not been well characterized in the control of the endothelial barrier. Endothelial cells have been reported to rely more on glycolysis for energy production than oxidative phosphorylation despite the fact that they are exposed to blood with high levels of oxygen [33]. Glycolysis has been proven preferable for ATP production in endothelial cells as it allows for fast ATP production during vessel sprouting or changing cytoskeletal dynamics that could be accomplished under hypoxic conditions independent of oxygen [34]. Glycolysis not only allows for ATP production, but also for generation of nucleotides via the pentose phosphate pathway [35]. Despite the significant contribution of glycolysis to endothelial function, few studies have addressed its role in endothelial barrier integrity.
In the current study we investigated the potential involvement of σ1 receptors in enhancing endothelial barrier function using both genetic and pharmacological approaches. As σ1 has been reported to promote mitochondrial integrity and improve cell survival in neurological models[36], we tested potential mitochondrial versus cytoplasmic bioenergetic pathways as potential mechanisms. In addition, we assessed the extent to which the σ1 agonist PRE-084 can protect against endothelial barrier dysfunction caused by impairment of mitochondrial function.
MATERIALS AND METHODS
Endothelial cell culture
HUVEC were used as an in vitro model of endothelial barrier. The cells were obtained from LifeLine Cell Technology (Frederick, MD, USA) and grown in 10-cm petri dishes in Vasculife cell culture medium (LifeLine) supplemented with fetal bovine serum, endothelial growth supplements and antimicrobial supplement gentamycin and amphotericin B, on 1.5% porcine gelatin matrix (Sigma-Aldrich, St. Louis, MO, USA) in an incubator at 37 degrees C and 5% CO2. HUVEC were used at passages 1–5.
Pharmacological agents
PRE-084 was obtained from Tocris Biotechne (Minneapolis, MN, USA). CCCP was purchased from Abcam (Cambridge, MA, USA). FITC-albumin and TRITC-dextran were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Endothelial Barrier Function
TER served as an indicator of barrier function of endothelial cell monolayers. TER was determined using an electrical cell-substrate impedance sensor (ECIS) (Applied Biophysics, Troy, NY,USA) as previously described [24,27]. This method allows for real-time detection of small, momentary changes in barrier function relating to nanometer-scale changes in junctional structures [37]. Briefly, the cells were sub-cultured onto gelatin coated 96w20idf ECIS arrays and left undisturbed to form confluent monolayers. On the day of the experiment, the media was changed to VBM at least 1 hour before the experiment and the arrays were attached to ECIS instrument in a 37 °C, 5% CO2 incubator and cell monolayers were allowed to reach a steady-state TER prior to experiments.
Transfection
Cells were grown to 80% confluence, trypsinized and pelleted, and 500,000 cells were re-suspended in 100 μL electroporation master mix containing electroporation solution (Mirus Bio, Madison, WI, USA) and 200 nM siRNA. σ1 siRNA (knockdown: GGCUUGAGCUCACCACCUA) or control RNA (Non-Targeting: UGGUUUACAUGUUUUCCUA) purchased from Dharmacon (Lafayette, CO, USA). Next, the mixture was transferred to transfection cuvettes for transfection in the Nucleofector II device (Lonza, Basel, Switzerland) using program A-034. Following transfection, 500 μL of warm complete medium was added to the cuvette immediately after transfection. Cells were then seeded onto either gelatin-coated 35-mm culture dishes for protein extraction, Transwell chambers for permeability assays or Seahorse arrays for bioenergetics analyses (see below).
For the cells used to record TER, cells were seeded into gold electrode arrays (96w20idf, Applied Biophysics, Troy, NY) at 60,000 cells per well, followed by immediate commencement of recording by the ECIS instrument, which continued over the course of 70 hours. Media was changed at 1 hour and again at 24 hours. To test the response of transfected cells to PRE-084, media was changed at 70 hours to serum-free media followed by a stabilization period of to achieve steady-state TER readings for at least one hour before before PRE-084 or vehicle treatment.
Automated Western Blot Assay
Protein lysates were isolated and concentration was determined as previously described [26]. To determine protein levels, we used the Protein Simple WES capillary western blot system (San Jose, CA, USA) according to manufacturer’s instructions. Total protein concentrations of 0.2 mg/ml was used based on prior titration. Primary antibodies were used at 1:50 dilution. The primary antibodies used were: Rabbit polyclonal anti sigma-1R NBP1–82479 from NovusBio (Centennial, CO, USA), and mouse monoclonal anti β-actin #3700 from Cell Signaling Technology (Boston, MA, USA). Anti-Rabbit and anti-mouse secondary antibodies were supplied with the WES detection module kits from Protein Simple and the manufacturer’s compass software was used for data analysis. Band images based upon the densitometry data were generated using the software for figures. The raw densitometry peaks were utilized for quantitative analyses.
Transwell permeability assays
Assays to determine the apparent solute permeability (Psolute) of HUVEC monolayers to FITC-albumin (66 kDa) and TRITC-dextran (4.4 kDa) were performed as previously described [26]. Briefly, HUVEC were seeded on the upper chamber of gelatin-coated Transwell membranes (0.4 μm pores; Corning, Corning, MA), and allowed to grow for 3 days. On the day of the experiment, the medium was changed to serum free Vasculife basal media (VBM). After the end of the treatment period (PRE-084 or vehicle for 3 hours), a stock concentration of the molecular tracers (1 mg/ml) was added to the upper chamber for 30 minutes. Then, samples of media were collected from both upper (luminal) and lower (abluminal) chambers for fluorometric analysis with a SpectraMax plate reader and Softmax Pro 6.2 software (Molecular Devices, San Jose, CA) to determine concentrations of the tracers. The permeability coefficient was then calculated using the equation Psolute = [(CA/t) × (1/A) × (V/CL)], where CA is the abluminal concentration, t is the time (in seconds), A is the area of the membrane (in cm2), V is the volume of the abluminal chamber, and CL is the luminal concentration.
Bio-energetic measurements using Seahorse extracellular flux analyzer:
The Seahorse XFP analyzer system from Agilent (Santa Clara, CA, USA) was used to assess glycolytic rate and ATP production, utilizing assay kits purchased from Agilent. All protocols were done according to manufacturer’s instructions. Briefly, HUVEC were seeded onto Seahorse cell culture 8-well plates at a density of 40,000 cells/well the day before the experiment (or longer for some experiments involving siRNA transfection). The following day, media was changed to Seahorse medium containing glucose, glutamine and pyruvate as well as HEPES buffer and cells were incubated in a non-CO2 incubator for an hour before the experiment. The Seahorse XFP analyzer determines glycolytic rates and ATP production rates using Extracellular Acidification Rates (ECAR) and Oxygen Consumption Rate (OCR) measurements. Glycolytic rate assay measurement depends on the fact that glucose is converted to pyruvate and lactate in the cytoplasm, or to CO2 and water within mitochondria. Conversion of glucose to lactate results in proton production in the assay medium. The assay workflow is as following, baseline recordings of OCR and ECAR are obtained for approximately 20 min, then a Rotenone/Antimycin A mixture at a concentration of 0.5 μM is injected to inhibit oxygen consumption and CO2 derived protons. Later, 2-deoxyglucoase (an inhibitor of glycolysis) is injected at a concentration of 50 mM to confirm that the measurement of proton efflux rate prior to the injection is due to glycolysis.
Similar procedures were used to measure ATP production rates. Baseline measurements of ECAR and OCR were obtained followed by injection of 1.5 μM of oligomycin then 0.5 μM of Rot/AA. In cells, both glycolysis and oxidative phosphorylation contribute to extracellular proton extrusion, therefore the analyzer determines ATP production rates using equations involving both ECAR and OCR. Data were obtained from Wave Controller software and the Seahorse XFP built-in report generators [38]. Following the conclusion of the experiments, cells were lysed and protein was isolated for protein quantification using BCA protein assay kit that was purchased from Thermo Scientific (Waltham, MA). All assay readouts were normalized to the total protein quantity in each sample.
Direct Measurement of ATP levels
Quantification of ATP levels in HUVEC monolayers was accomplished using a luminescent ATP detection assay kit purchased from Abcam (Cambridge, MA, USA). All procedures were done according to manufacturer’s instructions. Briefly, HUVEC cells were seeded onto white 96 well plates with clear, flat bottoms. Cells were grown to confluence and media was changed to VBM at least 1 hour before the experiment. Cells were treated with CCCP or vehicle in the presence or absence of PRE-084 or vehicle for 4 hours. Then, cells were lysed with the provided detergent. The supplied substrate was then added. The plate was dark-adapted for 10 minutes and luminescence was measured with a SpectraMax plate reader and Softmax Pro 6.2 software (Molecular Devices, San Jose, CA). ATP concentration was then calculated based on the equation generated from the ATP standard curve.
Immunofluorescence and confocal microscopy
To assess the impact of PRE-084 and CCCP on endothelial junctional proteins, cultured HUVECs were immunolabeled for β-catenin (L54E2, Cell signaling) and ZO-1 (61–7300, Themo Fisher Scientific) following treatment with vehicle + vehicle, CCCP + vehicle, CCCP + PRE-084 or PRE-084 + vehicle. PRE-084 (100 μM) was added 5 minutes before treatment with CCCP (10 μM). The cells were fixed for immunofluorescence labeling 4 hours after CCCP was added, to match the time point of the TER experiment. Briefly, HUVEC were seeded onto gelatin-coated glass coverslips (Fisher Scientific, Hampton, NH) and incubated in endothelial growth medium until confluent. The medium was changed to VBM and allowed to incubate for 1 hour before the treatment. After the various treatments, cells were fixed in 4% paraformaldehyde in PBS for 10 minutes, permeabilized in 0.1% Triton-X-100 in PBS for 10 minutes and blocked for nonspecific binding with 10% serum in PBS for 1 hour. Cells were incubated with primary antibodies overnight at 4 °C. They were then washed 3× for 10 minutes with 0.1% Tween-20 in PBS, followed by incubation for 1 hour with Alexa-488- and Alexa-647-conjugated secondary antibodies (Life Technologies, Carlsbad, CA, USA). The samples were washed 3× again, and the coverslips were mounted onto microscope slides with ProLong Gold antifade mounting medium with DAPI (Invitrogen, Carlsbad, CA). Images were collected with an Olympus FV1000 spectral inverted laser scanning confocal microscope (Olympus, Center Valley, PA, USA), and the images were analyzed with NIH ImageJ software [39]. Regions of interest drawn encompassing the junctional regions of adjacent endothelial cells were drawn as previously described [29], and the intensities within these regions were compared between groups.
Data Analysis
Summarized data are presented as mean ± SE. Differences between 2 groups were evaluated using unpaired t-tests. Differences between three or more groups were assessed by either by one-way or two-way ANOVA followed by appropriate post hoc tests for multiple comparisons. Significance was accepted when p<0.05. All analyses were performed with GraphPad Prism 8 software. Details of analysis methods are provided in each figure legend.
RESULTS
To test the impact of σ1 activation on endothelial barrier function, we performed a concentration-response study with the highly selective σ1 agonist, PRE-084, at concentrations of 1, 5, 10, 50, 100, 150, and 200 μM, based on previously reported data [6,40–42]. The results show a concentration-dependent increase in barrier enhancement (Fig. 1A,B). The enhancement of TER started approximately 1 h following application of PRE-084 and peaked after 3 h (Fig. 1A). At this time point, concentrations of PRE-084 that were equal to or greater than 50 μM caused a significant increase in barrier function compared to control (Fig. 1B). A maximum increase in TER occurred at a concentration of 100 μM PRE-084, with higher concentrations tested having a similar effect. Based on these findings, we chose to use 100 μM PRE-084 and the 3 h time point in subsequent experiments. To test whether the TER enhancement reflects a reduction in endothelial permeability, Transwell assay experiments were carried out to determine permeability of HUVEC monolayers to FITC-albumin (66 kDa) and TRITC-dextran (4.4 kDa). PRE-084 (100 μM) significantly decreased permeability to both tracers at 3 hours post-treatment (Fig. 1 C, D).
Fig. 1.
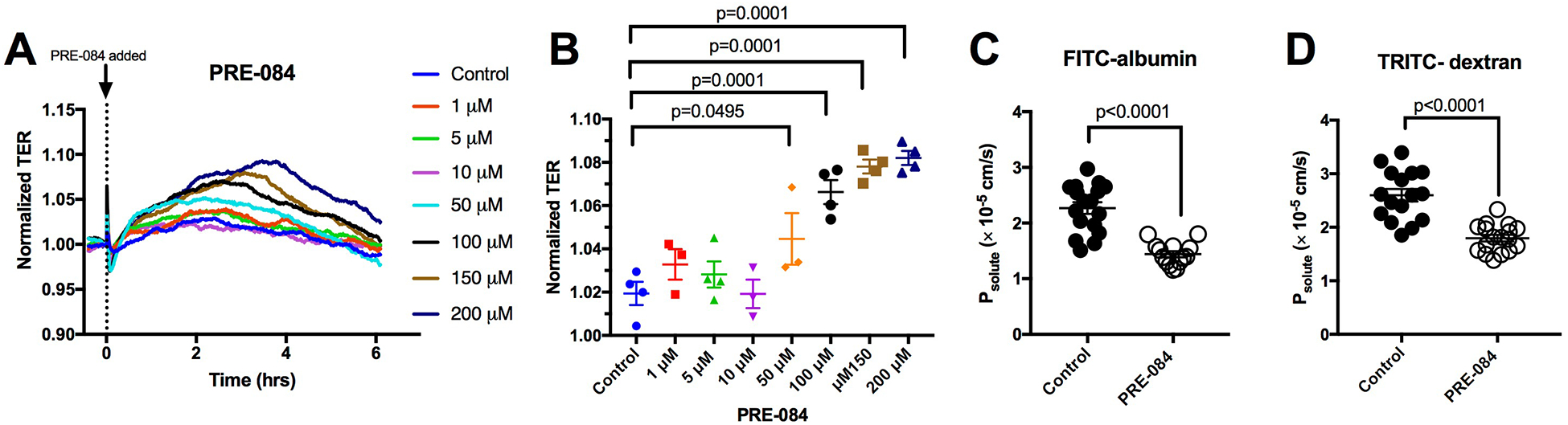
The σ1 agonist PRE-084 elevates barrier function and decreases permeability of HUVEC monolayers. A. Traces showing the time course of changes in TER of HUVEC treated with the shown concentrations of PRE-084. The TER values were normalized to the time point just prior to treatment with PRE-084 or vehicle control (time = 0 h). B. The individual and mean normalized TER values for each group after 3 hours of PRE-084 or vehicle control treatment are shown. One-Way ANOVA followed by Dunnett’s multiple comparison test was used for analysis. N=4 HUVEC monolayers per group. P values are shown for differences considered significant (p < 0.05). C. Scatter plot showing the difference in HUVEC monolayer permeability to FITC-albumin between 3-h vehicle and 3-h 100 μM PRE-084 treatment. N=17 for control and N=14 for PRE-084. D. Scatter plot showing the difference in permeability to TRITC-dextran between 3-h vehicle and 3-h 100 μM PRE-084 treatment. N=16 for control and N=17 for PRE-084. For panels C and D, unpaired t-tests were used for analysis and P values are shown for each comparison.
To investigate whether σ1, in the absence of exogenous activation, contributes to baseline endothelial barrier function, we used siRNA to selectively diminish σ1 expression in HUVEC monolayers. Immediately after transfection with siRNA directed against SIGMAR1 mRNA or non-targeting control RNA, HUVEC were seeded onto ECIS arrays and allowed to form confluent monolayers. Both siRNA-treated cells and control cells exhibited an initial rapid rise in TER in the first 10 h after seeding, reflecting attachment and spreading out on the electrode substratum (Fig 2A). However, when reaching the “elbow” of the trace when the rise in TER becomes more gradual, a difference between the two groups became evident starting at 10 h post-transfection (fig. 2A). The mean TER of endothelial monolayers treated with SIGMAR1 siRNA continued to significantly remain below the mean TER control group until the end of the experiment (Fig. 2A,B). We also assessed whether reducing expression of σ1 affects HUVEC monolayer permeability. The results show that HUVEC subjected to siRNA knockdown of σ1 for 70 h had significantly higher permeability to both FITC-albumin and TRITC-dextran when compared to cells transfected with non-targeting control RNA (Figure 2 C, D). We confirmed the siRNA-mediated knockdown of σ1 at 48 h and 70 h post-transfection using an automated capillary electrophoresis Western blotting system. Representative images generated by the system are shown for the 48 h and 70 h time points (Fig. 2E). The mean band intensity ratios for these time points showed 47% and 74% decreases in σ1 protein, at 48 and 70 h respectively, compared to controls (Fig. 2F).
Fig. 2.
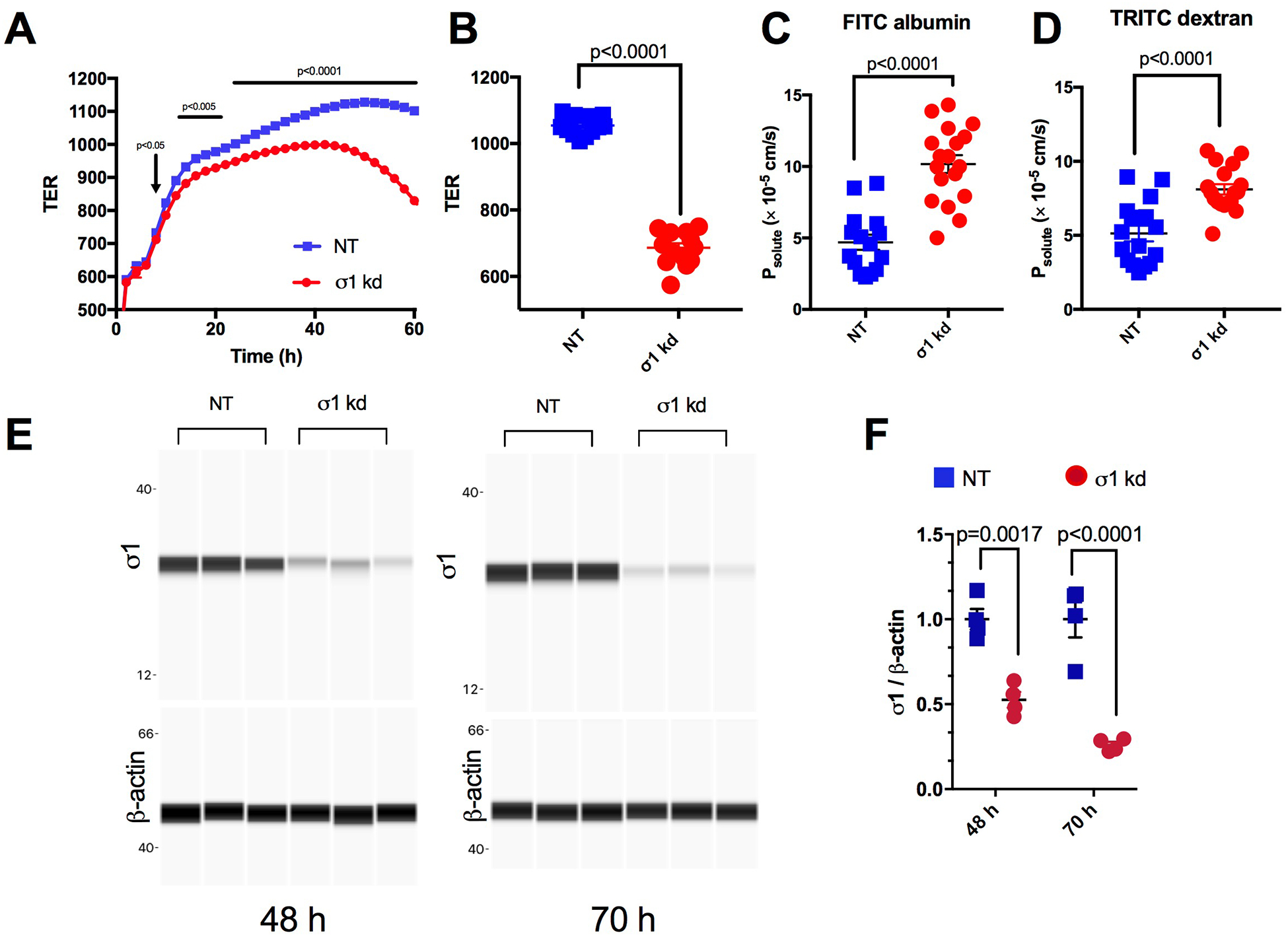
σ1 contributes in maintenance of baseline barrier function. A. Traces of the time course of the changes in mean TER starting immediately after HUVEC were transfected with either non-targeting RNA (NT) or SIGMAR1 siRNA (σ1 kd) and seeded onto ECIS electrodes. N = 14 wells per group. Analysis was done using repeated-measures ANOVA followed by Sidak’s multiple comparison test. B. The difference in mean TER between the two groups at the 70 h post-transfection time point showing all data points is expanded here. C. Scatter plot showing difference in permeability of FITC albumin between NT and σ1 kd HUVEC 70-h post transfection. N=15 for NT and N=18 for σ1 kd. An unpaired t-test was used for analysis. D. Scatter plot showing difference in permeability of TRITC dextran between NT and σ1 kd HUVEC 70-h post transfection. N=16 for NT and N=18 for σ1 kd. An unpaired t-test was used for analysis. E. Western blot confirming knockdown of σ1 in the cells transfected with SIGMAR1 siRNA compared to non-targeting RNA control at 48 h and 70 h. β-actin, which served as a loading control, is also shown. F. Mean band intensities of σ1 normalized to β actin for the σ1 kd and NT groups at 48 h and 70 h (n=4/group). Analysis was done by two-way ANOVA followed by Sidak’s multiple comparison test. P values are shown for differences considered significant (p < 0.05).
While PRE-084 has been reported to exert its action through selective activation of σ1 [43], to confirm that its barrier enhancing properties were indeed due to σ1 agonism and not a off-target effect, we tested the ability of PRE-084 to elicit endothelial barrier enhancement in cell monolayers with siRNA-mediated diminished σ1 expression. Given that knockdown of σ1 itself causes a decrease in TER (Fig. 2), we also used vehicle controls, producing four experimental groups in total: 1) non-targeting RNA + vehicle; 2) non-targeting RNA + 100 μM PRE-084; 3) SIGMAR1 siRNA + vehicle; and 4) SIGMAR1 siRNA + 100 μM PRE-084. While a noticeable increase in mean TER was evident in the non-targeting RNA groups after the addition of PRE-084 compared to vehicle, no increase was observed in the SIGMAR1 siRNA groups (Fig. 3A). Because SIGMAR1 siRNA reduced TER on its own (Fig. 3A), we constructed a time-course graph with TER is normalized to the time point just prior to the addition of PRE-084 or vehicle (t = 0 h). By normalizing the responses, we were able to more directly assess the specific impact of PRE-084 in each group (Fig. 3B). Comparison of the time-point at 3 h after the addition of PRE-084 shows that knockdown of σ1 abolishes the ability of PRE-084 to elevate TER (Fig. 3C,D). These findings highlight the requirement of σ1 for PRE-084-induced endothelial barrier enhancement.
Fig. 3.
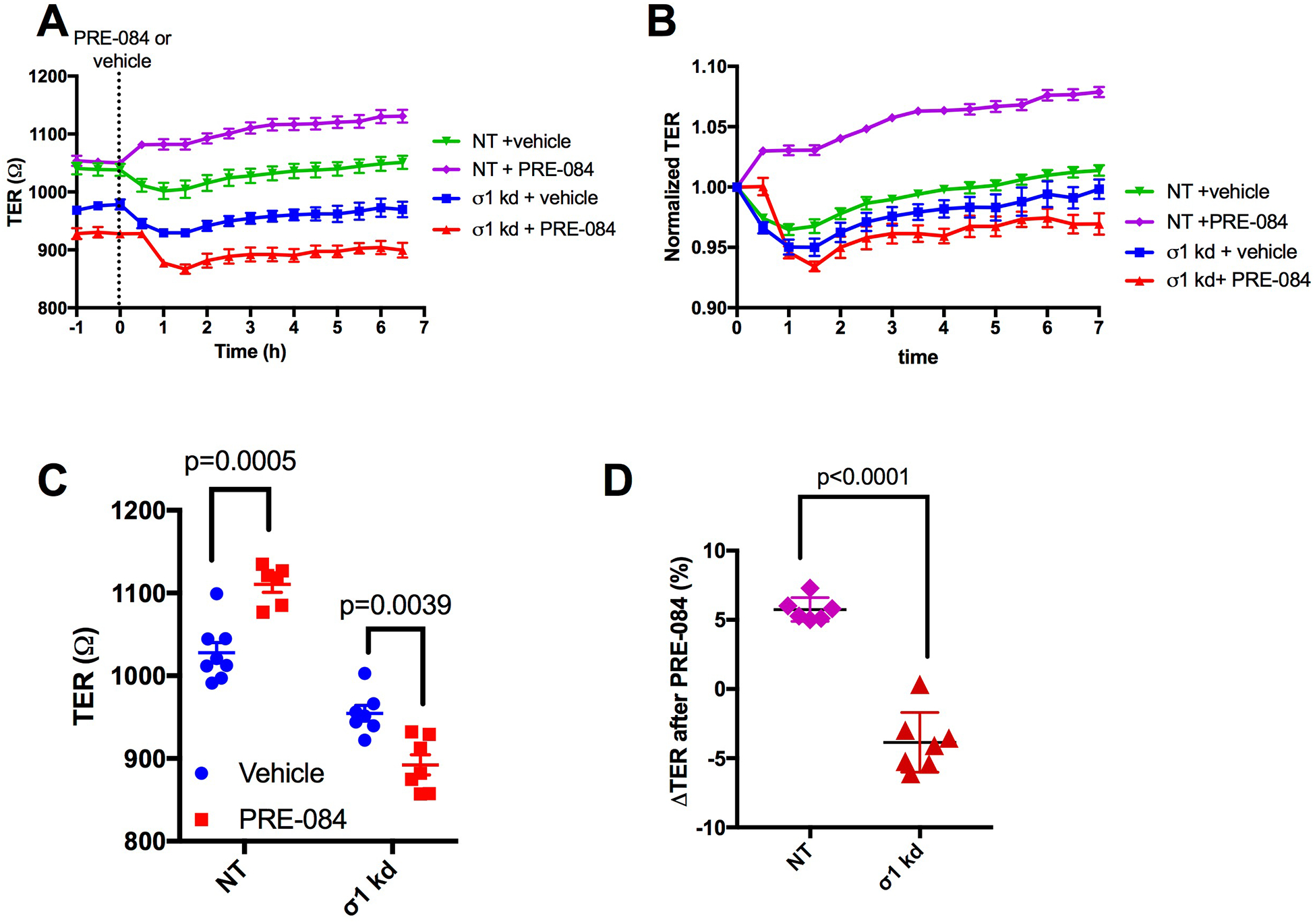
PRE-084 fails to enhance endothelial barrier function in the siRNA-mediated knockdown of σ1. A. The traces show changes in mean TER in the non-targeting RNA (NT) and SIGMAR1 siRNA-mediated knockdown (σ1 kd) groups, before after treatment with 100 μM PRE-084 or vehicle at time t=0 h. B. Time-dependent changes in barrier function in response to PRE-084 or vehicle for each group are also shown using normalized TER values. Normalized TER was calculated by dividing values at each time point by the TER value just prior to the addition of PRE-084 or vehicle (t = 0 h). C. Expanded individual and mean TER for each group is shown for the time point at 3 h after addition of vehicle or 100 μM PRE-084. Data were analyzed by two-way ANOVA followed by Sidak’s test. D. Expanded data for the change in TER (%) from t = 0 h for each group at 3 h after the addition of vehicle or PRE-084 is also shown. An unpaired t test was used for analysis. N= 6–8 HUVEC monolayers per group. P values are shown for all comparisons. Some time points in panel B appear not to have an error bar because the value of the SEM to be plotted was smaller than the size of the symbol.
Because σ1 is known to localize in the mitochondrial-associated ER membrane [1], and this receptor has been associated with the regulation of cellular bioenergetics [18], we investigated how activation of σ1 affects ATP production. HUVEC monolayers were treated with 100 μM PRE-084 for 3 h and then underwent a validated assay protocol to determine ATP production with the Agilent SeaHorse analyzer (Fig. 4A). During this protocol, the oxygen consumption rate (OCR; Fig. 4B) and extracellular acidification rate (ECAR; Fig. 4C) were determined before and after the cells were challenged with oligomycin to inhibit mitochondrial ATP production. A mixture of rotenone and antimycin-A were then added to discount the contribution of CO2 to total acidification. Cells treated with PRE-084 displayed a significantly elevated rate of ATP production due to glycolysis (Fig. 4D), while mitochondrial ATP production was significantly reduced (Fig. 4E). The net effect was a shift in the principal ATP production pathway, without a significant impact in total ATP production when comparing PRE-084 and control groups (Fig. 4F). The results suggest that PRE-084 promotes a shift in ATP production in endothelial cells. Therefore, we ran an additional assay protocol to determine the impact of PRE-084 (100 μM, 3 h) on endothelial glycolytic rate. In this protocol, the cells were treated with the mixture of rotenone and antimycin-A to inhibit all mitochondrial ATP production, and then with 2-deoxyglucose to inhibit glycolysis (Fig. 5A). Traces from the protocol show noticeable differences between PRE-084 and control for both GlycoPER (Fig. 5B) and OCR (Fig. 5C), and from these, we found that PRE-084 caused significant elevations in both basal glycolysis (Fig. 5D) and compensatory glycolysis (Fig. 5E). Related to these findings, PRE-084 significantly increased the basal PER (Fig. 5F) and decreased the ratio of mitochondrial OCR to glycoPER (Fig. 5G). This observation supports the findings described above in Fig. 4 that the glycolytic rate for ATP production is elevated in relation to mitochondrial ATP production. One additional finding was that endothelial monolayers treated with PRE-084 had a small, yet significant increase in the post-2-deoxyglucose acidification rate (Fig. 5H). The reason for this effect is currently unknown.
Fig. 4.
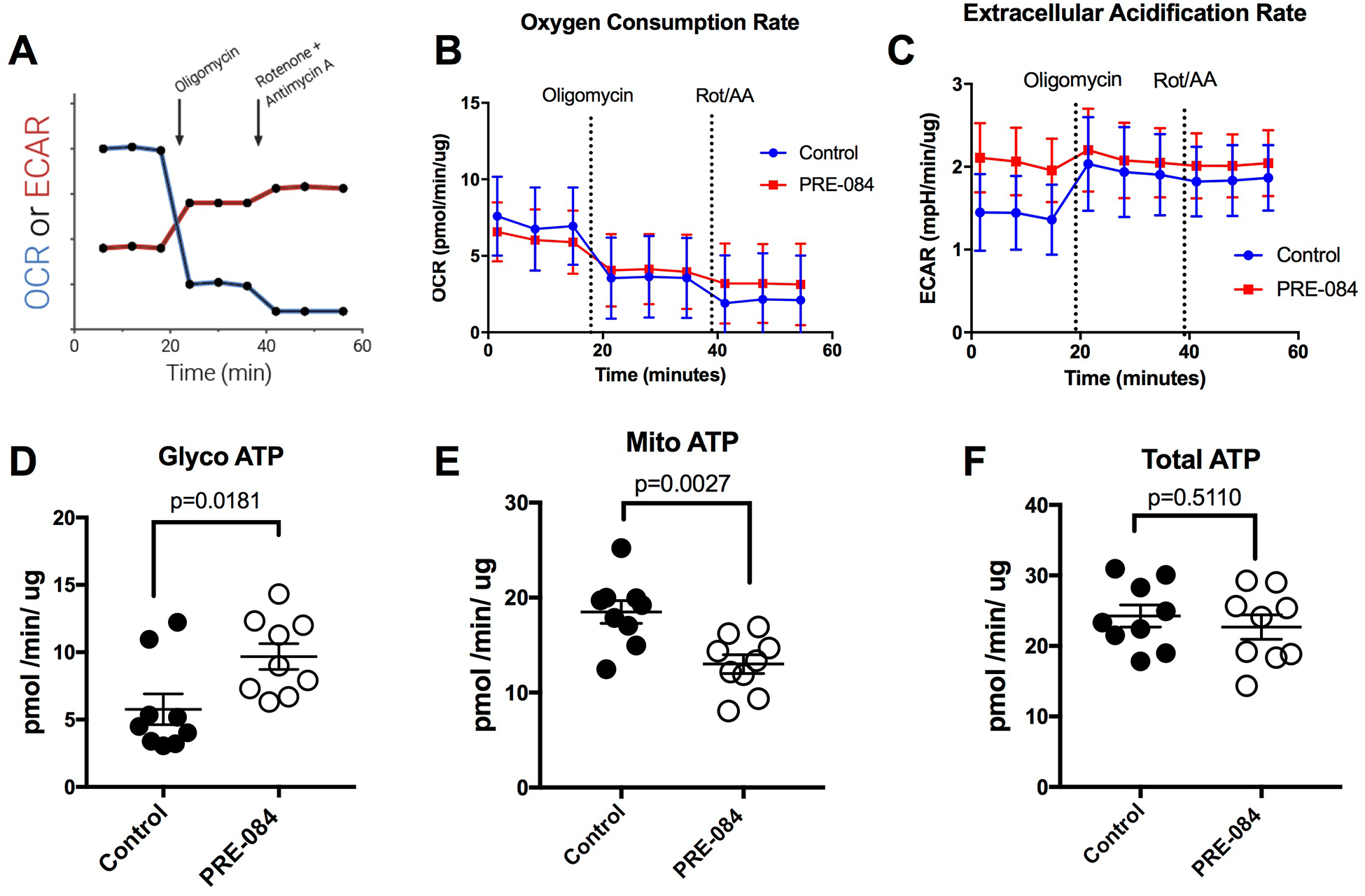
PRE-084 enhances glycolytic ATP production in HUVEC. Confluent HUVEC monolayers were treated either with 100 μM PRE-084 or vehicle control for 3 h and then subjected to the ATP production rate assays using the Agilent SeaHorse XFp system. The ATP production rate assay determines the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) before and after addition of oligomycin and later the rotenone/antimycin-A cocktail. A. Assay profile obtained from Agilent Technologies. © Agilent Technologies, Inc, Reproduced with Permission, Courtesy of Agilent Technologies, Inc. B. OCR, and C, ECAR of controls and PRE-084-treated cells over time during the protocol. Additional parameters calculated from the ATP production rate assay include the glycolytic ATP (D), mitochondrial ATP (E), and total ATP (F) production rates. For each parameter, the two groups were compared using an unpaired t-test. N=9 HUVEC monolayers for each group. P values are shown for all comparisons.
Fig. 5.
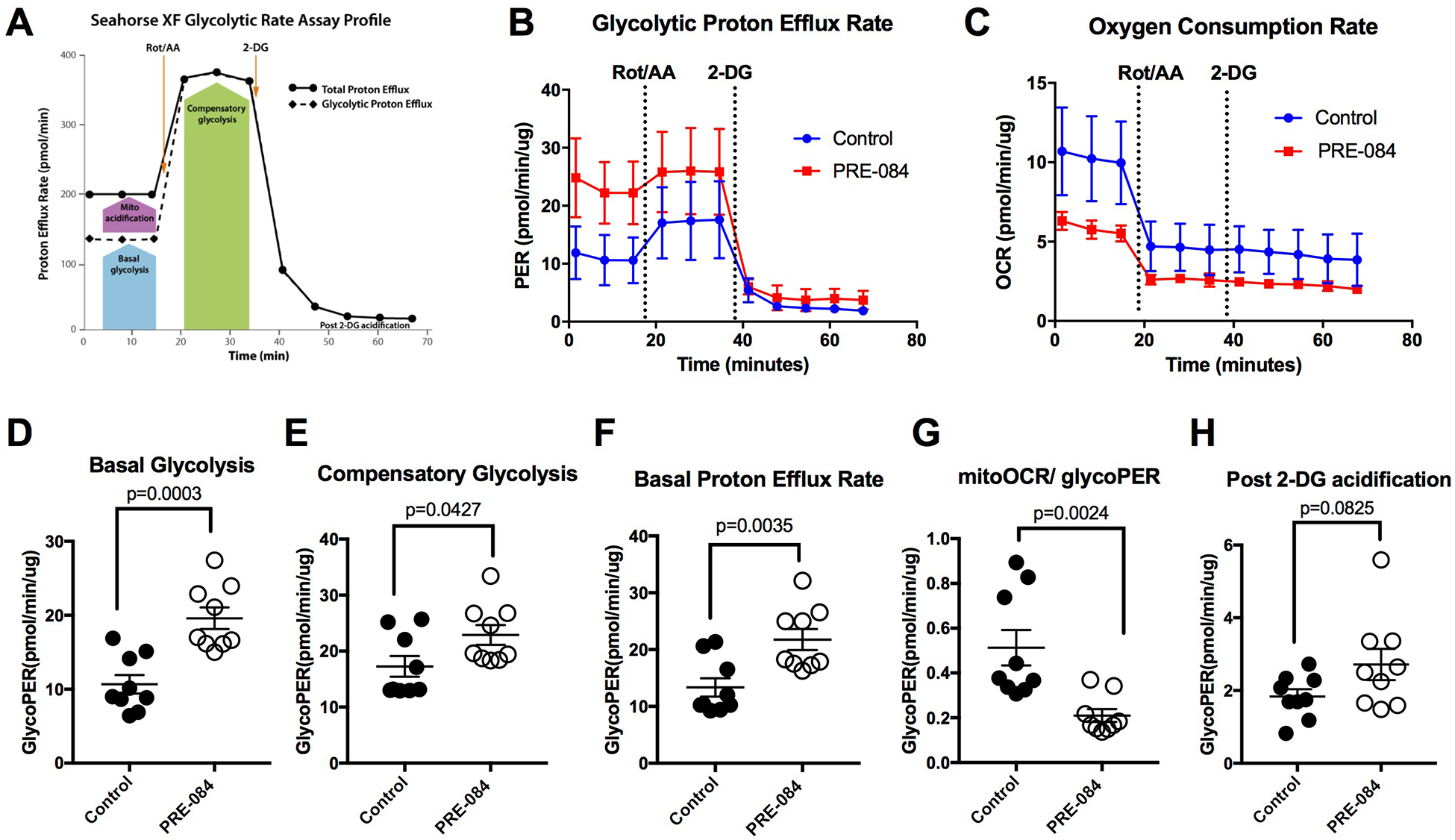
PRE-084 enhances glycolysis in HUVEC. Confluent HUVEC monolayers were treated either with 100 μM PRE-084 or vehicle control for 3 h and then subjected to the glycolytic rate using the Agilent SeaHorse XFp system. A. Assay profile of the glycolytic rate assay obtained from Agilent Technologies. © Agilent Technologies, Inc, Reproduced with Permission, Courtesy of Agilent Technologies, Inc. The glycolytic rate assay determines the glycolytic proton efflux rate (glycoPER, panel B) and the oxygen consumption rate (OCR, panel C) before and after addition of a rotenone/antimycin-A cocktail (Rot/AA) and then addition of 2-deoxyglucose (2-DG). Additional parameters calculated from the glycolytic rate assay include basal glycolysis (D), compensatory glycolysis (E), basal proton efflux rate (PER; panel F), the ratio of mitochondrial OCR to GlycoPER (G) and post 2-DG acidification (H). The two groups were compared using an unpaired t-test for each parameter. N=9 HUVEC monolayers for each group. P values are shown for all comparisons.
We also tested whether the PRE-084-induced enhancement of glycolysis is dependent upon σ1 by utilizing specific siRNA knockdown of σ1 expression. However, we first needed to test whether σ1 deficiency in absence of agonists has any effect on endothelial glycolysis. Therefore, the same glycolytic rate protocol as shown in Fig. 5A was utilized, but with HUVEC monolayers transfected with SIGMAR1 siRNA or non-targeting control RNA for 70 h. The results show a slight noticeable difference in the traces of glycoPER (Fig. 6A) and OCR (Fig. 6B) between the two groups. However, there were no significant difference between the two groups in any of the assay readouts including basal glycolysis, compensatory glycolysis, basal proton efflux rate, mitoOCR/glycoPER or post 2-DG acidification (Figure 6 C–G). Next, when we tested the effects of PRE-084 in cells with diminished expression of σ1, we found that the PRE-084 no longer caused any remarkable changes in the PER or OCR traces in comparison to vehicle-treated cells (Fig. 7A, B). Moreover, PRE-084 did not elicit any changes in the basal glycolysis, compensatory glycolysis, basal proton efflux rate and mitoOCR/glycoPER in comparison to control in cells with siRNA-mediated knockdown of σ1 (Fig. 7C–F). Interestingly, for currently unknown reasons, a small, yet significant decrease in the post-2-deoxyglucose acidification rate was observed in response to PRE-084 when σ1 expression was diminished in cultured HUVEC (Fig. 7G), which was different form normal HUVEC (Fig. 5H). Collectively, these findings indicate that the ability of PRE-084 to enhance glycolytic rate in endothelial cells is dependent upon σ1.
Fig. 6.
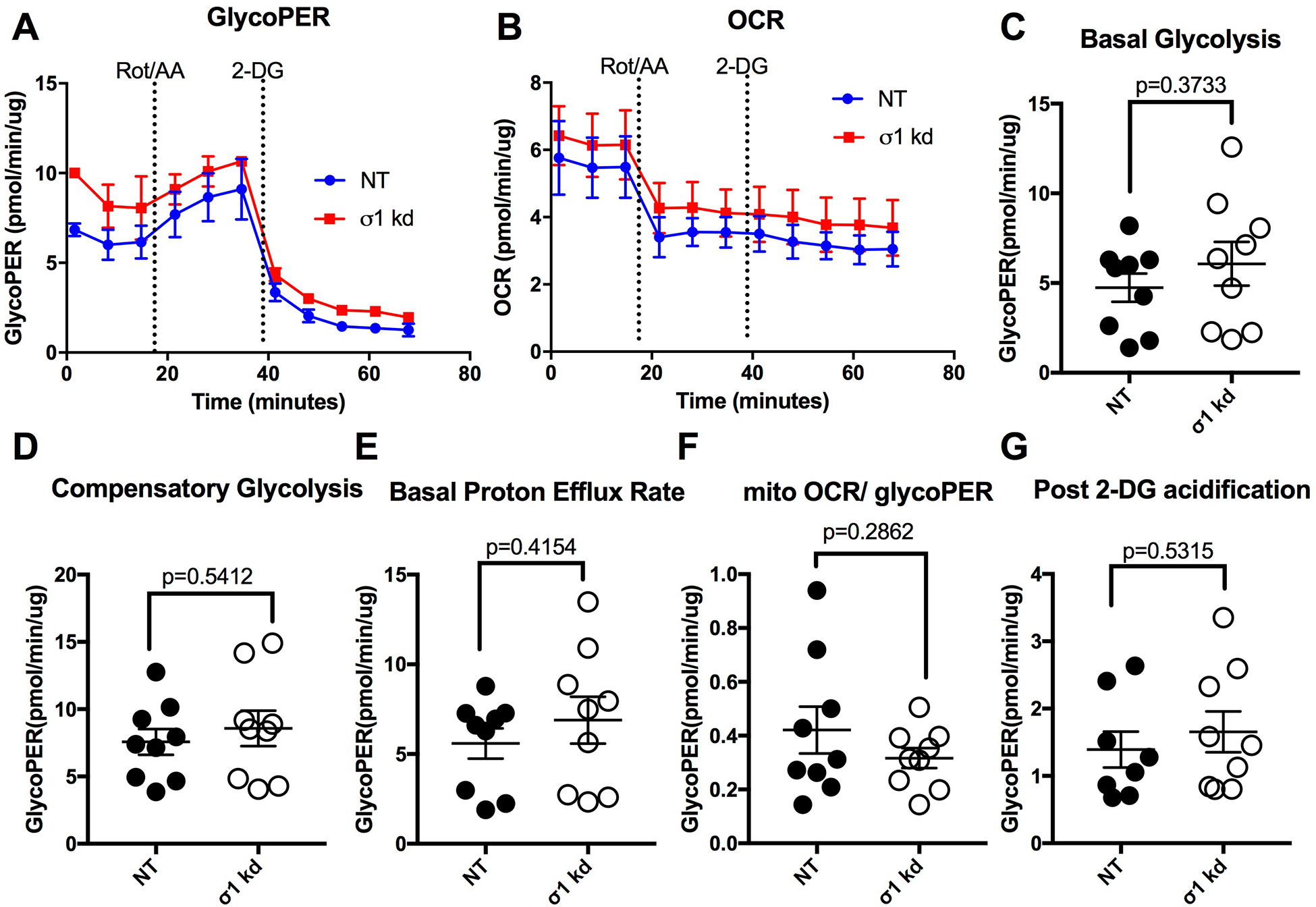
Bioenergetic profile of HUVEC with siRNA-mediated knockdown of σ1 at 70-h post transfection. A. Time course of glycolytic proton efflux rate (GlycoPER) of σ1 kd cells vs. NT control cells during the glycolytic rate assay, in which cells were treated with a rotenone/antimycin-A cocktail (Rot/AA) and later with 2-deoxyglucose (2-DG). B. Oxygen consumption rate (OCR) during the same assay. Additional parameters were calculated from the assay for basal glycolysis (C), compensatory glycolysis (D), basal protein efflux rate (E), the ratio of mitochondrial oxygen rate (mitoOCR) to GlycoPER (F), and post-2-deoxyglucose acidification (G). The two groups were compared with unpaired t-tests for each parameter. P values are shown for all comparisons. N=9 HUVEC monolayers per group.
Fig. 7.
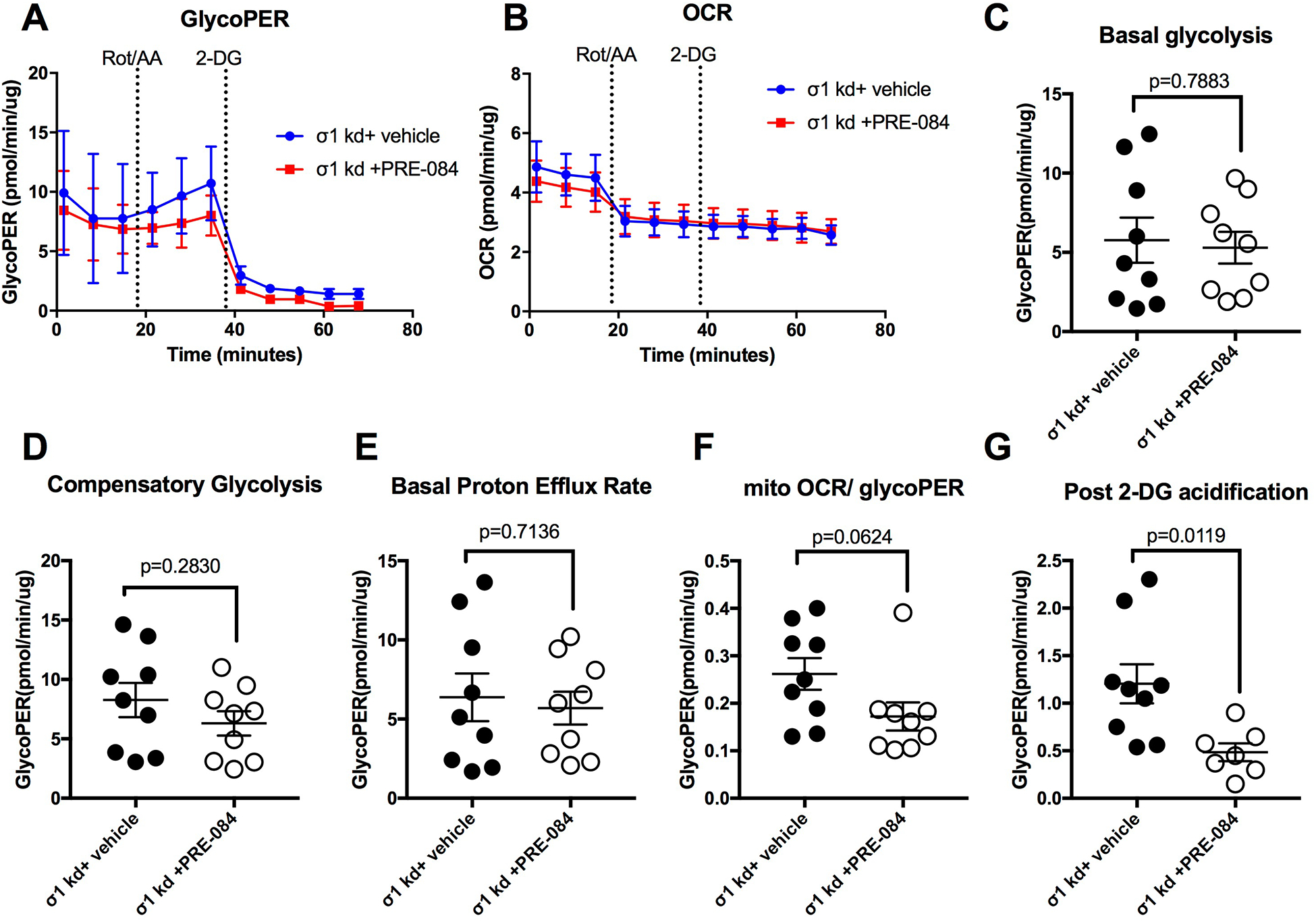
PRE-084 fails to enhance endothelial glycolytic rate after siRNA-mediated knockdown of σ1. At 70 h post-transfection, confluent HUVEC monolayers transfected with SIGMAR1 siRNA (σ1 kd) were treated with PRE-084 or vehicle for 3 h. A. Time course of glycolytic proton efflux rate (GlycoPER) of σ1 kd cells versus NT control cells during the glycolytic rate assay, in which cells were treated with a rotenone/antimycin-A cocktail (Rot/AA) and later with 2-deoxyglucose (2-DG). B. Oxygen consumption rate (OCR) during the same assay. Additional parameters were calculated from the assay for basal glycolysis (C), compensatory glycolysis (D), basal protein efflux rate (E), the ratio of mitochondrial oxygen rate (mitoOCR) to GlycoPER, and the post-2-deoxyglucose acidification (G). The two groups were compared with unpaired t-tests for each parameter. P values are shown for all comparisons. N=7–9 HUVEC monolayers per group.
Because mitochondrial dysfunction has been reported to contribute to increased microvascular permeability in various models of shock [28–30], we investigated whether PRE-084-induced enhancement of glycolytic ATP production could potentially serve as a barrier-protective mechanism. To accomplish this we modeled mitochondrial dysfunction in cultured endothelial cell monolayers using the mitochondrial uncoupler, carbonyl cyanide m-chlorophenyl hydrazone (CCCP). CCCP is a protonophore that is known to drive inward proton gradient across mitochondrial membrane causing disruption of mitochondrial depolarization and uncoupling oxygen consumption from ATP production [44–47]. CCCP caused a time dependent, sustained drop in TER (Fig. 8A). Pretreatment with PRE-084 for 5 min prior to the addition of CCCP did not prevent the initial drop in TER (Fig. 8A), however at later time points, the mean TER of cells treated with PRE-084 and CCCP were significantly higher than the mean TER of cells treated with CCCP alone (Fig. 8A,B). These findings suggest that activation of σ1 can at least partially compensate and help maintain endothelial barrier function when mitochondrial function is compromised.
Fig. 8.
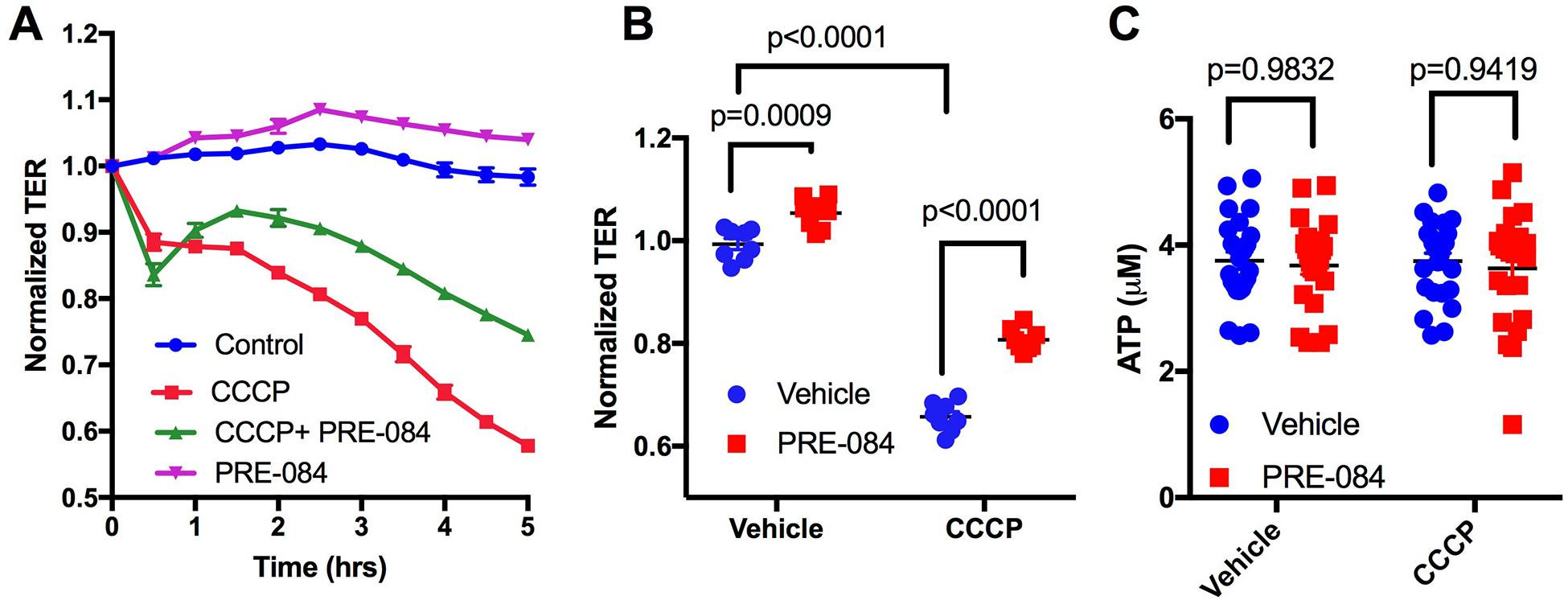
PRE-084 partially reduces endothelial barrier dysfunction caused by the mitochondrial oxidative phosphorylation uncoupler CCCP independent of total ATP level modulation. A. Traces of mean normalized TER over time in cells treated with 10 μM of CCCP in the presence or absence of 100 μM PRE-084 pretreatment for 5 minutes are shown. PRE-084 was added at time t=0 h and CCCP five minutes later. B. Mean normalized TER at 4 hours post treatment for each group. N=8 HUVEC monolayers for each group. Analysis was done by two-way ANOVA followed by Sidak’s multiple comparison test. P values are shown for all comparisons of interest. C. Scatter plot showing differences in total ATP levels between groups. N=24/group. Analysis was done by two-way ANOVA followed by Sidak’s multiple comparison test.
We next investigated whether the CCCP-induced endothelial barrier dysfunction may be by decreased production of ATP. Also, because we had observed that PRE-084 favors glycolytic ATP production rates while leaving total ATP production rates unchanged in Fig. 4, we conducted a more direct assessment of ATP level quantifications in this particular experiment. Consistent with the results in figure 4, PRE-084 alone did not change total levels of ATP (Figure 8C). However, we also found there was also no change in total ATP levels compared to control caused by CCCP or the combination of CCCP and PRE-084 (Figure 8C). These findings suggest that CCCP-induced endothelial barrier dysfunction, and the partial protection from CCCP-induced endothelial barrier dysfunction elicited by PRE-084 are not due to a change in overall ATP production in endothelial cells. Rather, the mechanism likely involves an ATP-independent aspect of mitochondrial dysfunction.
Because tight junctions and adherens junctions play a major role in endothelial barrier, we investigated to what extent CCCP disrupts the integrity of junctional protein architecture. We used immunofluorescence to label junctional proteins in HUVEC monolayers treated with 10 μM CCCP in presence or absence of 100 μM PRE-084. We chose β-catenin as an representative adherens junction protein and ZO-1 to represent tight junctions. CCCP caused apparent disruption of junctional β-catenin (green) and ZO-1 (red) in the form of junctional breakages compared to control (Fig. 9A). In the CCCP+PRE-084 group, there were still some junctional breakages but this was also accompanied by higher intensities of both β-catenin and ZO-1 at junctional regions. Cells treated with only PRE-084 did not appear to have different β-catenin or ZO-1 intensities along the junctions (Figure 9A). Using quantification of intensities of both proteins in junctional regions using imageJ, we observed that in cells that were exposed to CCCP, treatment with PRE-084 significantly increased both β-catenin and ZO-1 levels within junctions (Fig. 9B). These findings suggest that PRE-084 can enhance junctional protein architecture in the context of mitochondrial-disruption-induced endothelial barrier dysfunction.
Fig. 9.
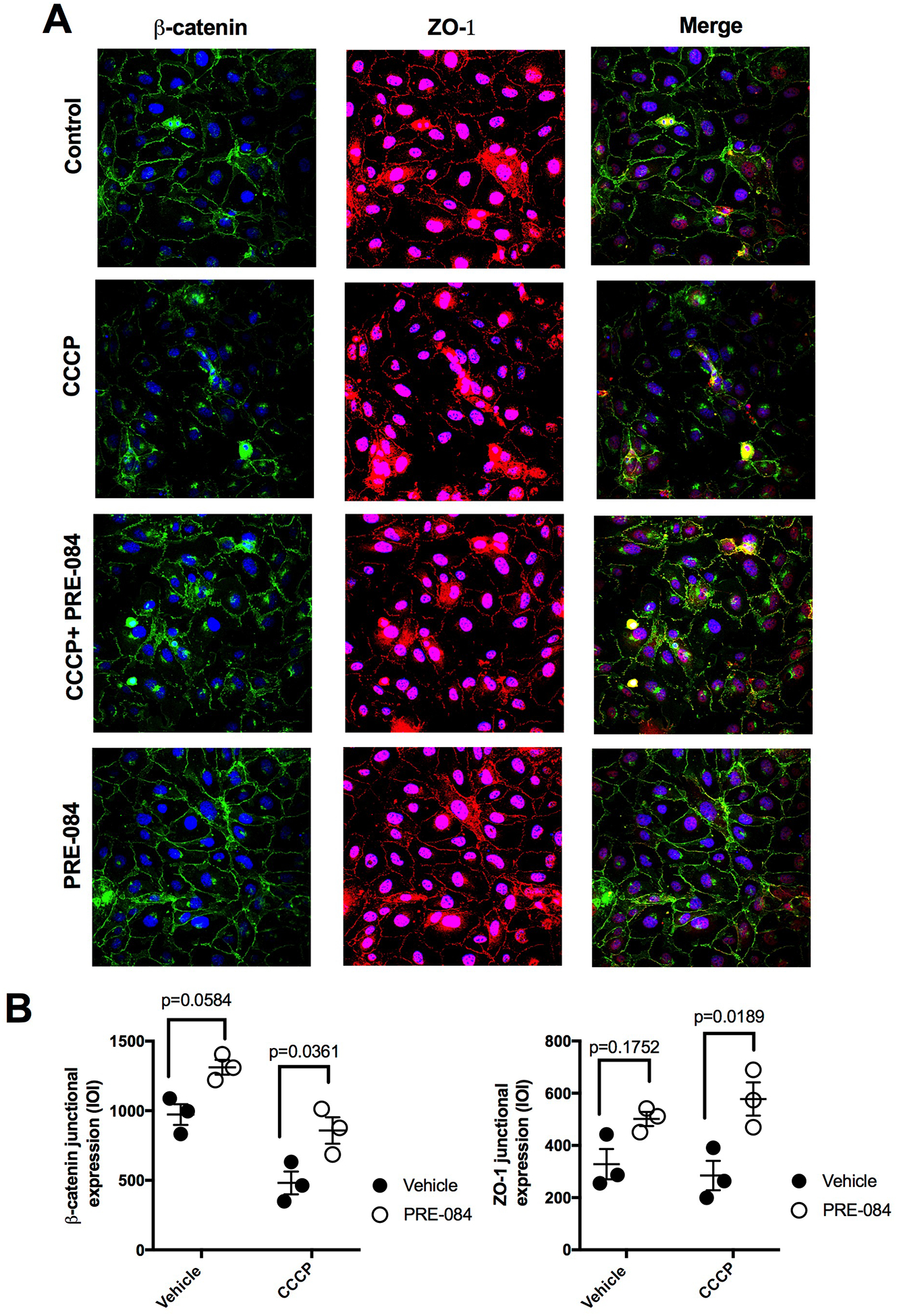
PRE-084 partially preserves endothelial junctional integrity after CCCP. A. Representative immunofluorescence images of HUVEC treated with CCCP 10 μM for 4 hours in the presence or absence of PRE-084 100 μM showing β-catenin (green), ZO-1 (red), Nuclei (blue). B. Scatter plots showing quantification of β-catenin and ZO-1 at paracellular junction regions. Analysis was done using 2-way AVOVA followed by Tukey’s multiple comparison test. N=3 monolayers/group.
DISCUSSION
This is the first study delineating the role of σ1 in endothelial barrier function from a mechanistic perspective, to the best of or knowledge. Barrier function is a critical property of the endothelium for normal blood-tissue exchange and homeostasis. The contribution of σ1 to endothelial barrier function was recently suggested in a study using global σ1 knockout mice, which reported that σ1 activation can protect against blood-brain barrier leakage [17]. However, whether the findings from that study reflected a true role for σ1 in controlling diffusive permeability versus filtration in endothelium in general was not clear. In the current study we tested the direct impact of σ1 activation on barrier function in a more tightly controlled model. We chose cultured endothelial cell monolayers for study in order to be able to attribute any findings about signaling mechanisms directly to the endothelium, and not other cell types.
The current work demonstrates that σ1 stimulation with PRE-084 elevates barrier function in cultured endothelial cell monolayers, and that, conversely, decreased expression of σ1 impairs endothelial barrier function. Thus, σ1 provides tonic regulation of barrier function and can further enhance barrier function upon activation. In addition, our results show for the first time that σ1 activation has a profound impact on the glycolysis in endothelial cells, increasing glycolytic ATP production while decreasing mitochondrial ATP production. Moreover, this mechanism may be targeted to enhance oxygen delivery to local tissues by reducing O2 consumption by the endothelium. Endothelial mitochondrial dysfunction has been reported in multiple models of microvascular hyperpermeability [28–30]. In the current study we modeled this scenario using CCCP, which is known to cause mitochondrial damage, depolarization and energy depletion [44,47–49]. We found that the σ1 agonist PRE-084 could partially reduce CCCP-induced endothelial barrier dysfunction, suggesting that σ1 could potentially serve as a useful therapeutic target to preserve endothelial barrier function in pathologies involving mitochondrial dysfunction.
Despite the fact that neither CCCP nor PRE-084 caused a significant change in total ATP levels, our finding that σ1 activation enhances glycolysis and glycolytic ATP production, suggests that σ1 activation may counteract barrier destabilization caused by CCCP, by a mechanism that may involve upregulation of glycolysis and subsequent preservation of junctional integrity (Fig. 5,9). This concept is consistent with a previously reported finding, in which agents that enhance glycolysis such as fructose could counteract CCCP-induced cell toxicity by glycolytic ATP formation rather than by preserving mitochondrial membrane potential [49]. Minimal or no effect of CCCP on total endothelial ATP levels was expected as it has been reported that endothelial cells are characterized by glycolytic formation of ATP accounting for up to 85% of total ATP production, with mitochondrial respiration considered as a secondary source [50,51]. Thus, the barrier disruption caused by CCCP in the form of impaired TER and junctional destabilization is likely to be caused by impairment of another aspect of mitochondrial function and/or signaling independent of ATP level changes. This assumption is consistent with a recent finding by Hough et al that endothelial mitochondrial depolarization caused endothelial barrier impairment by a signaling pathway that involves calcineurin-cofilin-actin axis [52]. At this stage, additional details of the ATP-independent mechanism remain elusive.
Zecchin et al. highlighted multiple advantages of using glycolysis by endothelial cells as the primary source of energy. In addition to sparing oxygen for usage by perivascular cells, a reduction in endothelial mitochondrial respiration decreases reactive oxygen species generation within the endothelium that could have detrimental effects on barrier function [51]. It has been shown that glycolysis regulatory enzymes such as phosphofructokinase associate with the endothelial actin cytoskeleton and lamellipodia causing cytoskeletal protrusions during vessel sprouting [34]. These enzymes would also presumably be available to provide energy for the cytoskeletal rearrangements that underlie local lamellipodia, which have been connected to maintenance of endothelial barrier function [26]. The role of glycolytic ATP production in such cellular rearrangements has also been determined through experimental and computational modeling [53]. Moreover, active spatiotemporal patterns of ATP-dependent signaling at the cell periphery are evident when endothelial cell monolayers are stimulated to enhance their barrier function [27].
Despite the correlation that we found between σ1-activation-induced barrier enhancement and glycolysis upregulation (Figs. 1, 4, and 5), we did not find a comparable negative correlation between barrier impairment of σ1-knockdown cells and their energetic profile (Fig. 2 and 6). Thus, at this stage we cannot conclude that an energetic failure/modulation is necessarily the primary cause for the barrier defect in endothelial cells with diminished expression of σ1. This issue will need additional investigation. However, we did observe that diminished expression of σ1 impaired the ability of PRE-084 to evoke enhancement of glycolysis and endothelia barrier function, suggesting the requirement of σ1 for these responses (Fig. 7). Related to this, an unexpected, yet interesting finding was that when σ1 expression was selectively diminished by siRNA treatment, the action of PRE-084 appeared to be reversed in some instances (Fig 3 C,D, Fig 7 G). While the explanation for this phenomenon is currently unknown, we speculate that there is a possibility that the absence or severe depletion of σ1 allows significantly more binding of PRE-084 to other molecules for which it has less affinity for in comparison to σ1. This possibility remains an avenue for future exploration.
In conclusion, this is the first study to our knowledge to show the significant contribution of σ1 receptors in maintenance of endothelial barrier function under basal conditions and after challenge with barrier disrupters. Our data show for the first time that σ1 activation leads to enhancement of glycolysis and subsequent glycolytic ATP production, which are tightly linked to enhancing endothelial barrier function. In contrast, σ1 deficiency leads to disruption of the barrier function. Future work investigating the therapeutic potential of σ1 activation in microvascular hyperpermeability in response to injury or under disease conditions is warranted.
PERSPECTIVES.
σ1 has gained considerable attention due to its neuroprotectisve and cardioprotective functions. Our results show a novel role for σ1receptors in the stabilization and enhancement of barrier function, as well as in the elevation of endothelial cell glycolytic ATP production. This significant finding suggests that σ1 agonists are potential therapeutic agents for the treatment of pathologies involving microvascular leakage.
ACKNOWLEDGEMENTS
Funding source: National Institutes of Health, National Institute of General Medical Sciences, R01 GM120774, Edith Wright Hartley graduate scholarship.
LIST OF ABBREVIATIONS
- σ1
The sigma-1 receptor
- ER
Endoplasmic reticulum
- IP3R
Inositol trisphosphate receptor
- HUVEC
Human umbilical vein endothelial cells
- TER
Transendothelial electrical resistance
- ECIS
Electrical cell-substrate impedance sensor
- VBM
Vasculife Basal Media
- ECAR
Extracellular Acidification Rate
- OCR
Oxygen Consumption Rate
- Rot/AA
Rotenone/Antimycin A
- 2-DG
2-Deoxy-Glucose
- σ1 kd
Sigma-1 knock down
- NT
Non targeting Transfected
- CCCP
Carbonyl cyanide m-chlorophenyl hydrazone
- GlycoPER
Glycolytic Proton Efflux Rate
- mitoOCR
Mitochondrial Oxygen Consumption Rate
- FITC
Fluorescein isothiocyanate
- TRITC
Tetramethylrhodamine
REFERENCES
- 1.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131: 596–610, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Wu Z, Bowen WD. Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: constitutive enhancement of calcium signaling in MCF-7 tumor cells. J Biol Chem 283: 28198–28215, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abraham MJ, Fleming KL, Raymond S, Wong AYC, Bergeron R. The sigma-1 receptor behaves as an atypical auxiliary subunit to modulate the functional characteristics of Kv1.2 channels expressed in HEK293 cells. Physiol Rep 7: e14147, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balasuriya D, D’Sa L, Talker R, Dupuis E, Maurin F, Martin P, Borgese F, Soriani O, Edwardson JM. A direct interaction between the sigma-1 receptor and the hERG voltage-gated K+ channel revealed by atomic force microscopy and homogeneous time-resolved fluorescence (HTRF(R)). J Biol Chem 289: 32353–32363, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Katnik C, Cuevas J. Sigma receptor activation inhibits voltage-gated sodium channels in rat intracardiac ganglion neurons. Int J Physiol Pathophysiol Pharmacol 2: 1–11, 2009. [PMC free article] [PubMed] [Google Scholar]
- 6.Katnik C, Guerrero WR, Pennypacker KR, Herrera Y, Cuevas J. Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical neurons during in vitro ischemia. J Pharmacol Exp Ther 319: 1355–1365, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi T, Su TP. Cholesterol at the endoplasmic reticulum: roles of the sigma-1 receptor chaperone and implications thereof in human diseases. Subcell Biochem 51: 381–398, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruscher K, Wieloch T. The involvement of the sigma-1 receptor in neurodegeneration and neurorestoration. J Pharmacol Sci 127: 30–35, 2015. [DOI] [PubMed] [Google Scholar]
- 9.Reddy DS, Kaur G, Kulkarni SK. Sigma (sigma1) receptor mediated anti-depressant-like effects of neurosteroids in the Porsolt forced swim test. Neuroreport 9: 3069–3073, 1998. [DOI] [PubMed] [Google Scholar]
- 10.Kamei H, Noda Y, Kameyama T, Nabeshima T. Role of (+)-SKF-10,047-sensitive sub-population of sigma 1 receptors in amelioration of conditioned fear stress in rats: association with mesolimbic dopaminergic systems. Eur J Pharmacol 319: 165–172, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Maurice T, Roman FJ, Su TP, Privat A. Beneficial effects of sigma agonists on the age-related learning impairment in the senescence-accelerated mouse (SAM). Brain Res 733: 219–230, 1996. [DOI] [PubMed] [Google Scholar]
- 12.Villard V, Espallergues J, Keller E, Alkam T, Nitta A, Yamada K, Nabeshima T, Vamvakides A, Maurice T. Antiamnesic and neuroprotective effects of the aminotetrahydrofuran derivative ANAVEX1–41 against amyloid beta(25–35)-induced toxicity in mice. Neuropsychopharmacology 34: 1552–1566, 2009. [DOI] [PubMed] [Google Scholar]
- 13.Katnik C, Garcia A, Behensky AA, Yasny IE, Shuster AM, Seredenin SB, Petrov AV, Seifu S, McAleer J, Willing A, Cuevas J. Treatment with afobazole at delayed time points following ischemic stroke improves long-term functional and histological outcomes. Neurobiol Dis 62: 354–364, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Tagashira H, Bhuiyan S, Shioda N, Hasegawa H, Kanai H, Fukunaga K. Sigma1-receptor stimulation with fluvoxamine ameliorates transverse aortic constriction-induced myocardial hypertrophy and dysfunction in mice. Am J Physiol Heart Circ Physiol 299: H1535–1545, 2010. [DOI] [PubMed] [Google Scholar]
- 15.Amer MS, McKeown L, Tumova S, Liu R, Seymour VA, Wilson LA, Naylor J, Greenhalgh K, Hou B, Majeed Y, Turner P, Sedo A, O’Regan DJ, Li J, Bon RS, Porter KE, Beech DJ. Inhibition of endothelial cell Ca(2)(+) entry and transient receptor potential channels by Sigma-1 receptor ligands. Br J Pharmacol 168: 1445–1455, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trujillo AN, Katnik C, Cuevas J, Cha BJ, Taylor-Clark TE, Breslin JW. Modulation of mesenteric collecting lymphatic contractions by sigma1-receptor activation and nitric oxide production. Am J Physiol Heart Circ Physiol 313: H839–H853, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu DY, Chi TY, Ji XF, Liu P, Qi XX, Zhu L, Wang ZQ, Li L, Chen L, Zou LB. Sigma-1 receptor activation alleviates blood-brain barrier dysfunction in vascular dementia mice. Exp Neurol 308: 90–99, 2018. [DOI] [PubMed] [Google Scholar]
- 18.Abdullah CS, Alam S, Aishwarya R, Miriyala S, Panchatcharam M, Bhuiyan MAN, Peretik JM, Orr AW, James J, Osinska H, Robbins J, Lorenz JN, Bhuiyan MS. Cardiac Dysfunction in the Sigma 1 Receptor Knockout Mouse Associated With Impaired Mitochondrial Dynamics and Bioenergetics. J Am Heart Assoc 7: e009775, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duran WN, Sanchez FA, Breslin JW. Microcirculatory Exchange Function In: Handbook of Microcirculation, edited by Tuma RF, Duran WN, Ley K. San Diego: Academic Press, 2008, p. 81–124. [Google Scholar]
- 20.Sun H, Breslin JW, Zhu J, Yuan SY, Wu MH. Rho and ROCK signaling in VEGF-induced microvascular endothelial hyperpermeability. Microcirculation 13: 237–247, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Aramoto H, Breslin JW, Pappas PJ, Hobson RW 2nd, Duran WN. Vascular endothelial growth factor stimulates differential signaling pathways in in vivo microcirculation. Am J Physiol Heart Circ Physiol 287: H1590–1598, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Breslin JW, Kurtz KM. Lymphatic endothelial cells adapt their barrier function in response to changes in shear stress. Lymphat Res Biol 7: 229–237, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adderley SP, Lawrence C, Madonia E, Olubadewo JO, Breslin JW. Histamine activates p38 MAP kinase and alters local lamellipodia dynamics, reducing endothelial barrier integrity and eliciting central movement of actin fibers. Am J Physiol Cell Physiol 309: C51–59, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alves NG, Yuan SY, Breslin JW. Sphingosine-1-phosphate protects against brain microvascular endothelial junctional protein disorganization and barrier dysfunction caused by alcohol. Microcirculation 26: e12506, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breslin JW, Daines DA, Doggett TM, Kurtz KH, Souza-Smith FM, Zhang XE, Wu MH, Yuan SY. Rnd3 as a Novel Target to Ameliorate Microvascular Leakage. J Am Heart Assoc 5: e003336, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breslin JW, Zhang XE, Worthylake RA, Souza-Smith FM. Involvement of local lamellipodia in endothelial barrier function. PLoS One 10: e0117970, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang XE, Adderley SP, Breslin JW. Activation of RhoA, but Not Rac1, Mediates Early Stages of S1P-Induced Endothelial Barrier Enhancement. PLoS One 11: e0155490, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Childs EW, Tharakan B, Byrge N, Tinsley JH, Hunter FA, Smythe WR. Angiopoietin-1 inhibits intrinsic apoptotic signaling and vascular hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol 294: H2285–2295, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Alves NG, Trujillo AN, Breslin JW, Yuan SY. Sphingosine-1-Phosphate Reduces Hemorrhagic Shock and Resuscitation-Induced Microvascular Leakage by Protecting Endothelial Mitochondrial Integrity. Shock 52: 423–433, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin B, Liu Y, Li T, Zeng K, Cai S, Zeng Z, Lin C, Chen Z, Gao Y. Ulinastatin mediates protection against vascular hyperpermeability following hemorrhagic shock. Int J Clin Exp Pathol 8: 7685–7693, 2015. [PMC free article] [PubMed] [Google Scholar]
- 31.Busija DW, Rutkai I, Dutta S, Katakam PV. Role of Mitochondria in Cerebral Vascular Function: Energy Production, Cellular Protection, and Regulation of Vascular Tone. Compr Physiol 6: 1529–1548, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katakam PV, Wappler EA, Katz PS, Rutkai I, Institoris A, Domoki F, Gaspar T, Grovenburg SM, Snipes JA, Busija DW. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol 33: 752–759, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res 116: 1231–1244, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154: 651–663, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Bierhansl L, Conradi LC, Treps L, Dewerchin M, Carmeliet P. Central Role of Metabolism in Endothelial Cell Function and Vascular Disease. Physiology (Bethesda) 32: 126–140, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goguadze N, Zhuravliova E, Morin D, Mikeladze D, Maurice T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox Res 35: 1–18, 2019. [DOI] [PubMed] [Google Scholar]
- 37.Giaever I, Keese CR. Micromotion of mammalian cells measured electrically. Proc Natl Acad Sci U S A 88: 7896–7900, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romero Rogers, Neilson Dranka. Quantifying Cellular ATP Production Rate Using Agilent Seahorse XF Technology. Agilent Technologies, Inc, Lexington, MA, USA, 2018. [Google Scholar]
- 39.WS. R. U.S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/. [Google Scholar]
- 40.Lopez OV, Gorantla S, Segarra AC, Andino Norat MC, Alvarez M, Skolasky RL, Melendez LM. Sigma-1 Receptor Antagonist (BD1047) Decreases Cathepsin B Secretion in HIV-Infected Macrophages Exposed to Cocaine. J Neuroimmune Pharmacol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu J, Zang S, Chen X, Jiang L, Gu A, Cheng J, Zhang L, Wang J, Xiao H. Involvement of the delayed rectifier outward potassium channel Kv2.1 in methamphetamine-induced neuronal apoptosis via the p38 mitogen-activated protein kinase signaling pathway. J Appl Toxicol 38: 696–704, 2018. [DOI] [PubMed] [Google Scholar]
- 42.Zhang K, Zhao Z, Lan L, Wei X, Wang L, Liu X, Yan H, Zheng J. Sigma-1 Receptor Plays a Negative Modulation on N-type Calcium Channel. Front Pharmacol 8: 302, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hyrskyluoto A, Pulli I, Tornqvist K, Ho TH, Korhonen L, Lindholm D. Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: involvement of calpastatin and the NF-kappaB pathway. Cell Death Dis 4: e646, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim ML, Minamikawa T, Nagley P. The protonophore CCCP induces mitochondrial permeability transition without cytochrome c release in human osteosarcoma cells. FEBS Lett 503: 69–74, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Zhang YQ, Shen X, Xiao XL, Liu MY, Li SL, Yan J, Jin J, Gao JL, Zhen CL, Hu N, Zhang XZ, Tai Y, Zhang LS, Bai YL, Dong DL. Mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone induces vasorelaxation without involving KATP channel activation in smooth muscle cells of arteries. Br J Pharmacol 173: 3145–3158, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganote CE, Armstrong SC. Effects of CCCP-induced mitochondrial uncoupling and cyclosporin A on cell volume, cell injury and preconditioning protection of isolated rabbit cardiomyocytes. J Mol Cell Cardiol 35: 749–759, 2003. [DOI] [PubMed] [Google Scholar]
- 47.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kowluru RA, Mohammad G, Santos JM, Tewari S, Zhong Q. Interleukin-1beta and mitochondria damage, and the development of diabetic retinopathy. J Ocul Biol Dis Infor 4: 3–9, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nieminen AL, Saylor AK, Herman B, Lemasters JJ. ATP depletion rather than mitochondrial depolarization mediates hepatocyte killing after metabolic inhibition. Am J Physiol 267: C67–74, 1994. [DOI] [PubMed] [Google Scholar]
- 50.Yetkin-Arik B, Vogels IMC, Nowak-Sliwinska P, Weiss A, Houtkooper RH, Van Noorden CJF, Klaassen I, Schlingemann RO. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci Rep 9: 12608, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zecchin A, Kalucka J, Dubois C, Carmeliet P. How Endothelial Cells Adapt Their Metabolism to Form Vessels in Tumors. Front Immunol 8: 1750, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hough RF, Islam MN, Gusarova GA, Jin G, Das S, Bhattacharya J. Endothelial mitochondria determine rapid barrier failure in chemical lung injury. JCI Insight 4, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cruys B, Wong BW, Kuchnio A, Verdegem D, Cantelmo AR, Conradi LC, Vandekeere S, Bouche A, Cornelissen I, Vinckier S, Merks RM, Dejana E, Gerhardt H, Dewerchin M, Bentley K, Carmeliet P. Glycolytic regulation of cell rearrangement in angiogenesis. Nat Commun 7: 12240, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]