

Abstract
Medicinal‐chemistry optimization follows strategies replacing functional groups and attaching larger substituents at a promising lead scaffold. Well‐established bioisosterism rules are considered, however, it is difficult to estimate whether the introduced modifications really match the required properties at a binding site. The electron density distribution and pK a values are modulated influencing protonation states and bioavailability. Considering the adjacent H‐bond donor/acceptor pattern of the hinge binding motif in a kinase, we studied by crystallography a set of fragments to map the required interaction pattern. Unexpectedly, benzoic acid and benzamidine, decorated with the correct substituents, are totally bioisosteric just as carboxamide and phenolic OH. A mono‐dentate pyridine nitrogen out‐performs bi‐dentate functionalities. The importance of correctly designing pK a values of attached functional groups by additional substituents at the parent scaffold is rendered prominent.
Keywords: bioisosterism, drug design, fragments, pKa shift, protein kinase A
Designed with the correct pK a value and appropriate electron withdrawing and pushing groups, benzoic acid and benzamidine are bioisosteric with respect to the binding to the hinge region of a kinase.
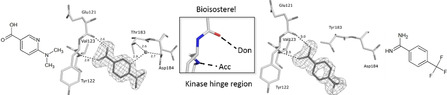
Lead optimization in medicinal chemistry usually follows a strategy modifying a promising lead scaffold by replacing functional groups and attaching larger substituents and moieties, which reveal the same biological response, according to well‐established bioisosterism rules.[ 1 , 2 , 3 , 4 , 5 ] Nowadays mostly reference is taken to structural data of the target protein and decorations of the parent scaffold are guided by molecular design regarding steric and interaction complementarity with the targeted protein subsite. Again, this step is inspired by bioisosterism concepts and supported by molecular modeling software which retrieves putative occupants binding to other proteins with similar pockets.[ 6 , 7 ] The steadily increasing plethora of crystal structures deposited in the publically available pdb databank endows such strategies with the required power. [8]
It is however, extremely difficult to estimate whether the selected functional group replacements or the scaffold‐expansions have the ideally suited properties for the given binding site. This decision concerns properties, such as the correct placement of a charged or strongly polarized group influencing the overall distribution of electron density across the attached moiety or the adjustment of an optimal pK a value affecting protonation and bioavailability.[ 9 , 10 , 11 , 12 ] Furthermore, the parent scaffold, to which the bioisosteric replacement is chemically connected, will also impose constraints depending on the chemical connectivity of the molecule. These constraints can be of steric or electronic nature. The latter will reshuffle the electron density distribution in the molecule depending on the selected attachment chemistry. In consequence, pK a values or the placement and directionality of interactions can be altered. [13] Therefore, it is hardly possible to predict whether, e.g., the required pK a properties of the replacing functional group will optimally match the interactions required by the protein's counter group. Likely, the protein environment will induce effects that re‐distribute the electron density and polarize the ligand. [14] As a result, medicinal chemists often have to realize that the planned bioisosteric replacement did not achieve the expected potency increase in the newly designed and laboriously synthesized target molecules.
Here, the following information would be highly inspiring: How would the group, planned for the replacement, bind to the protein if it was freed from all spatial and electronic constraints imposed by the attachment to the parent scaffold? Then, the optimal interaction pattern with the protein environment would be disclosed. One way could be, the use of sophisticated model calculations to answer this question. However, since reshuffling of electron density, polarization effects and in consequence modulations of protonation states are involved, only high‐level quantum chemical calculations will provide meaningful answers.[ 15 , 16 , 17 , 18 , 19 ] These are presently still far from routine and require long‐standing experience in setting up such calculations to generate relevant information. In addition, they still need enormous computational resources. Thus as an alternative, at least as long as such tools are desperately missing, reference could be taken to experimental data.
We recently learnt from neutron diffraction also providing reliable information about hydrogens that the amino group of aniline becomes protonated once bound to the S1 pocket of the serine protease trypsin. This pocket hosts at its far end the negatively charged carboxylate group of a deeply buried aspartate. Formally, the protonation of aniline corresponds to a shift of four orders of magnitude in the pK a value of aniline (pK a=4.6) in water considering the pH 7.5 applied in the crystallographic experiment. Surprisingly, the mutual distance between the fragment's ammonium group and the protein's carboxylate group amounts to 4 Å. [20]
Since this example shows that long‐distance polarization effects are difficult to predict and computational models need experimental information to embark on tool developments, we decided to follow an experimental approach to collect information about the modulation of binding properties of functional groups expected to exhibit bioisosteric properties. We wanted to see how such groups find their way to bind to a protein site lacking any spatial constraint imposed by the attachment to the parent scaffold of a larger ligand. For our study, we selected the hinge region of a kinase and investigated a series of small molecule probes, also named “fragments”, [21] with respect to their adopted binding poses next to the kinase hinge motif. We considered particularly pK a properties and modulation of the electron density distribution across the parent scaffold in dependence of the attached substituents.
A broad range of chemical motifs have been discovered over the last 30 years to complement the recognition pattern provided by the amino acids of the kinase hinge region.[ 22 , 23 ] Mono, bi and tridentate interaction motifs have been described to interact with the peptide stretch of the hinge. [24] Observed differences have been exploited to gain not only affinity but in particular selectivity for individual members of the kinome.[ 25 , 26 , 27 ]
We selected the well‐crystallizing and comprehensively studied cAMP‐dependent protein kinase (PKA). [28] It recognizes the adenine portion of the natural substrate ATP and uses adjacent donor and acceptor functionalities of the accessible hinge peptide bonds to hydrogen‐bond the cofactors. [29] Thus, an ideal motif to complement the given donor/acceptor pattern would be a carboxamide group of a ligand. We therefore started with benzamide as probe fragment. The adopted binding mode shows the expected dual ladder donor/acceptor H‐bonding pattern with the hinge. To our surprise, however, the introduction of a pyridine‐type nitrogen in the aromatic core, changes the binding pose entirely and the fragment prefers to use its pyridine nitrogen to interact with the hinge. Attaching an amino group as electron pushing substituent reveals the former expected binding pose. But already the attachment of a hydroxyl group alters the binding geometry and, at least in part, the hydroxyl group is used to interact with the hinge.
These quite surprising results, obtained with a series of very simple ligands, stimulated us to extend our study to a variety of other functional groups including benzoic acids and benzamidines. Quite unexpectedly, a benzoic acid and a benzamidine, decorated with the correct substituents, turned out to be fully bioisosteric, provided a matching protonation situation is given.
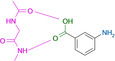
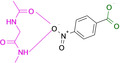
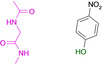
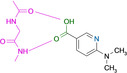
We succeeded to determine the crystal structures of the 19 fragments with a central aromatic moiety and attached substituents of deviating properties (Table 1). Their pKa values were measured in aqueous solution. All fragments bind in the active site next to the hinge region where usually the natural ligand ATP is bound. The individual binding modes of 1–19 are roughly depicted in Table 1 and displayed in Figure 1, Figure 2, and Figure 3. A detailed description of the individual binding modes with the individual interaction patterns is given in the Supporting Information along with the protocol applied to determine the pKa values.
Table 1.
Chemical formulas, schematic interaction pattern with hinge (magenta), measured pK a values in water (assignment color‐coded), pdb‐codes and crystallographic resolution of the fragments studied in this contribution.
|
No. |
Chemical formula (hinge to the left, magenta) |
pK a values |
pdb code |
Resolution [Å] |
|---|---|---|---|---|
|
1 |
|
<2 >12 |
6SNX |
1.40 |
|
|
|
|
|
|
|
2 |
|
<2 3.37 >12 |
5N3H |
1.36 |
|
|
|
|
|
|
|
3 |
|
<2 3.68 >12 |
5N3Q |
1.31 |
|
|
|
|
|
|
|
4 |
|
<2 8.36 >12 |
5N3S[a] |
1.14 |
|
|
|
|
|
|
|
5 |
|
9.99 [b] |
6Z44 |
1.38 |
|
|
|
|
|
|
|
6 |
|
1.33 10.43 |
5N33 |
1.43 |
|
|
|
|
|
|
|
7 |
|
<2 >12 |
6SPS |
1.65 |
|
|
|
|
|
|
|
8 |
|
<2 3.74 >12 |
6SPY |
1.60 |
|
|
|
|
|
|
|
9 |
|
<2 3.53 >12 |
6SOX |
1.38 |
|
|
|
|
|
|
|
10 |
|
4.01 |
6SNN |
1.82 |
|
|
|
|
|
|
|
11 |
|
3.48 |
6SPM |
1.37 |
|
|
|
|
|
|
|
12 |
|
3.17 4.56 |
6SPU |
1.39 |
|
|
|
|
|
|
|
13 |
|
3.20 |
5N3J |
1.12 |
|
|
|
|
|
|
|
14 |
|
6.91 |
6Z08 |
1.49 |
|
|
|
|
|
|
|
15 |
|
3.05 6.17 |
5N3E |
1.53 |
|
|
|
|
|
|
|
16 |
|
10.78 |
5N3D |
1.77 |
|
|
|
|
|
|
|
17 |
|
7.75 >12 |
6YPS |
1.35 |
|
|
|
|
|
|
|
18 |
|
11.32 |
5N3C |
1.77 |
|
|
|
|
|
|
|
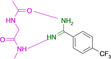
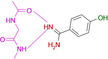
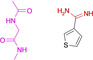
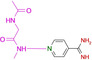
19 |
|
2.23 10.12 |
6ZN0 |
1.59 |
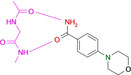
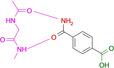
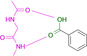
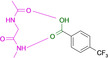
[a] second orientation with carboxamide to the hinge; [b] value taken from CRC Handbook of Chemistry and Physics. [30]
Figure 1.
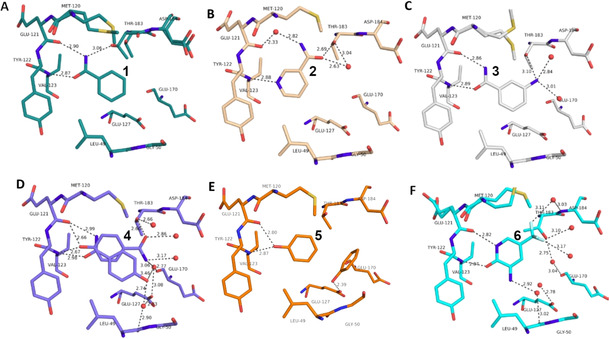
Overview of different ligands binding to the hinge region of PKA. A: Benzamide 1 (pdb‐code: 6SNX), B: Pyridine‐3‐carboxamide 2 (5N3H), C: 3‐Aminobenzamide 3 (5N3Q), D: 4‐Hydroxybenzamide 4 (5N3S), E: Phenol 5 (6Z44), F: 3‐Amino‐5‐(trifluoromethyl)‐1H‐pyridin‐2‐one 6 (5N33). Here and in the following images, heteroatoms type coded (N: blue, O: red, F: turquoise, S: yellow, water molecules red spheres, H‐bonds as dotted lines with distances in Å. Carbon atoms colored differently for the various complexes).
Figure 2.
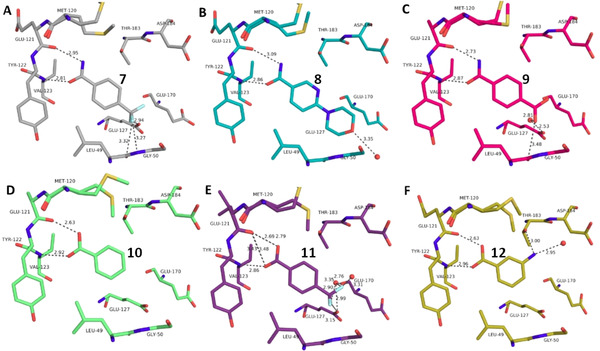
Overview of different ligands binding to the hinge region of PKA. A: 4‐(Trifluoromethyl)benzamide 7 (6SPS), B: 6‐(Morpholin‐4‐yl)pyridine‐3‐carboxamide 8 (6SPY), C: Terephtalic acid monoamide/ 4‐Carbamoylbenzoic acid 9 (6SOX), D: Benzoic acid 10 (6SNN), E: 4‐(Trifluoromethyl)benzoic acid 11 (6SPM), F: 3‐Aminobenzoic acid 12 (6SPU).
Figure 3.
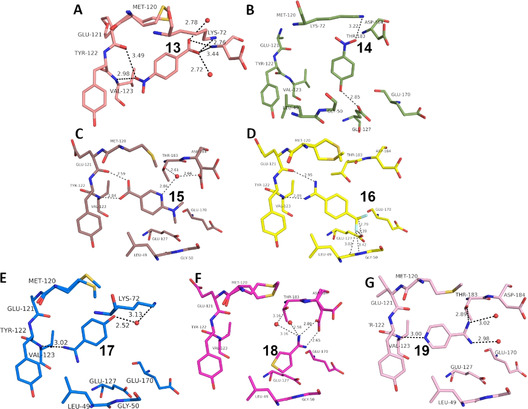
Overview of different ligands binding to the hinge region of PKA. A: 4‐Nitrobenzoic acid 13 (5N3J), B: 4‐Nitrophenol 14 (6Z08) C: 6‐Dimethylaminopyridine‐3‐carboxylic acid 15 (5N3E), D: 4‐(Trifluoromethyl)benzcarboximidamide 16 (5N3D), E: 4‐Hydroxy‐benzamidine 17 (6YPS), F: Thiophene‐3‐carboximidamide 18 (5N3C), G: Iso‐nicotinamidine 19 (6ZN0).
In kinases, the hinge region provides a typical recognition pattern that binds the natural substrate ATP. As drug development has been focused on the inhibition of the ATP binding site, putative lead candidates must incorporate functional groups that can respond to this pattern. The hinge region exposes the amide bonds of the peptide backbone to establish hydrogen bonds with a putative small molecule ligand. Thus, an alternating pattern of H‐bond donor and acceptor facilities seems on first sight ideal to respond to the features of the protein.
To map the binding properties of possible small molecule ligands with the required properties, we selected small aromatic fragments featuring carboxamide functionalities at or embedded into the considered aromatic ring system. As expected, a large portion of the tested fragments adopts exactly the estimated pattern of two nearly parallel hydrogen bonds with the hinge region (cf. fragments 1, 3, 4, 7, 8, 9, Figure 1 and Figure 2). Quite surprising is the binding pose of 2 as this fragment uses its pyridine‐type nitrogen to contact the hinge and the carboxamide group is embedded into a network of contact‐mediating water molecules. The pyridine‐type nitrogen with a pK a value of 3.37 in aqueous solution is obviously the better acceptor of an H‐bond provided by the hinge. Unlikely, the water‐embedded carboxamide is the determining factor leading to the altered binding pose.
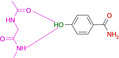
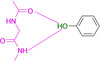
A second surprise is fragment 4 which adopts two orientations in the crystal structure, either the expected interaction to the hinge via its carboxamide group, but surprisingly also via its hydroxyl function located at the opposing end of the fragment. The OH group (pK a=8.36) seems capable to serve as both, either as H‐bond donor and acceptor to the hinge. This is confirmed by the binding mode of phenol 5 (Figure 1 E). Even the embedding of the carboxamide functionality into the six‐membered ring of 6 allows the usual bidentate addressing of the hinge. However, it requires the tautomer with broken aromaticity. Here, the pyridine‐type nitrogen (pK a=1.33) becomes protonated and serves as H‐bond donor for the hinge carbonyl oxygen.
Interestingly enough, also benzoic acid derivatives can serve as binders to the hinge, provided they adopt in neutral state the required H‐bond donor/acceptor pattern (10, 11, 12, 15, Figure 2 and Figure 3). Featured with this protonation state, they establish a binding pattern very similar to the carboxamide group. The binding geometry of the parent benzoic acid 10 (Figure 2 D) and benzamide 1 (Figure 1 A) are virtually identical. In aqueous solution, the pK a values of the acids tested here fall into a range between 3–4, making the neutral form in aqueous solution also the most likely form in the binding pocket.
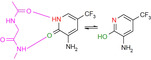
Surprisingly, p‐nitrobenzoic acid 13 (Figure 3 A) falls completely out of this pattern. Even though its pK a value is found in the same range (3.20), the fragment prefers to contact the hinge with its nitro group in mono‐dentate fashion. At first glance, this seems rather unusual, since the nitro group is known to be a worse H‐bond acceptor compared to the carboxylic acid. Nevertheless, quite a number of protein‐ligand complexes are known, where the nitro group is involved in H‐bonding. In the PDB 506 interactions of this type could be found (for details see SI), whereby one additional ligand apart from 13 binds to the hinge region of a kinase forming an H‐bond (pdb‐code: 2W7X). Here, the opposing carboxylate group of 13 is placed into the region where it can form a charge‐assisted interaction to the ammonium group of Lys72. We therefore assume deprotonation and in consequence the formation of the salt bridge to Lys72 is the driving force to shift the protonation state of this group to the deprotonated form. An energetically favored salt bridge is established. Based on this experience, we tested nitrophenol 14 (Figure 3 B). It features the nitro group but lacks an opposing functionality that can easily be deprotonated (pK a=6.91) to establish a charge‐assisted contact to Lys72. As a consequence, 14 avoids any contact to the hinge, quite different from 4 and 5. Instead, it places the nitro group next to Lys72, but thereby achieves only a very long hydrogen bond with non‐ideal geometry.
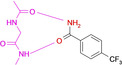
Perhaps the most surprising result is displayed by the binding mode of the benzamidine derivative 16 (Figure 3 D). Aromatic amidines are the typical groups used to interact with Asp or Glu in proteins, due to their high pK a values, well beyond 12. However, featured with electron withdrawing groups at the aromatic portion, these ligands can show lower pK a values, here of 10.78. This allows 16 to bind in neutral state at the amidino function and then it contacts the hinge with the required H‐bond donor/acceptor pattern. In this state, the benzamidine fragment adopts identical binding mode as the benzoic acid derivatives (e.g. 10–12, 15), a more than surprising and definitely unexpected bioisosteric replacement! An analysis of all available PDB structures showed that each of the amidino groups attached to an aromatic ring system served as a hydrogen‐bond donor while interacting with a protein (for details see SI).
This observation stimulated us to study additional aromatic amidines. Fragments 17 and 18 exhibit higher pK a values for the amidino function. 17 (Figure 3 E) contacts the hinge in mono‐dentate fashion, very similar to the p‐nitrobenzoic acid 13. Fragment 18 avoids entirely to interact with the hinge. Instead it recruits the carboxylate of Asp184 to form a salt bridge. We assume that this displays the driving force for the formation of the observed binding pose.
An interesting case is again fragment 19 as it features an amidino group, which is even less basic than the one present in 16 (pK a (19)=10.12; pK a (16)=10.78). Nevertheless, it does not contact the hinge via an H‐bond donor/acceptor pattern of the amidino group like 16, instead it uses its pyridine‐type nitrogen (pK a=2.23) to accept an H‐bond from the hinge, similarly to fragment 2. The actual protonation state of the amidino group is difficult to estimate. However, as the group forms only three rather long H‐bonds, the uncharged state appears likely. As for 2, the determining feature for the adopted binding pose of 19 is, supposedly, the interaction formed by the pyridine‐type nitrogen. In contrast, fragment 16, exhibiting a similar pK a value, features only the amidino group to establish H‐bonds. The preference of 19 for the pyridine‐type nitrogen to interact with the hinge matches with the situation found for 2. Interestingly enough, several drug developments resulted in the usage of a pyridine‐type nitrogen as sole group to accept a hydrogen bond from the hinge (e.g. Lapatinib, Gefitinib, Erlotinib, Vandetinib, Tanduinib, SB202190, SB203580).[ 31 , 32 ] Possibly, instead of crowded decoration with adjacent ligand functional groups to interact with the hinge, a single contact is favorable. In all cases, it should not be forgotten that a bidentate H‐bonding pattern with donor and acceptor functionalities on opposite sides of the interacting molecules always suffers from repulsive secondary interactions of the aligned hydrogen bonds. This feature has first been discussed by Zimmerman et al. [33] in the field of host‐guest chemistry. It matters, however, also in protein‐ligand interactions as we could show some time ago for ligand binding to a tRNA‐modifying enzyme. [34]
In conclusion, the current study tries to map the features of the ATP‐binding pocket of the well‐known kinase PKA by studying a series of fragments interacting with or close to the hinge region of the enzyme. The backbone stretch of the hinge provides a pattern of H‐bond donor and acceptor functionalities that can be addressed by a putative ligand. Thus, ligands exhibiting a feature of close‐by acceptor and donor groups, capable to respond to the H‐bonding pattern of the protein, appear ideal on first sight. Functional groups such as a carboxamide, carboxylic acid but even an amidino group, provided being present in uncharged state, satisfy the required pattern. The remaining molecule has only to be decorated with the required electron pushing or withdrawing groups to guarantee the necessary protonation state. However, such design requires neat control of the pK a properties of a molecule. Due to polarization effects in the binding pocket, pK a values, defined in aqueous solution, can be easily shifted by several orders of magnitude (cf. trypsin‐aniline case referred above [20] ). This makes such predictions extremely difficult. Furthermore, we observed that some surprising binding modes result from additional features given in a binding pocket and the functional groups assumed to be similar in terms of pK a values prefer to adopt deviating protonation states in a different local area of the pocket (e.g. 13, 18, 19). Particularly, with respect to the presence of a pyridine‐type nitrogen in a ligand surprising changes in the binding poses were observed. Possibly, the preference for a single hydrogen‐bond contact to the hinge established by the pyridine‐type nitrogen can be explained by the avoidance of repulsive secondary interactions, which automatically occur if bidentate hydrogen bonds are formed with a mutually opposing donor/acceptor pattern on the binding partners.
Since the correct prediction of protonation states, protein‐induced pK a shifts due to polarization effects at a binding site and the impact of secondary repulsive interactions are difficult to model, even by computational means, the alternative approach to map binding‐site features by multiple fragment crystal structures appears quite promising. With the enhancements of fast data collection at current synchrotrons, high‐throughput crystal structure analyses are easily feasible. The mapping by fragments has the important advantage that small molecular probes usually assemble at the hot spots of interaction and give insights how binding of a particular functional group occurs once released from the spatial constraints imposed by a larger embedding molecular scaffold given in a drug‐size molecule.
Accession Codes: PKA⋅Ligand complexes (ID, pdb code): Benzamide (1, 6SNX), Pyridine‐3‐carboxamide (2, 5N3 H), 3‐Aminobenzamid (3, 5N3Q), 4‐Hydroxy‐benzamide (4, 5N3S), Phenol (5, 6Z44), 3‐Amino‐5‐(trifluoromethyl)‐1H‐pyridin‐2‐one (6, 5N33), 4‐(Trifluoromethyl) benzamide (7, 6SPS), 6‐(Morpholin‐4‐yl) pyridine‐3‐carboxamide (8, 6SPY), 4‐Carbamoylbenzoic acid (9, 6SOX), Benzoic acid (10, 6SNN), 4‐(Trifluoromethyl) benzoic acid (11, 6SPM), 3‐Aminobenzoic acid (12, 6SPU), 4‐Nitrobenzoic acid (13, 5N3J), 4‐Nitrophenol (14, 6Z08), 6‐Dimethylaminopyridine‐3‐carboxylic acid (15, 5N3E), 4‐(Trifluoromethyl) benzcarboximidamide (16, 5N3D), 4‐Hydroxybenzamidine (17, 6YPS), Thiophene‐3‐carboximidamide (18, 5N3C), Iso‐nicotinamidine (19, 6ZN0).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would particularly like to acknowledge the help and support of the beamline scientists during diffraction data collection at BESSY II (HZB, Berlin, Germany), DESY, P11 (Hamburg, Germany) and ESRF, ID29 (ESRF, Grenoble, France). We would like to thank the HZB for financial support of travel costs. Open access funding enabled and organized by Projekt DEAL.
M. Oebbeke, C. Siefker, B. Wagner, A. Heine, G. Klebe, Angew. Chem. Int. Ed. 2021, 60, 252.
References
- 1. Keserű G. M., Makara G. M., Drug Discovery Today 2006, 11, 741–748. [DOI] [PubMed] [Google Scholar]
- 2. Patani G. A., LaVoie E. J., Chem. Rev. 1996, 96, 3147–3176. [DOI] [PubMed] [Google Scholar]
- 3. Lima L. M., Barreiro E. J., Curr. Med. Chem. 2005, 12, 23–49. [DOI] [PubMed] [Google Scholar]
- 4. Burger A., Prog. Drug Res. 1991, 37, 287–371. [DOI] [PubMed] [Google Scholar]
- 5. Olesen P. H., Curr. Opin. Drug Discovery Dev. 2001, 4, 471–478. [PubMed] [Google Scholar]
- 6. Ferreira L. G., Dos Santos R. N., Oliva G., Andricopulo A. D., Molecules 2015, 20, 13384–13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou Z., Felts A. K., Friesner R. A., Levy R. M., J. Chem. Inf. Model. 2007, 47, 1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burley S. K., Berman H. M., Christie C., Duarte J. M., Feng Z., Westbrook J., Young J., Zardecki C., Protein Sci. 2018, 27, 316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garton N. S., Barker M. D., Davis R. P., Douault C., Hooper-Greenhill E., Jones E., Lewis H. D., Liddle J., Lugo D., McCleary S., et al., Bioorg. Med. Chem. Lett. 2016, 26, 4606–4612. [DOI] [PubMed] [Google Scholar]
- 10. Waring M. J., Arrowsmith J., Leach A. R., Leeson P. D., Mandrell S., Owen R. M., Pairaudeau G., Pennie W. D., Pickett S. D., Wang J., et al., Nat. Rev. Drug Discovery 2015, 14, 475–486. [DOI] [PubMed] [Google Scholar]
- 11. Pennington L. D., Aquila B. M., Choi Y., Valiulin R. A., Muegge I., J. Med. Chem. 2020, 63, 8956—8976. [DOI] [PubMed] [Google Scholar]
- 12. Erlanson D. A., de Esch I. J. P., Jahnke W., Johnson C. N., Mortenson P. N., J. Med. Chem. 2020, 63, 4430–4444. [DOI] [PubMed] [Google Scholar]
- 13. Grunewald G. L., Seim M. R., Lu J., Makboul M., Criscione K. R., J. Med. Chem. 2006, 49, 2939–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brandsdal B. O., Smalås A. O., Aqvist J., Proteins Struct. Funct. Bioinf. 2006, 64, 740–748. [DOI] [PubMed] [Google Scholar]
- 15. Lin Y.-l., Lim C., J. Am. Chem. Soc. 2004, 126, 2602–2612. [DOI] [PubMed] [Google Scholar]
- 16. Gaspari R., Rechlin C., Heine A., Bottegoni G., Rocchia W., Schwarz D., Bomke J., Gerber H.-D., Klebe G., Cavalli A., J. Med. Chem. 2016, 59, 4245–4256. [DOI] [PubMed] [Google Scholar]
- 17. Phipps M. J. S., Fox T., Tautermann C. S., Skylaris C.-K., J. Chem. Theory Comput. 2017, 13, 1837–1850. [DOI] [PubMed] [Google Scholar]
- 18. Cavasotto C. N., Methods Mol. Biol. 2020, 2114, 257–268. [DOI] [PubMed] [Google Scholar]
- 19. Cavasotto C. N., Adler N. S., Aucar M. G., Front. Chem. 2018, 6, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schiebel J., Gaspari R., Sandner A., Ngo K., Gerber H.-D., Cavalli A., Ostermann A., Heine A., Klebe G., Angew. Chem. Int. Ed. 2017, 56, 4887–4890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4965–4969. [Google Scholar]
- 21. Erlanson D. A., Curr. Opin. Biotechnol. 2006, 17, 643–652. [DOI] [PubMed] [Google Scholar]
- 22. Urich R., Wishart G., Kiczun M., Richters A., Tidten-Luksch N., Rauh D., Sherborne B., Wyatt P. G., Brenk R., ACS Chem. Biol. 2013, 8, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu Y., Kunimoto R., Bajorath J., Chem. Biol. Drug Des. 2017, 89, 834–845. [DOI] [PubMed] [Google Scholar]
- 24. Zhang J., Yang P. L., Gray N. S., Nat. Rev. Cancer 2009, 9, 28–39. [DOI] [PubMed] [Google Scholar]
- 25. Mukherjee P., Bentzien J., Bosanac T., Mao W., Burke M., Muegge I., J. Chem. Inf. Model. 2017, 57, 2152–2160. [DOI] [PubMed] [Google Scholar]
- 26. Fang Z., Grütter C., Rauh D., ACS Chem. Biol. 2013, 8, 58–70. [DOI] [PubMed] [Google Scholar]
- 27. Turnham R. E., Scott J. D., Gene 2016, 577, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taylor S. S., Zhang P., Steichen J. M., Keshwani M. M., Kornev A. P., Biochim. Biophys. Acta Proteins Proteomics 2013, 1834, 1271–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Czodrowski P., Hölzemann G., Barnickel G., Greiner H., Musil D., J. Med. Chem. 2015, 58, 457–465. [DOI] [PubMed] [Google Scholar]
- 30. CRC Handbook Chem. Physics (Ed.: Lide D. R.), CRC Press, Boca Raton, 2004. [Google Scholar]
- 31. Fabian M. A., Biggs W. H., Treiber D. K., Atteridge C. E., Azimioara M. D., Benedetti M. G., Carter T. A., Ciceri P., Edeen P. T., Floyd M., et al., Nat. Biotechnol. 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- 32. Klebl B. M., Müller G., Exp. Opin. Therapeut. Targets 2005, 9, 975–993. [DOI] [PubMed] [Google Scholar]
- 33. Murray T. J., Zimmerman S. C., J. Am. Chem. Soc. 1992, 114, 4010–4011. [Google Scholar]
- 34. Neeb M., Czodrowski P., Heine A., Barandun L. J., Hohn C., Diederich F., Klebe G., J. Med. Chem. 2014, 57, 5554–5565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary