

Abstract
Background and purpose
Rhabdomyolysis is a medical emergency characterized by acute skeletal muscle breakdown with a sudden rise and subsequent fall of serum creatine kinase (CK) levels. Rhabdomyolysis events are provoked by exposure to external triggers, possibly in combination with an increased genetic susceptibility. We aimed to describe comprehensively the external triggers and potentially pathogenic genetic variants possibly implicated in increased rhabdomyolysis susceptibility.
Methods
We performed a retrospective single‐center study, including a total of 1302 patients with an acute CK level exceeding 2000 IU/l.
Results
Anoxia was the most frequently reported trigger (40%). A subset of 193 patients were clinically suspected of an underlying genetic disorder (recurrent episodes, a positive family history, very high or persistently increased CK levels). In 72 of these patients, an unequivocal genetic defect was identified. A total of 22 genes with pathogenic variants were identified, including 52 different variants. Of those, 11 genes have been previously associated with rhabdomyolysis (ACADVL, ANO5, CPT2, DMD, DYSF, FKRP, HADHA, PGM1, LPIN1, PYGM, RYR1). Eleven genes are probably implicated in increased susceptibility (including AGL, CAPN3, CNBP, DMPK, MAGT1, ACADM, SCN4A, SGCA, SGCG, SMPD1, TANGO2).
Conclusion
These findings suggest that the spectrum of genetic susceptibility for rhabdomyolysis has not yet been completely clarified. With the increasing availability of next‐generation sequencing in a diagnostic setting, we expect that in more cases a genetic defect will be identified.
Keywords: genetic susceptibility, hyperCKaemia, next‐generation sequencing, rhabdomyolysis
Rhabdomyolysis events are provoked by exposure to external triggers, possibly in combination with an increased genetic susceptibility. Anoxia was the most frequently reported external trigger (40%). A subset of 193 patients (14.8%) were clinically suspected of an underlying genetic disorder. A total of 22 genes with pathogenic variants were identified, of which 11 have been previously associated with rhabdomyolysis (ACADVL, ANO5, CPT2, DMD, DYSF, FKRP, HADHA, PGM1, LPIN1, PYGM, RYR1). Eleven genes are probably implicated in increased susceptibility (including AGL, CAPN3, CNBP, DMPK, MAGT1, ACADM, SCN4A, SGCA, SGCG, SMPD1, TANGO2).
Introduction
Rhabdomyolysis is a complex condition relevant to many medical disciplines, involving the rapid dissolution of damaged or injured skeletal muscle. This disruption of skeletal muscle integrity leads to the direct release of intracellular muscle components, including myoglobin, creatine kinase (CK), aldolase, lactate dehydrogenase and electrolytes, into the circulation and extracellular space. Clinical manifestations range from a largely asymptomatic illness with isolated serum CK level elevation (hyperCKaemia) to a life‐threatening condition with profound myoglobinuria, often leading to acute renal failure. Definitions vary, but rhabdomyolysis is most commonly defined as a clinical syndrome of severe myalgia, muscle weakness and swelling, with sudden elevation of CK levels, with or without the presence of myoglobinuria, and subsequent fall of CK levels.
Three retrospective cohort studies in a hospital setting, performed in 2005, 2009 and 2012, described the demographics and different causes of rhabdomyolysis. All three studies, performed when diagnostic genetic testing was not yet widely available, focused on external triggers as a cause for the rhabdomyolysis event and identified exogenous toxins as the most frequently reported trigger, followed by traumatic muscle injury [1, 2, 3]. However, recent reports suggest that rhabdomyolysis events can be attributed to a combination of certain environmental factors (e.g., strenuous exercise and/or febrile infection) and a predisposing genotype [4]. A combination of external triggers and genetic predisposition may thus be required for an individual to exceed the threshold for developing rhabdomyolysis (Fig. 1), and the majority of rhabdomyolysis events may therefore have to be considered multifactorial. To identify episodes of rhabdomyolysis suggestive of an underlying genetic susceptibility, we have introduced the acronym RHABDO (Box 1) [5, 6]. This acronym was developed based on an extensive literature review and has been adopted in a recent review on exertional rhabdomyolysis [7]. It aims to distinguish the patients in whom the external triggers are sufficient to explain the episode of rhabdomyolysis from those in whom the external triggers insufficiently explain the (severity of the) rhabdomyolysis event.
Figure 1.
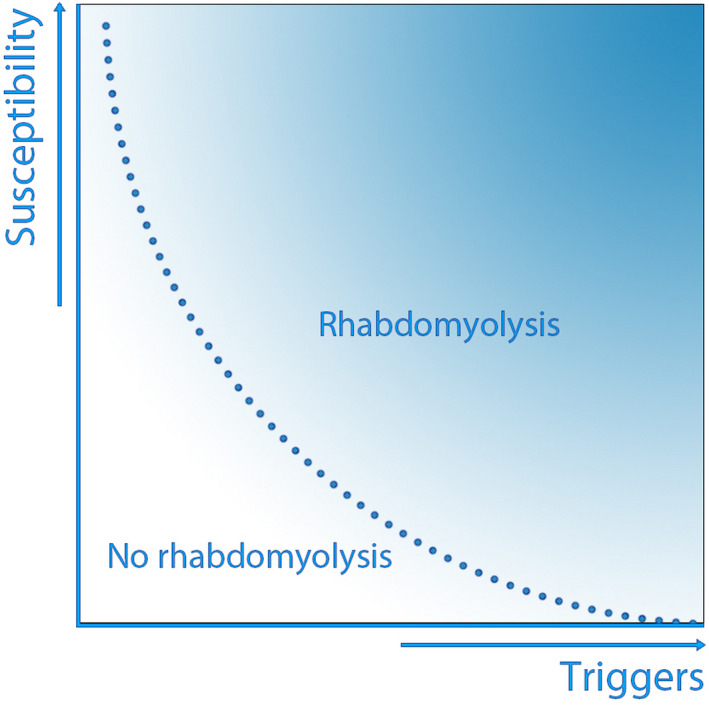
Graph showing that rhabdomyolysis events can be attributed to a combination of environmental factors (e.g., strenuous exercise and/or febrile infection) and a predisposing genotype. A combination of external triggers and genetic predisposition may thus be required for an individual to exceed the threshold for developing rhabdomyolysis. [Colour figure can be viewed at wileyonlinelibrary.com]
Box 1. A genetic defect increasing rhabdomyolysis susceptibility can be considered in cases fulfilling one or more ‘RHABDO’ criteria, described below 5 .
| R | ‐ Recurrent episodes of exertional rhabdomyolysis |
| H | ‐ HyperCKaemia persisting more than 8 weeks after event |
| A | ‐ Accustomed physical exercise |
| B | ‐ Blood creatine kinase (CK) >50 × upper limit of normal |
| D | ‐ Drug ingestion/medication/supplements or other exogenous and endogenous factors cannot sufficiently explain the rhabdomyolysis severity |
| O | ‐ Other family members affected / other exertional symptoms (e.g. cramps or myalgia) |
So far, a number of Mendelian genetic defects have been identified to increase rhabdomyolysis susceptibility, including a number of genes implicated in muscle metabolism and mitochondrial function (e.g., ACADVL, CPT2, PYGM or LPIN1), [8, 9, 10, 11] and various muscular dystrophies (e.g., Becker muscular dystrophy and limb girdle muscle dystrophy 2I [LGMD2I]) [12], or in certain congenital myopathies with specific defects in calcium homeostasis and excitation‐contraction coupling (e.g., RYR1) [13]. Such marked genetic heterogeneity poses a considerable diagnostic challenge for clinicians [5]. The awareness of specific genotype–phenotype correlations is of great importance, not only for establishing the correct genetic diagnosis, but also for effective personalized counseling. The means to detect a genetic contribution have increased markedly since the introduction of next‐generation sequencing; therefore, an observational study on genetic variants implicated in rhabdomyolysis susceptibility may be particularly useful.
In the present single‐center study we reviewed all patients with a serum CK level of ≥2000 IU/l, admitted or referred to our center between January 2014 and January 2019. First, we described the external triggers and epidemiological data in all patients. Next, those with a proven or presumed genetic susceptibility were further assessed by reviewing patient data records, resulting in an overview of all genetic variants and clinical characteristics in both subsets Box 1.
Materials and methods
Identification
A flow diagram illustrating the selection process is shown in Fig. 2. Medical records of patients admitted to the Radboud University Medical Center (Nijmegen, The Netherlands) between January 2014 and January 2019 were reviewed. Patients were selected if they (i) had the International Classification of Diseases discharge diagnosis code ‘rhabdomyolysis’ and (ii) had a serum CK value >2.000 IU/l. A recent study amongst 10 096 healthy US adults concluded that the upper limit of normal (ULN) of CK values may vary from 194 to 1001 IU/l, depending on sex, ethnicity and training levels [14]. Therefore, taking into account these baseline differences, a CK threshold of 2000 IU/l was determined, reducing the chance of including possibly healthy individuals. Lastly, patients were included if they (iii) were referred to the neurology outpatient department after an episode of rhabdomyolysis, for which they had been admitted elsewhere.
Figure 2.
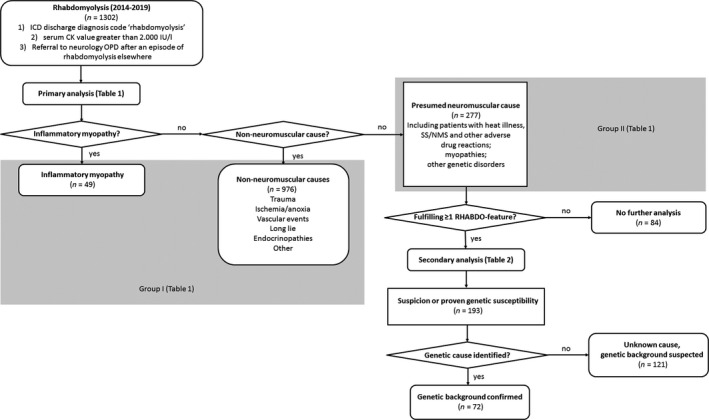
Flow diagram of the study selection process. CK, creatine kinase; ICD, International Classification of Diseases; NMS, neuroleptic malignant syndrome; OPD, outpatient department; SS, serotonin syndrome.
Triggers
First, for our primary analysis, triggers causing rhabdomyolysis events were analyzed and subdivided into two groups: (i) presumed neuromuscular causes and (ii) non‐neuromuscular causes or persisting hyperCKaemia due to muscular dystrophies. The second group was subdivided into: ischemia/anoxia (including vascular occlusion, thrombo‐embolism, shock, aortic dissections or asphyxia); traumatic muscle injury; ‘long lie’ (defined as having lain on the floor or ground for over an hour after falling) [15]; postoperative status; convulsions; endocrinopathies (including electrolyte disturbances, hypo‐ and hyperthyroidism or diabetic decompensation); acquired myopathies (including idiopathic inflammatory myopathies); ‘other, genetic’ or ‘other, acquired’.
The following patients were classified as having (presumed) neuromuscular disorders: those with muscle dystrophies, metabolic or mitochondrial myopathies, congenital myopathies or other inherited muscle diseases, those with heat‐illness (including heat cramps, heat syncope, heat exhaustion or heat stroke); and those with adverse drug reactions, subdivided into neuroleptic malignant syndrome (NMS), serotonin syndrome (SS) or ‘other adverse drug reactions’. NMS or SS was diagnosed if rhabdomyolysis was associated with exposure to medication known to cause these syndromes, and the patient had symptoms of tremor and rigidity (NMS), or if the patient fulfilled the Hunter criteria (SS) [16]. Finally, patients were classified as ‘other’ if they were diagnosed with another disease, not included in the above group (e.g. Prader‐Willi syndrome, G6PD deficiency).
Data extraction
After identifying the triggers and underlying susceptibilities, patients in the group with a (presumed) neuromuscular cause, fulfilling any of the RHABDO features (Box 1) were selected for our secondary analysis. These included patients with recurrent episodes of rhabdomyolysis, hyperCKaemia persisting 8 weeks after the event, CK levels >10 000 IU/l, or a positive family history of neuromuscular symptoms (including cramps, myalgia or proven neuromuscular diseases). The term ‘cramps’ will be used to indicate either cramps (neurogenic) or contractures (myopathic). Features ‘A’ (accustomed to exercise) and ‘D’ (drug ingestion insufficient to explain event of rhabdomyolysis) were omitted, since these data were not available through patient chart review. Information was extracted from each patient data record regarding: clinical characteristics (including sex, age, medical history, family history, a recurrent pattern of rhabdomyolysis); laboratory results (including peak CK level, hyperCKaemia lasting >8 weeks [only if CK levels were followed up]); symptoms of rhabdomyolysis (including myalgia, muscle cramp/muscle weakness/muscle stiffness, myoglobinuria or dark urine); and exposure to potentially myotoxic medication at first presentation; [17, 18] presence of specific triggers preceding the rhabdomyolysis episode or contributing to worsening of symptoms (including strenuous exercise, episodes of fever/infection, exposure to heat or cold, dehydration, illicit drug use, excessive alcohol intake (>10 units alcohol per week), long lie, prescription of potentially myotoxic medication, metabolic causes (hypo‐ or hyperthyroidism, vitamin D deficiency), seizures, or trauma, and outcome (alive or dead).
Subsequently, data were extracted regarding the extent of genetic testing performed. Whole‐exome sequencing was available in our hospital since 2013. Until then, conventional sequencing methods (e.g. Sanger sequencing) were used.
Genes in which variants were found were compared with the genes covered in a previously published review on genes associated with rhabdomyolysis [5]. For each additional potentially pathogenic variant found, a systematic search in PubMED was conducted, using the keywords ‘rhabdomyolysis’, ‘hyperCKaemia’, ‘myoglobinuria’ and the generic name of the gene potentially implicated, to identify any relevant literature. The detailed search strategy is outlined in Appendix 1.
Statistics
Categorical data were compared using the chi‐squared test or Fisher's exact test in case of fewer than five patients. Continuous variables were compared using the Mann–Whitney U‐test.
Results
Primary analysis
A total of 1302 patient records were selected (Table 1). Of these, 934 (72%) were male and 368 (28%) were female. The median (range) age was 53 (0–98) years. Death during the episode of increased CK level was reported in 289 patients (22%). The median (range) peak CK was 4423 (2008–5 013 800) IU/l. In all categories, male patients were affected more frequently than female, except for those in the long lie group (P < 0.001). Patient characteristics are described in Table 1.
Table 1.
Triggers and genetic factors underlying rhabdomyolysis in 1302 patients admitted to the Radboud University Medical Center between 2014 and 2019
| Category | Patients, n (%) (N = 1302) | Median (range) age, years | Male, n (%) | Death, n (%) | Mean (SD) CK, IU/l | Median (range) CK, IU/l |
|---|---|---|---|---|---|---|
| I: Persisting hyperCKaemia or non‐neuromuscular cause for event of rhabdomyolysis (n = 1025) | ||||||
| Ischemia/anoxia | 550 (42.2) | 53.0 (0–93) | 400 (72.7) | 167 (30.4) | 7788 (19 869) | 3820 (2017–313 250) |
| Traumatic muscle injury | 258 (19.8) | 42.5 (0–98) | 190 (73.6) | 48 (18.6) | 6576 (9729) | 3792 (2014–81 732) |
| Long lie | 67 (5.2) | 76 (23–98) | 32 (47.8) | 20 (29.8) | 9690 (18 300) | 5278 (2196–120 000) |
| Postoperative | 47 (3.6) | 59 (10–80) | 36 (76.6) | 10 (21.2) | 4255 (2948) | 3298 (2008–15 740) |
| Convulsion | 26 (2.0) | 40.5 (0–83) | 22 (84.6) | 6 (23.0) | 7525 (14 017) | 2944 (2118–73 850) |
| Endocrinopathies | 12 (0.9) | 49.5 (8–91) | 9 (75.0) | 2 (16.7) | 9234 (17 885) | 3930 (2817–65 890) |
| Idiopathic inflammatory myopathies | 49 (3.8) | 58 (8–85) | 28 (57.1) | 5 (10.2) | 7360 (5587) | 5456 (2091–27 100) |
| Other | 16 (1.2) | 64.5 (0–76) | 10 (55.5) | 7 (38.8) | 3836 (1485) | 3415 (2037–6833) |
| II: Presumed neuromuscular cause for event of rhabdomyolysis (n = 277) | ||||||
| Presumed genetic susceptibility | ||||||
| Unknown cause | 152 (11.7) | 41 (1–94) | 106 (69.7) | 17 (11.1) | 30 527 (126 140) | 12 281 (2046–1 500 000) |
| Heat illness | 13 (1.0) | 45 (23–75) | 11 (84.6) | 1 (7.7) | 7765 (7973) | 4825 (2238–24 950) |
| SS/NMS | 9 (0.7) | 32 (18–71) | 5 (55.6) | 2 (22.2) | 27 406 (32 001) | 9570 (4551–92 400) |
| Adverse drug reactions | 8 (0.6) | 41 (18–68) | 7 (87.5) | 1 (12.5) | 26 362 (63 761) | 3012 (2496–184 100) |
| Proven genetic susceptibility | ||||||
| Muscle dystrophy | 55 (4.2) | 13 (0–53) | 52 (94.5) | 2 (3.6) | 111 785 (12 643) | 5856 (2201–74 000) |
| Metabolic/ mitochondrial myopathy | 28 (2.2) | 25 (0–66) | 16 (55.1) | 1 (6.9) | 201 090 (926 615) | 9206 (2261–5 013 800) |
| RYR1 related disease | 7 (0.5) | 25 (16–43) | 6 (85.7) | 0 (0.0) | 159 794 (208 989) | 29 114 (4000–521 500) |
| Other inherited conditions | 5 (0.4) | 28 (14–50) | 4 (80.0) | 0 (0.0) | 19,993 (21,446) | 22 000 (2859–41 116) |
| Total | 1302 (100.0) | 53 (0–98) | 934 (71.7) | 289 (22.2) | 15 563 (146 332) | 4423 (2008–5 013 800) |
CK, creatine kinase; hyperCKaemia, isolated raised serum creatine kinase; NMS, neuroleptic malignant syndrome; SS, serotonin syndrome.
Other: neoplasm, acute necrotizing pancreatitis, burns patients, graft vs. host disease, transplant failure, G6PD deficiency, Prader‐Willi syndrome.
Patients are subdivided into two main categories (corresponding to I and II in Fig. 2): events of rhabdomyolysis with a non‐neuromuscular cause or persisting hyperCKemia, and rhabdomyolysis events with a presumed neuromuscular cause.
Of 1302 patients, 1025 (79%) had a non‐neuromuscular cause for the rhabdomyolysis event, 277 (21%) had a proven genetic susceptibility, or were suspected of having a neuromuscular condition at the time of the event (Table 1). Ischemia/anoxia was the most common trigger, followed by trauma; these two triggers were responsible for 42% and 20% of all cases, respectively.
Secondary analysis
Of the group of 277 patients with a (presumed) neuromuscular cause, 193 fulfilled at least one of the RHABDO features (Table 2) and were included in the secondary analysis. These involved 139 male (72%) and 54 female patients (28%) with a median (range) age of 31 (0–94) years. Recurrent episodes were present in 99 patients (63%) fulfilling at least one RHABDO feature, 45 patients (51%) had a CK level >1.5 times the ULN lasting longer than 8 weeks, 134 patients (69%) had a CK level of >10 000 IU/l and 45 patients (30%) had a family history of neuromuscular symptoms. Myoglobinuria was present in 54 patients (45%). In 88 patients (45.6%), multiple RHABDO features were present. Exercise was the most common trigger of rhabdomyolysis in 107 patients (55%), followed by 55 patients (28%) in whom one or more potentially myotoxic medication was prescribed during the episode, including statins (28 patients), opioids (25 patients), antipsychotic agents (12 patients) selective serotonin reuptake inhibitors (10 patients) and tricyclic antidepressants (four patients) [19]. In 92 patients (48%), multiple triggers could be identified. Death during the episode of rhabdomyolysis occurred in 17 patients (9%), and 20 patients were admitted to the intensive care unit (20%).
Table 2.
Characteristics of rhabdomyolysis events in patients with a genetically proven muscle disease and patients with a presumed genetic susceptibility, fulfilling at least one of the RHABDO features
| Proven genetic susceptibility | Presumed genetic susceptibility | Total | P | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (N = 72) | (N = 121) | (N = 193) | ||||||||
| Sex, n (%) | ||||||||||
| Male | 56 (77.8) | 83 (68.6) | 139 (72.0) | 0.169 | ||||||
| Female | 16 (22.2) | 38 (31.4) | 54 (28.0) | |||||||
| Age, years | ||||||||||
| Median (range) | 19.0 (0–62) | 40.0 (1–94) | 31.0 (0–94) | <0.001 | ||||||
| Mean (SD) | 22.4 (15.6) | 42.0 (19.2) | 34.7 (20.3) | |||||||
| Underlying cause, n (%) | ||||||||||
| Neuromuscular disorder | ||||||||||
| Muscular dystrophy | 36 (50.0) | ‐ | 36 (13.5) | |||||||
| Metabolic/mitochondrial | 25 (34.7) | ‐ | 25 (13.0) | |||||||
| RYR1‐related | 7 (9.7) | ‐ | 7 (3.6) | |||||||
| Other inherited | 4 (5.6) | ‐ | 4 (2.1) | |||||||
| Heat illness | ‐ | 3 (2.4) | 3 (1.6) | |||||||
| Medication | ||||||||||
| SS/NMS | ‐ | 4 (3.2) | 4 (2.1) | |||||||
| Other ADR | ‐ | 4 (3.2) | 4 (2.1) | |||||||
| Unknown | ‐ | 110 (88.0) | 110 (57.0) | |||||||
| Peak CK, IU/l | ||||||||||
| Median (range) | 10 870 (2231–5 013 800) | 17 870 (2100–1 500 000) | 16 201 (2100–5 013 800) | 0.019 | ||||||
| Mean (SD) | 104 608 (591 992) | 17 870 (140 800) | 64 346 (378 094) | |||||||
| RHABDO features, n (%) | Yes | No | NR | Yes | No | NR | Yes | No | NR | |
| Recurrent | 51 (89.5) | 6 (10.5) | 15 | 48 (47.5) | 53 (52.5) | 20 | 99 (62.7) | 59 (37.3) | 35 | <0.001 |
| HyperCKaemia > 8 weeks | 27 (87.1) | 4 (12.9) | 41 | 18 (31.0) | 40 (69.0) | 63 | 45 (50.6) | 44 (49.4) | 104 | <0.001 |
| Blood CK > 10 000 IU/l | 39 (54.2) | 33 (45.8) | ‐ | 95 (78.5) | 26 (21.5) | ‐ | 134 (69.4) | 59 (30.6) | ‐ | <0.001 |
| Other family members | 29 (47.5) | 32 (52.5) | 11 | 16 (17.6) | 75 (82.4) | 30 | 45 (29.7) | 107 (70.3) | 41 | <0.001 |
| Number RHABDO‐features, n (%) | ||||||||||
| 1 | 28 (38.9) | 77 (63.6) | 105 (54.5) | |||||||
| 2 | 22 (30.6) | 33 (27.3) | 55 (28.5) | |||||||
| 3 | 15 (20.8) | 10 (8.3) | 25 (13.0) | |||||||
| 4 | 7 (9.7) | 1 (0.8) | 8 (4.1) | |||||||
| Triggers, n (%) | ||||||||||
| Exercise | 45 (62.5) | 62 (51.2) | 107 (55.4) | 0.080 | ||||||
| Fever/ infection | 13 (18.1) | 26 (21.5) | 39 (20.2) | 0.642 | ||||||
| Heat | 0 | 13 (10.7) | 13 (6.7) | 0.005 | ||||||
| Cold | 1 (1.4) | 7 (5.8) | 8 (4.1) | 0.149 | ||||||
| Dehydration | 2 (2.8) | 5 (4.1) | 7 (3.6) | 0.655 | ||||||
| Illicit drug use and/or alcohol | 7 (9.7) | 35 (29.0) | 42 (21.7) | 0.002 | ||||||
| Long lie | 0 | 9 (7.4) | 9 (4.7) | 0.020 | ||||||
| Medication | 10 (13.9) | 45 (37.2) | 55 (28.5) | 0.007 | ||||||
| Hypothyroidism | 6 (8.3) | 11 (9.1) | 17 (8.8) | 0.911 | ||||||
| Vitamin D deficiency | 5 (6.9) | 1 (0.8) | 6 (3.1) | 0.025 | ||||||
| Direct trauma | 1 (1.4) | 1 (0.8) | 2 (1.0) | 1.000 | ||||||
| No trigger found | 16 (22.2) | 8 (6.6) | 22 (11.4) | 0.005 | ||||||
| Outcome, n (%) | ||||||||||
| Dead | 3 (4.2) | 14 (11.6) | 17 (8.8) | 0.079 | ||||||
| Alive | 69 (95.8) | 107 (88.4) | 176 (91.2) | |||||||
| # Triggers, n (%) | ||||||||||
| 0 | 14 (19.4) | 8 (6.6) | 22 (11.4) | |||||||
| 1 | 33 (45.8) | 46 (38.0) | 79 (40.9) | |||||||
| 2 | 19 (26.4) | 36 (29.8) | 55 (28.5) | |||||||
| 3 | 5 (7.0) | 23 (19.0) | 28 (14.5) | |||||||
| 4 | 1 (1.4) | 8 (6.6) | 9 (4.7) | |||||||
| >1 trigger, n (%) | ||||||||||
| Yes | 25 (34.7) | 67 (55.4) | 92 (47.7) | 0.005 | ||||||
| Symptoms, n (%) | Yes | No | NR | Yes | No | NR | Yes | No | NR | |
| Myalgia and/or muscle cramp | 53 (85.5) | 9 (14.5) | 10 | 85 (85.0) | 15 (15.0) | 21 | 138 (85.2) | 24 (14.8) | 31 | 0.401 |
| Muscle stiffness | 18 (72.0) | 7 (28.0) | 47 | 8 (53.3) | 7 (46.7) | 106 | 26 (38.8) | 14 (61.2) | 153 | 0.001 |
| Muscle weakness | 39 (78.0) | 11 (22.0) | 22 | 44 (59.5) | 30 (41.5) | 47 | 83 (66.9) | 41 (33.1) | 69 | 0.047 |
| Muscle swelling | 1 (16.7) | 5 (83.3) | 66 | 14 (63.6) | 8 (36.4) | 99 | 15 (53.6) | 13 (46.4) | 165 | 0.038 |
| Dark urine/ myoglobinuria | 27 (56.3) | 21 (43.7) | 24 | 27 (38.0) | 44 (62.0) | 50 | 54 (45.3) | 65 (54.7) | 74 | 0.076 |
| WES performed, n (%) | ||||||||||
| Yes | 16 (22.2) | 39 (32.2) | 55 (28.5) | |||||||
| No | 56 (77.8) | 82 (67.8) | 138 (71.5) | |||||||
ADR, adverse drug reaction; CK, creatine kinase; hyperCKaemia, isolated raised serum creatine kinase; NMS, neuroleptic malignant syndrome; NR, not reported; SS, serotonin syndrome; WES, whole‐exome sequencing.
Seventy‐two patients (37%) proved to have a pathogenic variant in a neuromuscular gene that explained the increased susceptibility to developing rhabdomyolysis. Variants were identified on genetic testing either before or at follow‐up after the rhabdomyolysis event. In 121 patients (63%) no genetic cause was identified.
Table 2 shows a comparison of patients with a proven genetic susceptibility (n = 72) with those in whom this was presumed based on the RHABDO features (n = 121). In both groups, a male preponderance was found. The mean age in the group with proven inherited muscle disease was 20 years younger (95% confidence interval [CI] 14.921–24869; P < 0.001) compared to the group without known cause. Furthermore, rhabdomyolysis was more frequently recurrent (90% vs. 48%; P < 0.001) and CK levels less frequently exceeded 10 000 IU/l (54% vs. 79%; P < 0.001). CK levels were lower in patients with a proven genetic susceptibility (median 10 870 IU/l) compared to those with a presumed susceptibility (median 17 870 IU/l, U = 3659; P = 0.019). Patients with a proven susceptibility less frequently had multiple triggers (P = 0.05). Death was reported more often in patients with a presumed susceptibility, but this was not statistically significant (4% vs. 12%; P = 0.079). Triggers that were found significantly more often in those with a presumed genetic susceptibility were exposure to heat (0 vs. 10%; P = 0.005), use of excessive alcohol or illicit drugs (7% vs. 35%; P = 0.002), long lie (0% vs. 7%; P = 0.001) and the prescription of possible myotoxic medication (7% vs. 22%; P = 0.007).
Analysis of genetic variants
In the group of patients with a proven pathogenic variant (n = 72, column 1, Table 2) 22 different genes were implicated, involving 56 different variants. In the group of patients suspected of having a specific genetic susceptibility (n = 121, column 2, Table 2), variants in 16 different genes were found, all considered to be variants of uncertain significance (Data S1; Table S1). Whole‐exome sequencing was performed in 16 patients (22%) in the group of 72 patients with a proven genetic susceptibility, and in 39 (32%) of the patients with a presumed genetic susceptibility. Results of genetic testing are listed in Table 3; variants in genes that have been previously implicated in events of rhabdomyolysis in a review by Scalco et al. [5] are underlined. Of the 22 different genes in which variants were found, 11 genes were not reviewed by Scalco et al. (AGL, ACADM, CAPN3, CNBP, DMPK, MAGT1, SCN4A, SGCA, SGCG, SMPD1 and TANGO2). However, more recently, TANGO2 has been implicated in events of rhabdomyolysis [20]. Information regarding the variants of uncertain significance, found in the group of 121 patients suspected of having a genetic susceptibility, is listed in Table S1. A thorough systematic literature search and description of these genes is provided in the Supporting information.
Table 3.
Gene–phenotype relationships found in a subset of 72 patients with a proven pathogenic variant
| Patient | Gene | Encodes | Phenotype | Zygosity | Variants |
|---|---|---|---|---|---|
| 1 | ACADVL | Very long‐chain Acyl‐CoA dehydrogenase | VLCAD deficiency | Heterozygous | c.589G> A (p.Val197Met) |
| 2–6 | VLCAD deficiency | NR | NR | ||
| 7 | AGL | Glycogen debranching enzyme | Glycogen storage disease type IIIa | Compound heterozygous | c.848T> C (p.Val283Ala) |
| 8 | ANO5 | Anoctamin V | LGMD2L | Heterozygous | c.191dup (p.Asn64fs)c.1733T> C (p.Phe578Ser) |
| 9 | LGMD2L | Heterozygous | c.191dup (p.Asn64fs)c.2272C> T (p.Arg 758Cys) | ||
| 10 | Asymptomatic hyperCKaemia | Homozygous | c.191dup (p.Asn64fs) | ||
| 11 | CAPN3 | Calpain III | LGMD2A | Heterozygous | c.509A> G (p.Tyr170Cys)c.554A> T (p.Tyr185Phe)c.2115 + 1_2115+2dup (r.spl?) |
| 12 | CNBP | Zinc finger protein IX | Myotonic dystrophy type II | Heterozygous | Repeat expansion (CCTG) intron 1 |
| 13 | Myotonic dystrophy type II | NR | NR | ||
| 14 | CPT2 | Carnitine palmitoyltransferase II | CPT2‐deficiency | Heterozygous | c.302C> T (p.Ala101Val) |
| 15 | CPT2‐deficiency |
Homozygous Heterozygous |
c.338C> T (p.Ser113Leu) c.679C> T (p.Leu227Phe) |
||
| 16 | DMD | Dystrophin | Duchenne muscular dystrophy | Hemizygous | Del 56‐78 |
| 17 | Becker muscular dystrophy | Hemizygous | Del(X)(p.21.1‐p. ? | ||
| 18 | Duchenne muscular dystrophy | Hemizygous | Del exon 45‐52 | ||
| 19 | Duchenne muscular dystrophy | Hemizygous | Del exon 10‐21 | ||
| 20‐21 | Duchenne muscular dystrophy | Hemizygous | Del exon 48‐54 | ||
| 22 | Duchenne muscular dystrophy | Hemizygous | Del exon 51 | ||
| 23 | Duchenne muscular dystrophy | Hemizygous | Del exon 48‐52 | ||
| 24 | Becker muscular dystrophy | Hemizygous | Del exon 45‐47 | ||
| 25‐27 | Becker muscular dystrophy | Hemizygous | Del exon 45‐48 | ||
| 28 | Duchenne muscular dystrophy | Hemizygous | Del exon 35‐43 | ||
| 29 | Female DMD carrier | Hemizygous | Del exon 61‐62 | ||
| 30 | Duchenne muscular dystrophy | Hemizygous | Del exon 75 | ||
| 31 | Becker muscular dystrophy | Hemizygous | Del exon 34 | ||
| 32 | Duchenne muscular dystrophy | Hemizygous | Del exon 11‐17 | ||
| 33 | Duchenne muscular dystrophy | Hemizygous | Insertion exon 37 Mutation exon 16 | ||
| 34 | Becker muscular dystrophy | Hemizygous | Del exon 45‐49 | ||
| 35 | Duchenne muscular dystrophy | Hemizygous | Split sice mutation exon 7 | ||
| 36 | Duchenne muscular dystrophy | Hemizygous | Dup exon 46‐47 | ||
| 37 | Duchenne muscular dystrophy | Hemizygous | Del exon 49‐50 | ||
| 38‐39 | Duchenne muscular dystrophy | Hemizygous | NR | ||
| 40 | DMPK | Dystrophia myotonica protein kinase | Myotonic dystrophy type I | NR | Repeat expansion CTG (>200) |
| 41 | DYSF | Dysferlin | LGMD2B | Homozygous | c.356T> C (p.Leu119Pro) |
| 42‐43 | LGMD2B | NR | NR | ||
| 44 | FKRP | Fukutin‐related protein | LGMD2I | Homozygous | c.826C> A (p.Leu276Ile) |
| 45 | HADHA | Mitochondrial trifunctional protein | LCHAD deficiency | NR | c.1528G> C c.2099delG |
| 46 | LCHAD deficiency | NR | NR | ||
| 47 | MAGT1 | Magnesium transporter type I | XMEN‐disease | Hemizygous | c.664delC (p.Met223Cysfs*40) |
| 48 | ACADM | Medium chain acyl‐CoA dehydrogenase | MCAD deficiency | Heterozygous | c.865.G> A |
| 49 | PGM1 | Phosphoglucomutase | Congenital glycosylation disorder type It | Heterozygous | c.988G> C (p.Gly330Arg)c.1258T> C (p.Tyr420His)c.1264 C> T (Arg422Trp) |
| 50 | Congenital glycosylation disorder type It | Heterozygous | c.419G> A (p.Gly140Asp) c.1597C> T (p.Arg533Trp) | ||
| 51 | LPIN | Lipin I | Acute recurrent autosomal recessive myoglobinuria | Homozygous | c.1471C> T |
| 52 | PYGM | Glycogen phosphorylase | McArdle disease | Heterozygous | c.148C> T (p.Arg50*)c.1700A> G (p.Gln567Arg) |
| 53‐58 | McArdle disease | Homozygous | c.148C> T (p.Arg50*) | ||
| 59 | McArdle disease | NR | NR | ||
| 60 | RYR1 | Ryanodine receptor 1 | Heterozygous | c.10219G> T (p.Ala3407Ser) | |
| 61 | Heterozygous | c.6961A> G (p.Ile2321Val)c.14545G> A (p.Val4849Ile) | |||
| 62 | Heterozygous | c.14545G> A (p.Val4849Ile) | |||
| 63 | NR | c.6617C> T | |||
| 64‐65 | Heterozygous | c.7300G> A (p.Gly2434Arg) | |||
| 66 | Heterozygous | c.7018T> C (p.Phe2340Leu) | |||
| 67 | SCN4A | Sodium channel, type IV, Alpha subunit | Congenital paramyotonia | Heterozygous | c.3917G> T (p.Gly1306Val) |
| 68 | SGCA | Sarcoglycan alpha | LGMD2D | NR | NR |
| 69 | SGCG | Sarcoglycan gamma | LGMD2C | NR | Exon 5: IVS5 + 2T>C |
| 70 | SMPD1 | Sphingomyelin phosphodiesterase 1 | Niemann‐Pick disease, type A/ B | Heterozygous | NR |
| 71 | TANGO2 | Transport and golgi organization II | Metabolic encephalomyopathy, recurrent rhabdomyolysis, cardiac arrhythmias and neurodegeneration | Homozygous | Del(22)(q11.21) |
| 72 | mtDNA | MELAS | m.3243A> G |
hyperCKaemia, isolated raised serum creatine kinase; LCAD, long‐chain acyl‐CoA dehydrogenase LGMD, Limb girdle muscle dystrophy; MCAD, medium‐chain acyl‐CoA dehydrogenase; MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke‐like episodes; NR, Not reported; VLCAD, very‐long‐chain acyl‐CoA dehydrogenase; XMEN, X‐linked immunodeficiency with magnesium defect, Epstein‐Barr virus infection, and neoplasia.
Genes that have been previously associated with rhabdomyolysis by Scalco et al. [5] are underlined.
Discussion
In the present single‐center study, we identified 1302 patients with rhabdomyolysis. Ischemia/anoxia was the most frequent trigger, followed by trauma. Of these patients, 193 had clinical characteristics suggestive of a genetic susceptibility, and, of those, 72 patients had a confirmed variant in a (putative) rhabdomyolysis‐associated gene. Pathogenic variants in 22 different genes were identified, likely to lead to increased rhabdomyolysis susceptibility. In 121 patients, a genetic susceptibility was suspected based on the RHABDO features, but no pathogenic genetic variant was identified. These involved 39 patients in whom genetic testing was performed (32%). In 19 of these patients, one or more variants of uncertain significance were identified (49%). Patients with a proven genetic susceptibility were younger than those with a presumed susceptibility. However, this observation probably reflects the presence of congenital diseases that are often diagnosed already in infancy or childhood.
The demographic distribution of our study population was similar to that observed in previous studies, in particular with regard to the male predominance, which was observed in all subsets, except for the long lie group [3, 4]. This male preponderance is a feature that has been observed in previous epidemiological studies on rhabdomyolysis [4]. In a study in patients with suspected malignant hyperthermia susceptibility, in vitro contracture testing was more often pathological in men, compared to women. [21]. This might be reflected by a sex‐dependent or hormonal variable. Regarding adverse drug reactions, men more often adhered to medication, and were more often prescribed antipsychotics [22]. In addition, a study on drug intoxication in six hospitals in the Netherlands showed that men had more frequently misused drugs (66% of all cases) [23]. A systematic review, performed in 2016, identified all cohort studies focusing on rhabdomyolysis published between January 2006 and 2016, reviewing 16 studies with epidemiological data [24]. Most studies focused on specific subsets of patients (i.e., pediatric patients, statin users, postoperative patients, military recruits, burns patients and those with drug‐related rhabdomyolysis). Three retrospective studies, performed in 2005, 2009 and 2012, conducted a hospital‐wide assessment of rhabdomyolysis patients [2, 3, 4]. Table 4 summarizes the results of these three previous retrospective studies and compares these with the results of the present study. Limited information regarding genetic testing was included in these previous studies, probably reflecting the minimal availability of genetic testing at the time. In 2017, Vivante et al. [25] employed whole‐exome sequencing in 21 unrelated patients with rhabdomyolysis, identifying disease‐causing variants in nine patients (43%). In our subgroup of 197 patients fulfilling the RHABDO criteria, we found proven genetic variants in 37% of the patients. Vivante et al. [25] described the genes associated with rhabdomyolysis and, corresponding to our approach, compared those with the genes summarized in the review by Scalco et al. [4] In the present study, we found an additional 11 genes possibly involved in an increased rhabdomyolysis susceptibility. A role in increased rhabdomyolysis susceptibility is plausible, considering that most of these genes are linked to metabolic and dystrophic neuromuscular conditions with similarities to other neuromuscular conditions with an increased rhabdomyolysis risk. To provide further supportive evidence for a potentially causative role of these genes, we conducted an extensive PubMED search, which is detailed in the Supporting information. This resulted in the notable observations described below.
Table 4.
Clinical characteristics and etiologies of rhabdomyolysis in previous hospital‐wide retrospective studies
| (n = 193) | 2019 Present study (n = 1302) | 2019 Present study Subgroup analysis (>1 RHABDO feature) | 2012 Herraez et al. [2] (n = 449) | 2008 Linares et al. [3](n = 106) | 2005 Melli et al. [4] (n = 475) |
|---|---|---|---|---|---|
| CK threshold, IU/l | >2000 | >2000 | >975 | >5000 | >975 |
| Male, % | 72 | 72 | 69 | 79 | 68 |
| Median (range) age, years | 53 (0–98) | 31 (0–94) | 66 ± 21a | 51 ± 20a | 47 (4–95) |
| Average (± SD) peak CK, IU/l | 15 563 ± 146 332 | 64 346 ± 378 094 | 14 355 (± NR) IU/l | 34 153 ± 76 456 | 168 052 (± NR) |
| Death, % | 22.2 | 8.8 | 18.7 | 12 | 3.4 |
| Etiological factors, % | |||||
| Ischemia/anoxia | 42 | 0 | 25 | 17 | NR |
| Direct muscle trauma | 20 | 1 | 24 | 23 | 9 |
| Immobility (‘long lie’) | 5 | 5 | 17 | 19 | 4 |
| Seizures | 2 | 0 | 2 | 8 | 7 |
| Muscle diseases | 11 | 37 | NR | NR | 10 |
| Illicit drugs/alcohol | ‐ | 22 | 8 | 28 | 34 |
| Exercise | ‐ | 55 | 4.3 | NR | 0.1 |
CK, creatine kinase; NR, not reported.
Disorders of glycogen metabolism
Unlike glycogen storage disease type V (McArdle's disease) and type VII, rhabdomyolysis is generally not considered a key feature in patients with glycogen storage disease type IIIa [26, 27]. A patient in the present study (patient 7, Table 3) experienced multiple episodes in which CK levels exceeded 1.5 times the ULN and subsequently returned to base levels. Rhabdomyolysis in patients with AGL variants has been described in the literature; however, only peak CK levels were mentioned and therefore the characteristic rise and fall in CK level was not reported, making it impossible to tell whether patients were experiencing episodes of rhabdomyolysis or, alternatively, had elevated baseline CK levels [30, 31].
Disorders of fatty acid metabolism
Scalco et al. [4] reviewed the association between rhabdomyolysis and deficiencies in long‐fatty‐acid oxidation (i.e., very‐long‐chain acyl‐CoA dehydrogenase [VLCAD] and long‐chain acyl‐CoA dehydrogenase [LCAD] deficiencies). In addition, we observed increased CK levels in one patient with medium‐chain acyl‐CoA dehydrogenase (MCAD) deficiency attributable to a pathogenic variant in ACADM. Rhabdomyolysis in patients with MCAD deficiency has been reported in various case reports and a more extensive clinical study [25, 28, 29].
Metabolic and mitochondrial disorders
The association between TANGO2 and rhabdomyolysis has been discovered more recently and therefore was not included in the review by Scalco et al. However, TANGO2 has been associated with events of rhabdomyolysis in more recently published previous studies [20].
Muscular dystrophies
In addition to the genes mentioned in Scalco et al. that are involved in the LGMD disorders (including ANO5, DYSF and FKRP), we found several other genes involving rhabdomyolysis in the context of LGMD disorders, including CAPN3 (LGMD2A), SGCA (LGMD2C) and SGCG (LGMD2E). In addition to the patients in our cohort, a literature search identified further studies reporting variants in these genes in patients with rhabdomyolysis or myoglobinuria, suggesting that these genes indeed play a role in increased rhabdomyolysis susceptibility [32, 37, 38]. However, in the above studies no longitudinal data regarding CK levels were provided, making the distinction between genuine rhabdomyolysis events and permanently elevated baseline CK levels impossible.
Other causes
We also identified variants in MAGT1 and SMPD1, however, no published reports could be identified implicating these genes in rhabdomyolysis previously. In addition, considering that in the present cohort only one patient each with variants in these genes was identified, a definite role of these genes remains uncertain.
To our knowledge, a comprehensive hospital‐wide assessment of rhabdomyolysis patients focusing on both external triggers and genetic factors has not yet been performed. Of the few single‐center studies reported, most focused on specific patient subsets (i.e., pediatrics, postoperative bariatric patients, patients in the emergency department or intensive care unit) rather than on rhabdomyolysis patients in their entirety [14]. The present study demonstrates and emphasizes the broad genetic heterogeneity that might increase susceptibility for developing rhabdomyolysis.
The present study has several limitations. Firstly, in a retrospective single‐center review, some details regarding symptoms, family history or presence of environmental triggers may not be thoroughly documented, likely leading to an underestimation of those factors. Secondly, in many patients, only one CK value was reported, making it impossible to differentiate between persisting hyperCKaemia and rhabdomyolysis, considering that the latter is defined as a rise and subsequent fall in CK values. Follow‐up could have taken place at a different hospital because the study setting was a tertiary referral center. For the same reasons, in many patients, it was also not possible to ascertain if elevated CK levels persisted beyond 8 weeks, one of the RHABDO criteria for increased genetic susceptibility. Also, we could have missed patients if their CK value was below our threshold at the time of measurement, but their CK values may nevertheless have been increasing. Thirdly, our study population consisted of a selection of patients identified through a laboratory search in a large academic hospital with a particular focus on muscle diseases, and therefore might not accurately reflect the rhabdomyolysis population presenting to a less‐specialized general hospital. Also, patients were included that were referred to our center for diagnostic testing, after admission elsewhere. This leads to a more comprehensive analysis, but may also have led to a selection bias, thus influencing the representativeness of our study population regarding epidemiological characteristics.
Three of the four RHABDO features were present more often in the subset of patients with a proven genetic susceptibility (P < 0.001); therefore, we would recommend genetic testing in rhabdomyolysis patients with at least one positive RHABDO feature, even if a trigger may seem clearly identifiable. For example, Dlamini et al. [13] describe a case of a father and daughter developing an episode of rhabdomyolysis during the same viral infection. Additional testing revealed a RYR1 variant that would have been dismissed as a viral myositis without the family history suggestive of a dominantly inherited neuromuscular disorder, prompting additional genetic testing. This example can be extrapolated to other clearly identifiable triggers, relevant for many medical disciplines (e.g., fever/infection, medication, exercise). In addition, some RHABDO features are probably more predictive than others, for example, an affected first‐degree relative.
The 11 additional genes identified in the present cohort that were not reported in the previous review by Scalco et al. emphasize the broad and probably expanding spectrum of genetic susceptibility underlying rhabdomyolysis. Further evaluation of this heterogeneity may result in specific recommendations for patients and subsequent genetic counseling of the family to prevent recurring possible life‐threatening events. With the increasing availability of next‐generation sequencing, we would recommend that genetic testing be performed in a diagnostic setting, after discussion with a tertiary neuromuscular center.
In conclusion, in the present single‐center study, we identified 1302 patients with increased CK levels suggestive of rhabdomyolysis. Ischemia/anoxia was the most frequently reported clearly identifiable trigger (40% of all cases), and 193 patients had clinical characteristics suggestive of an underlying genetic disorder, based on RHABDO criteria. Of those, 72 patients had a proven genetic susceptibility for the episode of rhabdomyolysis. Twenty‐two different genes were identified, involving 56 variants that were possibly involved in an increased susceptibility for developing rhabdomyolysis. Of those, 11 genes have been associated with rhabdomyolysis in a previously published review (ACADVL, ANO5, CPT2, DMD, DYSF, FKRP, HADHA, PGM1, LPIN1, PYGM, RYR1). In addition, 11 genes are probably implicated in increased susceptibility (including AGL, CAPN3, CNBP, DMPK, MAGT1, ACADM, SCN4A, SGCA, SGCG, SMPD1, TANGO2), expanding the wide genetic spectrum underlying rhabdomyolysis susceptibility.
Disclosure of conflicts of interest
The authors declare no financial or other conflicts of interest.
Supporting information
Table S1. Variants of uncertain significance.
Data S1. Genes involving proven pathogenic variants.
Appendix 1.
Search strategies
AGL
Glycogen storage disease type 3[MeSH] OR
Glycogen storage disease type 3[Tiab] OR
Glycogen storage disease type III[Tiab] OR
Glycogen debranching enzyme[MeSH] OR
Glycogen debranching enzyme[Tiab] OR
Glycogen debrancher enzyme[Tiab] OR
GDE[Tiab] OR
AGL[Tiab]
MCADL
Acyl coa dehydrogenase, medium chain[MeSH] OR
medium chain acyl coa[Tiab] OR
medium chain Acyl‐coa[Tiab] OR
MCACA‐dehydrogenase[Tiab] OR
MCAD[Tiab] OR
ACADM[Tiab]
CAPN3
LGMD2A[Tiab] OR
Limb girdle muscular dystrophy type 2A[Tiab] OR
Limb girdle muscular dystrophy 2A[Tiab] OR
Calpain 3[Tiab] OR
CANPL3[Tiab]
DMPK and CNBP
Myotonic dystrophy[MeSH] OR
Myotonic dystroph*[Tiab] OR
Dystrophia myotonica[Tiab] OR
Steinert[Tiab] OR
PROMM*[Tiab] OR
DMPK[Tiab] OR
CNBP[Tiab] OR
Myotonin‐protein kinase[Tiab] OR
ZNF9[Tiab] OR
Zinc finger protein 9[Tiab]
MAGT1
MAGT1[Tiab] OR
Magnesium transporter 1[Tiab] OR
XMEN[Tiab]
SCN4A
NAV1.4 Voltage‐Gated Sodium Channel[MeSH] OR
NAV1.4 Voltage‐Gated Sodium Channel[Tiab] OR
Sodium channel alpha subunit[Tiab] OR
SCN4A[Tiab]
SGCA & SGCG
Alpha sarcoglycan*[Tiab] OR
LGMD2D[Tiab] OR
Gamma sarcoglycan*[Tiab] OR
LGMD2C[Tiab]
SMPD1
Sphingomyelin phosphodiesterase[MeSH] OR
Sphingomyelin phosphodiesterase[Tiab] OR
Sphingomyelin cholinephosphohydrolase[Tiab] OR
Sphingomyelinase[Tiab]
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Herraez Garcia J, Torracchi Carrasco AM, Antoli‐Royo AC, de la Fuente Blanco R, Santos Jimenez MT. Rhabdomyolysis. A descriptive study of 449 patients. Med Clin (Barc). 2012; 139: 238–242. [DOI] [PubMed] [Google Scholar]
- 2. Linares LA, Golomb BA, Jaojoco JA, Sikand H, Phillips PS. The modern spectrum of rhabdomyolysis: drug toxicity revealed by creatine kinase screening. Curr Drug Saf 2009; 4: 181–187. [DOI] [PubMed] [Google Scholar]
- 3. Melli G, Chaudhry V, Cornblath DR. Rhabdomyolysis: an evaluation of 475 hospitalized patients. Medicine 2005; 84: 377–385. [DOI] [PubMed] [Google Scholar]
- 4. Scalco RS, Gardiner AR, Pitceathly RD, et al Rhabdomyolysis: a genetic perspective. Orphanet J Rare Dis. 2015; 10: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scalco RS, Snoeck M, Quinlivan R, et al Exertional rhabdomyolysis: physiological response or manifestation of an underlying myopathy? BMJ Open Sport Exerc Med 2016; 2: e000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Voermans NC. TW 6.3.3 Exercise‐induced rhabdomyolysis: diagnostic guidelines and RYR1‐related cases. 13th ICNMD conference Nice F, 2014.
- 7. Fernandes PM, Davenport RJ. How to do it: investigate exertional rhabdomyolysis (or not). Pract Neurol 2019; 19: 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andreu AL, Nogales‐Gadea G, Cassandrini D, Arenas J, Bruno C. McArdle disease: molecular genetic update. Acta Myol 2007; 26: 53–57. [PMC free article] [PubMed] [Google Scholar]
- 9. Antunes AP, Nogueira C, Rocha H, Vilarinho L, Evangelista T. Intermittent rhabdomyolysis with adult onset associated with a mutation in the ACADVL gene. J Clin Neuromuscul Dis 2013; 15: 69–72. [DOI] [PubMed] [Google Scholar]
- 10. Thuillier L, Rostane H, Droin V, et al Correlation between genotype, metabolic data, and clinical presentation in carnitine palmitoyltransferase 2 (CPT2) deficiency. Hum Mutat 2003; 21: 493–501. [DOI] [PubMed] [Google Scholar]
- 11. Michot C, Hubert L, Brivet M, et al LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum Mutat 2010; 31: E1564–E1573. [DOI] [PubMed] [Google Scholar]
- 12. Lindberg C, Sixt C, Oldfors A. Episodes of exercise‐induced dark urine and myalgia in LGMD 2I. Acta Neurol Scand 2012; 125: 285–287. [DOI] [PubMed] [Google Scholar]
- 13. Dlamini N, Voermans NC, Lillis S, et al Mutations in RYR1 are a common cause of exertional myalgia and rhabdomyolysis. Neuromuscul Disord 2013; 23: 540–548. [DOI] [PubMed] [Google Scholar]
- 14. George MD, McGill NK, Baker JF. Creatine kinase in the U.S. population: Impact of demographics, comorbidities, and body composition on the normal range. Medicine 2016; 95: e4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fleming J, Brayne C. Inability to get up after falling, subsequent time on floor, and summoning help: prospective cohort study in people over 90. BMJ 2008; 337: a2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96: 635–642. [DOI] [PubMed] [Google Scholar]
- 17. Zimmerman JL, Shen MC. Rhabdomyolysis. Chest 2013; 144: 1058–1065. [DOI] [PubMed] [Google Scholar]
- 18. Nance JR, Mammen AL. Diagnostic evaluation of rhabdomyolysis. Muscle Nerve 2015; 51: 793–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Torres PA, Helmstetter JA, Kaye AM, Kaye AD. Rhabdomyolysis: pathogenesis, diagnosis, and treatment. Ochsner J 2015; 15: 58–69. [PMC free article] [PubMed] [Google Scholar]
- 20. Dines JN, Golden‐Grant K, LaCroix A, et al TANGO2: expanding the clinical phenotype and spectrum of pathogenic variants. Genet Med 2019; 21: 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Islander G, Rydenfelt K, Ranklev E, Bodelsson M. Male preponderance of patients testing positive for malignant hyperthermia susceptibility. Acta Anaesthesiol Scand 2007; 51: 614–620. [DOI] [PubMed] [Google Scholar]
- 22. Aleman A, Kahn RS, Selten JP. Sex differences in the risk of schizophrenia: evidence from meta‐analysis. Arch Gen Psychiatry 2003; 60: 565–571. [DOI] [PubMed] [Google Scholar]
- 23. Duineveld C, Vroegop M, Schouren L, et al Acute intoxications: differences in management between six Dutch hospitals. Clin Toxicol (Phila) 2012; 50: 120–128. [DOI] [PubMed] [Google Scholar]
- 24. Chavez LO, Leon M, Einav S, Varon J. Beyond muscle destruction: a systematic review of rhabdomyolysis for clinical practice. Crit Care 2016; 20: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vivante A, Ityel H, Pode‐Shakked B, et al Exome sequencing in Jewish and Arab patients with rhabdomyolysis reveals single‐gene etiology in 43% of cases. Pediatr Nephrol 2017; 32: 2273–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kishnani PS, Austin SL, Arn P, et al Glycogen storage disease type III diagnosis and management guidelines. Genet Med 2010; 12: 446–463. [DOI] [PubMed] [Google Scholar]
- 27. Tarnopolsky MA. Myopathies related to glycogen metabolism disorders. Neurotherapeutics 2018; 15: 915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ruitenbeek W, Poels PJ, Turnbull DM, et al Rhabdomyolysis and acute encephalopathy in late onset medium chain acyl‐CoA dehydrogenase deficiency. J Neurol Neurosurg Psychiatry 1995; 58: 209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feillet F, Steinmann G, Vianey‐Saban C, et al Adult presentation of MCAD deficiency revealed by coma and severe arrythmias. Intensive Care Med 2003; 29: 1594–1597. [DOI] [PubMed] [Google Scholar]
- 30. El‐Karaksy H, Anwar G, El‐Raziky M, et al Glycogen storage disease type III in Egyptian children: a single centre clinico‐laboratory study. Arab J Gastroenterol 2014; 15: 63–67. [DOI] [PubMed] [Google Scholar]
- 31. Crushell E, Treacy EP, Dawe J, Durkie M, Beauchamp NJ. Glycogen storage disease type III in the Irish population. J Inherit Metab Dis 2010; 33(Suppl 3): S215–218. [DOI] [PubMed] [Google Scholar]
- 32. Lahoria R, Milone M. Rhabdomyolysis featuring muscular dystrophies. J Neurol Sci 2016; 361: 29–33. [DOI] [PubMed] [Google Scholar]
- 33. Merlini L, Sabatelli P, Columbaro M, et al Hyper‐CK‐emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve 2005; 31: 764–767. [DOI] [PubMed] [Google Scholar]
- 34. Li FY, Chaigne‐Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein‐Barr virus. Blood 2014; 123: 2148–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Osch T, Stunnenberg BC, Sternberg D, Kerklaan BJ. Prolonged attacks of weakness with hypokalemia in SCN4A‐related paramyotonia congenita. Muscle Nerve 2018; 58: E27–E28. [DOI] [PubMed] [Google Scholar]
- 36. Ceravolo F, Messina S, Rodolico C, Strisciuglio P, Concolino D. Myoglobinuria as first clinical sign of a primary alpha‐sarcoglycanopathy. Eur J Pediatr 2014; 173: 239–242. [DOI] [PubMed] [Google Scholar]
- 37. Tarnopolsky M, Hoffman E, Giri M, Shoffner J, Brady L. Alpha‐sarcoglycanopathy presenting as exercise intolerance and rhabdomyolysis in two adults. Neuromuscul Disord 2015; 25: 952–954. [DOI] [PubMed] [Google Scholar]
- 38. Pena L, Kim K, Charrow J. Episodic myoglobinuria in a primary gamma‐sarcoglycanopathy. Neuromuscul Disord 2010; 20: 337–339. [DOI] [PubMed] [Google Scholar]
- 39. Kremer LS, Distelmaier F, Alhaddad B, et al Bi‐allelic truncating mutations in TANGO2 cause infancy‐onset recurrent metabolic crises with encephalocardiomyopathy. Am J Hum Genet 2016; 98: 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Variants of uncertain significance.
Data S1. Genes involving proven pathogenic variants.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.