

Abstract
Encephalopathy of prematurity (EoP) is a common cause of long‐term neurodevelopmental morbidity in extreme preterm infants. Diffuse white matter injury (dWMI) is currently the most commonly observed form of EoP. Impaired maturation of oligodendrocytes (OLs) is the main underlying pathophysiological mechanism. No therapies are currently available to combat dWMI. Intranasal application of mesenchymal stem cells (MSCs) is a promising therapeutic option to boost neuroregeneration after injury. Here, we developed a double‐hit dWMI mouse model and investigated the therapeutic potential of intranasal MSC therapy. Postnatal systemic inflammation and hypoxia‐ischemia led to transient deficits in cortical myelination and OL maturation, functional deficits and neuroinflammation. Intranasal MSCs migrated dispersedly into the injured brain and potently improved myelination and functional outcome, dampened cerebral inflammationand rescued OL maturation after dWMI. Cocultures of MSCs with primary microglia or OLs show that MSCs secrete factors that directly promote OL maturation and dampen neuroinflammation. We show that MSCs adapt their secretome after ex vivo exposure to dWMI milieu and identified several factors including IGF1, EGF, LIF, and IL11 that potently boost OL maturation. Additionally, we showed that MSC‐treated dWMI brains express different levels of these beneficial secreted factors. In conclusion, the combination of postnatal systemic inflammation and hypoxia‐ischemia leads to a pattern of developmental brain abnormalities that mimics the clinical situation. Intranasal delivery of MSCs, that secrete several beneficial factors in situ, is a promising strategy to restore myelination after dWMI and subsequently improve the neurodevelopmental outcome of extreme preterm infants in the future.
Keywords: diffuse white matter injury, encephalopathy of prematurity, mesenchymal stem cells, microglia, oligodendrocytes, preterm birth, regenerative medicine
Main Points
Intranasal MSC therapy potently restores myelination after encephalopathy of prematurity.
MSCs modify their secretome in situ to support OL maturation by upregulating valuable, beneficial growth factors and/or anti‐inflammatory cytokines.
1. INTRODUCTION
Preterm birth is a major cause of life‐long neurodevelopmental sequelae in neonates, ranging from motor impairments and cognitive deficits to behavioral problems (Larroque et al., 2008; Linsell et al., 2018; MacKay, Smith, Dobbie, & Pell, 2010; Moster, Lie, & Markestad, 2008). These impairments are believed to be the consequence of encephalopathy of prematurity (EoP), a collective term used to delineate a variety of anomalies that result from disturbances in brain development due to an (extreme) premature birth (Volpe, 2009a). Diffuse white matter injury (dWMI), characterized by global hypomyelination in absence of cystic lesions, is a prominent hallmark of EoP (Back, 2017; Volpe, 2009b, 2017). Peri‐ and postnatal insults associated with preterm birth, such as inflammation and respiratory problems, are thought to negatively impact oligodendrocyte (OL) lineage development, resulting in insufficient myelination and subsequently dWMI (Lee, 2017; van Tilborg et al., 2016; Volpe, Kinney, Jensen, & Rosenberg, 2011). Despite the significant morbidity associated with dWMI, at present only supportive treatment is available for preterm infants. Therefore, novel treatment strategies to combat dWMI, tested in clinically relevant animal models of EoP, are urgently needed.
Mesenchymal stem cell (MSC) therapy has been shown to effectively improve functional outcome and aid in endogenous brain repair in numerous experimental models of adult and neonatal brain injury (Paul & Anisimov, 2013; van Velthoven, Kavelaars, van Bel, & Heijnen, 2010a; Volkman & Offen, 2017; Wagenaar et al., 2018). While these models often do not represent the pattern of dWMI observed in human patients, these studies do provide support of the potential of MSCs to protect and regenerate the white matter (Vaes et al., 2019). MSCs are believed, through paracrine signaling, to directly stimulate OL survival and maturation whilst attenuating neuroinflammation thereby favoring a cerebral milieu permissive for repair and (re)myelination (Cunningham, Redondo‐Castro, & Allan, 2018; Vaes et al., 2019). To non‐invasively target the brain with MSCs without loss of cells in the periphery, we and others have successfully applied stem cells via the nasal route (Danielyan et al., 2009; Donega et al., 2014; van Velthoven, Kavelaars, van Bel, & Heijnen, 2010b).
In the present study, we investigated neurodevelopmental outcome in a novel mouse model of dWMI, in which two clinically relevant hits, that is, postnatal hypoxia/ischemia and inflammation, were combined during a cerebral developmental stage corresponding with human preterm birth. Moreover, we aimed to determine the therapeutic potential of intranasal MSC therapy in this novel dWMI mouse model. To elucidate the mechanisms underlying the potential regenerative effect of MSCs, ex vivo MSC secretome analysis and primary cell cultures were used to assess the cell‐specific effects of MSCs on OL maturation, and microglia activation.
2. MATERIALS AND METHODS
2.1. MSC culture
GIBCO® mouse (C57BL/6) bone marrow‐derived MSCs were purchased (Invitrogen, S1502‐100; Carlsbad, CA) and cultured according to the manufacturer's instructions. Cells were passaged once (from P2 to P3) before in vivo administration or in in vitro experiments.
2.2. In vivo mouse model of dWMI
All animal experiments were carried out according to the Dutch and European guidelines (Directive 86/609, ETS 123, Annex II) and were approved by the Experimental Animal Committee Utrecht (Utrecht University, Utrecht, Netherlands) and the Central Authority for Scientific Procedures on Animals (The Hague, The Netherlands). The animals were kept under standard housing conditions with food and water available ad libitum, a 12‐hr light/dark cycle, and in a temperature‐controlled environment. Both sexes were included in all described experiments and randomly assigned to experimental groups with an equal distribution across groups. We used an adaptation of the mouse model described by Shen, Plane, and Deng (2010). Hypoxia‐ischemia (HI) was induced at postnatal day 5 (P5) in C57BL/6j mouse pups (in‐house breeding) by permanent unilateral occlusion of the right common carotid artery under isoflurane anesthesia (5–10 min; 5% induction, 3–4% maintenance with flow O2: air 1:1). After a recovery period of 75 min pups were exposed to 35 min of hypoxia (6% O2) in a temperature‐controlled hypoxic incubator (35.8–36.0°C). Directly following hypoxia, pups were i.p. injected with 1 mg/kg LPS (List Biological Laboratories, Campbell, CA) dissolved in 0.9% NaCl. Sham‐control littermates underwent surgical incision only, without occlusion of the carotid artery, hypoxia, or LPS injection. Afterward all animals were returned to their dams. Prior to MSC administration (on P8 [i.e., D3 after induction of dWMI], P11[i.e., D6] or P15 [i.e., D10]), 2 dosages of 2 μl Hyaluronidase (12.5 U/μl in total, Sigma‐Aldrich, St. louis, MO) dissolved in H2O were administered in each nostril (total of 8 μl) and inhaled spontaneously. Hyaluronidase is commonly used to improve permeability of the nasal mucosa and thereby facilitate intranasal cell delivery (Danielyan et al., 2009; van Velthoven et al., 2010b). Thirty minutes after hyaluronidase treatment, mice received intranasally MSCs (different dosages; total of 0.1 × 106, 0.5 × 106, 1.0 × 106, or 2.0 × 106 MSCs) in dPBS (Thermo‐Fisher, 14190‐169, Waltham, MA) in 2 dosages of 2 μl in each nostril (total of 8 μl). dPBS was used as vehicle solution. Mice were euthanized at various time points (P8 [i.e., D3 after induction of dWMI], P11 [i.e., D6], P19 [i.e., 2 weeks], P26 [i.e., 3 weeks], and P33 [i.e., 4 weeks]) for different experiments by an i.p. overdose of pentobarbital (250 mg/kg).
2.3. MSC tracing
2.3.1. MSC labeling
A detailed description of the synthesis of gold core‐mesoporous and lipid coated silica nanoparticles (AuNP‐MSN‐LIP) can be found in Figure S1. Two hours after cell passaging, MSCs were incubated with 25 μg/ml AuNP‐MSN‐LIP in culture medium during 48 hr. Following labeling, 0.5 × 106 MSCs in PBS were administered at P8 as described in the previous paragraph to both dWMI and sham‐controls.
2.3.2. Cell tracing in mouse tissue
Twelve hours after MSC administration, mice were sacrificed by overdose pentobarbital followed by decapitation. Brains were collected, the cerebrum was divided in ipsilateral and contralateral hemispheres and frozen separately in liquid nitrogen, as well as cerebellum and olfactory bulbs. Spleens, lungs, and livers were also collected and frozen in liquid nitrogen. Inductively coupled plasma mass spectrometry (ICP‐MS) was performed for quantitative assessment of the biodistribution of MSCs by detection of gold in mouse tissue homogenates. Snap frozen tissue sections were weighed in 2 ml plastic eppendorfs. Each sample was lyophilized overnight and reweighed. Each sample was then transferred to graduated glass vials (VWR). Next, freshly prepared aqua regia (HCl 30% and HNO3 60%, VWR) was added to each tissue section for wt/vol; 1 mg/50 μl. The samples were disintegrated overnight at 40°C using an ultrasonic bath (Bransonic®, Thermo Scientific). The samples were further homogenized by microwaving (5 × 30 seconds, 600 W) until the solutions were transparent and free of particulates. Next, freshly prepared 1% HNO3 was functionalized with 20 ppb of ruthenium (VWR) to form the matrix solution. All homogenized tissue and cell samples were diluted 1:10 in the as prepared matrix (typically 100:900 μl). Additionally, a gold standard curve ranging from 1 ng/L to 100 μg/L was made by dilution of a gold stock solution (VWR) in as‐prepared matrix. ICP‐MS was measured using an iCAP™ RQ ICP‐MS (Thermo Scientific). Gold content measured at mass 197 will have no interference from other ions. The detection limit was 1 ng/L.
2.4. Behavioral assessments
All behavioral paradigms were videotaped and scored by researchers blinded to the experimental conditions. Between all runs all setups were cleaned with soapy water and ethanol to eliminate smells.
2.4.1. Cylinder rearing test
To evaluate motor impairment the cylinder rearing test (CRT) was used at P19, P26, and P33. Animals were individually placed in a transparent cylinder (80 mm diameter and 300 mm height) and videotaped for a minimum of 3 min. At least 10 full weight‐bearing rearings against the cylinder wall were recorded for each animal. Mice that did not perform 10 rearings within the timeframe were retested ~30 min later. Forepaw usage was scored as left (impaired), right (non‐impaired), or both. Preference to use the non‐impaired forepaw was calculated as ((non‐impaired – impaired)/(non‐impaired + impaired + both))x100%.
2.4.2. Delayed spontaneous alternation in T‐maze
Executive memory functioning was assessed by measuring correct alternating behavior in a T‐maze at P26. Each trial consisted of two runs in a T‐shaped maze, a sample run, and a choice run. During the sample run, animals were placed in the starting arm of the T‐maze. The investigator then waited (max 2 min) for the animal to enter one of either goal arms, followed by the closing of the chosen arm, forcing the animal to remain in the same spot for 30 s. Animals were then returned to the home cage for 2 min, and put back into the starting arm for the choice run. If the animal would first enter the unexplored arm, this was scored as a correct alternation, while exploration of the previously explored arm was scored as an incorrect alternation. Animals performed two trials a day, during three consecutive days (i.e., 6 trials in total). Percentage of correct alternations was calculated as (the total number of choice runs choosing the alternate/unexplored arm of the six total trials) × 100%.
2.4.3. Open field test
Anxiety‐related behavior was assessed using the open field paradigm. Animals were placed in an arena (52.5 cm × 35.5 cm × 40.5 cm) during 30 min and locomotion was automatically tracked using Ethovision software (Noldus, Wageningen, The Netherlands). The arena was divided in two sections, the outer rim, and the inner zone. Time spent in both zones, the number of entries into the inner zone, and the time spent in the inner zone were recorded.
2.5. Electron microscopy
After anesthesia mice (n = 3, SHAM; n = 4 dWMI) were perfused transcardially with HBSS (for flushing) followed by fixative containing 4% formaldehyde (Sigma, G5882), 2.5% glutaraldehyde (EM grade, Sigma, 16005), and 0.5% NaCl in phosphate buffer pH 7.4 according to Karlsson and Schultz as described previously (Weil, Ruhwedel, Meschkat, Sadowski, & Möbius, 2019). Brains were dissected and postfixed in the same fixative overnight. Vibratome sections of 300 μm thickness were prepared in sagittal direction and area of interest, the caudal corpus callosum of the ipsilateral hemisphere, were punched out using a hollow biopsy punch. After postfixation in 2% OsO4 (Science Services, München, Germany) in 0.1 M phosphate buffer pH 7.3 pieces of tissue were embedded in EPON (Serva) after dehydration with acetone. Ultrathin sections were prepared using a Leica UC7 ultramicrotome (Leica, Vienna, Austria) and stained with UranylLess™ (Science Services, Munich, Germany). EM overview images were obtained with a Zeiss LEO912 electron microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany) at ×6300 magnification with an on‐axis 2 k CCD camera (TRS, Moorenweis, Germany). Image analysis was performed using ImageJ (Fiji, Version 2.0.0‐rc‐69/1.52). For g‐ratio analysis ~150 axons were chosen in a systematic random sampling fashion by overlaying the image with a line grid and selecting axons hit by crossing lines. G‐ratios (axonal diameter divided by the fiber diameter including the myelin sheath) were calculated from circular areas equivalent to the measured areas of axons and myelin sheath including the axon.
2.6. Postmortem MRI
2.6.1. Image acquisition
After a lethal dose of pentobarbital animals were transcardially perfused with PBS and 4% PFA in PBS at P26. Before scanning, brains with intact skulls fixed in 4% PFA were kept in PBS with sodium azide during a minimum of 7 days. High‐resolution structural MRI scans were performed on a 9.4 T horizontal bore MR system (Varian, Palo Alto, CA) equipped with a 6 cm internal diameter (ID) gradient insert with gradients up to 1 T/m. Three perfusion‐fixed mouse brains collected with intact skulls and immersed in Galden PFPE (PerFluoroPolyEther) ®D05 were positioned within a custom made solenoid coil with an internal diameter of 2.6 cm. Diffusion tensor imaging (DTI) was performed using a 3D diffusion‐weighted spin‐echo sequence with an isotropic spatial resolution of 150 μm, where the read‐ and phase‐ encode directions were acquired using 8‐shot Echo Planar Imaging (EPI)‐encoding, while the second phase direction was linearly phase‐encoded (repetition time (TR)/echo time (TE) 500/33 ms, 160 × 144 × 140 matrix, field‐of‐view (FOV) 24 × 21.6 × 20 mm3, Δ/δ 8.83/5.5 ms, b 1,000, 2,498, 3,999 s/mm2, 30 diffusion‐weighted images in non‐collinear directions for each diffusion gradient and six images without diffusion‐weighting (b = 0), number of averages per image 1, total number of images 96).
2.6.2. Morphometric analysis of the DTI images
All images were Fourier‐transformed and the complete data set was fit using the dtifit routine from the FMRIB's Diffusion Toolbox (Behrens et al., 2003; Behrens, Berg, Jbabdi, Rushworth, & Woolrich, 2007; Jbabdi, Sotiropoulos, Savio, Grana, & Behrens, 2012). This resulted in fractional anisotropy (FA) and mean diffusivity (MD) maps and a b0 image of the brains. The brain image on the b0‐image was extracted from the surrounding skull and tissue signal using the brain extraction tool BET2 of FSL on the b0‐image. All FA images were then registered to one of the sham mouse brains using FMRIB's Linear Image Registration Tool (FLIRT) and FMRIB's nonlinear registration tool (FNIRT) (Jenkinson, Bannister, Brady, & Smith, 2002; Jenkinson & Smith, 2001). The median of all nonlinear registered FA images from control mouse brains was used to obtain a new reference FA image, which was used for nonlinear co‐registration of the FA images from all mouse brains. The FA and MD maps and the b0 images of the control mouse brains were transformed to the reference FA image, and new median images were obtained for the FA, MD and b0. These were used as DTI reference files.
The Waxholm mouse brain T2* weighted image was downloaded from the NeuroImaging Tools and Resources Collaboratory (NITRC) website (https://www.nitrc.org/projects/incfwhsmouse) and converted to a gray/white matter‐ and CSF‐segmented image. This image was non‐linearly registered to a similarly segmented b0 reference image. In this manner, the regions of interest (hippocampus, lateral ventricle, internal capsule, anterior commissure, cerebellum) drawn on the Waxholm mouse brain were converted to the b0 reference image. Some manual adjustments had to be made to optimize the overlay of the ROIs on the reference image. Not all ROIs were available in the downloaded Waxholm atlas. Additional ROIs (e.g., the secondary motor cortex [M2]) were manually drawn based on the segmentation in the Paxinos and Watson atlas (M2). ROIs overlaid on the reference anatomical image were inversely registered to the individual mice FA and MD maps, and the ROI volume was determined. In addition, the pixels in the ROI were assigned to CSF, white matter, or gray matter based on their FA and MD values. Pixels with FA >0.3 were assigned to be white matter, pixels with an FA < 0.2 and an MD > 0.4 × 10−3 mm3/s were assigned to CSF, and remaining pixels were assigned to gray matter.
2.7. Immunohistochemistry
Following a lethal dose of pentobarbital animals were transcardially perfused with PBS and 4% PFA in PBS at P19, P26, and P33. Brains were postfixed during 24 hr in 4% PFA and dehydrated followed by embedment in paraffin. Coronal sections (8 μm) were cut at hippocampal level (−1.80 mm from bregma in adult mice). To assess hippocampal size, paraffin sections were stained with hematoxylin and eosin (HE). For immunofluorescent stainings sections were deparaffinized in xylene, and subsequently rehydrated in decreasing ethanol concentrations. For antigen retrieval, sections were heated to 95°C in sodium citrate buffer (0.01 M, pH 6). After cooling down and PBS (+ 0.05–0.1% Tween20) washings, sections were blocked with 5–10% normal species‐specific serum in PBS‐Tween20 or 2% bovine serum albumin (BSA)/0.1% saponin in PBS and incubated overnight with primary antibodies (Table 1). The following day, sections were washed in PBS and incubated with alexafluor‐594 and ‐488 conjugated secondary antibodies (Life technologies, Carlsbad, CA; 1:200–500) for 1–2 hr at room temperature, followed by DAPI (1:5000) counterstaining and embedment in Fluorsave (Merck Millipore, 345789). For 3,3′‐Diaminobenzidine (DAB) staining, sections were deparaffinized in xylene and 100% ethanol. Sections were incubated with 3% H2O2 to block endogenous peroxidase and then hydrated using decreasing ethanol concentrations. For antigen retrieval, sections were heated to 95°C in sodium citrate buffer (0.01 M, pH 6). After cooling down and PBS washings, sections were blocked with 5% normal horse serum in PBS, followed by overnight incubation with mouse anti‐MAP2 (M4403, Sigma; 1:1000) in 2% normal horse serum in PBS. The following day, sections were washed with PBS and incubated with biotinylated horse‐anti‐mouse (BA‐2000, Vector Laboratories; 1:100), followed by washing in PBS. Biotin was HRP‐labeled using a vectastain ABC kit (Vector laboratories) according to manufacturer's protocol, followed by 0.05 M Tris‐HCl (pH: 7.6) washing. Sections were incubated in 0.5 mg/ml DAB (Sigma) in 0.05 M Tris‐HCl with 0.009% H2O2 for staining. Sections were dehydrated in ethanol and embedded.
TABLE 1.
Overview of primary antibodies used in the study
| Antigen | Species (host) | Company productcode | Dilution |
|---|---|---|---|
| Anti‐MBP |
Rat Mouse |
Merck Millipore MAB386 Biolegend SMI‐94 |
1:500 1:1000 |
| Anti‐NF200 | Rabbit | Sigma N‐4142 | 1:400 |
| Anti‐Iba1 | Rabbit | Wako 019‐19741 | 1:500 |
| Anti‐GFAP | Mouse | Origene BM2287 | 1:200 |
| Anti‐Ki67 | Rabbit | Abcam 15580 | 1:300 |
| Anti‐Olig2 |
Rabbit Mouse |
Merck Millipore AB9610 Merck Millipore MABN50 |
1:500 1:500 |
| Anti‐APC/CC1 | Mouse | Calbiochem OP80 | 1:300 |
| Anti‐Cleaved Caspase 3 | Rabbit | Cell signal 9664 | 1:800 |
2.7.1. Microscopy
Investigators were blinded for experimental conditions during image acquisition. In HE‐ and MAP2‐stained sections a ×2.5 magnification was used to visualize both hemispheres using a light microscope (Zeiss, Oberkochen, Germany) with an AxioCam ICc 5 camera (Zeiss). For immunofluorescent stainings, images of both hemispheres were acquired using a Cell Observer microscope with an AxioCam MRm camera (Zeiss, Oberkochen, Germany). For MBP/NF200 stainings ×2.5 images were taken to visualize the cortex. In addition, three adjacent ×40 images were taken at a fixed distance from the external capsule into the cortex (layer III/IV), superjacent to the ipsi‐ and contralateral hippocampal areas. For exact locations, we refer to van Tilborg et al. (2017). For both Iba1 and GFAP analyses, three ×40 images were acquired in the corpus callosum. Moreover, one ×40 image of the CA1 region of the hippocampus (dorsal from the dentate gyrus) was obtained for GFAP area analyses. For CC1/Olig2, Ki67/Olig2, and Caspase3/Olig2 stainings two ×20 micrographs were taken in the corpus callosum and the cortex, directly dorsal from the external capsule.
2.7.2. Analyses
All image analyses were performed in a blinded fashion. Hippocampal areas in the contra‐ and ipsilateral hemispheres were measured on images of HE‐stained sections using Zen software (Zeiss, Oberkochen, Germany). Ipsilateral hippocampal area loss was expressed as the ratio of ipsilateral to contralateral hippocampal areas. Microglial morphology was evaluated using the particle analysis function of ImageJ v1.47 (Schneider, Rasband, & Eliceiri, 2012) as described by Zanier, Fumagalli, Perego, Pischiutta, and De Simoni (2015). Microglia were manually selected and morphological description parameters were measured. Morphological parameters of microglia were normalized for sham‐control values. GFAP (×40) and NF200 (×40) threshold analyses to calculate the positive area of staining, were carried out using ImageJ software. GFAP+ area measurements were normalized for sham‐control values. Following MBP/NF200 staining, the extent of cortical myelination and the microstructural integrity of myelinated axons was assessed as described earlier (van Tilborg et al., 2017). Values of all acquired images were averaged per animal. Cell counts of proliferation (Ki67), microglia (Iba1), apoptotic (cleaved caspase 3), and OL lineage (CC1/Olig2) markers were performed manually using Zeiss software (Axiovision and Zen; Zeiss, Oberkochen, Germany) and corrected for the measured area. Cells with a clear DAPI+ nucleus in combination with clear Ki67+/Olig2+, CC1+/Olig2+, cleaved caspase3+/Olig2+, or Iba1+ staining were counted. Images with excessive background staining or large artifacts were excluded.
2.8. MSC gene expression profiles
2.8.1. Brain extracts
At P8 (i.e., D3 after induction of dWMI) mice were sacrificed by overdose pentobarbital, decapitated, brains were collected and hemispheres were separately frozen in liquid nitrogen. The ipsilateral hemispheres of sham‐operated (n = 4) and dWMI mice (n = 4) were ground with a mortar and pestle chilled on liquid nitrogen. The tissue pieces were then weighed and homogenized at 150 mg/mL in knock‐out DMEM (Thermo Fisher, 10829018) containing a protease inhibitor cocktail (1:50 dilution; Invitrogen) using a potter tissue homogenizer (10 strokes), followed by centrifugation for 10 min at 10,000g at 4°C. The supernatant was collected as “brain extract” and protein concentration was measured with a protein assay (Biorad) with BSA as standard. Brain extracts were aliquoted and kept at −80°C until use.
2.8.2. PCR arrays
To evaluate the response of MSCs to the milieu encountered in the brain after induction of dWMI, MSCs were cultured and seeded in six‐well plates at 2.0 × 105 cells per well in standard MSC medium. After 24 hr, the culture medium was replenished with knock‐out DMEM containing either dWMI of sham‐operated brain extract at a concentration of 1 mg protein/ml. After 72 hr in culture, brain extract‐enriched medium was completely removed, wells were washed with ice‐cold PBS and total RNA of the MSCs was isolated using the RNeasy minikit (Qiagen). RNA quantity and quality was assessed by spectrophotometry (NanoDrop 2000, Thermo‐scientific, Waltham, MS) at 260 mm, and OD 260/280 ratio was determined to evaluate quality. cDNA transcription was performed using the RT2 first‐strand synthesis kit (Qiagen) according to the manufacturer's protocol. The cDNA of MSCs exposed to sham‐operated or dWMI brain extracts were pooled per condition and expression profiles of 168 growth factor‐ and cytokine‐ related genes were assessed by commercially available PCR arrays (Qiagen; PAMM‐041Z and PAMM‐150Z). PCR array analysis was carried out once following manufacturer's protocol with the RT2 Real‐Time SYBR green PCR Master Mix (Qiagen) on the Biorad iQ5. Data were normalized using multiple housekeeping genes provided within the PCR array and analyzed by comparing 2‐ΔCt using Qiagen software. Gene expression changes in MSCs exposed to dWMI brain extracts were calculated relative to MSCs exposed to brain extracts from sham‐operated mice. A fold regulation threshold of 2.0 was considered as either down‐ or upregulation.
2.8.3. Quantitative PCR validation
PCR array results were validated by quantitative PCR analysis on individual cDNA samples for selected genes. Real time RT‐PCR was performed using the iQ5 Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA) using SYBR green supermix (Biorad, 1708887, Hercules, CA). Primer sequences can be found in Table 2. Mean expression of GAPDH and β‐actin were used to normalize the data.
TABLE 2.
Overview of primer sequences used in validation qPCR
| Gene | Forward (FW) and reverse (REV) primer sequences |
|---|---|
| Gapdh |
FW: TGAAGCAGGCATCTGAGGG REV: CGAAGGTGGAAGAGTGGGAG |
| β‐actin |
FW: AGAGGGAAATCGTGCGTGAC REV: CAATAGTGATGACCTGGCCGT |
| CXCL12 |
FW: CGGTGTCCTCTTGCTGTCC REV: CTCTGGCGATGTGGCTCTC |
| LIF |
FW: CTTCTCCCTCTGGTCTCCAA REV: GGGTCAGGATGTTTCAGCAC |
| IL11 | FW: CTGCACAGATGAGAGACAAATTCC REV: GAAGCTGCAAAGATCCCAATG |
| IL10 |
FW: GCACCCACTTCCCAGTCG REV: GCATTAAGGAGTCGGTTAGCAG |
| IGF1 |
FW: CACATCATGTCGTCTTCACACC REV: GGAAGCAACACTCATCCACAATG |
| EGF |
FW: GTCCGTCTTATCAGGCATCAA REV: TGAGTAGAAGATCCGATCACCAA |
| CSF3 (GCSF) |
FW: AGTGTTCCCAAACTGGGTTCT REV: TTAGGGACTTCGTTCCTGTGA |
2.9. Cerebral expression of trophic factors following MSC treatment
To investigate the effect of MSC treatment on cerebral mRNA expression changes of trophic factors, dWMI mice that received vehicle or 0.5 × 106 MSCs intranasally at D3 after injury, were sacrificed by overdose pentobarbital followed by decapitation at 12 hr after intranasal treatment. Brains were collected, hemispheres were isolated and divided into a rostral and caudal part and frozen separately in liquid nitrogen. The cerebellum was frozen separately. The ipsilateral brain parts and cerebellum of vehicle‐treated (n = 3) and MSC‐treated mice (n = 4) were ground with a mortar and pestle chilled on liquid nitrogen. All tissue parts were lysated in RLT lysis buffer using a TissueLyser LT Adapter and stainless‐steel beads (all Qiagen, Hilden, DE), at 50 Hz for 2 minu following the supplier's protocol. RNA isolation was carried out using the RNeasy minikit (Qiagen), including on‐column DNase digestion with the RNase‐free DNase set (79254, Qiagen). RNA quantity and quality were assessed by spectrophotometry (NanoDrop 2000, Thermo‐scientific, Waltham, MS) at 260 mm, and OD 260/280 ratio was determined to evaluate quality. cDNA transcription was performed using the iScript reverse transcription supermix (Bio‐rad, Hercules, CA). Real‐time RT‐PCR for a selection of genes was carried out using the QuantStudio 3 (Applied Biosystems, Foster City, CA) using SYBR select master mix (4472903, Applied Biosystems). Primer sequences can be found in Table 2. Mean expression of GAPDH and β‐actin were used to normalize the data.
2.10. In vitro models of dWMI
2.10.1. Primary rat cultures
A mixed glial culture was obtained from P0‐2 Sprague Dawley rat pup cortices, as described in detail by Chen et al. (2007) with minor adaptations. In short, brains were isolated and cortices were dissected, followed by removal of the meninges. Cortices were pooled, minced, and subsequently dissociated using DNase I (10 μg/L, Sigma Aldrich, D5025) and Trypsin solution (0.01%, Sigma Aldrich, T1426) in HBSS. Tissue suspensions were dissociated by pipetting and filtered through a 0.70 μm filter twice. Cells were plated in poly‐D‐lysine‐coated (0.1 mg/ml, Sigma Aldrich, P6407) T75 culture flasks. For details on the media used, please see the paper. After a minimum of 10 DIV, microglia and oligodendrocyte precursor cells (OPCs) were harvested by mechanical shaking on an orbital shaker. Cultures were kept for a remaining 7 DIV for a second harvest.
2.10.2. Production of MCM
The flasks were shaken for 1 hr at 200 rpm at 37°C to collect microglia. Culture medium with detached microglia was centrifuged (10 min at 1200 rpm at RT) and microglia were counted. Microglia were plated at 0.5 × 106 cells per well on poly‐L‐ornithine (Sigma Aldrich, P3655)‐coated 24 wells plates. Microglia‐conditioned‐medium (MCM) was produced to induce a maturational arrest in primary OLs. Activated microglia produce pro‐inflammatory cytokines, mimicking in vivo inflammation, one of the etiological hallmarks of dWMI. To this means, 24 hr after plating, microglia were cultured in Basal Defined Medium (BDM) as described by Chen et al. (2007), with or without 50 ng/ml LPS (Sigma, L4616) during 24 hr at 37°C, producing respectively “MCM + LPS” or “MCM‐LPS”. MCM was collected, filter sterilized (0.20 μm), and stored at −80°C until use.
After removal of detached microglia from the culture flasks, flasks were filled with fresh medium and shaken for an additional 20 hr at 200 rpm at 37°C to isolate OPCs. The cell suspension was passed through a 20 μm sterile screening pouch (Merck Millipore, NY2004700, Burlington, MS) to remove possible remaining/detached microglia and astrocytes. The OPCs were collected by centrifugation (100 g during 10 min RT) and plated at 4.0 × 104 cells/well on poly‐D,L‐ornithine (Sigma Aldrich, P0421)‐coated 24‐wells plates in OPC medium (BDM with PDGF‐aa [Peprotech, 100‐13A] and bFGF [Peprotech, 100‐18B]). OPCs were cultured for 4 days before starting the experiments. Cells were fed with a complete OPC medium change every other day.
2.10.3. Primary mouse cultures
Primary cultures of cortical microglia were prepared from P1 C57BL/6 mice. In short, after dissection, meninges were removed and cortices were minced and incubated with 0.25% trypsin (Sigma, T4799) in Gey's balanced salt solution (GBSS) containing 100 U/ml penicillin, 100 μg/ml streptomycin, and 30 mM D (+)‐glucose during 15 min. The dissociated cell suspension was resuspended until homogenous and cultured in poly‐L‐ornithine‐coated flasks (Sigma, P3655) (at a concentration of cortices of 1 animal/flask) in DMEM/HamF10 (1:1) (Gibco, 41965‐039 and 31550‐023) supplemented with 10% FCS, 2 mM glutamine and antibiotics (see above). After 10–12 DIV culture flasks were shaken during 20–22 hr (130–135 rpm, 37°C) to detach microglia. The microglia were collected by centrifugation (120 rpm during 10 minutes RT), counted and seeded in poly‐L‐ornithine‐coated 24‐wells plates at a density of 1.0–2.0 × 105 cells per well. Microglia were cultured for 1 day before starting the experiments. After shaking, flasks were directly replenished with new culture medium and new microglia were harvested with a second shake after another 10 DIV.
2.10.4. Noncontact MSC‐glia cocultures
To study the effect of the MSCs' secretome on glial cells, MSCs were embedded in gel inserts to establish noncontact cocultures between MSCs and primary OPCs or microglia. This culture system enables cells to remain in their own medium, ensuring optimal cell viability. For these experiments, MSCs were cultured as described above. One day before the start of cocultures MSCs were embedded in Hydromatrix gel (Sigma, A6982) inserts (Merck Millipore, MCHT24H48) according to manufacturer's protocol in different cell densities (2.0–8.0 × 104 cells per insert).
At day 4 after OPC plating, when the majority of OLs displayed an immature pre‐OL morphology (i.e., a round cell body with multiple extensions), OPC medium containing PDGF‐AA and bFGF was replaced with either MCM + LPS or MCM‐LPS containing NAC (Sigma, A8199), CNTF (Peprotech, 450–50) and T3 (Sigma, T2752), leading to either induction of OL maturation (MCM‐LPS) or OL maturational arrest (MCM + LPS). Aside from MCM, we used TNFα as an alternative strategy to induce OL maturational arrest, as described previously (Bonora et al., 2014). For these experiments BDM containing TNFα (10 ng/ml, Peprotech, rat recombinant 400–19) and the differentiation growth factors (NAC [Sigma, A8199], CNTF [Peprotech, 450‐50] and T3 [Sigma, T2752]) was added to wells 4 days after OL plating. Concurrently, transwell inserts containing MSCs or hydromatrix gel without MSCs as a control, were added to the wells. After 72 hr of coculturing, inserts were removed and OLs were fixated.
Microglia were cultured and plated as described above. After 24 hr microglia were stimulated with 50 ng/ml LPS (Sigma, L4515). Concurrently, transwell inserts containing MSCs (4.0 × 104 cells per insert) were placed in each well. After 48 hr of coculture, inserts were removed and microglia supernatant was collected, aliquoted, and stored at −80C until ELISA.
2.10.5. Growth or immunomodulatory factor administration
At day 4 following OPC plating, when cells displayed a pre‐OL morphology, OPC differentiation or maturational arrest was induced using MCM or TNFα (see above). To the OPC cultures, the following selected factors were added: Noggin (murine recombinant, Peprotech 250‐38), IL10 (rat recombinant, Peprotech 400‐19), IL11 (murine recombinant, Peprotech 220‐11), LIF (murine recombinant, Peprotech 250‐02), CXCL12 (murine recombinant, Peprotech 250‐20a), GCSF (murine recombinant, Peprotech 250‐05), IGF1 (murine recombinant, Peprotech 250‐19) and EGF (murine recombinant, Peprotech 250‐09). Factors were added simultaneously with MCM + LPS or MCM‐LPS or 10 ng/ml TNFα to study their potential to boost OPC differentiation. All experiments were terminated after 72 hr. Optimal concentrations of the selected factors (Noggin 250 ng/ml, IL10 25 ng/ml, IL11 100 ng/ml, LIF 100 ng/ml, CXCL12 100 ng/ml, GCSF 100 ng/ml, IGF1 100 ng/ml, EGF 20 ng/ml) were obtained in dose–response experiments in which MBP expression by matured OLs was the final read‐out (Figure S2).
2.10.6. ELISA
TNFα concentrations in the microglia supernatant were measured using an ELISA kit for murine TNFα (Ucytech, Utrecht, The Netherlands) according to manufacturer's protocol. Because TNFα levels varied slightly between independent experiments, TNFα data were normalized to positive control conditions (50 ng/ml LPS; no insert).
2.10.7. Immunocytochemistry
Oligodendrocyte cultures were fixated with 4% PFA in PBS during 10 min. Subsequently 2% BSA and 0.1% saponin in PBS was used to block nonspecific binding. Plates were incubated with primary antibodies (rabbit‐anti‐Olig2, Merck Millipore AB9610; 1:1000 and mouse‐anti‐MBP, Biolegend SMI‐94, 1:1000) overnight at 4°C, washed with PBS, followed by incubation with alexafluor‐594 and ‐488 conjugated secondary antibodies (Life technologies; 1:1000) for 1 hr at room temperature. Cell nuclei were counterstained with Hoechst 33342 (Sigma) and wells were embedded in Fluorsave (Merck Millipore, 345789).
For each well, six adjacent fields were photographed (×10), starting at a fixed distance of the well edges. Olig2‐ and Hoechst‐positive cells were counted automatically using the analyze particles function in ImageJ v1.47. MBP area was determined using manual threshold analyses in ImageJ software v1.47. In order to reliably compare independent experiments, all results were normalized for the positive control (MCM + LPS; insert without MSCs/ 0 ng factor).
3. STATISTICS
All data are presented as mean ± SEM. Statistics were performed with Graphpad Prism 8.3. Comparison of two groups was tested using unpaired t tests, or in the event of unequal variances, non‐parametric Mann–Whitney tests. When comparing >2 groups, one‐way analysis of variance (ANOVA) with Bonferroni posthoc tests was carried out. A non‐parametric Kruskal‐Wallis test with Dunn's post hoc correction was used for comparison of multiple groups with unequal variances. p‐values <.05 were considered statistically significant. Specific sample sizes are mentioned in the figure captions.
4. RESULTS
4.1. A double‐hit mouse model of dWMI
4.1.1. Two postnatal hits cause transient myelination deficits without cortical neuronal/axonal loss
To mimic preterm birth‐related hits newborn P5 mouse pups were subjected to two hits: postnatal hypoxia‐ischemia plus inflammation. To investigate the effects of these double hits on myelination later in life, brain sections were stained for expression of myelin basic protein (MBP) at P19, P26, and P33 (i.e., 2, 3, and 4 weeks after induction of dWMI, respectively). In comparison to sham‐operated control littermates, we observed a significant decrease in cortical myelination, indicated by a reduction in MBP+ cortical coverage in the ipsilateral hemisphere, at P19 (p < .001) and P26 (p < .001) in animals subjected to dWMI, indicating myelination failure (Figure 1a,b). At P33 no significant differences in cortical myelination were observed anymore in dWMI animals compared with sham‐operated controls (p = .395). More detailed examination of the myelin microstructure by segmentation analysis showed that dWMI animals display a less complex organization of myelinated axons in the cortical white matter of the ipsilateral hemisphere, indicated by a significant reduction in the number of intersections (P19 p = .013; P26 p = .010) and fiber length (P26 p = .013), up to 3 weeks after injury induction (Figure 1c–e). Both hits were required to induce deficits in myelin microstructure; only LPS injection in sham‐operated animals or induction of HI without LPS injection did not result in significant myelin deficits (data not shown). In line with the cortical myelination data (Figure 1a), at P33 no significant differences in microstructural parameters (intersections p = .110 and fiber length p = .485) were observed between dWMI and control animals (Figure 1c–e). To assess more in depth whether myelination at P33 was endogenously recovered, we measured myelin sheath thickness using electron microscopy. We did not observe any significant differences in myelin g‐ratio, representing the relationship between axon size and myelin thickness, between dWMI and control animals (p = .759) (Figure 1f,g). Taken together, these results indicate that our double‐hit model induces a delay in myelination, rather than irreversible long lasting changes in white matter development. We did not observe any myelin deficits in the contralateral hemisphere of dWMI animals compared with both hemispheres of sham‐control littermates (data not shown), thus, from here onwards data will be shown from the ipsilateral hemisphere only.
FIGURE 1.
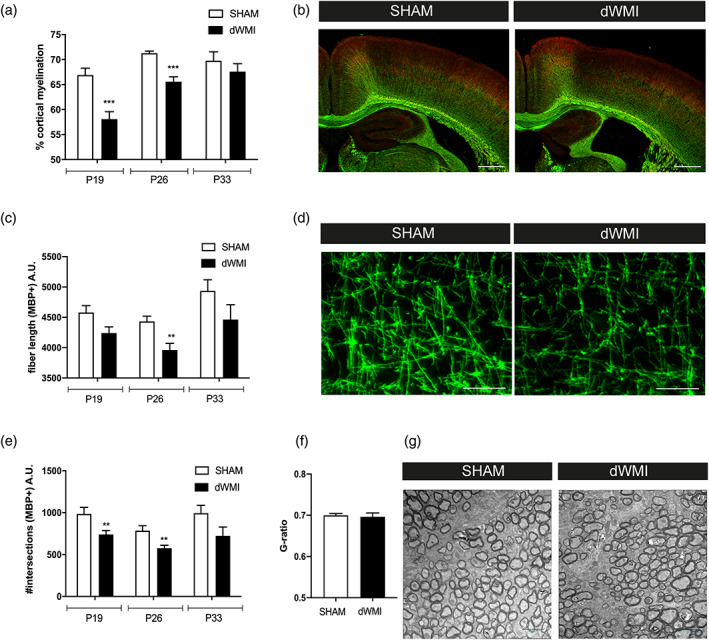
The combination of postnatal hypoxia/ischemia and systemic inflammation at P5 (dWMI model) causes a delay in myelination in neonatal mice. (a) Mice exposed to the double‐hit model displayed a transient reduction in cortical myelination (P19 SHAM n = 11 dWMI n = 11, P26 n = 7 in both groups, P33 SHAM n = 4 dWMI n = 5). (b) Representative fluorescent images (×2.5) of the ipsilateral cortex of a sham‐operated control mouse (left) and dWMI mouse (right) stained for axonal marker NF200 (red) and myelin marker MBP (green). Scale bars: 500 μm. (c, e) Microstructural changes in MBP+ fibers following dWMI induction were observed at P19 and P26, assessed by measuring fiber length (c) and number of intersections (e) (P19 SHAM n = 11 dWMI n = 11, P26 n = 8 in both groups, P33 SHAM n = 4 dWMI n = 5). (d) Representative fluorescent images (×40) of MBP+ axons in the ipsilateral cortex of a sham‐operated control mouse (left) and dWMI mouse (right). Scale bars: 100 μm (f) G‐ratio analyses (axonal diameter divided by the fiber diameter including the myelin sheath) reveal no changes in myelin enwrapment at P33 in SHAM (n = 3) and dWMI (n = 4) animals. (g) Representative electron microscopy images of the caudal corpus callosum in SHAM and dWMI animals at P33. Scale bars: 2 μm. **p < .01; ***p < .001 sham‐operated control versus dWMI animals at the specified timepoint [Color figure can be viewed at wileyonlinelibrary.com]
Besides studying myelination, we also assessed possible effects of our model on cerebral gray matter integrity by using HE, NF200, and MAP2 stainings to look at the hippocampal area, axonal integrity, and neuronal loss in cortical areas, respectively. Area measurements on HE sections revealed a unilateral reduction in ipsilateral hippocampal area in dWMI animals, stable over time and persistent up to 4 weeks after injury induction (P19 p = .004; P26 p = .003; P33 p = .004) (Figure 2a,b). We did not observe any loss of MAP2 staining in the cortex as a measure for acute neuronal damage at P19 (Figure 2e). Additionally, we did not detect any axonal damage in cortical regions as determined by NF200 stainings at P19‐P33 (Figure 2c,d). These findings indicate that gray matter area loss (i.e., reduced hippocampal area) observed in dWMI animals is not the result of acute neuronal loss or axonal damage, but rather due to maldevelopment of the gray matter structure secondary to myelination failure.
FIGURE 2.
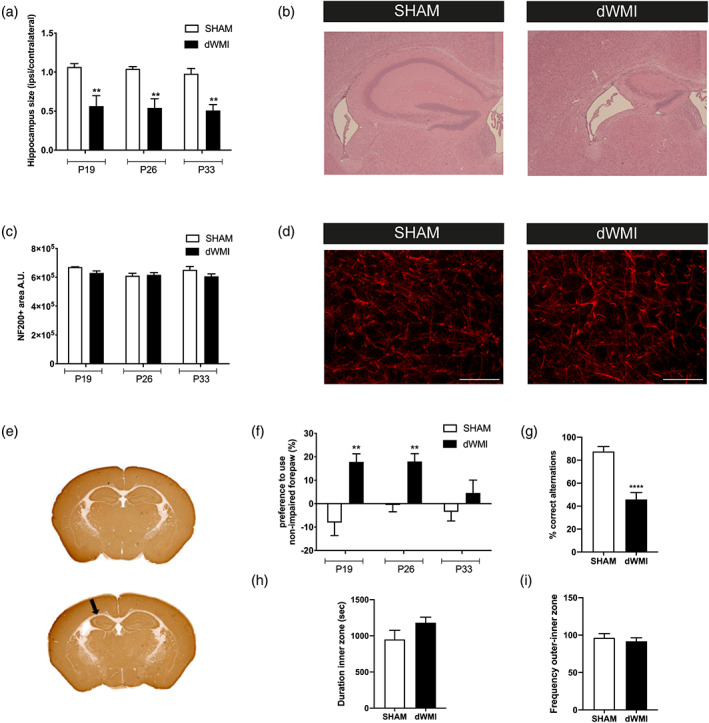
dWMI induction causes functional impairments in absence of cortical axonal deficits or acute neuronal loss. (a) Mice with dWMI displayed a persistent unilateral reduction in ipsilateral hippocampal size compared with sham‐control mice (P19/26 SHAM n = 7 dWMI = 7, P33 SHAM n = 4 dWMI n = 5). (b) Representative images of the ipsilateral HE‐stained hippocampus of a sham‐operated control mouse (left) and dWMI mouse (right). (c) No significant changes in NF200+ area were observed in the ipsilateral cortex of sham‐control (P19 n = 4, P26 n = 7, P33 n = 4) versus dWMI (P19 n = 4, P26 n = 7, P33 n = 5) animals. (d) Representative fluorescent images (×40) of NF200+ axons in the ipsilateral cortex of a sham‐control mouse (left) and dWMI mouse (right). Scale bars: 100 μm (e) Representative whole brain images stained for MAP2 at P19 showing reduced hippocampal area (arrow) but no further indications of overt neuronal loss. (f) dWMI animals (P19 n = 6, P26 n = 7, and P33 n = 7) performed worse compared with sham‐controls (P19 n = 8, P26 n = 8, and P33 n = 5) animals in the cylinder rearing test up to P26, indicating unilateral motor impairment. (g) Compared with sham‐control animals (n = 8), dWMI mice (n = 8) made less correct spontaneous alterations in the T‐maze at P26. (h,i) No changes in time spent in the inner zone (h) or frequency of inner zone entry (i) between sham‐control (n = 11) and dWMI (n = 11) were observed in the open field test at P26. **p < .01;****p < .0001 sham‐operated control versus dWMI animals at the specified timepoint [Color figure can be viewed at wileyonlinelibrary.com]
4.1.2. dWMI‐induced functional deficits include motor impairment and cognitive deficits
The functional consequences of dWMI were assessed by the cylinder rearing test (CRT) for motor function, open field for anxiety‐related behavior, and T‐maze for executive memory functioning. dWMI animals demonstrated a significant increase in non‐impaired forelimb preference in the CRT, indicating unilateral motor impairment corresponding to unilateral myelination deficits, up to P26 (P19/26 p = .003; P33 p = .299) (Figure 2f). Sham‐operated littermates did not show a significant preference to use either the left or right forepaw in the CRT. Executive memory and anxiety‐related behavior were assessed at P26 as the tests were not reliably performable with animals younger than this age and most prominent histological deficits of dWMI were observed up to P26. T‐maze performance of sham‐operated mice was very accurate with ~90% correct alternations, indicating high executive memory functioning under control conditions. In contrast, dWMI animals displayed executive memory deficits, with significantly less correct alternations (even <50%) in the T‐maze (p < .0001) (Figure 2g). Using the open field task, we did not find any indication for anxiety‐related behavior in dWMI animals as we did not detect differences in entries into, or time spent, in the inner zone of the arena between dWMI and control animals (p = .133) (Figure 2h,i).
4.1.3. dWMI induction is associated with microglia activation and astrogliosis
To study neuroinflammation, an important etiological hallmark of dWMI, brain sections of dWMI, and control mice were stained for the microglia marker Iba1 and astrocyte marker GFAP at P19, P26, and P33. In the corpus callosum of dWMI animals a higher number of Iba1‐positive cells were observed both at P19 and P26 after induction of injury (P19 p = .037; P26 p = .015) (Figure 3a). At P33 Iba1‐positive cell numbers in dWMI animals were reduced to sham‐operated control levels. Detailed analyses of numerous microglial morphological aspects (e.g., cell circularity and perimeter), revealed a more amoeboid, active morphology of microglia in dWMI animals compared with controls at P19 and P26 (Figure 3b–e). These morphological activation hallmarks of Iba1‐positive cells were restored at P33 after dWMI. An increase in GFAP+ area was observed in the corpus callosum and hippocampus of dWMI animals compared with sham‐controls at P19, with the most pronounced changes observed at P26 (CC p = .048 and HC p = .029). The increase in GFAP+ area in both brain areas was restored to the control level at P33 (Figure 3f–i). Collectively, these data indicate that the double‐hits applied at P5 in newborn mice induce a potent but transient neuro‐inflammatory response.
FIGURE 3.
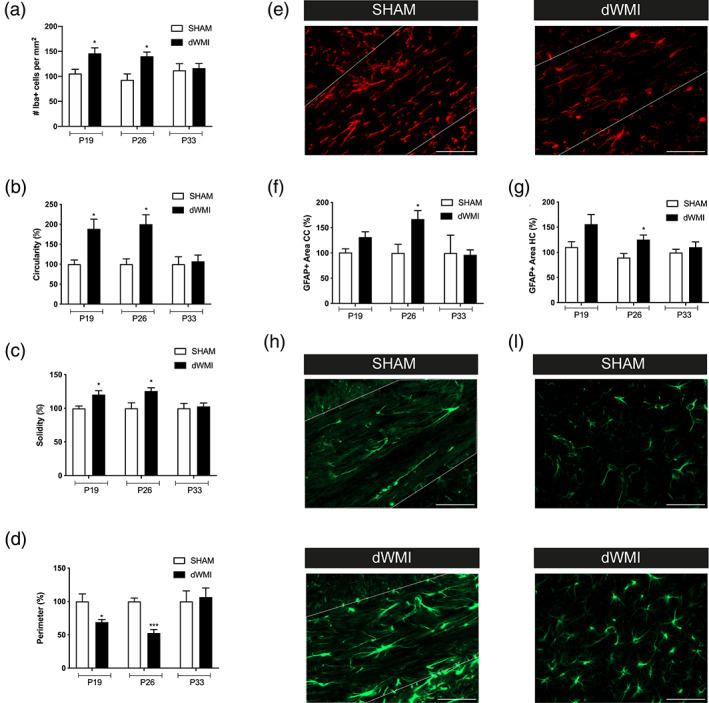
Microglia and astrocyte activity is transiently increased in dWMI animals. (a) dWMI induction leads to a transient rise in microglia (Iba+) numbers in the corpus callosum (P19 SHAM n = 6 dWMI n = 9, P26 SHAM n = 6 dWMI n = 10, P33 SHAM n = 7 dWMI n = 8). (b–d) Microglia in the corpus callosum of dWMI animals (P19/26/33 n = 8) demonstrate a more pro‐inflammatory phenotype with increased cell circularity (b) solidity (c), and decrease in cell perimeter (d) up to P26 compared with sham‐control (P19/26 n = 5, P33 n = 7, normalized to control values) mice. (e) Representative fluorescent images (×40) of Iba + cells in the corpus callosum (white outline) in a sham‐control (left) and dWMI animal (right). Scale bars: 100 μm (f, g) Quantification of GFAP+ area revealed increased astrocyte reactivity in dWMI animals (P19 n = 14, P26 n = 8, P33 n = 5, normalized to control values) compared with sham‐controls (P19 n = 10, P26 n = 7, P33 n = 4) in the corpus callosum and hippocampus at P26. (h,i). Representative fluorescent images (×40) of GFAP+ staining in the corpus callosum (h) and hippocampus (i) in a sham‐control (upper) and dWMI (lower) animal. Scale bars: 100 μm. *p < .05; ***p < .001 sham‐operated control versus dWMI animals at the specified timepoint [Color figure can be viewed at wileyonlinelibrary.com]
4.1.4. dWMI leads to white matter volume loss measured by postmortem MRI
Over the years, multiple human MR studies have identified long‐term region‐specific reductions in brain volumes of preterm infants compared with term peers. These volumetric changes have been associated with a poorer functional outcome (Keunen et al., 2016; Lind et al., 2011; Ment et al., 2009; Peterson et al., 2000). Using postmortem MRI, we measured the volumes of a range of predefined ROIs in both gray and white matter areas in sham‐control and dWMI mice at P26. The double‐hit model was associated with a reduction in total cerebral volume (p = .001) and borderline significant reduction in global WM volume (p = .052) compared with sham‐control animals (Figure 4a). Specific analyses of important WM structures showed a reduction in volumes of the corpus callosum (p = .001), internal capsule (trend, p = .060) and anterior commissure (p = .006) in dWMI animals compared with sham (Figure 4b–d). Moreover, dWMI animals displayed enlargement of the lateral ventricles (p = .046), a prominent hallmark of dWMI in preterm infants (Keunen et al., 2016; Lind et al., 2011) (Figure 4e). In line with findings in human studies (Keunen et al., 2016; Volpe, 2009a), dWMI animals also showed a loss of global gray matter volume (p = .008), along with volumetric deficits of gray matter structures, such as the hippocampus (p = .003) and secondary motor cortex (M2) (p = .004) (Figure 4g–i). Additionally, dWMI animals exhibited volumetric deficits of the cerebellum (p = .013) compared with sham‐controls (Figure 4j).
FIGURE 4.
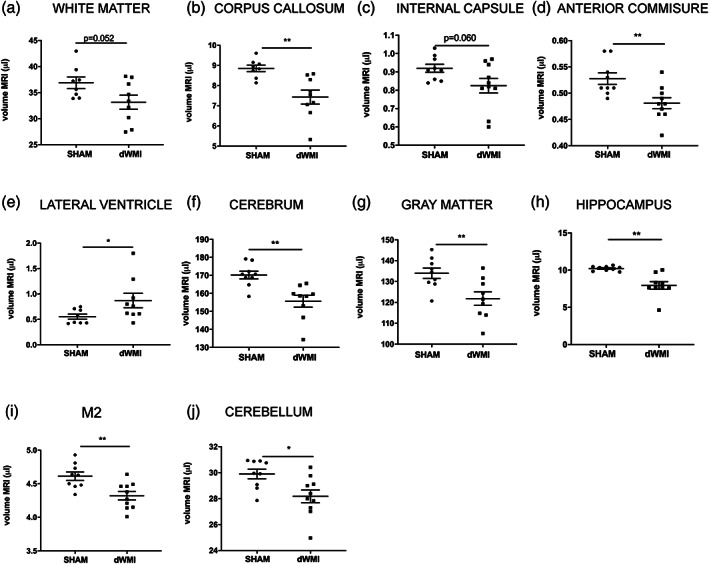
dWMI induction leads to global volumetric deficits on postmortem MRI. (a–j) dWMI animals (n = 9) displayed a reduction in volume (μl) of multiple white and gray matter structures compared with sham‐controls (n = 9) at P26. *p < .05; **p < .01. Nearly significant p values are indicated in (a) and (c)
4.1.5. dWMI is associated with disturbed oligodendrocyte lineage maturation
Multiple studies have suggested that myelination failure in dWMI is the result of arrested maturation of the oligodendrocyte (OL) lineage (Billiards et al., 2008; Buser et al., 2012). Double‐stainings using the proliferative marker Ki67 (indicative for immature, proliferative OL precursors) or the mature OL‐specific marker CC1, in combination with the nuclear OL marker Olig2 were used to investigate development of the OL lineage in animals exposed to the double‐hit model. Interestingly, at P8 (i.e., 3 days after induction of dWMI) an increase in the number of proliferating Ki67+/Olig2+ cells was observed in the cortex of dWMI animals compared with sham‐controls (p = .041) (Figure 5a,b). No significant differences in Ki67+/Olig2+ cell numbers were observed in the corpus callosum at P8 (p = .932) (Figure 5c). At P19, increased numbers of Ki67+/Olig2+ cells were observed in corpus callosum, but not the cortex, of dWMI animals compared with controls (p = .008 and p = .936, respectively) (Figure 5d–f). Importantly, the number of mature CC1+/Olig2+ cells was significantly reduced in cortical areas in dWMI animals compared with sham‐controls (p = .047), whereas CC1+/Olig2+ cell numbers in the corpus callosum were unchanged (p = .873) (Figure 5g–i). We were unable to detect any differences in mature (or immature) OLs between dWMI animals and sham‐controls at P26 (data not shown). CC1 analyses were not performed on P8, as this marker was hardly expressed at this time point.
FIGURE 5.
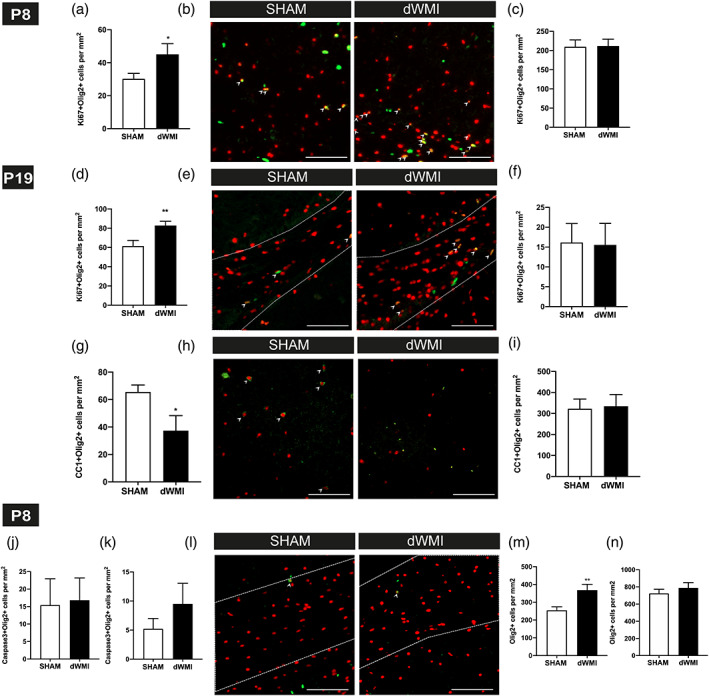
dWMI is associated with an early proliferative response of the OL lineage, followed by maturation arrest. (a,c) dWMI induction leads to an early increase (P8) of Ki67+/Olig2+ cells in the cortex (a), indicative of a proliferative response of the OL lineage. This proliferative response is absent in the corpus callosum (c) (SHAM n = 10, dWMI n = 7). (b) Representative fluorescent images of the cortex of a sham‐control (left) and dWMI (right) animal at P8, double‐stained for Ki67 (green) and Olig2 (red). Double‐positive cells are marked with an arrowhead. Scale bars: 100 μm. (d,f) dWMI mice (n = 12) showed a higher quantity of Ki67 + Olig2+ cells in the corpus callosum (d) compared with sham‐controls (n = 12) at P19, indicative of increased OL proliferation. No differences in Ki67 + Olig2+ cells were found in the cortex (f). (e) Representative fluorescent images (x20) of the corpus callosum (white outline), double‐stained for Ki67 (green) and Olig2 (red), of a sham‐control (left) and dWMI (right) mouse at P19. Double‐positive cells are marked with an arrowhead. Scale bars: 100 μm. (g,i) At P19, a lower quantity of mature (CC1+/Olig+) OLs was observed in the cortex (g) of dWMI animals (n = 5), compared with sham‐control animals (n = 5). We did not detect differences in the number of mature CC1+/Olig2+) OLs in the corpus callosum (i). (h) Representative fluorescent images of the P19 cortex, double‐stained for CC1(green) and Olig2 (red), of a sham‐control (left) and dWMI (right) mouse. Double‐positive cells are marked with an arrowhead. Scale bars: 100 μm. (j, k) At P8, no significant changes in the number of cleaved caspase3+/Olig2+ cells were observed in the corpus callosum (j) or cortex (k) of dWMI (n = 9) versus sham‐control (n = 10) mice. (l) Representative fluorescent images of the corpus callosum (white outline), double‐stained for cleaved caspase 3 (green) and Olig2 (red) of a sham‐control (left) and dWMI (right) animal at P8. Double‐positive cells are marked with an arrowhead. Scale bars: 100 μm. (m, n) dWMI induction is associated with an increase in Olig2+ cells, representing the total OL population, in the cortex (m), but not in the corpus callosum (n) (SHAM n = 10, dWMI n = 7). *p < .05; **p < .01 sham‐operated control versus dWMI animals [Color figure can be viewed at wileyonlinelibrary.com]
To assess the contribution of OL cell death to the observed reduced number of mature oligodendrocytes, we performed double‐stainings for cleaved caspase 3 and Olig2 on sections of dWMI and control mice at P8. Very low numbers of cleaved caspase3+/Olig2+ cells were observed in both the corpus callosum and cortex at P8 and no significant differences in numbers were observed between sham‐control and dWMI groups (cortex p = .279; corpus callosum p = .889) (Figure 5j–l). To check for possible cell death of OL progenitors before P8, we also assessed the total number of Olig2+ cells at P8. In line with the observed rise in proliferating cortical OLs, an increase in total Olig2+ cell numbers was detected at P8 in the cortex of dWMI animals compared with sham‐controls (p = .007) (Figure 5m). No significant differences in total Olig2+ numbers were observed in the corpus callosum at P8 between dWMI and sham‐control animals (p = .435) (Figure 5n).
In sum, our data show an early and persistent proliferative response of the OL lineage after dWMI induction, with a concurrent reduction of mature cortical OLs, without clear evident OL cell death or loss. These data together imply that in this model myelination failure is likely the result of arrested oligodendrocyte maturation rather than extensive loss of (immature) OLs.
4.2. Intranasal MSC treatment to repair dWMI
4.2.1. MSCs widely distribute throughout the injured brain following intranasal administration
To study MSC distribution, mesoporous silica‐coated gold nanoparticles (AuNP‐MSN) were synthesized (Figure S1a–d) and used to label MSCs. The labeling was most efficient after 48 hr of incubation and remained stable after cell concentrating (Figure S1e). Nanoparticle labeled MSCs were administered intranasally at D3 after dWMI induction (i.e., at P8) to investigate cell distribution in our model. MSC distribution could be measured by detecting the gold signal in tissue homogenates using ICP‐MS. Twelve hours after MSC administration, the majority of the measured gold was found in the brain of dWMI animals and a significantly lower amount of gold in peripheral organs (i.e., lungs, liver, and spleen) (p = .0005; p = .002; p = .0006 respectively) (Figure 6a). In contrast, in sham‐controls the highest mass of gold was observed in the lungs and liver after intranasal MSC administration (Figure 6b). To investigate cerebral distribution of MSCs, the brain was divided into ipsi‐ and contralateral rostral and caudal cerebrum, cerebellum, and bulbi. An even distribution of the total amount of gold was observed in both ipsi‐ and contralateral cerebrum and cerebellum of dWMI animals (Figure 6c) indicating a dispersed distribution throughout the diffusely affected brain.
FIGURE 6.
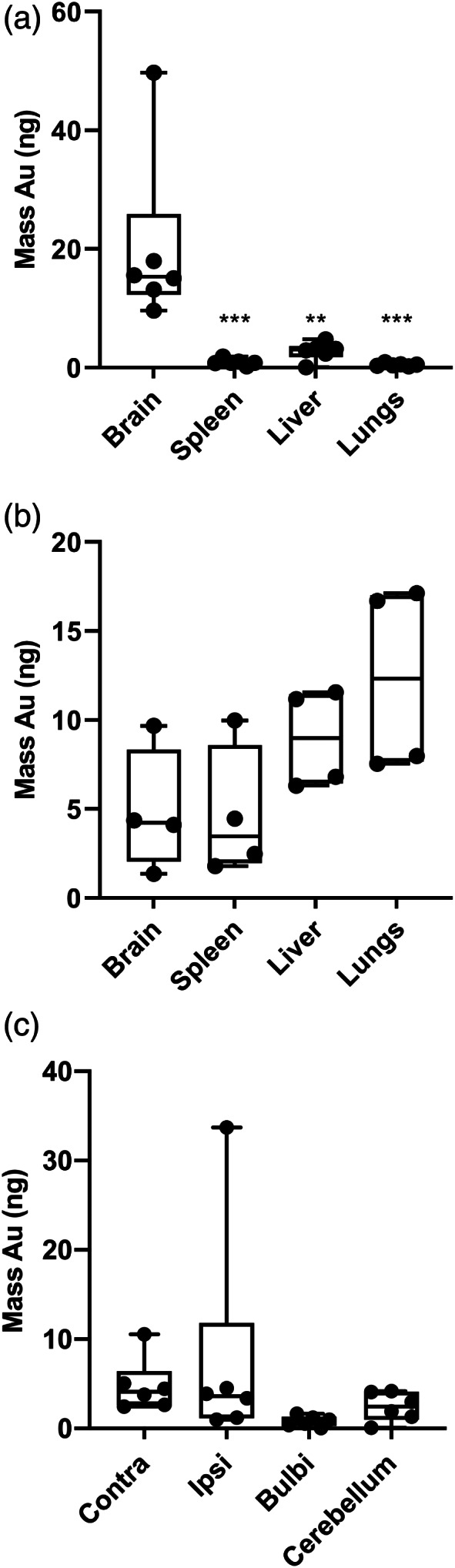
Intranasally administered silica coated gold nanoparticle‐labeled MSCs evenly distribute throughout the brain after dWMI induction. (a,b) After intranasal administration of nanoparticle‐labeled MSCs at D3 (i.e., P8) after injury induction or sham operation, the highest mass of gold was observed in the brain for dWMI animals (n=6) (a) versus peripheral organs in sham‐control animals (n=6) (b). (c) Gold nanoparticles, used to label MSCs, were evenly distributed throughout the brain parts after dWMI **p < .01; ***p < .001 peripheral organs versus brain
4.2.2. Intranasal administration of MSCs improves myelination and functional deficits
To investigate the potential of intranasal MSC therapy on myelin deficits, dWMI animals were intranasally treated with different dosages of MSCs at 3 days after induction of brain injury. At P19, vehicle‐treated dWMI animals displayed a significant reduction in cortical myelination compared with sham‐control animals (p = .0002) which could be rescued by intranasal treatment with 0.5 × 106, 1.0 × 106, or 2.0 × 106 MSCs, as indicated by increased cortical MBP+ coverage (p = .001, p = .0008, and p = 0.0316 dWMI‐veh vs. dWMI‐MSC dosis respectively) (Figure 7a). A lower dose of 0.1 × 106 MSCs did not significantly enhance cortical myelination after dWMI and was therefore not assessed further in this study (data not shown). Microstructural MBP analyses showed similar results: complexity of myelin fibers was negatively affected by dWMI (fiber length p = .048, intersections p = .0026), however, an intranasal dose of 0.5 × 106 and 1.0 × 106 MSCs completely restored the number of intersections (p = .0003 and p = .0039 vs. veh, respectively) and fiber length (p = .0048 and p = .0171 vs. veh, respectively) to control level. The highest dose of 2.0 × 106 MSCs provided partial recovery of myelin complexity after dWMI: increasing the number of intersections (p = .0349) without significantly boosting fiber length (p = .421) (Figure 7b,c). Intranasal treatment with MSCs did not significantly restore developmental gray matter deficits after dWMI, with persistent hippocampal area loss in HE‐stained sections after MSC treatment (Figure 7d).
FIGURE 7.
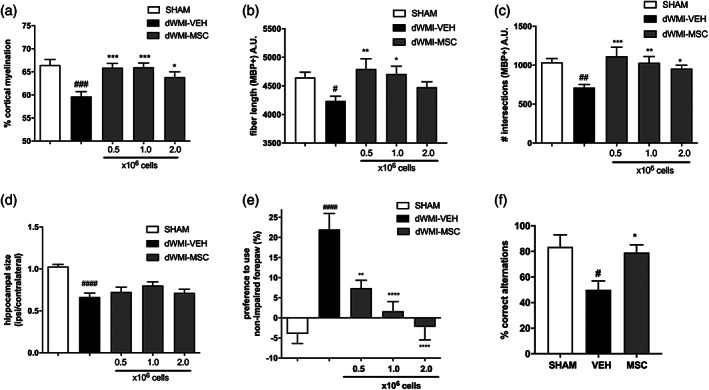
Intranasal MSC treatment boosts myelination and rescues motor and cognitive impairments in dWMI mice. (a) Intranasal administration of 0.5 × 106 (n = 11), 1.0 × 106 (n = 11), and 2.0 × 106 (n = 14) MSCs restores MBP+ coverage of the cortex up to sham‐control levels (n = 13), when compared with vehicle treatment (n = 12). (b,c) An intranasal dose of 0.5 × 106 and 1.0 × 106 MSCs completely restored myelin complexity assessed by fiber length (b) and the number of intersections (c), to sham‐control level (SHAM n = 13, VEH n = 16, 0.5 × 106 MSCs n = 12, 1.0 × 106 MSCs n = 13, and 2.0 × 106 MSCs n = 13). (d) Intranasal MSC treatment does not restore hippocampal size (SHAM n = 14, VEH n = 14,0.5 × 106 MSCs n = 18, 1.0 × 106 MSCs n = 18, and 2.0 × 106 MSCs n = 16). (e) Motor performance measured with the cylinder rearing test improved after MSC treatment (SHAM n = 14, VEH n = 14, 0.5 × 106 MSCs n = 12, 1.0 × 106 MSCs n = 15, and 2.0 × 106 MSCs n = 11). (f) Intranasal treatment with 0.5 × 106 MSCs restores the percentage of correct alternations in the T‐maze up to sham‐control levels in dWMI mice (SHAM n = 4, VEH n = 4, and 0.5 × 106 MSCs n = 8). #p < .05; ##p < .01; ###p < .001; ####p < .0001 vehicle‐treated dWMI animals versus sham‐controls; *p < .05; **p < .01; ***p < .001; ****p < .0001 MSC‐treated dWMI animals versus vehicle‐treated dWMI animals
To examine the effect of MSC treatment on motor outcome after dWMI, animals were subjected to the CRT at P19. Intranasal treatment with 0.5 × 106, 1.0 × 106, and 2.0 × 106 MSCs at 3 days after dWMI potently improved motor outcome by reducing forepaw preference (p = .003, p < .0001 and p < .0001, respectively). In line with our histological findings on myelination, the motor outcome did not improve after treatment with 0.1 × 106 MSCs (data not shown). Based on the histological and functional outcome parameters as shown in Figure 7a–e, we determined 0.5 × 106 MSCs to be the lowest effective dose ensuring optimal recovery after dWMI. The effect of 0.5 × 106 MSCs on executive memory at P26 was assessed next. dWMI animals treated with vehicle demonstrated impairments in executive memory determined by a reduction in correct alternation in the T‐maze compared with sham‐controls. Intranasal MSC treatment showed a potent favorable effect on executive functioning by restoring levels of correct alternations up to sham‐control levels (p = .029) (Figure 7f).
4.2.3. MSC treatment attenuates microglia and astrocyte activation following dWMI
In line with our previous findings, an increase in the number of Iba1+ cells was observed in the corpus callosum of vehicle‐treated animals compared with sham‐control animals at P19 (p = .031). Intranasal MSC treatment at 3 days after induction of injury successfully reduced the number of Iba1+ cells to levels observed in sham‐control animals (p = .045) (Figure 8a). More detailed analyses of microglial morphology revealed an amoeboid (activated) microglia morphology in vehicle‐treated dWMI mice, while MSC treatment significantly reduced the activation state of Iba1+ cells (Figure 8b–d). In addition, we assessed astrocyte reactivity in the corpus callosum and hippocampus. In line with the microglia data, intranasal MSC treatment strongly decreased GFAP+ area compared with vehicle‐treatment in both brain regions (CC p = .026 and HC p = .001) (Figure 8e–f).
FIGURE 8.
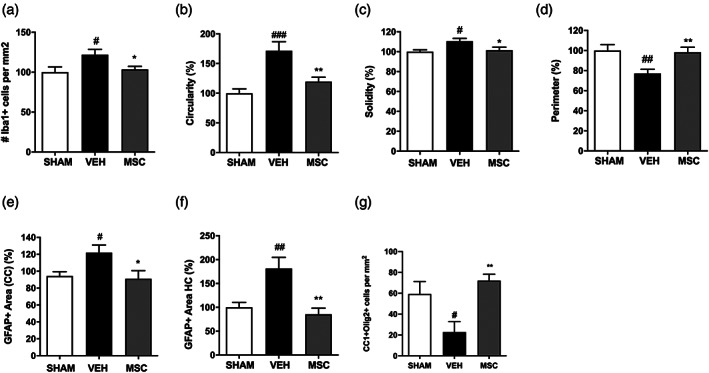
Intranasal MSC treatment dampens the neuro‐inflammatory response and restores OL maturation following dWMI (a) Quantification of microglia density in the corpus callosum revealed a reduction of Iba1 + cells after MSC treatment compared with vehicle treatment (SHAM n = 9, VEH n = 10, MSC n = 14). (b–d) Microglia morphology analyses assessed by cell circularity (b), solidity (c), and perimeter (d), showed a less pro‐inflammatory phenotype following MSC treatment compared with vehicle treatment (SHAM n = 9, VEH n = 11, MSC n = 12, normalized to control values). (e,f) A reduction in GFAP+ area in the corpus callosum (e) and hippocampus (f) was observed following intranasal MSC treatment compared with vehicle‐treatment (SHAM n = 8, VEH n = 8, MSC n = 8, normalized to control values). G. Intranasal administration of 0.5 × 106 MSCs restored CC1+/Olig2+ cells numbers up to sham‐control levels in dWMI animals, indicating a boost in OL lineage maturation (SHAM n = 6, VEH n = 6, MSC n = 13). #p < .05; ##p < .01; ###p < .001 vehicle‐treated dWMI animals versus sham‐controls; *p < .05; **p < .01 MSC‐treated dWMI versus vehicle‐treated dWMI animals
4.2.4. Treatment with intranasal MSCs boosts OL maturation
To determine if the observed recovery of myelination after MSC treatment corresponds with an increase in mature OLs, double‐stainings for CC1 and Olig2 were performed on brain sections of vehicle‐ and MSC‐treated animals at P19. dWMI induced a reduction in cortical CC1+/Olig2+ (p = .032), which was rescued by intranasal treatment with 0.5 × 106 MSCs at P8 (i.e., 3 days after injury induction) (p = .001), indicating recovery of mature OL numbers by MSCs after dWMI (Figure 8g).
4.2.5. Intranasal MSC treatment for dWMI: Starting earlier is better
To gain insight in the treatment window of intranasal MSC therapy, we delayed the timing of MSC administration from 3 days to 6 or 10 days after induction of dWMI. The lowest effective dose of MSCs (0.5 × 106 cells) was administered and complexity of cortical myelination was assessed using segmentation analyses at P26. Figure 9a shows that the dWMI‐induced reduction in fiber length (p = .0076 compared with sham) was restored in dWMI animals that received intranasal MSC treatment at 3 days after injury induction (p = .039 vs. veh). Delay in MSC treatment to 6 or 10 days after induction of brain injury led to a strong reduction in treatment efficacy (D6/10 p > .999 vs. veh) (Figure 9a).
FIGURE 9.
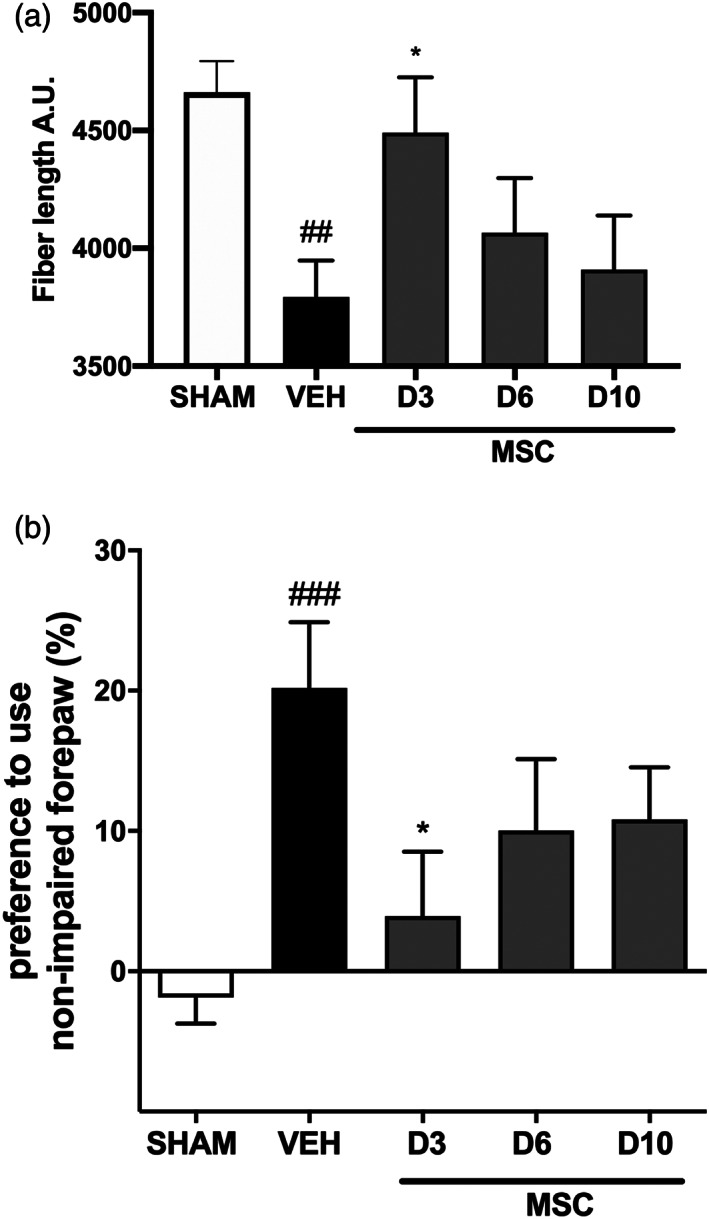
Delayed intranasal administration reduces the regenerative potential of MSCs. (a). Fiber length, a microstructural myelin parameter, was restored in dWMI animals that received MSC treatment at 3 days after injury induction. Delay in MSC treatment to 6 or 10 days led to a reduction in treatment efficacy (SHAM n = 13, VEH n = 13, MSC‐day 3 n = 14, MSC‐day 6 n = 9, MSC‐day 10 n = 9). (b) The beneficial effect of MSC treatment on motor performance, measured with the cylinder rearing test, was partially lost when MSC treatment was postponed to day 6 or day 10 after dWMI induction (SHAM n = 18, VEH n = 14, MSC‐day 3 n = 7, MSC‐day 6 n = 11, MSC‐day 10 n = 9). ##p < .01; ###p < .001 vehicle‐treated dWMI animals versus sham‐controls; *p < .05; MSC‐day three treated dWMI versus vehicle‐treated dWMI animals
Assessment of motor outcome supported these histological findings. The beneficial effect of D3 MSC treatment (p = .046) was partially lost when treatment was postponed to D6 or D10 after dWMI (Figure 9b). Collectively, these results indicate a relative limited time‐window for intranasal MSC treatment in our double‐hit mouse model of dWMI.
4.2.6. MSCs boost OL maturation and attenuate microglia activation in noncontact cocultures
Previous studies have shown that the regenerative potential of MSC treatment is primarily mediated by paracrine signaling, without engraftment of stem cells (Cunningham et al., 2018; Vaes et al., 2019; van Velthoven et al., 2010a). To investigate whether paracrine signaling by MSCs can act on OLs directly, we subjected primary cultured pre‐OLs to medium of LPS‐stimulated microglia (MCM + LPS), in a noncontact coculture assay with MSCs, and measured MBP+ area as maturational read‐out. Pre‐OLs exposed to MCM + LPS demonstrated a strong reduction in MBP+ area (p = .002), indicating impaired maturation, compared with pre‐OLs cultured in the medium of non‐stimulated microglia (MCM‐LPS). Subsequent coculture with 2, 4, 8 × 104 embedded MSCs led to a significant increase in MBP+ area in the OL cultures exposed to MCM + LPS ([borderline] p = .084, p = .001, and p < .0001, respectively) (Figure 10a,b).
FIGURE 10.
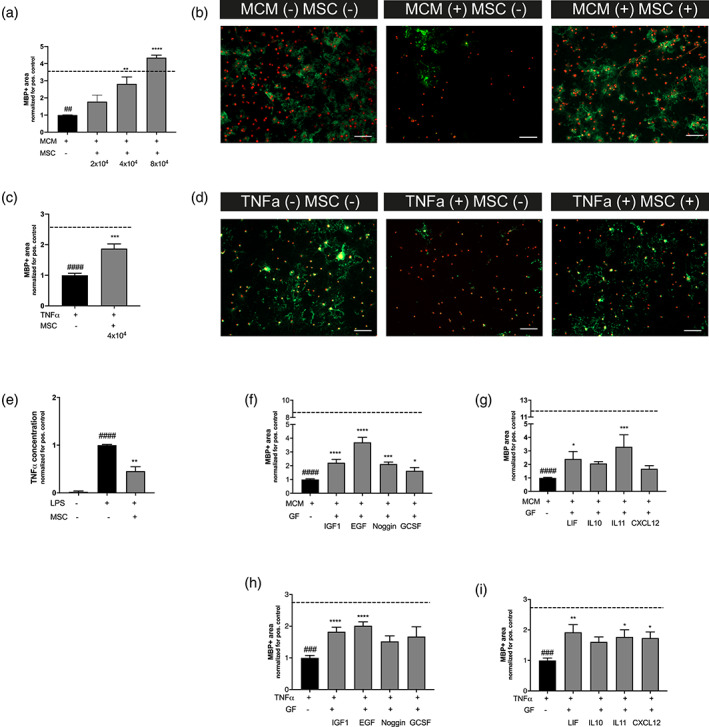
The MSCs secretome boosts OL maturation and attenuates microglia activation in vitro. (a) MCM + LPS causes a reduction in MBP+ area (dashed line represents MBP+ area in MCM‐LPS control condition), MSC treatment with 4 × 104 or 8 × 104 MSCs in a noncontact co‐culture significantly improves OL maturation (n = 3 independent experiments, 3–4 observations per experiment normalized for the positive control, for example, cells exposed to MCM + LPS). (b) Representative fluorescent images (×10) of primary cultured oligodendrocytes stained for oligodendrocyte marker Olig2 (red) and myelin component MBP (green). Cells were exposed to MCM‐LPS (MCM−) or MCM + LPS (MCM+) and 4 × 104 MSCs (MSC+) in a noncontact gel‐insert. Scale bars: 100 μm. (c) Exposure to 10 ng/ml TNFα leads to a reduction in MBP production (dashed line represents MBP+ area in medium without TNFα), MSC treatment with 4x104 MSCs in a noncontact co‐culture significantly boosts OL maturation (n = 2 independent experiments, 3–4 observations per experiment, normalized for the positive control, for example, cells exposed to TNFα). (d) Representative fluorescent images (×10) of primary cultured oligodendrocytes stained for oligodendrocyte marker Olig2 (red) and myelin component MBP (green). Cells were exposed to medium with (+) or without (−) 10 ng/ml TNFα and 4 × 104 MSCs (MSC+) in noncontact gel‐insert. Scale bars: 100 μm. (e). Treatment with 4 × 104 MSCs in a noncontact gel‐insert attenuates microglial TNFα production (n = 2 independent experiments, two observations per experiment, normalized for the positive control, for example, cells exposed to LPS). (f,g) Addition of IGF1, EGF, Noggin, GCSF, LIF, and IL11 but not IL10 and CXCL12 significantly improves MBP+ area by primary cultured oligodendrocytes following MCM + LPS exposure (dashed line represents MBP+ area in MCM‐LPS control condition) (n = 2 independent experiments, 3–4 observations per experiment normalized for the positive control, for example, cells exposed to MCM + LPS). (h,i) Addition of IGF1, EGF, LIF, IL11, and CXCL12 but not Noggin, G‐CSF, and IL10 boosts OL maturation after TNFα‐induced OL maturational arrest (dashed line represents MBP+ area in medium without TNFα) (n = 2 independent experiments, 3–4 observations per experiment, normalized for the positive control, for example, cells exposed to TNFα). ##p < .01; ###p < .001; ####p < .0001 MCM+ or TNF+ condition (black bars) versus MCM‐ or TNFα‐ control (dashed line), respectively; *p < .05; **p < .01; ***p < .001; ****p < .0001 factor‐exposed MCM+ or TNF+ condition (gray bars) versus MCM+ or TNF+ control condition (black bars) [Color figure can be viewed at wileyonlinelibrary.com]
MCM + LPS is a stringent stimulus to mimic dWMI in vitro, leading to both reduced OL maturation and reduced OL survival, and varies slightly per batch of LPS‐stimulated microglia. To be in line with our in vivo findings of OL maturation arrest in absence of pronounced OL apoptosis, we repeated the experiments with a low dose of TNFα as stimulus. Exposure of pre‐OLs to TNFα reduced MBP+ area (p < .0001), without affecting Olig2+ cell numbers (p = .758), implying a true OL maturational arrest (Figure S2). Pre‐OLs cocultured in the presence of 4 × 104 embedded MSCs displayed a significant increase in MBP+ area compared with cells cocultured without MSCs (p = .0002) (Figure 10c,d).
To study the paracrine effects of MSCs directly on microglia, we exposed primary cultured microglia to 50 ng/ml LPS and cocultured with 4 × 104 MSCs. LPS stimulation strongly increased the production of TNFα by microglia in the supernatant compared with nonstimulated cells (p < .0001). Coculture with MSCs led to a significant decrease in TNFα secretion, indicating attenuation of microglial activation (p = .0093) (Figure 10e). Taken together, these results indicate that MSCs can act on both oligodendrocytes and microglia directly through paracrine signaling.
4.2.7. Changes in MSC gene expression profile in dWMI environment
To identify candidate factors in the secretome of MSCs that could underlie the regenerative capacity after encountering the dWMI brain milieu, we cultured MSCs in presence of brain protein extracts from either dWMI or sham‐control animals obtained at 3 days after injury (i.e., the timepoint of MSC treatment in vivo). Gene expression changes in MSCs were examined using PCR arrays focused on growth factors and cyto/chemokines. Using a cut‐off of two‐fold change, 56 differentially regulated genes were identified (Table S3). To investigate the potency of individual MSC‐secreted factors to boost OL maturation and subsequent myelination, we selected candidate factors from the PCR arrays that have been associated with white matter development or repair in the literature (i.e., IGF1, EGF, GCSF[CSF3], IL10, IL11, LIF, and CXCL12). IGF1 and CXCL12 expression by MSCs seemed negatively impacted by the dWMI milieu, however, both these factors have been shown to play a role in OL lineage development (Masters, Werner, Roberts Jr., LeRoith, & Raizada, 1991; Maysami et al., 2006; Patel, McCandless, Dorsey, & Klein, 2010; Wilson, Onischke, & Raine, 2003). Moreover, Noggin, an inhibitor of factors strongly associated with OL maturation arrest (i.e., the BMP family), was added to the panel of factors to mimic BMP4 downregulation by MSCs. PCR array results for these seven factors were confirmed by real‐time RT‐PCR on the individual samples, with a downregulation of CXCL12 and IGF1 and an upregulation of LIF, IL11, and GCSF (CSF3). IL10 and EGF were both upregulated in MSCs exposed to dWMI extracts, but did not reach the two‐fold cut‐off (Table S3). Interestingly, EGF expression seemed to be exclusively induced by the dWMI milieu, as MSCs cultured with sham‐control brain extracts did not express any EGF.
4.2.8. MSC treatment regulates expression of trophic factors after dWMI
To investigate whether MSC treatment changes the cerebral gene expression of the selected trophic factors (i.e., IGF1, EGF, GCSF (CSF3), IL10, IL11, LIF, and CXCL12) in dWMI animals in vivo, we compared mRNA expression levels of the factors between vehicle‐ and MSC‐treated dWMI animals at 12 hr after intranasal treatment. MSC‐treated dWMI animals displayed an upregulation of EGF, GCSF and LIF mRNA in the ipsilateral hemisphere compared with vehicle‐treated dWMI animals, though mean expression levels varied between tissue parts (Table 3). Expression of IL11 showed a modest upregulation following MSC administration, however restricted to the caudal part of the ipsilateral hemisphere. IGF1 and CXCL12 expression was (slightly) downregulated in dWMI animals that received MSC treatment (Table 3). IL10 expression was not detected in any of the tissue parts.
TABLE 3.
Trophic gene expression changes in dWMI animals after MSC treatment versus vehicle treatment
| Gene symbol | Fold regulation 2‐(ΔΔCt) dWMI‐MSC vs. dWMI‐VEH |
|---|---|
| IGF1 | |
| Rostral | 1.16 |
| Caudal | 0.99 |
| Cerebellum | 0.85 |
| EGF | |
| Rostral | 4.40 |
| Caudal | 0.56 |
| Cerebellum | 0.69 |
| IL11 | |
| Rostral | 0.70 |
| Caudal | 1.63 |
| Cerebellum | 0.67 |
| LIF | |
| Rostral | 1.96 |
| Caudal | 2.05 |
| Cerebellum | 1.34 |
| GCSF | |
| Rostral | 3.69 |
| Caudal | 2.48 |
| Cerebellum | 1.32 |
| CXCL12 | |
| Rostral | 1.00 |
| Caudal | 0.95 |
| Cerebellum | 0.66 |
4.2.9. MSC‐secreted factors boost myelin production in vitro
The selected factors were tested for their potential to boost maturation in primary OL cultures challenged with inflammatory stimuli. MCM + LPS as a stimulus led to a strong decrease in MBP+ area compared with pre‐OLS cultured under non‐inflammatory (MCM‐LPS) conditions (p < .0001) as observed earlier (Figure 10f,g). Optimal concentrations of all selected factors on OL maturation were determined in dose–response experiments (Figure S2). Addition of IGF1, EGF, Noggin, GCSF, LIF, or IL11 significantly increased maturation in the OL cultures determined by MBP+ area (p < .0001, p < .0001, p = .0005, p = .0176, p = .048, p = .0002, respectively) (Figure 10f,g). Addition of CXCL12 and IL10 failed to significantly boost OL differentiation in this assay (p = .934 and p = .629, respectively). Moreover, to mimic OL maturation arrest without significant loss of cells, pre‐OLs were exposed to TNFα. Addition of IGF1, EGF, LIF, IL11, and CXCL12 significantly increased MBP+ area in OLs challenged with 10 ng/ml TNFα, indicating that these factors help OLs to overcome their maturational arrest (p < .0001, p < .0001, p = .002, p = .011, p = .023, respectively) (Figure 10h,i). Moreover, GCSF therapy was associated with a borderline significant increase in MBP+ area (p = .056). Addition of IL10 and Noggin did not significantly rescue maturation of pre‐OLs after TNFα exposure (Figure 10h,i).
5. DISCUSSION
Here we investigated a novel double‐hit model of dWMI in mouse pups using behavioral paradigms, postmortem MRI, electron microscopy, and immunostainings. We show that combined postnatal inflammation and hypoxia/ischemia in P5 mice induces transient myelination deficits, neuroinflammation, a maturational arrest in OLs, behavioral impairments and global volumetric deficits of white and gray matter structures. Moreover, we explored the regenerative potential of intranasal MSC treatment after dWMI. Intranasally administered MSCs were dispersedly distributed throughout the brain in dWMI animals compared with sham‐controls. We report that intranasal administration of MSCs restores OL maturation and myelination, dampens the neuroinflammatory response, and improves functional outcome. Furthermore, we demonstrate that MSCs modify their secretome dependent on the cerebral environment. Exposure to dWMI milieu leads to an increased expression of beneficial growth factors and/or anti‐inflammatory cytokines, valuable in boosting OL differentiation. Intranasal treatment with MSCs modulates availability of beneficial factors in vivo, promoting a cerebral milieu more permissive for repair. Moreover, we show in vitro that MSCs can act on both oligodendrocytes and microglia directly, boosting OL maturation under pro‐inflammatory conditions, and attenuating microglia activation. Additionally, MSC‐produced factors, identified in our PCR array and associated with white matter repair in literature, were able to rescue maturation of OLs individually in vitro. Collectively, these results imply that intranasal MSC therapy is a potent treatment strategy to restore myelination in dWMI, and that the neuroregenerative properties of MSCs are likely mediated by their secretome.
Clinically relevant animal models of EoP are essential for translation of potential novel treatment options and further elucidation of underlying pathophysiological mechanisms. Key translational aspects for relevant animal models for EoP are (multiple) etiological factors relevant for the target patient, induction at a developmental brain stage comparable with the (extreme) preterm infant and a diffuse pattern of WMI that resembles EoP in the human patient. Our mouse model incorporates two postnatal insults, both unequivocally linked to EoP pathophysiology. Both hypoxia/ischemia and inflammation have been linked to OL maturation arrest and insufficient myelination in EoP, and are believed to work synergistically (Rezaie & Dean, 2002; Zhao, Chen, Xu, & Pi, 2013). Inflammation in the preterm brain is believed to be triggered by maternal inflammation/infection and postnatal infections, such as neonatal sepsis (Back & Miller, 2014; Bennet et al., 2018). Moreover, disturbances in cerebral oxygenation (either hypoxia or hyperoxia) as a result of an underdeveloped respiratory system and mechanical ventilation, are often encountered by the human preterm infant (Brown & DiBlasi, 2011; Stoll et al., 2015). Unlike a number of previously proposed single‐hit models (reviewed in [van Tilborg et al., 2016]), our model incorporated two postnatal hits, reflecting more closely the multi‐factorial etiology of EoP (Deng, 2010; Khwaja & Volpe, 2008). In our model, mice were subjected to two hits at P5, a developmental time‐window roughly corresponding to white matter development at 24–28 weeks of human gestation, with a peak in immature non‐myelinating OLs (Salmaso, Jablonska, Scafidi, Vaccarino, & Gallo, 2014; Semple, Blomgren, Gimlin, Ferriero, & Noble‐Haeusslein, 2013). We did not observe myelination deficits following a single hit of hypoxia‐ischemia or systemic inflammation. Although the method of hypoxia/ischemia is also used in older postnatal models to induce acute gray matter loss often accompanied by cystic lesions spreading into the white matter tracts, we did not observe (cortical) neuronal loss. Instead, our double‐hit model at P5 induced brain injury closely resembling human post‐mortem and imaging data in preterm dWMI, including a reduction in cortical myelination and myelin complexity in absence of (macroscopic) cystic lesions (Back, 2017; Back & Miller, 2014; Volpe, 2017). These differences in brain injury patterns can mainly be explained by the developmental stage of the rodent brain at P5 versus, for example, P9 and the depth and duration of the systemic hypoxia (Semple et al., 2013).
As proposed in human studies (Billiards et al., 2008; Buser et al., 2012; Verney et al., 2012), hampered OL maturation is likely to underlie the observed insufficient myelination, indicated by the early and persistent rise in immature OLs, with reduction of mature OL numbers and without evident OL cell death. In line with these human studies, in our mouse model we observed an early proliferative response of OPCs in the cortex at P8. Subsequently, at P19, we detected a proliferative response in the corpus callosum, but not the cortex. The increase in proliferation was accompanied by a deficit in mature OLs in the cortex, while the number of mature OLs in the corpus callosum seemed unaffected. Importantly, at P8 we did not find any evidence of extensive OL loss, measured by the number of cleaved caspase3+ OLs. Overt OL cell death prior to P8 also seems unlikely, as dWMI animals did not show Olig2+ cell loss in the corpus callosum, and even an increase in cortical Olig2+ cells, possibly reflecting the early surge in proliferation. These results are also in line with previous observations in our rat double‐hit model of fetal inflammation and postnatal hypoxia (van Tilborg et al., 2018). The spatial differences in immature and mature OLs in the cortex and corpus callosum could be the result of temporal differences in OL migration and myelination. Previous studies have shown that myelination of the corpus callosum precedes that of the somatosensory cortex in rodents (Downes & Mullins, 2013; Vincze, Mázló, Seress, Komoly, & Ábrahám, 2008). Therefore, it is possible that injury induction at P5 mainly targets a population of vulnerable immature cortical OLs, leading to increased proliferation at P8, followed by a deficit in cortical mature OLs and myelination at P19. The observed proliferative response in the corpus callosum at P19 could indicate an endogenous (regenerative) response aimed at restoring cortical myelination deficits of local quiescent OPCs in the corpus callosum, or cells derived from other progenitor pools, such as the subventricular zone (Bonfanti et al., 2017; Hughes, Kang, Fukaya, & Bergles, 2013; van Tilborg et al., 2018; Viganò et al., 2016; Xing et al., 2014). This suggestion is supported by observations in a multiple sclerosis model, with temporal differences in OPC repopulation and maturation between the cortex and corpus callosum (Baxi et al., 2017). Interestingly, at P26 we were unable to detect any differences in mature (or immature) OLs between dWMI animals and sham‐controls, followed by endogenous restoration of cortical myelination at P33. White matter deficits were confirmed using postmortem MRI, with volumetric changes in important white matter structures and enlargement of the lateral ventricles, similar to imaging observations in human extreme preterm infants (Keunen et al., 2016; Lind et al., 2011). In line with the hypothesis of EoP as a global dysmaturation disorder, affecting all developing cell types, including interneurons, and structures in the immature brain, we also observed a reduction in gray matter volumes on MRI and histological hippocampal size, without overt neuronal loss (Keunen et al., 2016; Volpe, 2009a). In depth investigation of potential interneuron disturbances in our model should be considered in the future. Moreover, in line with human postmortem studies, we observed a potent neuroinflammatory response after induction of dWMI (Billiards et al., 2008; Buser et al., 2012; Haynes et al., 2003; Kadhim et al., 2001; Verney et al., 2012) illustrated by increased microglia numbers (likely the consequence of increased proliferation or migration [Umekawa, Osman, Han, Ikeda, & Blomgren, 2015]), with an activated, pro‐inflammatory morphology (Davis, Foster, & Thomas, 1994). Microglia activation after dWMI was accompanied by an increase in astrocyte reactivity, characterized by hypertrophy of the soma (Khakh & Sofroniew, 2015; Schmidt‐Kastner, Wietasch, Weigel, & Eysel, 1993). On a behavioral level, dWMI animals displayed impaired motor performance and cognitive impairments. Several clinical studies have found an association between preterm birth and (mild) cognitive disability (Johnson et al., 2009; Linsell et al., 2018; Nosarti et al., 2007). Aside from motor or cognitive impairments, clinical follow‐up studies often report a higher prevalence in psychiatric disorders, including anxiety disorders and autism spectrum disorder (ASD), in preterm‐born children and adolescents (Johnson & Marlow, 2011). In our mouse model we did not find an indication for anxiety‐like behavior in the open field test, in contrast to (mild) indications of anxiety previously described in our rat double‐hit model (van Tilborg, Achterberg, et al., 2018). In addition, van Tilborg, Achterberg, et al. (2018) identified ASD‐like behavioral deficits following dWMI induction. To study whether these deficits are also applicable to our mouse model of postnatal inflammation and hypoxia/ischemia more behavioral tasks should be performed in the future. Taken together, the double‐hit model of postnatal inflammation and hypoxia/ischemia leads to a clinically relevant pattern of developmental brain injury in neonatal mice, with regard to anatomical, microstructural, and functional outcome.
In our model, the majority of observed deficits were most pronounced up to 3 weeks following injury induction, with evidence of (endogenous) recovery of myelination, neuroinflammation and motor behavior at 4 weeks (i.e., P33) after dWMI. EM analyses confirmed that at 4 weeks after dWMI myelination differences were absent, illustrated by a comparable thickness of the myelin sheath between dWMI and sham animals at P33. This could imply that our double‐hit model induces a developmental delay in the immature rodent brain rather than irreversible long‐lasting changes. Similarly, endogenous recovery of histological myelin deficits has been reported in other rodent models of dWMI (van Tilborg, Achterberg, et al., 2018; van Tilborg et al., 2016). Such findings are in contrast to data from clinical imaging studies of preterm infants, in whom changes on structural and diffusion‐weighted MRI persist into adulthood (Allin et al., 2011; Eikenes, Løhaugen, Brubakk, Skranes, & Håberg, 2011; Ment et al., 2009; Nagy et al., 2009; Nosarti et al., 2002; Nosarti et al., 2008). This discrepancy could be the result of a relatively higher regenerative capacity of the rodent brain compared with humans, limiting the possibility for long‐term follow‐up in rodent models of dWMI (Kaplan, Mishra, & Kohn, 2015; van Tilborg et al., 2016). Interestingly, despite apparent histological recovery of myelination, some follow‐up studies do report long‐lasting alterations in myelin microstructure measured with DTI or behavioral functioning (Chahboune et al., 2009; Favrais et al., 2011; Scafidi et al., 2014; Schmitz et al., 2011; van Tilborg, Achterberg, et al., 2018). When evaluating our model, some considerations should be taken into account. First, we cannot exclude spatial differences in the dWMI brains, in other words the possibility that myelination in other brain regions does not completely restore up to sham‐control levels. Additionally, a delay in white matter development could influence maturation of brain connectivity and other linked brain regions/cell types, including interneurons, still leading to long‐lasting functional impairments (Benamer, Vidal, & Angulo, 2020; Volpe, 2009a; Zonouzi et al., 2015). As example, we did not observe an endogenous recovery of loss of hippocampal area in our model.
Previously, we showed that intranasal MSC treatment was able to improve functional outcome and reduce gray and white matter deficits in a rodent model of term hypoxia‐ischemia, by promoting endogenous repair mechanisms and dampening cerebral inflammation (Donega et al., 2013; Donega et al., 2014). Additionally, we showed that MSC treatment is safe, as MSCs do not engraft (Donega et al., 2015; van Velthoven, Kavelaars, van Bel, & Heijnen, 2011). Moreover, a profound body of evidence supports the regenerative potential of MSC therapy in a multitude of white matter pathologies, and was recently summarized by our group (Vaes et al., 2019). In this study we showed via gold nanoparticle cell labeling and ICP‐MS that intranasally applied MSCs dispersedly migrate throughout the brain after dWMI induction, with minimal loss of gold signal (i.e., labeled MSCs) to peripheral organs, in clear contrast to what was observed in sham‐control animals. Though these data imply that MSCs favor migration toward the injured brain after intranasal administration, the potential beneficial role of MSCs in the periphery remains unclear in this model (Jellema et al., 2013). The widespread cerebral distribution of MSCs might be the result of systemic inflammation and hypoxia leading to diffuse patterns of myelin deficits, though additional studies are needed to determine the timing of migration and final location of MSCs after intranasal treatment in our model. In the field of EoP, several studies have reported beneficial effects of MSC therapy on OL lineage maturation and survival, subsequent myelination and modulation of the inflammatory response in the CNS and periphery (Vaes et al., 2019). In line with these studies, we report here increased numbers of mature OLs, recovery of cortical myelination and myelin microstructure, dampening of neuroinflammation and improved functional outcome after intranasal MSC administration in our dWMI model. Even though large differences in pathophysiological hallmarks (i.e., prominent cell death of pre‐OLs in other models) and methodology (i.e., MSC source and/or route/timing of administration) exist between other studies and ours, these concurrent findings strengthen the promising role of MSCs to restore dWMI in the preterm brain. In contrast to the beneficial effects of MSC treatment on myelination and neuroinflammation, MSCs failed to restore hippocampal area loss after dWMI. We suggest that the loss of hippocampal area in this model possibly is the result of interference with normal development and construction of certain gray matter tracts, for example due to changes in synaptic input, leading to long‐lasting hippocampal volume loss. Interestingly, MSC treatment did improve cognitive functioning in the spontaneous alternation T‐maze. Although this spatial memory task does (in part) rely on proper hippocampal functioning, other brain regions, such as the prefrontal cortex, thalamus and cerebellum are also required for optimal spatial memory performance (Lalonde, 2002). Thus, the observed improvement in T‐maze performance after MSC therapy is likely the result of restoration of myelination in other brain areas involved in executive functioning or improved functioning of the remaining hippocampal tissue.
One of the challenges for future clinical application of MSCs is determination of the optimal treatment protocol. Here, we propose the lowest effective dose of 0.5 × 106 cells BM‐MSCs applied intranasally based on histological and functional outcome parameters. A similar dosage has shown to be effective in a P2‐3 rat model of EoP using intranasal delivery of cells (Oppliger et al., 2016). Intracranial administration in a P3‐4 mouse model of EoP, an invasive but direct route with minimal loss of cells during migration, warranted a lower effective dose of 0.25 × 106 cells (Mueller et al., 2017).
Apart from the dosage, the optimal window of MSC treatment in EoP is still unclear. Here, we observed a relative limited treatment window, measured by myelination parameters and functional outcome when intranasal treatment was postponed until D6 or D10 after dWMI compared with treatment at D3. A similar reduction in MSC treatment efficacy over time has been observed in other preclinical studies in the field of adult ischemic stroke, severe intraventricular hemorrhage and neonatal stroke (Kim et al., 2012; Park et al., 2016; Wang et al., 2014). In contrast, earlier work from our group showed a treatment window of at least 10 days in a mouse model of term hypoxic–ischemic brain injury (van Velthoven et al., 2010a).The reduction in MSC treatment efficacy could be due to reduced homing of cells following intranasal administration when damage patterns are less severe or more diffuse in nature, or could be the result of a limited regenerative capacity of MSCs in later stages of EoP pathophysiology. In the latter option, optimization of MSC therapy, using preconditioning or genetic modification strategies, could be explored to prolong the treatment window. A limited treatment window of MSCs would underline the need for early identification (i.e., biomarkers) of preterm infants at risk for EoP to ensure timely treatment.
MSCs are believed to exert their regenerative properties by adaptation of their secretome, stimulating endogenous repair mechanisms through paracrine signaling (Kassis, Vaknin‐Dembinsky, & Karussis, 2011; Liang, Ding, Zhang, Tse, & Lian, 2014; Paton et al., 2017; van Velthoven et al., 2010a). However, the exact mechanisms that underlie the regenerative effect of MSCs on white matter damage in vivo remain unclear. On the one hand, the MSCs' secretome, containing growth factors and cytokines, as well as microvesicles and exosomes, could act directly on OL precursors, thereby supporting OL maturation and subsequent myelination. However, the effect of MSC‐secreted factors could also be mediated by attenuation of neuroinflammation (i.e., microglia activation), leading indirectly to a more favorable milieu for OL maturation. Here, we demonstrate in vitro that the MSCs' secretome can directly promote maturation of OPCs under pro‐inflammatory conditions, as well as attenuate microglia activation. Moreover, MSC treatment induced mRNA expression changes of the selected beneficial factors in brains of dWMI mice, when compared with vehicle‐treated dWMI brains. Although these in vivo changes closely resembled the observed patterns of up‐ and downregulation in the secretome when MSCs were exposed to the dWMI milieu ex vivo, the current data do not indisputably prove which cells are responsible for the expression changes. Aside from a direct effect of the MSCs' secretome in situ, the observed effects on beneficial factor expression levels could also reflect an indirect effect of MSCs on factor production by for example microglia and/or astrocytes, indirectly contributing to a more repair‐promoting milieu in the brain. This theory likely applies to EGF, as expression of this trophic factor was upregulated in MSC‐treated dWMI brains in vivo, whereas we did not observe any expression changes in the dWMI‐exposed MSCs ex vivo. Taken together, these results indicate that the observed potent regenerative effects of MSCs in vivo on OL maturation and dampening of neuroinflammation are likely mediated through direct paracrine signaling on multiple glial cell types.
To get more insight in which individual MSC‐secreted factors are potent to boost OL maturation in vitro, we selected promising growth factors and cytokines that are differentially expressed in MSCs encountering dWMI milieu ex vivo. Addition of IGF1, EGF, GCSF, LIF, and IL11 restored MBP+ area of primary OLs up to control levels following inflammation in vitro. Interestingly, Noggin, a BMP4 inhibitor, failed to significantly restore OL differentiation in the TNFα maturation arrest assay, suggesting that Noggin predominately plays a role in OL survival and to a lesser extent in OL maturation. The contrary could be proposed for CXCL12. These findings imply a potentially promising future role for trophic factor (mono)therapy or combination therapy with MSCs, in the treatment of EoP. However, additional in vivo studies, including back‐to‐back comparison of efficacy of MSCs, that can deliver a more constant mixture of trophic factors, are needed to substantiate these statements.
In summary, this study shows that intranasal MSC treatment is a potent new strategy to support myelination and functional outcome in a mouse model of EoP. Our results indicate that MSCs potently modify their secretome in situ to support OL maturation by upregulating a mixture of beneficial factors valuable in underpinning proper OL maturation and subsequent myelination of the developing brain at risk.
Supporting information
Figure S1 AuNP‐MSN nanoparticle characterization and rMSC nanoparticle uptake analysis. A)Transmission electron microscopy (TEM) image of AuNP (left) and AuNP‐MSN (right)_ Scale bar is 200 nm_ B) Size analysis of AuNP (black) and AuNP‐MSN (red) by dynamic light scattering (DLS)_ C) UV–Vis spectroscopy of AuNP (black) and AuNP‐MSN (red) showing a slight shift in absorbance after silica coating. D) Size analysis of 20 particles using TEM images by imageJ; AuNP (left) and AuNP‐MSN (right). E) internalization of AuNP‐MSN‐LIP nanoparticles in rMSCs after 2, 6, 24 and 48 hours of incubation, analysed by flow cytometry. F) Internalization of AuNP‐MSN‐LIP after 48 h incubation with rMSCs and concentration to 62.5x108 clls/mL.
Figure S2 MSC‐secreted factors boost myelin production in a dose‐dependent manner in primary OL cultures. A. Exposure to 10 ng/mL TNFα does not lead to OL death indicated by a similar amount Olig2+ cells in medium with TNFα+ compared with medium without TNFα (n = 3 independent experiments, 3–4 observations per experiment; normalized for the positive control, for example, cells exposed to TNFα). B‐I. Addition of IGF1 (B), IL11 (C), CXCL12 (D), EGF (E),LIF (G), IL10 (H) and Noggin (I), but not GCSF (F), significantly improved myelin production by primary cultured oligodendrocytes in an dose‐dependent manner following MCM + LPS exposure (dashed line represents MBP+ area in MCM‐LPS control condition) (n = 2 independent experiments, 2–3 observations per experiment, normalized for the positive control, for example, cells exposed to MCM + LPS without addition of a factor). ####: p < 0.0001 MCM+ (black bars) vs. MCM‐ (dashed line) respectively; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001 factor‐exposed MCM+ condition (gray bars) vs. MCM+ control condition (black bars).
Table S3 Gene expression profile of MSCs after culture with SHAM or dWMI brain extracts
ACKNOWLEDGMENTS
This work was funded by Brain Foundation the Netherlands. We would like to thank Rik van Vliet, Michiel van der Lelie and Angela Vandersteen for their technical assistance.
Vaes JEG, van Kammen CM, Trayford C, et al. Intranasal mesenchymal stem cell therapy to boost myelination after encephalopathy of prematurity. Glia. 2021;69:655–680. 10.1002/glia.23919
Caren M. van Kammen and Chloe Trayford contributed equally to this manuscript and share the second authorship
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Allin, M. P. , Kontis, D. , Walshe, M. , Wyatt, J. , Barker, G. J. , Kanaan, R. A. , … Nosarti, C. (2011). White matter and cognition in adults who were born preterm. PLoS One, 6(10), e24525 10.1371/journal.pone.0024525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back, S. A. (2017). White matter injury in the preterm infant: Pathology and mechanisms. Acta Neuropathologica, 134(3), 331–349. 10.1007/s00401-017-1718-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back, S. A. , & Miller, S. P. (2014). Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Annals of Neurology, 75(4), 469–486. 10.1002/ana.24132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxi, E. G. , DeBruin, J. , Jin, J. , Strasburger, H. J. , Smith, M. D. , Orthmann‐Murphy, J. L. , … Calabresi, P. A. (2017). Lineage tracing reveals dynamic changes in oligodendrocyte precursor cells following cuprizone‐induced demyelination. Glia, 65(12), 2087–2098. 10.1002/glia.23229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, T. E. , Berg, H. J. , Jbabdi, S. , Rushworth, M. F. , & Woolrich, M. W. (2007). Probabilistic diffusion tractography with multiple fibre orientations: What can we gain? NeuroImage, 34(1), 144–155. 10.1016/j.neuroimage.2006.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, T. E. , Woolrich, M. W. , Jenkinson, M. , Johansen‐Berg, H. , Nunes, R. G. , Clare, S. , … Smith, S. M. (2003). Characterization and propagation of uncertainty in diffusion‐weighted MR imaging. Magnetic Resonance in Medicine, 50(5), 1077–1088. 10.1002/mrm.10609 [DOI] [PubMed] [Google Scholar]
- Benamer, N. , Vidal, M. , & Angulo, M. C. (2020). The cerebral cortex is a substrate of multiple interactions between GABAergic interneurons and oligodendrocyte lineage cells. Neuroscience Letters, 715, 134615 10.1016/j.neulet.2019.134615 [DOI] [PubMed] [Google Scholar]
- Bennet, L. , Dhillon, S. , Lear, C. A. , van den Heuij, L. , King, V. , Dean, J. M. , … Gunn, A. J. (2018). Chronic inflammation and impaired development of the preterm brain. Journal of Reproductive Immunology, 125, 45–55. 10.1016/j.jri.2017.11.003 [DOI] [PubMed] [Google Scholar]
- Billiards, S. S. , Haynes, R. L. , Folkerth, R. D. , Borenstein, N. S. , Trachtenberg, F. L. , Rowitch, D. H. , … Kinney, H. C. (2008). Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathology, 18(2), 153–163. 10.1111/j.1750-3639.2007.00107.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfanti, E. , Gelosa, P. , Fumagalli, M. , Dimou, L. , Viganò, F. , Tremoli, E. , … Abbracchio, M. P. (2017). The role of oligodendrocyte precursor cells expressing the GPR17 receptor in brain remodeling after stroke. Cell Death & Disease, 8(6), e2871–e2871. 10.1038/cddis.2017.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora, M. , De Marchi, E. , Patergnani, S. , Suski, J. M. , Celsi, F. , Bononi, A. , … Pinton, P. (2014). Tumor necrosis factor‐alpha impairs oligodendroglial differentiation through a mitochondria‐dependent process. Cell Death and Differentiation, 21(8), 1198–1208. 10.1038/cdd.2014.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M. K. , & DiBlasi, R. M. (2011). Mechanical ventilation of the premature neonate. Respiratory Care, 56(9), 1298–1313. 10.4187/respcare.01429 [DOI] [PubMed] [Google Scholar]
- Buser, J. R. , Maire, J. , Riddle, A. , Gong, X. , Nguyen, T. , Nelson, K. , … Back, S. A. (2012). Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Annals of Neurology, 71(1), 93–109. 10.1002/ana.22627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahboune, H. , Ment, L. R. , Stewart, W. B. , Rothman, D. L. , Vaccarino, F. M. , Hyder, F. , & Schwartz, M. L. (2009). Hypoxic injury during neonatal development in murine brain: Correlation between in vivo DTI findings and behavioral assessment. Cerebral Cortex, 19(12), 2891–2901. 10.1093/cercor/bhp068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Balasubramaniyan, V. , Peng, J. , Hurlock, E. C. , Tallquist, M. , Li, J. , & Lu, Q. R. (2007). Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nature Protocols, 2(5), 1044–1051. 10.1038/nprot.2007.149 [DOI] [PubMed] [Google Scholar]
- Cunningham, C. J. , Redondo‐Castro, E. , & Allan, S. M. (2018). The therapeutic potential of the mesenchymal stem cell secretome in ischaemic stroke. Journal of Cerebral Blood Flow and Metabolism, 38(8), 1276–1292. 10.1177/0271678x18776802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielyan, L. , Schafer, R. , von Ameln‐Mayerhofer, A. , Buadze, M. , Geisler, J. , Klopfer, T. , … Frey, W. H., 2nd. (2009). Intranasal delivery of cells to the brain. European Journal of Cell Biology, 88(6), 315–324. 10.1016/j.ejcb.2009.02.001 [DOI] [PubMed] [Google Scholar]
- Davis, E. J. , Foster, T. D. , & Thomas, W. E. (1994). Cellular forms and functions of brain microglia. Brain Research Bulletin, 34(1), 73–78. 10.1016/0361-9230(94)90189-9 [DOI] [PubMed] [Google Scholar]
- Deng, W. (2010). Neurobiology of injury to the developing brain. Nature Reviews Neurology, 6(1759–4766), 328–336. 10.1038/nrneurol.2010.53 [DOI] [PubMed] [Google Scholar]
- Donega, V. , Nijboer, C. H. , Braccioli, L. , Slaper‐Cortenbach, I. , Kavelaars, A. , van Bel, F. , & Heijnen, C. J. (2014). Intranasal administration of human MSC for ischemic brain injury in the mouse: in vitro and in vivo neuroregenerative functions. PLoS One, 9(11), e112339–e112339. 10.1371/journal.pone.0112339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donega, V. , Nijboer, C. H. , van Tilborg, G. , Dijkhuizen, R. M. , Kavelaars, A. , & Heijnen, C. J. (2014). Intranasally administered mesenchymal stem cells promote a regenerative niche for repair of neonatal ischemic brain injury. Experimental Neurology, 261, 53–64. 10.1016/j.expneurol.2014.06.009 [DOI] [PubMed] [Google Scholar]
- Donega, V. , Nijboer, C. H. , van Velthoven, C. T. , Youssef, S. A. , de Bruin, A. , van Bel, F. , … Heijnen, C. J. (2015). Assessment of long‐term safety and efficacy of intranasal mesenchymal stem cell treatment for neonatal brain injury in the mouse. Pediatric Research, 78(5), 520–526. 10.1038/pr.2015.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donega, V. , van Velthoven, C. T. , Nijboer, C. H. , van Bel, F. , Kas, M. J. , Kavelaars, A. , & Heijnen, C. J. (2013). Intranasal mesenchymal stem cell treatment for neonatal brain damage: Long‐term cognitive and sensorimotor improvement. PLoS One, 8(1), e51253–e51253. 10.1371/journal.pone.0051253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes, N. , & Mullins, P. (2013). The development of myelin in the brain of the juvenile rat. Toxicologic Pathology, 42(5), 913–922. 10.1177/0192623313503518 [DOI] [PubMed] [Google Scholar]
- Eikenes, L. , Løhaugen, G. C. , Brubakk, A. M. , Skranes, J. , & Håberg, A. K. (2011). Young adults born preterm with very low birth weight demonstrate widespread white matter alterations on brain DTI. NeuroImage, 54(3), 1774–1785. 10.1016/j.neuroimage.2010.10.037 [DOI] [PubMed] [Google Scholar]
- Favrais, G. , van de Looij, Y. , Fleiss, B. , Ramanantsoa, N. , Bonnin, P. , Stoltenburg‐Didinger, G. , … Gressens, P. (2011). Systemic inflammation disrupts the developmental program of white matter. Annals of Neurology, 70(4), 550–565. 10.1002/ana.22489 [DOI] [PubMed] [Google Scholar]
- Haynes, R. L. , Folkerth, R. D. , Keefe, R. J. , Sung, I. , Swzeda, L. I. , Rosenberg, P. A. , … Kinney, H. C. (2003). Nitrosative and oxidative injury to Premyelinating oligodendrocytes in periventricular leukomalacia. Journal of Neuropathology & Experimental Neurology, 62(5), 441–450. 10.1093/jnen/62.5.441 [DOI] [PubMed] [Google Scholar]
- Hughes, E. G. , Kang, S. H. , Fukaya, M. , & Bergles, D. E. (2013). Oligodendrocyte progenitors balance growth with self‐repulsion to achieve homeostasis in the adult brain. Nature Neuroscience, 16(6), 668–676. 10.1038/nn.3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jbabdi, S. , Sotiropoulos, S. N. , Savio, A. M. , Grana, M. , & Behrens, T. E. (2012). Model‐based analysis of multishell diffusion MR data for tractography: How to get over fitting problems. Magnetic Resonance in Medicine, 68(6), 1846–1855. 10.1002/mrm.24204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellema, R. K. , Wolfs, T. G. , Lima Passos, V. , Zwanenburg, A. , Ophelders, D. R. , Kuypers, E. , … Kramer, B. W. (2013). Mesenchymal stem cells induce T‐cell tolerance and protect the preterm brain after global hypoxia‐ischemia. PLoS One, 8(8), e73031 10.1371/journal.pone.0073031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson, M. , Bannister, P. , Brady, M. , & Smith, S. (2002). Improved optimization for the robust and accurate linear registration and motion correction of brain images. NeuroImage, 17(2), 825–841. 10.1016/s1053-8119(02)91132-8 [DOI] [PubMed] [Google Scholar]
- Jenkinson, M. , & Smith, S. (2001). A global optimisation method for robust affine registration of brain images. Medical Image Analysis, 5(2), 143–156. 10.1016/s1361-8415(01)00036-6 [DOI] [PubMed] [Google Scholar]
- Johnson, S. , Fawke, J. , Hennessy, E. , Rowell, V. , Thomas, S. , Wolke, D. , & Marlow, N. (2009). Neurodevelopmental disability through 11 years of age in children born before 26 weeks of gestation. Pediatrics, 124(2), e249–e257. 10.1542/peds.2008-3743 [DOI] [PubMed] [Google Scholar]
- Johnson, S. , & Marlow, N. (2011). Preterm birth and childhood psychiatric disorders. Pediatric Research, 69(8), 11–18. 10.1203/PDR.0b013e318212faa0 [DOI] [PubMed] [Google Scholar]
- Kadhim, H. , Tabarki, B. , Verellen, G. , De Prez, C. , Rona, A. M. , & Sébire, G. (2001). Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology, 56(10), 1278–1284. 10.1212/wnl.56.10.1278 [DOI] [PubMed] [Google Scholar]
- Kaplan, H. M. , Mishra, P. , & Kohn, J. (2015). The overwhelming use of rat models in nerve regeneration research may compromise designs of nerve guidance conduits for humans. Journal of Materials Science: Materials in Medicine, 26(8), 226 10.1007/s10856-015-5558-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassis, I. , Vaknin‐Dembinsky, A. , & Karussis, D. (2011). Bone marrow mesenchymal stem cells: Agents of immunomodulation and neuroprotection. Current Stem Cell Research & Therapy, 6(1), 63–68. 10.2174/157488811794480762 [DOI] [PubMed] [Google Scholar]
- Keunen, K. , Išgum, I. , van Kooij, B. J. M. , Anbeek, P. , van Haastert, I. C. , Koopman‐Esseboom, C. , … Benders, M. J. N. L. (2016). Brain volumes at term‐equivalent age in preterm infants: Imaging biomarkers for neurodevelopmental outcome through early school age. The Journal of Pediatrics, 172, 88–95. 10.1016/j.jpeds.2015.12.023 [DOI] [PubMed] [Google Scholar]
- Khakh, B. S. , & Sofroniew, M. V. (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nature Neuroscience, 18(7), 942–952. 10.1038/nn.4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja, O. , & Volpe, J. J. (2008). Pathogenesis of cerebral white matter injury of prematurity. Archives of Disease in Childhood. Fetal and Neonatal Edition, 93(2), F153–F161. 10.1136/adc.2006.108837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, E. S. , Ahn, S. Y. , Im, G. H. , Sung, D. K. , Park, Y. R. , Choi, S. H. , … Park, W. S. (2012). Human umbilical cord blood–derived mesenchymal stem cell transplantation attenuates severe brain injury by permanent middle cerebral artery occlusion in newborn rats. Pediatric Research, 72, 277 10.1038/pr.2012.71 [DOI] [PubMed] [Google Scholar]
- Lalonde, R. (2002). The neurobiological basis of spontaneous alternation. Neuroscience and Biobehavioral Reviews, 26(1), 91–104. 10.1016/s0149-7634(01)00041-0 [DOI] [PubMed] [Google Scholar]
- Larroque, B. , Ancel, P. Y. , Marret, S. , Marchand, L. , Andre, M. , Arnaud, C. , … Kaminski, M. (2008). Neurodevelopmental disabilities and special care of 5‐year‐old children born before 33 weeks of gestation (the EPIPAGE study): A longitudinal cohort study. The Lancet, 371(9615), 813–820. 10.1016/s0140-6736(08)60380-3 [DOI] [PubMed] [Google Scholar]
- Lee, Y. A. (2017). White matter injury of prematurity: Its mechanisms and clinical features. Journal of Pathology and Translational Medicine, 51(5), 449–455. 10.4132/jptm.2017.07.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X. , Ding, Y. , Zhang, Y. , Tse, H. F. , & Lian, Q. (2014). Paracrine mechanisms of mesenchymal stem cell‐based therapy: Current status and perspectives. Cell Transplantation, 23(9), 1045–1059. 10.3727/096368913x667709 [DOI] [PubMed] [Google Scholar]
- Lind, A. , Parkkola, R. , Lehtonen, L. , Munck, P. , Maunu, J. , Lapinleimu, H. , … Group, P. S . (2011). Associations between regional brain volumes at term‐equivalent age and development at 2 years of age in preterm children. Pediatric Radiology, 41(8), 953–961. 10.1007/s00247-011-2071-x [DOI] [PubMed] [Google Scholar]
- Linsell, L. , Johnson, S. , Wolke, D. , O'Reilly, H. , Morris, J. K. , Kurinczuk, J. J. , & Marlow, N. (2018). Cognitive trajectories from infancy to early adulthood following birth before 26 weeks of gestation: A prospective, population‐based cohort study. Archives of Disease in Childhood, 103(4), 363–370. 10.1136/archdischild-2017-313414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay, D. F. , Smith, G. C. , Dobbie, R. , & Pell, J. P. (2010). Gestational age at delivery and special educational need: Retrospective cohort study of 407,503 schoolchildren. PLoS Medicine, 7(6), e1000289 10.1371/journal.pmed.1000289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters, B. A. , Werner, H. , Roberts, C. T., Jr. , LeRoith, D. , & Raizada, M. K. (1991). Insulin‐like growth factor I (IGF‐I) receptors and IGF‐I action in oligodendrocytes from rat brains. Regulatory Peptides, 33(2), 117–131. 10.1016/0167-0115(91)90207-W [DOI] [PubMed] [Google Scholar]
- Maysami, S. , Nguyen, D. , Zobel, F. , Pitz, C. , Heine, S. , Hopfner, M. , & Stangel, M. (2006). Modulation of rat oligodendrocyte precursor cells by the chemokine CXCL12. Neuroreport, 17(11), 1187–1190. 10.1097/01.wnr.0000227985.92551.9a [DOI] [PubMed] [Google Scholar]
- Ment, L. R. , Kesler, S. , Vohr, B. , Katz, K. H. , Baumgartner, H. , Schneider, K. C. , … Reiss, A. L. (2009). Longitudinal brain volume changes in preterm and term control subjects during late childhood and adolescence. Pediatrics, 123, 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moster, D. , Lie, R. T. , & Markestad, T. (2008). Long‐term medical and social consequences of preterm birth. New England Journal of Medicine, 359(3), 262–273. 10.1056/NEJMoa0706475 [DOI] [PubMed] [Google Scholar]
- Mueller, M. , Oppliger, B. , Joerger‐Messerli, M. , Reinhart, U. , Barnea, E. , Paidas, M. , … Schoeberlein, A. (2017). Wharton's jelly mesenchymal stem cells protect the immature brain in rats and modulate cell fate. Stem Cells and Development, 26(4), 239–248. 10.1089/scd.2016.0108 [DOI] [PubMed] [Google Scholar]
- Nagy, Z. , Ashburner, J. , Andersson, J. , Jbabdi, S. , Draganski, B. , Skare, S. , … Lagercrantz, H. (2009). Structural correlates of preterm birth in the adolescent brain. Pediatrics, 124(5), e964–e972. 10.1542/peds.2008-3801 [DOI] [PubMed] [Google Scholar]
- Nosarti, C. , Al‐Asady, M. H. , Frangou, S. , Stewart, A. L. , Rifkin, L. , & Murray, R. M. (2002). Adolescents who were born very preterm have decreased brain volumes. Brain, 125(Pt 7), 1616–1623. 10.1093/brain/awf157 [DOI] [PubMed] [Google Scholar]
- Nosarti, C. , Giouroukou, E. , Healy, E. , Rifkin, L. , Walshe, M. , Reichenberg, A. , … Murray, R. M. (2008). Grey and white matter distribution in very preterm adolescents mediates neurodevelopmental outcome. Brain, 131(Pt 1), 205–217. 10.1093/brain/awm282 [DOI] [PubMed] [Google Scholar]
- Nosarti, C. , Giouroukou, E. , Micali, N. , Rifkin, L. , Morris, R. G. , & Murray, R. M. (2007). Impaired executive functioning in young adults born very preterm. Journal of the International Neuropsychological Society, 13(4), 571–581. 10.1017/S1355617707070725 [DOI] [PubMed] [Google Scholar]
- Oppliger, B. , Joerger‐Messerli, M. , Mueller, M. , Reinhart, U. , Schneider, P. , Surbek, D. V. , & Schoeberlein, A. (2016). Intranasal delivery of umbilical cord‐derived mesenchymal stem cells preserves myelination in perinatal brain damage. Stem Cells and Development, 25(16), 1234–1242. 10.1089/scd.2016.0027 [DOI] [PubMed] [Google Scholar]
- Park, W. S. , Sung, S. I. , Ahn, S. Y. , Sung, D. K. , Im, G. H. , Yoo, H. S. , … Chang, Y. S. (2016). Optimal timing of mesenchymal stem cell therapy for neonatal intraventricular hemorrhage. Cell Transplantation, 25(6), 1131–1144. 10.3727/096368915x689640 [DOI] [PubMed] [Google Scholar]
- Patel, J. R. , McCandless, E. E. , Dorsey, D. , & Klein, R. S. (2010). CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proceedings of the National Academy of Sciences of the United States of America, 107(24), 11062–11067. 10.1073/pnas.1006301107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton, M. C. B. , McDonald, C. A. , Allison, B. J. , Fahey, M. C. , Jenkin, G. , & Miller, S. L. (2017). Perinatal brain injury as a consequence of preterm birth and intrauterine inflammation: Designing targeted stem cell therapies. Frontiers in Neuroscience, 11, 200 10.3389/fnins.2017.00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, G. , & Anisimov, S. V. (2013). The secretome of mesenchymal stem cells: Potential implications for neuroregeneration. Biochimie, 95(12), 2246–2256. 10.1016/j.biochi.2013.07.013 [DOI] [PubMed] [Google Scholar]
- Peterson, B. S. , Vohr, B. , Staib, L. H. , Cannistraci, C. J. , Dolberg, A. , Schneider, K. C. , … Ment, L. R. (2000). Regional brain volume abnormalities and long‐term cognitive outcome in preterm infants. JAMA, 284(15), 1939–1947. 10.1001/jama.284.15.1939 [DOI] [PubMed] [Google Scholar]
- Rezaie, P. , & Dean, A. (2002). Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology, 22(3), 106–132. 10.1046/j.1440-1789.2002.00438.x [DOI] [PubMed] [Google Scholar]
- Salmaso, N. , Jablonska, B. , Scafidi, J. , Vaccarino, F. M. , & Gallo, V. (2014). Neurobiology of premature brain injury. Nature Neuroscience, 17(3), 341–346. 10.1038/nn.3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafidi, J. , Hammond, T. R. , Scafidi, S. , Ritter, J. , Jablonska, B. , Roncal, M. , … Gallo, V. (2014). Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature, 506(7487), 230–234. 10.1038/nature12880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt‐Kastner, R. , Wietasch, K. , Weigel, H. , & Eysel, U. T. (1993). Immunohistochemical staining for glial fibrillary acidic protein (GFAP) after deafferentation or ischemic infarction in rat visual system: Features of reactive and damaged astrocytes. International Journal of Developmental Neuroscience, 11(2), 157–174. 10.1016/0736-5748(93)90076-P [DOI] [PubMed] [Google Scholar]
- Schmitz, T. , Ritter, J. , Mueller, S. , Felderhoff‐Mueser, U. , Chew, L.‐J. , & Gallo, V. (2011). Cellular changes underlying Hyperoxia‐induced delay of white matter development. The Journal of Neuroscience, 31(11), 4327 10.1523/JNEUROSCI.3942-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, C. A. , Rasband, W. S. , & Eliceiri, K. W. (2012). NIH image to ImageJ: 25 years of image analysis. Nature Methods, 9(7), 671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple, B. D. , Blomgren, K. , Gimlin, K. , Ferriero, D. M. , & Noble‐Haeusslein, L. J. (2013). Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Progress in Neurobiology, 106‐107, 1–16. 10.1016/j.pneurobio.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y. , Plane, J. M. , & Deng, W. (2010). Mouse models of periventricular leukomalacia. Journal of Visualized Experiments, 39(e1951), 1–4. 10.3791/1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll, B. J. , Hansen, N. I. , Bell, E. F. , Walsh, M. C. , Carlo, W. A. , Shankaran, S. , … Higgins, R. D. (2015). Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993‐2012. JAMA, 314(10), 1039–1051. 10.1001/jama.2015.10244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umekawa, T. , Osman, A. M. , Han, W. , Ikeda, T. , & Blomgren, K. (2015). Resident microglia, rather than blood‐derived macrophages, contribute to the earlier and more pronounced inflammatory reaction in the immature compared with the adult hippocampus after hypoxia‐ischemia. Glia, 63(12), 2220–2230. 10.1002/glia.22887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaes, J. E. G. , Vink, M. A. , de Theije, C. G. M. , Hoebeek, F. E. , Benders, M. , & Nijboer, C. H. A. (2019). The potential of stem cell therapy to repair white matter injury in preterm infants: Lessons learned from experimental models. Frontiers in Physiology, 10, 540 10.3389/fphys.2019.00540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilborg, E. , Achterberg, E. J. M. , van Kammen, C. M. , van der Toorn, A. , Groenendaal, F. , Dijkhuizen, R. M. , … Nijboer, C. H. A. (2018). Combined fetal inflammation and postnatal hypoxia causes myelin deficits and autism‐like behavior in a rat model of diffuse white matter injury. Glia, 66(1), 78–93. 10.1002/glia.23216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilborg, E. , de Theije, C. G. M. , van Hal, M. , Wagenaar, N. , de Vries, L. S. , Benders, M. J. , … Nijboer, C. H. (2018). Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia, 66(2), 221–238. 10.1002/glia.23256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilborg, E. , Heijnen, C. J. , Benders, M. J. , van Bel, F. , Fleiss, B. , Gressens, P. , & Nijboer, C. H. (2016). Impaired oligodendrocyte maturation in preterm infants: Potential therapeutic targets. Progress in Neurobiology, 136(1873–5118) [Electronic], 28–49. 10.1016/j.pneurobio.2015.11.002 [DOI] [PubMed] [Google Scholar]
- van Tilborg, E. , van Kammen, C. M. , de Theije, C. G. M. , van Meer, M. P. A. , Dijkhuizen, R. M. , & Nijboer, C. H. (2017). A quantitative method for microstructural analysis of myelinated axons in the injured rodent brain. Scientific Reports, 7(1), 16492–16492. 10.1038/s41598-017-16797-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velthoven, C. T. , Kavelaars, A. , van Bel, F. , & Heijnen, C. J. (2010a). Mesenchymal stem cell treatment after neonatal hypoxic‐ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain, Behavior, and Immunity, 24(3), 387–393. 10.1016/j.bbi.2009.10.017 [DOI] [PubMed] [Google Scholar]
- van Velthoven, C. T. , Kavelaars, A. , van Bel, F. , & Heijnen, C. J. (2010b). Nasal administration of stem cells: A promising novel route to treat neonatal ischemic brain damage. Pediatric Research, 68(5), 419–422. 10.1203/PDR.0b013e3181f1c289 [DOI] [PubMed] [Google Scholar]
- van Velthoven, C. T. , Kavelaars, A. , van Bel, F. , & Heijnen, C. J. (2011). Mesenchymal stem cell transplantation changes the gene expression profile of the neonatal ischemic brain. Brain, Behavoir and Immunity, 25(7), 1342–1348. 10.1016/j.bbi.2011.03.021 [DOI] [PubMed] [Google Scholar]
- Verney, C. , Pogledic, I. , Biran, V. , Adle‐Biassette, H. , Fallet‐Bianco, C. , & Gressens, P. (2012). Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. Journal of Neuropathology and Experimental Neurology, 71(3), 251–264. 10.1097/NEN.0b013e3182496429 [DOI] [PubMed] [Google Scholar]
- Viganò, F. , Schneider, S. , Cimino, M. , Bonfanti, E. , Gelosa, P. , Sironi, L. , … Dimou, L. (2016). GPR17 expressing NG2‐glia: Oligodendrocyte progenitors serving as a reserve pool after injury. Glia, 64(2), 287–299. 10.1002/glia.22929 [DOI] [PubMed] [Google Scholar]
- Vincze, A. , Mázló, M. , Seress, L. , Komoly, S. , & Ábrahám, H. (2008). A correlative light and electron microscopic study of postnatal myelination in the murine corpus callosum. International Journal of Developmental Neuroscience, 26(6), 575–584. 10.1016/j.ijdevneu.2008.05.003 [DOI] [PubMed] [Google Scholar]
- Volkman, R. , & Offen, D. (2017). Concise review: Mesenchymal stem cells in neurodegenerative diseases. Stem Cells, 35(8), 1867–1880. 10.1002/stem.2651 [DOI] [PubMed] [Google Scholar]
- Volpe, J. J. (2009a). Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurology, 8(1), 110–124. 10.1016/s1474-4422(08)70294-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe, J. J. (2009b). The encephalopathy of prematurity–brain injury and impaired brain development inextricably intertwined. Seminars in Pediatric Neurology, 16(4), 167–178. 10.1016/j.spen.2009.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe, J. J. (2017). Confusions in nomenclature: "periventricular leukomalacia" and "white matter injury"‐identical, distinct, or overlapping? Pediatric Neurology, 73, 3–6. 10.1016/j.pediatrneurol.2017.05.013 [DOI] [PubMed] [Google Scholar]
- Volpe, J. J. , Kinney, H. C. , Jensen, F. E. , & Rosenberg, P. A. (2011). The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. International Journal of Developmental Neuroscience, 29(4), 423–440. 10.1016/j.ijdevneu.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar, N. , de Theije, C. G. M. , de Vries, L. S. , Groenendaal, F. , Benders, M. , & Nijboer, C. H. A. (2018). Promoting neuroregeneration after perinatal arterial ischemic stroke: Neurotrophic factors and mesenchymal stem cells. Pediatric Research, 83(1–2), 372–384. 10.1038/pr.2017.243 [DOI] [PubMed] [Google Scholar]
- Wang, L. Q. , Lin, Z. Z. , Zhang, H. X. , Shao, B. , Xiao, L. , Jiang, H. G. , … Jin, K. L. (2014). Timing and dose regimens of marrow mesenchymal stem cell transplantation affect the outcomes and neuroinflammatory response after ischemic stroke. CNS Neuroscience & Therapeutics, 20(4), 317–326. 10.1111/cns.12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil, M. T. , Ruhwedel, T. , Meschkat, M. , Sadowski, B. , & Möbius, W. (2019). Transmission electron microscopy of oligodendrocytes and myelin. Methods in Molecular Biology, 1936, 343–375. 10.1007/978-1-4939-9072-6_20 [DOI] [PubMed] [Google Scholar]
- Wilson, H. C. , Onischke, C. , & Raine, C. S. (2003). Human oligodendrocyte precursor cells in vitro: Phenotypic analysis and differential response to growth factors. Glia, 44(2), 153–165. 10.1002/glia.10280 [DOI] [PubMed] [Google Scholar]
- Xing, Y. L. , Röth, P. T. , Stratton, J. A. , Chuang, B. H. , Danne, J. , Ellis, S. L. , … Merson, T. D. (2014). Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. Journal of Neuroscience, 34(42), 14128–14146. 10.1523/jneurosci.3491-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanier, E. R. , Fumagalli, S. , Perego, C. , Pischiutta, F. , & De Simoni, M.‐G. (2015). Shape descriptors of the "never resting" microglia in three different acute brain injury models in mice. Intensive Care Medicine Experimental, 3(1), 39–39. 10.1186/s40635-015-0039-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , Chen, Y. , Xu, Y. , & Pi, G. (2013). Effect of intrauterine infection on brain development and injury. International Journal of Developmental Neuroscience, 31(7), 543–549. 10.1016/j.ijdevneu.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Zonouzi, M. , Scafidi, J. , Li, P. , McEllin, B. , Edwards, J. , Dupree, J. L. , … Gallo, V. (2015). GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nature Neuroscience, 18(5), 674–682. 10.1038/nn.3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 AuNP‐MSN nanoparticle characterization and rMSC nanoparticle uptake analysis. A)Transmission electron microscopy (TEM) image of AuNP (left) and AuNP‐MSN (right)_ Scale bar is 200 nm_ B) Size analysis of AuNP (black) and AuNP‐MSN (red) by dynamic light scattering (DLS)_ C) UV–Vis spectroscopy of AuNP (black) and AuNP‐MSN (red) showing a slight shift in absorbance after silica coating. D) Size analysis of 20 particles using TEM images by imageJ; AuNP (left) and AuNP‐MSN (right). E) internalization of AuNP‐MSN‐LIP nanoparticles in rMSCs after 2, 6, 24 and 48 hours of incubation, analysed by flow cytometry. F) Internalization of AuNP‐MSN‐LIP after 48 h incubation with rMSCs and concentration to 62.5x108 clls/mL.
Figure S2 MSC‐secreted factors boost myelin production in a dose‐dependent manner in primary OL cultures. A. Exposure to 10 ng/mL TNFα does not lead to OL death indicated by a similar amount Olig2+ cells in medium with TNFα+ compared with medium without TNFα (n = 3 independent experiments, 3–4 observations per experiment; normalized for the positive control, for example, cells exposed to TNFα). B‐I. Addition of IGF1 (B), IL11 (C), CXCL12 (D), EGF (E),LIF (G), IL10 (H) and Noggin (I), but not GCSF (F), significantly improved myelin production by primary cultured oligodendrocytes in an dose‐dependent manner following MCM + LPS exposure (dashed line represents MBP+ area in MCM‐LPS control condition) (n = 2 independent experiments, 2–3 observations per experiment, normalized for the positive control, for example, cells exposed to MCM + LPS without addition of a factor). ####: p < 0.0001 MCM+ (black bars) vs. MCM‐ (dashed line) respectively; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001 factor‐exposed MCM+ condition (gray bars) vs. MCM+ control condition (black bars).
Table S3 Gene expression profile of MSCs after culture with SHAM or dWMI brain extracts
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.