

Abstract
Coiled coils (CCs) are powerful supramolecular building blocks for biomimetic materials, increasingly used for their mechanical properties. Here, we introduce helix‐inducing macrocyclic constraints, so‐called staples, to tune thermodynamic and mechanical stability of CCs. We show that thermodynamic stabilization of CCs against helix uncoiling primarily depends on the number of staples, whereas staple positioning controls CC mechanical stability. Inserting a covalent lactam staple at one key force application point significantly increases the barrier to force‐induced CC dissociation and reduces structural deformity. A reversible His‐Ni2+‐His metal staple also increases CC stability, but ruptures upon mechanical loading to allow helix uncoiling. Staple type, position and number are key design parameters in using helical macrocyclic templates for fine‐tuning CC properties in emerging biomaterials.
Keywords: coiled coil, lactam, metal coordination, peptide stapling, single-molecule force spectroscopy
Staple it! A terminal helical turn in heterodimeric coiled coils was stapled with a covalent lactam or a reversible metal‐coordination bond. The staples provide resistance to helix uncoiling and increase coiled coil stability to thermal denaturation and external mechanical forces.

Coiled coils (CCs) are naturally occurring, intertwined helical structures in proteins important for gene expression (e.g. transcription factors) and mechanical function (e.g. extracellular matrix and cytoskeleton proteins). [1] CCs are supercoils formed from two to seven α‐helices with a highly repetitive sequence motif of seven amino acids (heptads), designated abcdefg (Figure 1 A). [2] Amino acids at positions a and d typically form a hydrophobic core, while charged amino acids at e and g positions direct CC formation via ionic interactions. The solvent‐exposed positions b, c and f are well‐suited for modifications without affecting the overall CC structure. [3] Synthetic CC sequences and structurally related helical assemblies serve as tunable scaffolds for condensation reactions [4] or functional group transfer [5] as well as for molecular switches that sense receptor binding [6] or the local ionic strength. [7] In recent years, the mechanical properties of CCs have enabled their application as building blocks for biomimetic hydrogels, [8] protein origami structures, [9] artificial membrane fusion domains [10] and molecular force sensors. [11] To tune the mechanical properties of synthetic CCs, a detailed understanding of their sequence‐structure‐mechanical relationships is crucial. We have recently shown that the helix propensity of the solvent‐exposed amino acids affects the mechanical stability of a 4‐heptad CC heterodimer when applying a shear force at two opposing termini. [12] Further, molecular dynamics simulations have shown that CC heterodimers and heterotrimers respond to the applied shear force with helix uncoiling. [13] Simulations with constrained dihedral angles caused the CC chains to dissociate in helical conformations. As the helices remained folded, chain dissociation occurred at smaller extensions, but required higher forces. [13b] Here, we show that introducing helix constraints (Figure 1), so‐called staples, can also stabilize CCs in experiments and provide control over their thermodynamic, kinetic and mechanical properties.
Figure 1.
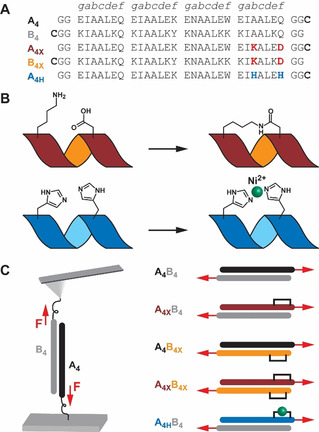
Coiled coil design. A) Peptide sequences. For immobilization, Cys was inserted at the C‐terminus of the A4 peptides and the N‐terminus of the B4 peptides. B) Staple design. A helix‐inducing macrocycle was formed via amide bond formation between Lys and Asp side chains (X) or via Ni2+ coordination to two His residues (H). C) AFM‐SMFS experiments. Each peptide was coupled to the surface via Cys and the dimeric coiled coil was subjected to shear force (F).
Peptide stapling is a well‐established strategy for inducing a stable α‐helical structure in short single‐chain peptides.[ 14 , 15 ] Stapling utilizes ionic interactions, for example, Glu‐Lys, [16] metal‐coordination, for example, His‐[Metal]2+, [17] or more robust helix‐stabilizing covalent bridges.[ 14 , 15 , 18 ] The participating amino acids are often introduced in an i→i+4 spacing to efficiently constrain one α‐helical turn.[ 14c , 15a , 15e , 16 , 17 , 18b , 18d , 18f , 18g ] Stapled peptides were initially developed for investigating α‐helix nucleation, folding and stability.[ 16 , 17 , 18 ] Peptide stapling has started to be used for drug development, as helical constraints improve target binding, resistance to proteases and, in some cases, cell‐penetration.[ 3a , 3c , 15b , 15c , 15d ] While stapling is well‐established for individual helices, it has been applied much less to helix assemblies. A few examples show a positive effect of stapling on the thermodynamic stability of CCs[ 3 , 8d , 10 , 18c , 18e , 19 ] and other helix bundles. [20] Here, we establish that controlled stapling enhances CC stability to mechanical load. We initially introduced a covalent lactam staple into a CC heterodimer and characterized the effect of stapling with atomic force microscope (AFM)‐based single‐molecule force spectroscopy (SMFS). Specifically, we focused on varying the staple position with respect to one force application point (Figure 1) and compared the extent of mechanical and thermodynamic stabilization. We further examined the consequence of replacing the lactam staple with a reversible metal coordination bond to obtain molecular insights into the response of stapled CCs to mechanical load.
As a model system, we used the well‐established heterodimeric CC A4B4 (Figure 1). A4B4 displays a high thermodynamic stability (T m=81 °C and K D <1.0×10−10 M at 20 °C [21] ) and a low dissociation rate (k off=3.2×10−4 s−1 at 25 °C [13a] ). Despite high overall stability, A4B4 has a region of low helix propensity at the C‐terminus of the A4 chain [22] (Figure S1). With the goal of maximizing CC stability, we inserted a covalent Lysi→Aspi+4 lactam staple[ 14g , 18d ] (Figure 1 B; Figures S2, S3) in the solvent‐exposed b and f positions of the C‐terminal heptad of A4 (A4X). Another motivation for stapling at the C‐terminus was the observation that C‐ rather than N‐terminal staples have a larger effect on helix nucleation and thermodynamic stability.[ 10 , 18f ] The stapled CC was mechanically probed in a shear loading geometry, choosing the stapled C‐terminal heptad of A4X as one force application point in combination with the N‐terminal heptad of B4. To define the force application points, we introduced a Cys residue [23] at the C‐terminus of A4X and at the N‐terminus of B4 (Figure 1). In the A4XB4 loading geometry, the staple is expected to display the largest effect on CC mechanical stability. As the least helical part of the CC is stabilized directly at the force application point, we expect increased resistance to uncoiling and chain separation. To investigate design principles for heptad stapling, we relocated the staple to the C‐terminus of B4 (Figures S4, S5) while maintaining the force application points (A4B4X). Further, we investigated the possible synergy of two stapled termini (A4XB4X). Lastly, we replaced the covalent lactam staple with a reversible His‐Ni2+‐His bridge[ 8d , 17a , 17b ] across the b and f positions of the C‐terminal heptad of A4 (A4HB4; Figure 1 B; Figure S6).
To examine the effect of stapling CC heptads, we first characterized the structure and thermodynamic stability of all stapled and non‐stapled CC‐forming peptides, as well as the resulting heterodimeric CCs with circular dichroism (CD) spectroscopy. CD spectra showed higher helicity for the individual lactam‐stapled peptides A4X and B4X (88 % and 85 %) than for the corresponding unstapled peptides A4 and B4 (31 % and 65 %; Figure S7A). A4 was largely unfolded with a minimum at 205 nm, shifting to prominent ellipticity minima at 208 and 222 nm after stapling in A4X. B4 showed typical minima for α‐helical CCs, indicating a tendency to form homodimers,[ 8c , 21 ] which was further enhanced upon C‐terminal stapling (B4X).
Inserting the lactam staple also increased helix stabilization of the CCs, although less dramatically than for individual chains (Figure 2 A). For the parent CC A4B4 and all stapled CCs the ratio of the mean residue molar ellipticity [Θ]MRE at 222 nm and 208 nm is greater than 1 (Table 1), suggesting formation of stable CCs. [24] At 23 °C, a single lactam staple in either the A or B chain led to a 4 % increase in overall helicity, while combining them fortified the CC bundle helicity by about 8 %. Covalent stapling also affected the thermodynamic stability. For singly stapled CCs, A4XB4 and A4B4X, the melting temperature T m increased from 77 °C (A4B4) to 81 °C and 83 °C (Table 1; Figure S10; Table S1), respectively. When both CC‐forming peptides were stapled (A4XB4X), T m increased from 77 °C to >95 °C, suggesting that two staples have more than an additive effect. The reversible His‐Ni2+‐His staple induced higher structural stability in the A4H peptide, increasing helicity by ≈10 % upon addition of Ni2+ (Figure S8A). However, the helicity and T m values for the Ni2+‐complexed CC A4HB did not increase beyond that of the parent CC A4B4 (Figure 2 B, Table 1, Figure S10). It should be noted that A4HB4 was measured in non‐coordinating PIPPS‐buffered saline (PIPPS‐BS), whereas phosphate‐buffered saline (PBS) was used for the lactam‐stapled CCs. The parent CC A4B4 showed only small differences in the helicity and T m when measured in these different buffers (Table 1; Figures S9, S10).
Figure 2.
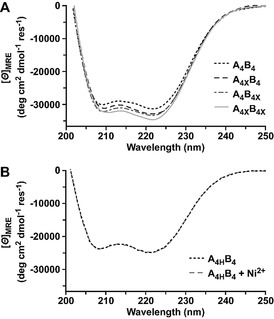
Circular dichroism spectra of stapled coiled coils. A) Spectra of the lactam‐stapled CCs A4XB4, A4B4X and A4XB4X as well as the parent CC A4B4. All spectra were measured in PBS (pH 7.4, 23 °C). B) Spectrum of the His‐containing CC A4HB4 in the absence and presence of Ni2+ (150 μM NiCl2) in PIPPS‐BS (pH 7.4, 23 °C). The reducing agent tris(2‐carboxyethyl)phosphine (TCEP) was added in all measurements to prevent disulfide bond formation.
Table 1.
Thermodynamic, kinetic and mechanical parameters obtained using CD spectroscopy and AFM‐based SMFS.
|
Coiled coil |
[Θ]MRE_222/ [Θ]MRE_208 |
T m (°C)[a] |
F (pN)[b] |
Δx (nm)[c] |
k off (s−1)[c] |
|---|---|---|---|---|---|
|
A4B4 (PBS) |
1.05 |
76.9±0.9 |
28.4 |
1.77 |
1.08×10−3 |
|
A4XB4 (PBS) |
1.09 |
81.1±0.0 |
41.9 |
1.38 |
1.04×10−4 |
|
A4B4X (PBS) |
1.05 |
83.1±1.0 |
33.4 |
1.53 |
5.96×10−4 |
|
A4XB4X (PBS) |
1.11 |
>95.0 |
40.2 |
1.50 |
4.36×10−5 |
|
A4B4 (PIPPS‐BS) |
1.01 |
80.3±0.2 |
24.5 |
1.77 |
1.46×10−3 |
|
A4HB4 + Ni2+ (PIPPS‐BS)[d] |
1.04 |
78.9±0.7 |
34.0 |
1.90 |
3.08×10−5 |
[a] Mean ± standard error of the mean (n=3 independent measurements). [b] Most probable rupture force for one example data set, measured at a retract speed of 400 nm s−1. All values for the extracted most probable rupture forces and their corresponding loading rates are shown in Table S2. [c] Δx and k off obtained from a Bell‐Evans fit to all data points (n=3; statistical analysis in SI). [d] For CD, 150 μM NiCl2; for SMFS, ≥500 μM NiCl2.
To investigate the mechanical stability of stapled CCs, SMFS was performed (Figure 1 C). As CC chain separation in shear geometry is loading rate‐dependent,[ 8d , 12 , 13 ] force‐extension curves were measured at six different cantilever retract speeds in three independent experiments. Representative force‐extension curves (Figure S11) and rupture force histograms (Figure 3 A), measured at 400 nm s−1, show that all lactam‐stapled CCs have a higher rupture force than the parent CC A4B4. Insertion of the staple directly at the point of force application (A4XB4) increased the rupture force by 15 pN (Table S2). When the staple was located in the complementary helix (A4B4X), only a 7 pN increase was observed. If both helices were stapled (A4XB4X), the increase in rupture force was comparable to A4XB4 (13 pN). This was the first indication that heptad stapling affects thermodynamic and mechanical stability of CCs differently, and that synergistic effects of multiple staples may be overruled by local helix stabilities at the point of force application.
Figure 3.
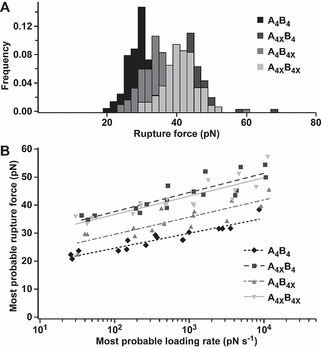
AFM‐based SMFS of lactam‐stapled coiled coils. A) Rupture force histograms, obtained at a retract speed of 400 nm s−1. B) Dynamic SMFS data, from three independent experiments, were fitted to the Bell‐Evans model (solid and dashed lines). All measurements were performed in PBS (pH 7.4, 23 °C).
To obtain the force‐free dissociation rate k off and the distance to the transition state Δx, the most probable rupture forces F and the most probable loading rates r=dF/dt were extracted (Figures S12–S15; Table S2) and the data were fitted to the Bell‐Evans model [25] (Figure 3 B; Table 1). All lactam‐stapled CCs displayed a significantly different response to the applied force when compared to A4B4, as determined by a Peacock test (Table S3; Figure S18). Specifically, the presence of a lactam staple decreased k off while simultaneously making the structure less deformable (reduced Δx). This is consistent with our earlier simulation results with constrained dihedral angles, where we observed higher rupture forces at the cost of reduced extensibility. [13b] Comparing the two CCs with one lactam staple, the position of the staple appears to be crucial. The effect of the staple was larger when it was inserted directly at the force application point (A4XB4 vs. A4B4X). However, the significant stabilizing contribution of stapling the complementary helix (A4B4X) highlights that local helix stability is allosterically transmitted to the partner helix across the hydrophobic interface. Stapling both peptides (A4XB4X) did not further increase the mechanical stability (Table S3; Figure S18). In combination, these results reveal critical differences between thermodynamic and mechanical consequences of heptad stapling. While thermodynamic stability was insensitive to staple location in either the A4 or B4 helix, mechanical stabilization was larger when the staple was located at the force application point. Interestingly, insertion of two staples synergistically increased the thermodynamic stability of the CC, but did not further enhance mechanical stabilization. While these results validate our rational design approach for the specific CC chosen, limitations are that the observed amount of stabilization at the force application point may depend on peptide sequence and staple position. In this work, the helix stabilizing Lysi→Aspi+4 lactam bridge was used at the C‐terminus, as it is the terminus with the lower helix propensity (Figure S1). Further, the lactam staple is more effective at the C‐terminus than at the N‐terminus [18f] due to its unique helix stabilizing mechanism. [18g] To generalize our finding that a staple is most efficient when inserted directly at the force application point, different CC sequences and more effective N‐terminal staples could also be tested.
For the specific sequence used here, the reduction of Δx supports our hypothesis that A4B4 chain separation in shear geometry initiates with helix uncoiling at the C‐terminal force application point, followed by dissociation of partially uncoiled chains perpendicular to the force axis. [13a] Inserting the lactam staple at the weakest point shifts the location of initial uncoiling to a different, originally more stable, position in the structure. This directly increases the energy barrier to dissociation (lower k off), accompanied with a smaller tolerance to deformation (reduced Δx). To increase the versatility of CC stapling and to further test this hypothesis, we replaced the covalent lactam stapled Lysi and Aspi+4 residues in A4 with metal‐stapled Hisi and Hisi+4 residues (A4H). In non‐coordinating PIPPS‐BS, the rupture force of A4HB4 increased by ≈7 pN in the presence of Ni2+ when compared to the parent CC A4B4 (Figure 4 A; Figure S16, S17; Table S2). A decrease in k off was observed, similar to the lactam‐stapled CC, A4XB4 (Figure 4 B; Figure S19). In contrast to A4XB4, however, Δx was not reduced when compared to the non‐stapled parent CC A4B4 (Table 1; Table S4). These results highlight that the metal staple most likely breaks as a result of the applied force. Forces between 40–300 pN were reported for interactions between Ni2+‐NTA and His6‐tags, [26] but less force is required here as only one imidazole‐Ni2+ bond needs to be broken. Forced rupture of the reversible staple explains the lower k off, as additional energy is required to break a metal‐His bond. Once the staple is open, CC uncoiling can proceed as in the absence of the staple. This interpretation is consistent with previous experiments where two His‐Ni2+‐His staples were inserted into A4B4, one at each force application point. These previous experiments also showed a reduced k off, combined with a small, insignificant reduction in Δx. [8d]
Figure 4.
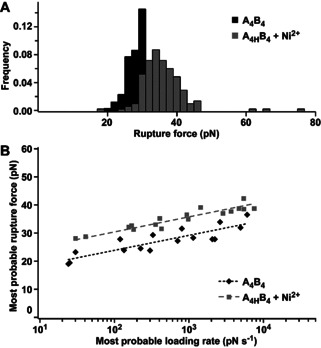
AFM‐based SMFS of the His‐Ni2+‐His stapled coiled coil in PIPPS‐BS. A) Rupture force histogram (retract speed 400 nm s−1). B) Dynamic SMFS data (n=3) fitted to the Bell‐Evans model (dashed lines). Ni2+: ≥500 μM NiCl2.
In summary, staple insertion is a robust strategy to enhance the thermodynamic, kinetic and mechanical stability of heterodimeric CCs. While the presence of two staples synergistically increased the thermodynamic stability of the stapled CC, no such effect was observed when the CC was exposed to mechanical force. Considering the previously established force‐induced chain separation mechanism, we propose that the applied force probes the weakest point in the CC structure. Constraining the least stable part increased the rupture force and reduced the force‐free dissociation rate by one order of magnitude. Both the lactam staple and the reversible metal staple increased the energy barrier to uncoiling‐assisted dissociation to a similar extent, but the force‐induced chain dissociation pathway was different. The His‐Ni2+‐His staple opened up when force was applied and allowed helix uncoiling from the force application point, whereas the covalent staple most likely shifted CC uncoiling to a different position in the CC structure. In the future, testing different staple positions with respect to the force application point, comparing effective staples at the C‐ versus N‐terminus and inserting multiple staples into the same helix, can help to further define the effect of helix stabilization on CC mechanical stability. From a biological perspective, these findings make it conceivable that ionic interactions located in i→i+4 spacing also increase local helix stabilities in naturally occurring CCs and may define the regions that uncoil with higher probability in response to axial forces. Helix‐inducing staples appear to be excellent tools for tuning the mechanical stability of CCs to enable a diverse range of applications. In particular, metal staples can be reversibly formed in situ and may thus be employed for tuning CC nanomechanical building blocks, for example as crosslinks in biomimetic hydrogels.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Ana Vila Verde and Angelo Valleriani for inspiring discussions about CC simulations and SMFS data analysis. This work was supported by the Max Planck Society (K.G.B., A.E.B.‐P.), the International Max Planck Research School (IMPRS) on Multiscale Bio‐Systems (P.L.G., I.T.), the Australian Research Council (D.P.F.; DP180103244), the ARC Centre of Excellence (D.P.F., A.D.dA; CE200100012) and the NHMRC (D.P.F.; SPRF fellowship 1117017). Open access funding enabled and organized by Projekt DEAL.
P. López-García, A. D. de Araujo, A. E. Bergues-Pupo, I. Tunn, D. P. Fairlie, K. G. Blank, Angew. Chem. Int. Ed. 2021, 60, 232.
Contributor Information
Prof. Dr. David P. Fairlie, Email: d.fairlie@uq.edu.au.
Dr. Kerstin G. Blank, Email: kerstin.blank@mpikg.mpg.de.
References
- 1.
- 1a. Kammerer R. A., J. Int. Soc. Matrix Biol. 1997, 15, 555–565; [DOI] [PubMed] [Google Scholar]
- 1b. Mason J. M., Arndt K. M., ChemBioChem 2004, 5, 170–176. [DOI] [PubMed] [Google Scholar]
- 2. Lupas A. N., Bassler J., Trends Biochem. Sci. 2017, 42, 130–140. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Rao T., Ruiz-Gómez G., Hill T. A., Hoang H. N., Fairlie D. P., Mason J. M., PLOS ONE 2013, 8, e59415; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Drobnak I., Gradišar H., Ljubetič A., Merljak E., Jerala R., J. Am. Chem. Soc. 2017, 139, 8229–8236; [DOI] [PubMed] [Google Scholar]
- 3c. Baxter D., Perry S. R., Hill T. A., Kok W. M., Zaccai N. R., Brady R. L., Fairlie D. P., Mason J. M., ACS Chem. Biol. 2017, 12, 2051–2061. [DOI] [PubMed] [Google Scholar]
- 4. Severin K., Lee D. H., Kennan A. J., Ghadiri M. R., Nature 1997, 389, 706–709. [DOI] [PubMed] [Google Scholar]
- 5. Reinhardt U., Lotze J., Zernia S., Mörl K., Beck-Sickinger A. G., Seitz O., Angew. Chem. Int. Ed. 2014, 53, 10237–10241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10402–10406. [Google Scholar]
- 6. Mueller C., Grossmann T. N., Angew. Chem. Int. Ed. 2018, 57, 17079–17083; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 17325–17329. [Google Scholar]
- 7. Liu B., Poolman B., Boersma A. J., ACS Chem. Biol. 2017, 12, 2510–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Petka W. A., Harden J. L., McGrath K. P., Wirtz D., Tirrell D. A., Science 1998, 281, 389–392; [DOI] [PubMed] [Google Scholar]
- 8b. Wang C., Stewart R. J., Kopeček J., Nature 1999, 397, 417–420; [DOI] [PubMed] [Google Scholar]
- 8c. Dånmark S., Aronsson C., Aili D., Biomacromolecules 2016, 17, 2260–2267; [DOI] [PubMed] [Google Scholar]
- 8d. Tunn I., de Léon A. S., Blank K. G., Harrington M. J., Nanoscale 2018, 10, 22725–22729; [DOI] [PubMed] [Google Scholar]
- 8e. Tunn I., Harrington M. J., Blank K. G., Biomimetics 2019, 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Boyle A. L., Bromley E. H. C., Bartlett G. J., Sessions R. B., Sharp T. H., Williams C. L., Curmi P. M. G., Forde N. R., Linke H., Woolfson D. N., J. Am. Chem. Soc. 2012, 134, 15457–15467; [DOI] [PubMed] [Google Scholar]
- 9b. Ljubetič A., Lapenta F., Gradišar H., Drobnak I., Aupič J., Strmšek Ž., Lainšček D., Hafner-Bratkovič I., Majerle A., Krivec N., Benčina M., Pisanski T., Veličković T. Ć., Round A., Carazo J. M., Melero R., Jerala R., Nat. Biotechnol. 2017, 35, 1094–1101. [DOI] [PubMed] [Google Scholar]
- 10. Crone N. S. A., Kros A., Boyle A. L., Bioconjugate Chem. 2020, 31, 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Wang X., Ha T., Science 2013, 340, 991–994; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Goktas M., Blank K. G., Adv. Mater. Interfaces 2017, 4, 1600441. [Google Scholar]
- 12. López-García P., Goktas M., Bergues-Pupo A. E., Koksch B., Varón Silva D., Blank K. G., Phys. Chem. Chem. Phys. 2019, 21, 9145–9149. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Goktas M., Luo C., Sullan R. M. A., Bergues-Pupo A. E., Lipowsky R., Vila Verde A., Blank K. G., Chem. Sci. 2018, 9, 4610–4621; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Bergues-Pupo A. E., Blank K., Lipowsky R., Vila Verde A., Phys. Chem. Chem. Phys. 2018, 20, 29105–29115. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Felix A. M., Heimer E. P., Wang C.-T., Lambros T. J., Fournier A., Mowles T. F., Maines S., Campbell R. M., Wegrzynski B. B., Toome V., Fry D., Madison V. S., Int. J. Pept. Protein Res. 1988, 32, 441–454; [DOI] [PubMed] [Google Scholar]
- 14b. Blackwell H. E., Grubbs R. H., Angew. Chem. Int. Ed. 1998, 37, 3281–3284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 3469–3472; [Google Scholar]
- 14c. Schafmeister C. E., Po J., Verdine G. L., J. Am. Chem. Soc. 2000, 122, 5891–5892; [Google Scholar]
- 14d. Taylor J. W., Pept. Sci. 2002, 66, 49–75; [Google Scholar]
- 14e. Walensky L. D., Kung A. L., Escher I., Malia T. J., Barbuto S., Wright R. D., Wagner G., Verdine G. L., Korsmeyer S. J., Science 2004, 305, 1466–1470; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14f. Shepherd N. E., Abbenante G., Fairlie D. P., Angew. Chem. Int. Ed. 2004, 43, 2687–2690; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2741–2744; [Google Scholar]
- 14g. Shepherd N. E., Hoang H. N., Abbenante G., Fairlie D. P., J. Am. Chem. Soc. 2005, 127, 2974–2983. [DOI] [PubMed] [Google Scholar]
- 15.Reviews:
- 15a. Hill T. A., Shepherd N. E., Diness F., Fairlie D. P., Angew. Chem. Int. Ed. 2014, 53, 13020–13041; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13234–13257; [Google Scholar]
- 15b. Walensky L. D., Bird G. H., J. Med. Chem. 2014, 57, 6275–6288; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Lau Y. H., de Andrade P., Wu Y., Spring D. R., Chem. Soc. Rev. 2015, 44, 91–102; [DOI] [PubMed] [Google Scholar]
- 15d. Robertson N., Jamieson A., Rep. Org. Chem. 2015, 5, 65–74; [Google Scholar]
- 15e. Fairlie D. P., Dantas de Araujo A., Biopolymers 2016, 106, 843–852. [DOI] [PubMed] [Google Scholar]
- 16. Marqusee S., Baldwin R. L., Proc. Natl. Acad. Sci. USA 1987, 84, 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Ghadiri M. R., Choi C., J. Am. Chem. Soc. 1990, 112, 1630–1632; [Google Scholar]
- 17b. Ruan F., Chen Y., Hopkins P. B., J. Am. Chem. Soc. 1990, 112, 9403–9404; [Google Scholar]
- 17c. Kelso M. J., Hoang H. N., Appleton T. G., Fairlie D. P., J. Am. Chem. Soc. 2000, 122, 10488–10489; [Google Scholar]
- 17d. Kelso M. J., Beyer R. L., Hoang H. N., Lakdawala A. S., Snyder J. P., Oliver W. V., Robertson T. A., Appleton T. G., Fairlie D. P., J. Am. Chem. Soc. 2004, 126, 4828–4842. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Jackson D. Y., King D. S., Chmielewski J., Singh S., Schultz P. G., J. Am. Chem. Soc. 1991, 113, 9391–9392; [Google Scholar]
- 18b. Kim Y.-W., Kutchukian P. S., Verdine G. L., Org. Lett. 2010, 12, 3046–3049; [DOI] [PubMed] [Google Scholar]
- 18c. Zhang F., Timm K. A., Arndt K. M., Woolley G. A., Angew. Chem. Int. Ed. 2010, 49, 3943–3946; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4035–4038; [Google Scholar]
- 18d. de Araujo A. D., Hoang H. N., Kok W. M., Diness F., Gupta P., Hill T. A., Driver R. W., Price D. A., Liras S., Fairlie D. P., Angew. Chem. Int. Ed. 2014, 53, 6965–6969; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7085–7089; [Google Scholar]
- 18e. Ali A. M., Forbes M. W., Woolley G. A., ChemBioChem 2015, 16, 1757–1763; [DOI] [PubMed] [Google Scholar]
- 18f. Hoang H. N., Driver R. W., Beyer R. L., Hill T. A., de Araujo A. D., Plisson F., Harrison R. S., Goedecke L., Shepherd N. E., Fairlie D. P., Angew. Chem. Int. Ed. 2016, 55, 8275–8279; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8415–8419; [Google Scholar]
- 18g. Hoang H. N., Wu C., Hill T. A., de Araujo A. D., Bernhardt P. V., Liu L., Fairlie D. P., Angew. Chem. Int. Ed. 2019, 58, 18873–18877; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 19049–19053. [Google Scholar]
- 19.
- 19a. M. E. Houston, Jr. , Gannon C. L., Kay C. M., Hodges R. S., J. Pept. Sci. 1995, 1, 274–282; [DOI] [PubMed] [Google Scholar]
- 19b. Haney C. M., Horne W. S., Chem. Eur. J. 2013, 19, 11342–11351; [DOI] [PubMed] [Google Scholar]
- 19c. Lathbridge A., Mason J. M., ACS Chem. Biol. 2019, 14, 1293–1304; [DOI] [PubMed] [Google Scholar]
- 19d. Sadek J., Wuo M. G., Rooklin D., Hauenstein A., Hong S. H., Gautam A., Wu H., Zhang Y., Cesarman E., Arora P. S., Nat. Commun. 2020, 11, 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang J., Zhang J., Sun X., Liu C., Li X., Chen L., Chem. Biol. Drug Des. 2019, 94, 1292–1299. [DOI] [PubMed] [Google Scholar]
- 21. Thomas F., Boyle A. L., Burton A. J., Woolfson D. N., J. Am. Chem. Soc. 2013, 135, 5161–5166. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Muñoz V., Serrano L., Nat. Struc. Biol. 1994, 1, 399–409; [DOI] [PubMed] [Google Scholar]
- 22b. Pace C. N., Scholtz J. M., Biophys. J. 1998, 75, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zimmermann J. L., Nicolaus T., Neuert G., Blank K., Nat. Protoc. 2010, 5, 975–985. [DOI] [PubMed] [Google Scholar]
- 24. Lau S. Y., Taneja A. K., Hodges R. S., J. Biol. Chem. 1984, 259, 13253–13261. [PubMed] [Google Scholar]
- 25.
- 25a. Evans E., Ritchie K., Biophys. J. 1997, 72, 1541–1555; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Bergues-Pupo A. E., Goktas M., Tunn I., Lopez-Garcia P., Verde A. V., Blank K. G., Valleriani A., J. Chem. Phys. 2018, 149, 244120. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Conti M., Falini G., Samori B., Angew. Chem. Int. Ed. 2000, 39, 215–218; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 221–224; [Google Scholar]
- 26b. Schmitt L., Ludwig M., Gaub H. E., Tampé R., Biophys. J. 2000, 78, 3275–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary