

Abstract
We herein report on new synthetic strategies for the preparation of pyridine and imidazole substituted 2,2’‐dihalo biphenyls. These structures are pre‐ligands suitable for the preparation of respective stannoles. The latter can successfully be transmetalated to K[AuCl4] forming non‐palindromic [(C^C^D)AuIII] pincer complexes featuring a lateral pyridine (D=N) or N‐heterocyclic carbene (NHC, D=C’) donor. The latter is the first report on a pincer complex with two formally anionic sp2 and one carbenic carbon donor. The [(C^C^D)AuIII] complexes show intense phosphorescence in solution at room temperature. We discuss the developed multistep strategy and touch upon synthetic challenges. The prepared complexes have been fully characterized including X‐ray diffraction analysis. The gold(III) complexes’ photophysical properties have been investigated by absorption and emission spectroscopy as well as quantum chemical calculations on the quasi‐relativistic two‐component TD‐DFT and GW/Bethe–Salpeter level including spin–orbit coupling. Thus, we shed light on the electronic influence of the non‐palindromic pincer ligand and reveal non‐radiative relaxation pathways of the different ligands employed.
Keywords: gold complexes, luminescence, photophysics, pincer ligand, quantum chemistry
Capture the gold(fish)! Donor‐substituted dihalobiphenyls are suitable pre‐ligands for the preparation of luminescent, non‐palindromic [(C^C^D)AuIII] complexes with a lateral pyridine or carbene donor. Like an anglerfish's rod enmeshes its prey, the lateral donor tunes the gold‘s electronic properties which is hold by the rigid biphenyl mouth. Synthesis, spectroscopy and quantum chemistry draw a comprehensive picture about the new complexes prepared.
Introduction
Phosphorescent emitters based on gold(III) [1] are far less studied in the context of phosphorescent organic light‐emitting diodes (PhOLEDs) [2] than systems incorporating other heavy metals, for example, iridium(III), [3] ruthenium(II) [4] or platinum(II). [5] However, there is increasing interest in gold(III)‐based systems mainly employing a 2,6‐diphenylpyridine (C^N^C)‐based pincer ligand. [6] The (C^N^C) pincer diminishes radiationless relaxation pathways of excited complexes due to its rigid nature [7] and—in combination with an additional strong donor ligand—results in high ligand field splitting thereby shifting metal‐centred d‐states to higher energies. The latter avoids population of these metal centred states regularly seen to be responsible for radiationless relaxation pathways. [8]
The first example of [(C^N^C)AuIII] complexes luminescent in frozen solution at 77 K was reported by Chi‐Ming Che and co‐workers in 1998. [9] In 2005, Vivian Wing‐Wah Yam and co‐workers combined the (C^N^C) motif with alkynyl ligands to obtain AuIII complexes which show phosphorescence in solution at room temperature. [10] Later on, the field of luminescent [(C^N^C)AuIII] complexes was further broadened by employing carbenes, [11] alkyl donors [12] or thiolates [13] as ancillary ligands. The pincer's structure was modified as well, for example, by substituting the central pyridine by pyrazine, thus, affecting emission quantum yields and wavelengths. [14]
We note that highly emissive tetradentate AuIII complexes reported only recently may outperform many tridentate systems in this regard, [15] however, complexes with tetradentate ligands are less attractive for possible applications in chemical or catalytic transformations because all coordination sites of the central AuIII atom are occupied. That [(C^N^C)AuIII] complexes are valuable candidates for the latter applications could impressively be shown by Bochmann and co‐workers who reported on the synthetic value of [(C^N^C)AuIII] hydroxides [16] and hydrides. [17] In addition, they prepared [(C^N^C)AuIII] olefin [18] and alkyne complexes [19] thereby showing the suitability of these complexes for C−C bond forming reactions. Finally, some [(C^N^C)AuIII] complexes are investigated in the realm of anticancer drug research [20] with a very recent study about photo‐activatable cytotoxic [(C^N^C)AuIII] hydrides. [21]
The (C^N^C) motif is introduced by treating a mono‐cyclometalated mercury compound like 1 with gold(III) salts (Scheme 1). In 2015, Nevado and co‐workers reported on the synthesis of the non‐palindromic [22] [(C^C^N)AuIII] analogue which exhibits exchanged positions of the central pyridine ring and one lateral phenyl donor (4 in Scheme 1). [23] The latter is prepared without need for toxic mercury compounds in an elegant way by means of two successive, microwave‐assisted C−H activations, using pyridine substituted terphenyls designed to allow only one kind of twofold cyclometalated products.
Scheme 1.
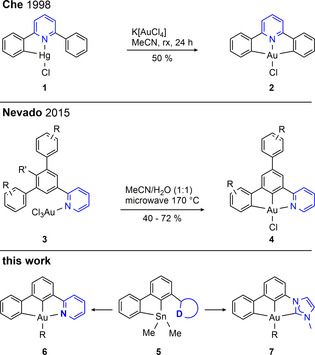
Preparation of palindromic (C^N^C) and non‐palindromic (C^C^N) AuIII complexes reported in the literature and the synthetic approach presented in this study. D: Donor.
The nonpalindromic (C^C^N) pincer was shown to exhibit high emission quantum yields making its gold(III) complexes particularly interesting for OLED fabrication. [24] This was assigned to a higher ligand field splitting of these complexes compared to the palindromic (C^N^C) congeners which was investigated by TDDFT.[ 8b , 25 ]
The central phenyl donor of the (C^C^N) ligand exhibits a stronger trans influence than the (C^N^C)’s central pyridine. This made the preparation of stable AuIII fluorides [23] and formates [26] possible and finds expression in notably different NMR shifts of the respective hydrides. [17b] Moreover, the trans influence affects the complexes’ reactivity: [(C^C^N)AuIII] carboxylates favour thermal decarbonylation reactions [27] whereas the (C^N^C) based carboxylates show elimination of CO2. [28]
There are no reports in the literature on gold(III) pincer complexes with two aryl donors and a third donor other than pyridine (D=N), for example, carbene. However, bidentate, (C^C’) cyclometalated (C’=NHC carbon donor) AuIII complexes were reported by von Arx et al. [29] and Crespo et al. [30]
In the present study, we report on the development of a synthetic access to 5,5‐dimethyl‐5H‐dibenzo[b,d]stannoles (5) with a pyridine (D=N) or imidazolium (D=C’) substituent in 4‐position, which are suitable precursors for the new AuIII complexes 6 and 7 (Scheme 1). On the one hand, this synthetic approach constitutes a complementary variant of the synthesis developed by Nevado and co‐workers to prepare non‐palindromic [(C^C^N)AuIII] complexes. On the other hand, it is the first example of a transition metal pincer complex with one NHC and two formally anionic phenyl donors. Both pincer complexes are combined with phenylethynyl or pentafluorophenyl ligands resulting in compounds highly phosphorescent in solution at room temperature. The photophysical properties are investigated experimentally and by means of TDDFT and the Bethe–Salpeter methodology including spin–orbit coupling described by us only recently, [31] thereby showing the utility of these methods in the field of photophysics and photochemistry.
Results and Discussion
Retrosynthetic considerations
Our synthetic approach for the preparation of non‐palindromic [(C^C^D)AuIII] complexes (D=pyridine, NHC) is based on the synthesis of biphenyldiyl gold(III) dimer 10 by transmetalation of the stannole 9 with gold(III) salts, already described by Usón et al. in 1980 (Scheme 2). [32]
Scheme 2.
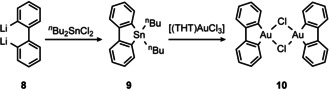
Preparation of biphenyldiyl gold(III) dimer 10 according to Usón et al. [32]
Direct transmetalation of dilithiobiphenyls to AuIII is only possible for very electron‐poor biphenyls like perfluorinated ones [33] or special AuIII precursors [34] due to the tendency of AuIII to become reduced by organometallic reagents. Besides the findings of Toste, [35] Bourissou [36] and Bertrand [37] on the oxidative addition of biphenylene to several AuI species, the transmetalation of stannoles is the method of choice for the preparation of cyclometalated biphenyldiyl AuIII complexes.[ 34 , 38 ] Thus, we anticipated the corresponding 4‐substituted stannoles 5 would be equally useful for the preparation of non‐palindromic (C^C^D) complexes of gold.
Dilithiobiphenyl 8 may be prepared by reaction of biphenyl with two equivalents of nBuLi, [39] however, this variant suffers from low yields and regioselectivity when employing substituted biphenyls. Therefore, we focused on the synthesis of 2,2’‐dihalobiphenyls 13 and 16 with bromo or iodo substituents (Scheme 3), which should serve as suitable pre‐ligands for the preparation of the pursued stannoles 5 (Scheme 1).
Scheme 3.
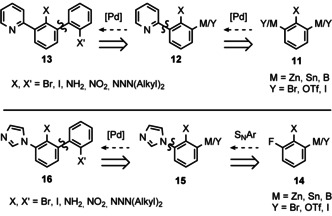
Retrosynthetic aspects of the preparation of dihalobiphenyls with pyridine (C^C^N) (13) or NHC (C^C^C’) (16) donor. [Pd]: Pd catalysed C−C cross coupling.
The anticipated dihalobiphenyls 13 and 16 should be accessible by introducing a 2‐halophenyl to phenylpyridine 12 and phenyl imidazole 15 by means of palladium catalysed C−C cross‐couplings. The halogens may be obtained by transformation of suitable N‐based functional groups, that is, anilines (X, X’=NH2), nitrobenzenes (X, X’=NO2) or triazenes (X, X’=NNNR2), by means of sandmeyer‐type reactions, [40] eventually after reduction of NO2. The latter must be performed after building the core pincer structure to ensure selectivity of the C−C cross couplings. Phenylpyridine 12 might be obtained by C−C cross coupling as well starting with 11 having (pseudo)halogen or metal substituents in 2‐ and 6‐position. Imidazole (15) should be introducible by means of nucleophilic aromatic substitution at 2‐fluoro nitrobenzene 14.
Pre‐ligand synthesis
With the above‐mentioned strategy at hand, we first coupled commercially available nitroaniline 17 with 2‐(tributylstannyl)pyridine [41] followed by diazotization‐iodination to obtain iodine substituted phenylpyridine 19 in excellent yield.
Iodide 19 was then coupled with 2‐(aminophenyl)boronic acid by means of a Suzuki–Miyaura coupling.[ 42 , 43 ] Unfortunately, diazotization‐iodination of 20 failed, although there are reports on the successful diazotization‐iodination of 2′‐nitro‐2‐phenyl‐anilines (Scheme 4). [44]
Scheme 4.
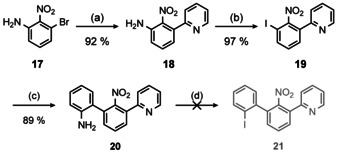
Attempt to obtain biphenyl 21. Conditions: (a) 2‐(Tributylstannyl)pyridine, LiCl, [Pd(PPh3)4] (4 mol %), xylene, rx, 6 h; b) 1) HCl, NaNO2, H2O, 0 °C, 30 min 2) KI, 0 °C→rt, 12 h; (c) 2‐(aminophenyl)boronic acid, K3PO4, XPhos Pd G3 (3 mol %), [42] 1,4‐dioxane/H2O (4:1), 70 °C, 90 min; (d) 1) HCl, NaNO2, H2O, 0 °C, 30 min 2) KI, 0 °C→rt, 12 h. The structure of 20 in the solid state could be determined by X‐ray diffraction analysis (Figure S9, Supporting Information).
Beside the classical variant using HCl/NaNO2/KI, we also tried DMSO‐based reagents [45] or modifications employing CuBr2. [46] Substitution of NH2 by Me3Sn according to a diazotization‐stannylation protocol led only to unidentified decomposition products. [47] Attempts to isolate the corresponding diazonium salt employing H[BF4]/NaNO2 or [NO][PF6] were unsuccessful as well. Reduction of the nitro‐group of 20 and subsequent double diazotization‐iodination of the resulting amines is not effective, because 2,2’‐diaminobiphenyls cyclize to the corresponding benzo[c]cinnolines under diazotization conditions. [48] We also directly introduced 2‐bromophenyl to 19, however, this approach had unacceptably low yields (Section S 1.2, Supporting Information).
We then turned our attention to a different approach based on the triazene 23 as key structure which was described by Knochel and co‐workers in the context of carbazole syntheses. [49] Magnesiation using turbo‐Grignard, [50] subsequent transmetalation with ZnBr2 and reaction with 2‐bromopyridine by means of a Negishi‐coupling[ 40 , 51 ] gave 24 in 87 % isolated yield (Scheme 5).
Scheme 5.
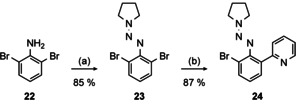
Synthesis of triazene 24. Conditions: (a) 1) HCl, NaNO2, H2O, 0 °C, 30 min. 2) K2CO3, pyrrolidine, MeCN, 0 °C→rt, 2 h; (b) 1) iPrMgCl⋅LiCl, THF, −40 °C→−15 °C, 4 h. 2) ZnBr2, −20 °C→5 °C, 1 h. 3) 2‐bromopyridine, [Pd(PPh3)4] (10 mol %), reflux, 16 h.
Then, we prepared the corresponding Negishi‐reagent of 24 and coupled with 1,2‐diiodobenzene to yield 25 (Scheme 6).
Scheme 6.
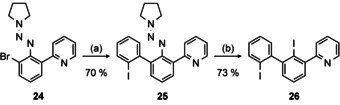
Synthesis of diiodo biphenyl 26. Conditions: (a) 1) nBuLi (pre‐cooled to −78 °C), −100 °C, 90 min. 2) 1 m ZnBr2 in THF (pre‐cooled to −78 °C), −100 °C→−78 °C, 1 h, rt, 1 h. 3) 1,2‐diiodobenzene, [Pd(PPh3)4] (5 mol %), rx, 16 h. (b) Amberlyst 15®, KI, MeCN, 70 °C, 10 min. The structure of 25 in the solid state could be determined by X‐ray diffraction analysis (Figure S9, Supporting Information).
The latter was only successful under meticulous temperature control: 24 was lithiated at −98 °C with precooled (−78 °C) nBuLi in hexanes and subsequently transmetalated with ZnBr2 to obtain a Negishi‐reagent suitable for coupling with 1,2‐diiodobenzene. Prior to that, we tried magnesiation of 24 using turbo‐Grignard which did not occur even at elevated temperatures (THF, 80 °C) leaving the bromide unaffected. The same holds for attempts to directly zincate 24 using elementary zinc prepared by the method of Rieke [52] or in the presence of LiCl. [53] This is probably rooted in the electron‐rich nature of 24 in contrast to the substrates reported by the group of Knochel [53a] who used only electron‐deficient triazenes for direct zinc insertions. Treating 24 with elementary magnesium led only to decomposition products even in the presence of ZnCl2. Consequently, we tried to lithiate 24 and transmetalate with ZnBr2. Under standard conditions (nBuLi or two eq. tBuLi −78 °C) we detected only decomposition products, maybe due to decomposition by α‐deprotonation at the pyrrolidine which is known to readily happen at higher temperatures. [54]
The triazene 25 decomposes in the presence of HI [55] to the diiodo biphenyl 26. We identified a variation employing proton exchange resin and sodium iodide, that is, in situ formed HI, in dry acetonitrile to be the most effective. [56] This procedure avoids shortcomings of other variations, for example, reactions at the pyridine ring. [57]
For the preparation of a (C^C^C’) pre‐ligand we started with commercially available 1‐bromo‐3‐fluoro‐2‐nitrobenzene (27) (Scheme 7).
Scheme 7.
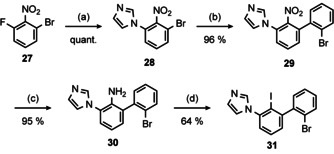
Synthesis of (C^C^C’) pre‐ligand 31. Conditions: (a) Imidazole, NaOH, DMSO, rt, 2 h; (b) 2‐bromphenyl boronic acid, [Pd(PPh3)4] (5 mol %), K2CO3, THF/H2O (2:1), rx, 18 h; (c) Fe, EtOH/HOAc (1:1), rx, 3 h; (d) 1) 6 m HCl, NaNO2, 0 °C, 30 min 2) KI, 0 °C→rt, 3 h. The structure of 29 in the solid state could be determined by X‐ray diffraction analysis (Figure S9, Supporting Information).
After introduction of imidazole by means of a nucleophilic aromatic substitution [58] (28), we introduced 2‐bromophenyl (29) and subsequently reduced the nitro group to obtain the aniline 30. The latter tends to cyclize with triazine formation (Section S 2.1, Supporting Information), however, under strongly acidic conditions the dihalobiphenyl 31 can be obtained in 64 % yield, corresponding to a good overall yield of 58 % over four steps. The presented route is especially attractive, since all intermediates may be used without purification.
Although several synthetic steps are necessary, the dihalobiphenyls 26 and 31 are easily prepared on a 10 to 20 g scale.
Complex synthesis and characterization
The diiodo biphenyl 26 was doubly lithiated and treated with Me2SnCl2 to obtain the stannole 32, which slowly decomposes upon chromatography; thus, analytically pure samples were obtained with moderate yields only (43 %). However, the crude stannole may be reacted with K[AuCl4] to obtain the non‐palindromic [(C^C^N)AuIII] complex 33 in 81 % yield (Scheme 8).
Scheme 8.
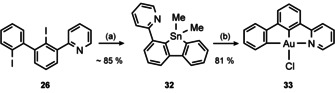
Synthesis of [(C^C^N)AuIIICl] (33). Conditions: (a) 1) tBuLi, Et2O, −78 °C, 2 h. 2) Me2SnCl2, −78 °C → rt, 16 h; (b) K[AuCl4], MeCN, 0 °C → rt, 1 h.
Complex 33 was obtained as colourless solid which dissolves readily in dichloromethane and toluene. However, after a couple of hours, it precipitates in form of yellow blocks which are suitable for X‐ray diffraction analysis. Furthermore, 33 crystallizes from CH2Cl2/n‐hexane with one molecule of n‐hexane in the unit cell (Figure S9, Supporting Information). The yellow blocks only modestly dissolve in hot organic solvents, thus, NMR analysis was performed in [D6]DMSO at 100 °C. Due to the poor solubility, chromatographic purification of 33 (instead of crystallization) results in notably reduced yields (62 %), however, the yellow blocks are fully suitable for further transformations. We note that 33 — if not already precipitated — decomposes in solution after three days forming elemental gold when exposed to light.
Thus, we treated 33 with lithium phenylacetylide or [(MeCN)AgC6F5] [59] to obtain the [(C^C^N)AuIII] complexes 34 and 35 in 86 % and 87 % yield, respectively (Scheme 9).
Scheme 9.
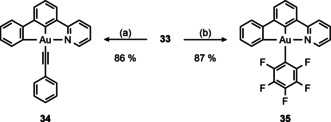
Synthesis of [(C^C^N)AuIII] complexes. (a) LiCCPh, Et2O/toluene (1:1), rt, 3 h; (b) [(MeCN)AgC6F5], CH2Cl2, rt, 16 h.
In a similar manner, we prepared stannole 36 and its methyl imidazolium salt 37 (Scheme 10).
Scheme 10.
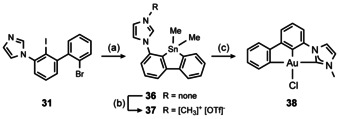
Synthesis of [(C^C^C’)AuIII] complex 38. Conditions: (a) 1) tBuLi, Et2O, −78 °C, 2 h. 2) Me2SnCl2, −78 °C→rt, 1 h; (b) MeOTf, CH2Cl2, rt, 2 h. (c) K[AuCl4], MeCN, K2CO3, rt, 2 h. The stannoles 36, 37 and [(C^C^C’)AuIII] complex 38 could not be isolated in pure form, thus, yields are not given.
Unfortunately, we were not able to obtain the stannoles 36 and 37 in pure form: Both are unstable during chromatography and did not crystallize. Nevertheless, we could prove their formation by NMR spectroscopy, APCI MS (36) and ESI MS (37) analyses of the crude reaction mixtures. The [(C^C^C’)AuIII] complex 38 which we obtained by treating 37 with K[AuCl4] in the presence of base rapidly decomposes in solution and in the presence of moisture too, which made the isolation of pure samples impossible as well.
However, crude 38 could be treated with lithium phenylacetylide or [(MeCN)AgC6F5] [59] to obtain the [(C^C^C’)AuIII] complexes 39 and 40 in 43 % and 49 % yield (Scheme 11), respectively, over four steps starting from dihalobiphenyl 31. The latter two complexes are stable towards moisture, air and chromatography.
Scheme 11.
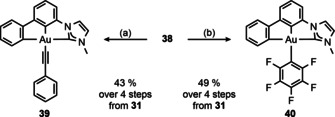
Synthesis of [(C^C^C’)AuIII] complexes 39 and 40. Conditions: (a) LiCCPh, Et2O/toluene (1:1), rt, 3 h; (b) [(MeCN)AgC6F5], CH2Cl2, rt, 16 h.
Crystals suitable for X‐ray diffraction analyses could be obtained from the pentafluorophenyl complexes 35 and 40 as well as the PhCC derivative 39 (Figure 1). Overall, the structural parameters of the investigated complexes are very similar. The Au−C17 bond of 39 has the same length as its [(C^C^N)AuIII] pendant, [23] but the Au−C17 bond of 40 (206.2(14) pm) is about 5 pm shorter compared to the respective bond of its (C^C^N) congener 35 (Au−C18: 211.2(10) pm). The latter may be a result of the better π acceptor properties of the lateral carbene as compared to the pyridine donor‐ligand. The molecules are rather closely packed in the solid state with distances between the planes spanned by the pincers ranging between 345 pm and 360 pm. The pentafluorophenyl complexes 35 and 40 are aligned in two different planes notably tilted against each other in the solid state, whereas the molecules of 39 are packed in a nearly parallel manner (Figure S10, Supporting Information).
Figure 1.
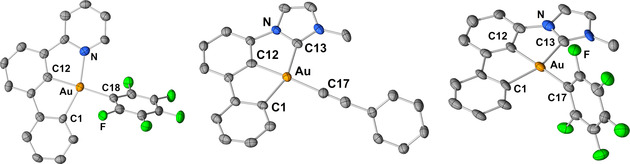
Solid state molecular structure of [(C^C^D)AuIII] complexes 35 (left), 39 (middle) and 40 (right). Thermal ellipsoids are set at 30 % probability. Hydrogen atoms are omitted for clarity. Only one molecule of the asymmetric unit of 35 is shown. The asymmetric unit of 35 contains half a molecule hexane (not shown). Selected bond lengths (pm) and angles (°), 35: Au‐C18 211.2 (10), Au‐C1 209.4(10), Au‐C12 200.7(10), Au‐N 212.4(9), C1‐Au‐C12 78.9(4), C12‐Au‐N 80.1(4), C1‐Au‐C18‐F 62.6(9); 39: Au‐C17 204.5(8), Au‐C1 206.4(7), Au‐C12 197.9(8), Au‐C13 207.4(8), C1‐Au‐C12 79.6(4), C12‐Au‐C13 78.2(3); 40: Au‐C17 206.2(14), Au‐C1 206.6(12), Au‐C12 200.3(12), Au‐C13 209.1(12), C1‐Au‐C12 78.9(6), C12‐Au‐C13 78.7(4), C1‐Au‐C17‐C 97.9(1).
All complexes adopt structures with the aryl entities of the ancillary ligands tilted against the plane spanned by the donor atoms of the pincer ligand. They are not perpendicular aligned to the pincer's plane, however, the preference for the tilted arrangement is important with respect to photophysical considerations. Thus, for the following discussion, we want to rely on an idealized situation of perfectly perpendicular aryl entities. The structural feature of tilted/perpendicular aryl entities are also found for most palindromic (C^N^C) analogues, either crystallographically [10a] or by means of DFT calculations. [10b] Nonetheless, the role of the tilted PhCC ligand is somewhat overlooked in the literature: Some (TD)DFT investigations are based on structures with a phenylethinyl ligand being in coplanar arrangement with the pincer moiety.[ 8b , 25 ] This may be misleading with respect to the interpretation of photophysical properties. The [(C^N^C)AuIII] motif belongs to the C 2v point group. Thus, πCNC→π*CNC intraligand transitions (IL) are of B1 symmetry, that is, dipole allowed. The same holds for the non‐palindromic [(C^C^D)AuIII] structure, whose symmetry is Cs (A’). The character of πethinyl→π*CNC interligand transitions (LL'CT) depends on the orientation of the PhCC: In a coplanar arrangement, LL'CT is of A1 symmetry (A’ in Cs), that is, an allowed transition; a tilted arrangement changes the π→π* LL'CT to A2, which is dipole forbidden in C 2v. In Cs, the latter is dipole allowed in one direction (A’’). The transition intensities are strongly affected by these symmetry properties, thus, being important for a sound discussion of photophysics.
Absorption and emission properties
Absorption and emission spectra of complexes 34, 35, 39 and 40 are shown in Figure 2. The photophysical properties are summarized in Table 1. We note that we did not observe any degradation of these complexes in the presence of light over the course of a couple of days neither in toluene nor in CH2Cl2.
Figure 2.
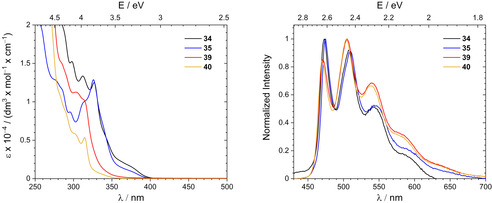
Left: UV/Vis spectra of [(C^C^N)AuIIICCPh)] (34), [(C^C^N)AuIIIC6F5)] (35), [(C^C^C’)AuIIICCPh)] (39) and [(C^C^C’)AuC6F5)] (40). Right: Emission spectra in solution. All spectra were recorded at 293 K in dry and degassed CH2Cl2. The excitation wavelength was chosen to match the respective first absorption maximum.
Table 1.
Photophysical data of gold complexes 34, 35, 39 and 40 in solution[a] at 293 K.
|
Complex |
|
|
|
|
|
|
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
34 |
298, 312, 327, 376 |
1.52, 1.34, 1.25, 0.15 |
473, 507, 543, 588 |
0.03 |
0.8 |
0.38 |
12.13 |
|||||||
|
35 |
278, 285, 295, 314, 326, 370 |
1.27, 1.16, 0.87, 1.00, 1.29, 0.12 |
475, 410, 546, 589 |
0.09 |
4.1 |
0.22 |
2.22 |
|||||||
|
39 |
284, 303, 314 |
1.51, 1.13, 1.02 |
471, 505, 540, 581 |
0.03 |
1.0 |
0.30 |
0.97 |
|||||||
|
40 |
302, 314, 336 |
0.58, 0.54, 0.06 |
470, 504, 539, 582 |
0.10 |
0.5 |
2.0 |
1.80 |
[a] Dried and deoxygenated dichloromethane. [b] Absolute quantum yields, determined by use of an integrating sphere (c=10−4 mol dm−3). [c] Phosphorescence lifetime. [d] Rate constant phosphorescence, . [e] Rate constant radiationless relaxation, .
Like the (C^C^N) complexes reported by Nevado et al., [23] the absorption bands of 34 and 35 are blue shifted compared to the palindromic (C^N^C) analogues.[ 10a , 16 ] The vibronically structured bands between 330 and 280 nm (ϵ ≈ 5000–15 000 dm3 mol−1 cm−1) with peak distances between 1200 cm−1 and 1400 cm−1 being typical for the pincer's ligand breathing modes probably arise from π→π* IL transitions.[ 10a , 23 , 60 ] LL'CT transitions responsible for the first absorption bands can be ruled out because in this case the absorption profile of phenylethynyl (34) and pentafluorophenyl (35) derivate should considerably differ.
The (C^C^C’) complexes 39 and 40 exhibit further blue‐shifted absorptions. Obviously, the lateral carbene donor leads to electronic states shifted to higher energies compared to the (C^C^N) congeners. This is rooted in the higher energy levels of imidazole π* orbitals [61] as well as the stronger ligand field splitting causing higher lying gold centred d‐orbitals. Thus, the absorption spectra of 39 and 40 clearly render the electron rich nature of the goldIII atom entailed by the (C^C^C’) pincer.
The investigated complexes show intense luminescence in solution at room temperature with emission lifetimes in the micro‐ and sub‐microsecond region indicating phosphorescence. The complexes are not emissive in the solid state maybe due to triplet–triplet annihilation [62] facilitated by the close proximity of the complex molecules in the solid state: for instance, the distance of the planes spanned by the pincer ligand between two molecules of 40 in the solid state is found to be only 347 pm (Figure S10, Supporting Information).
The emission pattern of all complexes is very similar. The vibronic structure with band distances of about 1300 cm−1 indicates pincer π*→π centred transitions. Interestingly, the carbene donor does not alter the emission profile. The emission wavelengths of the (C^C^C’) complexes 39 and 40 are negligibly shifted to higher energies. In addition, the PhCC or C6F5 ligands do not have any impact on the emission profile, thus, the transition is detached from these ligands. The emission spectra resemble the ones of other [(C^C^N)AuIII] [23] and [(C^N^C)AuIII] complexes[ 10 , 16 ] as well as biphenyl‐based [(C^C)AuIII] [63] or cyclometalated pyridine [64] and carbene [(C^C’)AuIII] complexes. [29] Thus, the π orbital of the emissive π→π* 3IL state is almost exclusively centred at the (bi)phenyl unit of the respective pincer ligand.
Emission quantum yields Φ in dichloromethane solution at room temperature range between 3 and 10 % being higher than the yields of many (C^N^C) analogues. [10] Obviously, the quantum yields are not affected by the lateral donor of the pincer, but the ligand besides the pincer: The C6F5 substituted systems 39 and 40 phosphoresce up to three times more efficient than the PhCC analogues 34 and 35. This is especially noteworthy, since the opposite finding was reported by Venkatesan and co‐workers about the luminescence of cyclometalated [(C^N)AuIII] [64] and [(C^C’)AuIII] [29] complexes. The authors found higher emission quantum yields for phenylethinyl substituted complexes than for pentafluorophenyl substituted ones. Furthermore, Bochmann and co‐workers have shown for palindromic [(C^N^C)AuIII] phenyl complexes that the phosphorescence quantum yield increases when going from pentafluorophenyl [(C^N^C)AuIIIC6F5] [65] to less fluorinated variants like [(C^N^C)AuIIIC6F4H] (Φ=0.6 %) and [(C^N^C)AuIIIC6H4F] (Φ=1.3 %). [16] This may be understood from the weaker donor strength of (per)fluorinated aryl ligands resulting in smaller ligand field splitting of the gold(III) atom. In contrast, the PhCC ligand is a sufficiently strong donor to enable phosphorescence.[ 10 , 66 ] Obviously, the non‐palindromic pincer motifs reported in the present study overcompensate the weaker ligand‐field splitting, that is, the ligand field splitting of 35 and 40 is strong enough despite the weak pentafluorophenyl donor. However, this does not explain why the latter complexes outperform the phenylethynyl 34 and 39 regarding phosphorescence quantum yields. In order to shed some more light on these aspects, we performed quantum chemical calculations.
Quantum chemical calculations
To investigate the phosphorescence properties of the four complexes, and especially the different quantum yields observed for the differently substituted complexes, GW/Bethe–Salpeter and time‐dependent density‐functional theory (TD‐DFT) calculations using the TPSSh functional were performed at the quasi‐relativistic two‐component level including spin–orbit coupling.
Absorption spectra were found to be in good agreement with experimental data. When optimizing the first excited triplet state, we find a shift of the emission lines for all four complexes to a nearly constant value. Calculated 0←0 triplet emission energies are found at 619 nm/2.00 eV (34), 620 nm/2.00 eV (35), 623 nm/1.99 eV (39), and 626 nm/1.98 eV (40) while a natural‐transition‐orbital (NTO) analysis confirms the π←π* triplet intraligand (3IL) character of this excitation as described in the previous section. The TD‐DFT (TPSSh) and GW/BSE calculations are in good mutual agreement. GW/BSE excitation and emission energies are slightly red shifted by approximately 0.15 eV.
Even though there is good agreement between the predicted and observed spectra, the quantum yield needs further investigation, as the difference between the PhCC and C6F5‐ligated systems cannot be explained by the emission spectra alone. A main difference between these complexes is given by their ability to rotate about the Au‐C axis. For the phenylethynyl complexes 39 and 34, we have obtained rotational barriers of only 2.0 kJ mol−1 and 1.1 kJ mol−1, respectively, and therefore assume a rapid rotation of the ligand at room temperature, as it was done by Yam et al. [8b] Due to steric hindrance, however, no rotation is observed for the bulkier pentafluorophenyl ligand, and a stable transition‐state geometry could not be located. In the vicinity of the transition‐state geometry, where the phenylethinyl and pincer ligands are nearly coplanar, the first triplet excited state changes its character from intra‐ (IL) to interligand charge‐transfer (LL'CT, Figure 3). This allows for an efficient pathway to release the excess energy of the excited state, suppressing the phosphorescence of complexes 34 and 39. In the tilted geometry, this LL'CT state is also present, but only as a higher‐lying excited state. Therefore, for the bulkier −C6F5 ligand, this non‐radiative relaxation pathway is closed as the overlap between the π systems is never sufficiently high at any reasonable geometry to yield a significant transition dipole moment.
Figure 3.
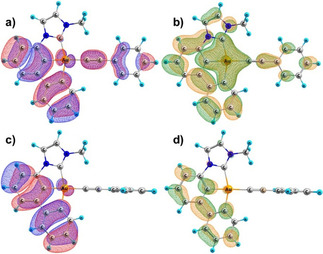
Natural transition orbitals of the hole (red/blue); particle (green/orange) pairs of the first triplet excitation in the planar (a, b) and tilted (c, d) configuration of complex 39.
But even for the PhCC complexes, the overall transition probability is still not high in the coplanar configuration, explaining why only diminished emission quantum yields are observed instead of full quenching. The different transition dipole moment's magnitude for tilted (LL'CT: A’’) and coplanar geometry (LL'CT: A’) nicely correspond to a qualitative picture based on group theory considerations.
We note that rotation is also possible for alkyl ligands, for which high phosphorescence quantum yields have been observed. [12] However, the non‐radiative LL'CT relaxation pathway does not exist for alkyl ligands due to the lack of an adjacent π system.
For a detailed overview of the methods used, excitation and emission spectra including spin‐orbit coupling, and a detailed analysis of the character of the corresponding relevant excitations, we refer to the Supporting Information.
Conclusions
In summary, we have described the development of a synthetic access to donor substituted 2,2’‐dihalo biphenyls which are suitable pre‐ligands for the preparation of highly luminescent [(C^C^D)AuIII] (D=pyridine, NHC) complexes by means of a transmetalation sequence. This synthetic methodology expands the possibilities for gold(III) pincer complex synthesis and constitutes a complementary variant to the approach of Nevado and co‐workers. In addition, we report on the first example of a pincer complex comprising two anionic, sp2‐hybridized and one neutral l‐type carbon donors.
We investigated the prepared pincer complexes by means of X‐ray diffraction analysis, UV/Vis and emission spectroscopy and supported our experimental findings by quantum chemical calculations on the TD‐DFT and GW/Bethe‐Salpeter level of theories. We could show the outstanding electronic properties of the non‐palindromic pincer motif compared to its palindromic congeners. A strong ligand field splitting is reflected in blue shifted absorptions and high emission quantum yields in solution at room temperature. In addition, we could clarify non‐radiative relaxation pathways and support our description by group theoretical arguments.
Upcoming studies currently being performed in our group are focused on modified procedures to introduce functional groups into the pincer moieties to systematically investigate and tune the chemical and photophysical properties of derived complexes. Furthermore, the dihalobiphenyls are examined with regard to their applicability to other transition metal complexes and main group elements probably opening up a rich chemistry with possible applications in catalysis, photophysics and ‐chemistry as well as material or pharmaceutical sciences.
Crystallographic data
Deposition Numbers 1986483, 1986486, 2009470, 2009476, 2009473, 2009475, 2009471, 2009477, 1986481, 2009474, 2009472, 1991871 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Waldemar Konrad for valuable support with chromatographic separations. We thank Prof. Joachim Podlech for proofreading the manuscript. W.F. gratefully acknowledges the Carl‐Zeiss Stiftung for a PhD scholarship as well as the Studienstiftung des deutschen Volkes for general support. Financial support by the Collaborative Research Centre CRC/Transregio 88, “Cooperative effects in homo‐ and heterometallic complexes (3MET)” is gratefully acknowledged (Projects B4 and C1). Open access funding enabled and organized by Projekt DEAL.
W. Feuerstein, C. Holzer, X. Gui, L. Neumeier, W. Klopper, F. Breher, Chem. Eur. J. 2020, 26, 17156.
Contributor Information
Prof. Dr. Wim Klopper, Email: klopper@kit.edu.
Prof. Dr. Frank Breher, Email: breher@kit.edu.
References
- 1. Tang M. C., Chan A. K., Chan M. Y., Yam V. W., Top. Curr. Chem. 2016, 374, 46. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Baldo M. A., O'Brien D. F., Thompson M. E., Forrest S. R., Phys. Rev. B 1999, 60, 14422–14428; [Google Scholar]
- 2b. Yang X., Neher D., Hertel D., Däubler T. K., Adv. Mater. 2004, 16, 161–166; [Google Scholar]
- 2c. Tao Y., Yang C., Qin J., Chem. Soc. Rev. 2011, 40, 2943–2970; [DOI] [PubMed] [Google Scholar]
- 2d. D'Andrade B. W., Thompson M. E., Forrest S. R., Adv. Mater. 2002, 14, 147–151; [Google Scholar]
- 2e. Baldo M. A., Thompson M. E., Forrest S. R., Nature 2000, 403, 750–753. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Lamansky S., Djurovich P., Murphy D., Abdel-Razzaq F., Lee H. E., Adachi C., Burrows P. E., Forrest S. R., Thompson M. E., J. Am. Chem. Soc. 2001, 123, 4304–4312; [DOI] [PubMed] [Google Scholar]
- 3b. Kawamura Y., Goushi K., Brooks J., Brown J. J., Sasabe H., Adachi C., Appl. Phys. Lett. 2005, 86, 071104; [Google Scholar]
- 3c. Bolink H. J., Coronado E., García Santamaria S., Sessolo M., Evans N., Klein C., Baranoff E., Kalyanasundaram K., Graetzel M., Nazeeruddin M. K., Chem. Commun. 2007, 3276–3278; [DOI] [PubMed] [Google Scholar]
- 3d. Lai P. N., Brysacz C. H., Alam M. K., Ayoub N. A., Gray T. G., Bao J., Teets T. S., J. Am. Chem. Soc. 2018, 140, 10198–10207. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Rudmann H., Shimada S., Rubner M. F., J. Am. Chem. Soc. 2002, 124, 4918–4921; [DOI] [PubMed] [Google Scholar]
- 4b. Welter S., Brunner K., Hofstraat J. W., De Cola L., Nature 2003, 421, 54–57; [DOI] [PubMed] [Google Scholar]
- 4c. Chi Y., Chou P. T., Chem. Soc. Rev. 2007, 36, 1421–1431. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Baldo M. A., O'Brien D. F., You Y., Shoustikov A., Sibley S., Thompson M. E., Forrest S. R., Nature 1998, 395, 151–154; [Google Scholar]
- 5b. Hissler M., McGarrah J. E., Connick W. B., Geiger D. K., Cummings S. D., Eisenberg R., Coord. Chem. Rev. 2000, 208, 115–137; [Google Scholar]
- 5c. Kui S. C., Hung F. F., Lai S. L., Yuen M. Y., Kwok C. C., Low K. H., Chui S. S., Che C. M., Chem. Eur. J. 2012, 18, 96–109; [DOI] [PubMed] [Google Scholar]
- 5d. Turner E., Bakken N., Li J., Inorg. Chem. 2013, 52, 7344–7351; [DOI] [PubMed] [Google Scholar]
- 5e. Cebrián C., Mauro M., Beilstein J. Org. Chem. 2018, 14, 1459–1481; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5f. Lee C., Zaen R., Park K.-M., Lee K. H., Lee J. Y., Kang Y., Organometallics 2018, 37, 4639–4647. [Google Scholar]
- 6.
- 6a. Wong K. M., Zhu X., Hung L. L., Zhu N., Yam V. W., Kwok H. S., Chem. Commun. 2005, 2906–2908; [DOI] [PubMed] [Google Scholar]
- 6b. Au V. K., Wong K. M., Tsang D. P., Chan M. Y., Zhu N., Yam V. W., J. Am. Chem. Soc. 2010, 132, 14273–14278; [DOI] [PubMed] [Google Scholar]
- 6c. Au V. K., Tsang D. P., Wong K. M., Chan M. Y., Zhu N., Yam V. W., Inorg. Chem. 2013, 52, 12713–12725; [DOI] [PubMed] [Google Scholar]
- 6d. Tang M. C., Tsang D. P., Chan M. M., Wong K. M., Yam V. W., Angew. Chem. Int. Ed. 2013, 52, 446–449; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 464–467; [Google Scholar]
- 6e. Tang M. C., Tsang D. P., Wong Y. C., Chan M. Y., Wong K. M., Yam V. W., J. Am. Chem. Soc. 2014, 136, 17861–17868; [DOI] [PubMed] [Google Scholar]
- 6f. Lee C. H., Tang M. C., Wong Y. C., Chan M. Y., Yam V. W., J. Am. Chem. Soc. 2017, 139, 10539–10550; [DOI] [PubMed] [Google Scholar]
- 6g. To W. P., Zhou D., Tong G. S. M., Cheng G., Yang C., Che C. M., Angew. Chem. Int. Ed. 2017, 56, 14036–14041; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14224–14229; [Google Scholar]
- 6h. Lee C. H., Tang M. C., Cheung W. L., Lai S. L., Chan M. Y., Yam V. W., Chem. Sci. 2018, 9, 6228–6232; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6i. Tang M. C., Leung M. Y., Lai S. L., Ng M., Chan M. Y., Wing-Wah Yam V., J. Am. Chem. Soc. 2018, 140, 13115–13124; [DOI] [PubMed] [Google Scholar]
- 6j. Li L.-K., Tang M.-C., Lai S.-L., Ng M., Kwok W.-K., Chan M.-Y., Yam V. W.-W., Nature Photonics 2019, 13, 185–191. [Google Scholar]
- 7.
- 7a. Ballhausen C. J., Bjerrum N., Dingle R., Eriks K., Hare C. R., Inorg. Chem. 1965, 4, 514–518; [Google Scholar]
- 7b. Andrews L. J., J. Phys. Chem. 1979, 83, 3203–3209. [Google Scholar]
- 8.
- 8a. Bronner C., Wenger O. S., Dalton Trans. 2011, 40, 12409–12420; [DOI] [PubMed] [Google Scholar]
- 8b. Lam E. S., Lam W. H., Yam V. W., Inorg. Chem. 2015, 54, 3624–3630. [DOI] [PubMed] [Google Scholar]
- 9. Wong K.-H., Cheung K.-K., Chan M. C.-W., Che C.-M., Organometallics 1998, 17, 3505–3511. [Google Scholar]
- 10.
- 10a. Yam V. W.-W., Wong K. M.-C., Hung L.-L., Zhu N., Angew. Chem. Int. Ed. 2005, 44, 3107–3110; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3167–3170; [Google Scholar]
- 10b. Wong K. M., Hung L. L., Lam W. H., Zhu N., Yam V. W., J. Am. Chem. Soc. 2007, 129, 4350–4365. [DOI] [PubMed] [Google Scholar]
- 11. Au V. K., Wong K. M., Zhu N., Yam V. W., J. Am. Chem. Soc. 2009, 131, 9076–9085. [DOI] [PubMed] [Google Scholar]
- 12. To W. P., Tong G. S. M., Cheung C. W., Yang C., Zhou D., Che C. M., Inorg. Chem. 2017, 56, 5046–5059. [DOI] [PubMed] [Google Scholar]
- 13. Currie L., Fernandez-Cestau J., Rocchigiani L., Bertrand B., Lancaster S. J., Hughes D. L., Duckworth H., Jones S. T., Credgington D., Penfold T. J., Bochmann M., Chem. Eur. J. 2017, 23, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fernandez-Cestau J., Bertrand B., Blaya M., Jones G. A., Penfold T. J., Bochmann M., Chem. Commun. 2015, 51, 16629–16632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee C. H., Tang M. C., Kong F. K., Cheung W. L., Ng M., Chan M. Y., Yam V. W., J. Am. Chem. Soc. 2020, 142, 520–529. [DOI] [PubMed] [Google Scholar]
- 16. Roşca D. A., Smith D. A., Bochmann M., Chem. Commun. 2012, 48, 7247–7249. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Roşca D. A., Smith D. A., Hughes D. L., Bochmann M., Angew. Chem. Int. Ed. 2012, 51, 10643–10646; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10795–10798; [Google Scholar]
- 17b. Rocchigiani L., Fernandez-Cestau J., Chambrier I., Hrobarik P., Bochmann M., J. Am. Chem. Soc. 2018, 140, 8287–8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Savjani N., Rosca D. A., Schormann M., Bochmann M., Angew. Chem. Int. Ed. 2013, 52, 874–877; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 908–911. [Google Scholar]
- 19. Rocchigiani L., Fernandez-Cestau J., Agonigi G., Chambrier I., Budzelaar P. H. M., Bochmann M., Angew. Chem. Int. Ed. 2017, 56, 13861–13865; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14049–14053. [Google Scholar]
- 20. Li C. K., Sun R. W., Kui S. C., Zhu N., Che C. M., Chem. Eur. J. 2006, 12, 5253–5266. [DOI] [PubMed] [Google Scholar]
- 21. Luo H., Cao B., Chan A. S. C., Sun R. W. Y., Zou T., Angew. Chem. Int. Ed. 2020, 59, 3131–3136; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3155–3160. [Google Scholar]
- 22. Peris E., Crabtree R. H., Chem. Soc. Rev. 2018, 47, 1959–1968. [DOI] [PubMed] [Google Scholar]
- 23. Kumar R., Linden A., Nevado C., Angew. Chem. Int. Ed. 2015, 54, 14287–14290; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14495–14498. [Google Scholar]
- 24. Beucher H., Kumar S., Merino E., Hu W.-H., Stemmler G., Cuesta-Galisteo S., González J. A., Jagielski J., Shih C.-J., Nevado C., Chem. Mater. 2020, 32, 1605–1611. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Ming Tong G. S., Chan K. T., Chang X., Che C. M., Chem. Sci. 2015, 6, 3026–3037; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Yang B. Z., Zhou X., Liu T., Bai F. Q., Zhang H. X., J. Phys. Chem. A 2009, 113, 9396–9403. [DOI] [PubMed] [Google Scholar]
- 26. Kumar R., Krieger J. P., Gomez-Bengoa E., Fox T., Linden A., Nevado C., Angew. Chem. Int. Ed. 2017, 56, 12862–12865; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13042–13045. [Google Scholar]
- 27. Beucher H., Merino E., Genoux A., Fox T., Nevado C., Angew. Chem. Int. Ed. 2019, 58, 9064–9067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9162–9165. [Google Scholar]
- 28. Roşca D. A., Fernández-Cestau J., Morris J., Wright J. A., Bochmann M., Sci. Adv. 2015, 1, e1500761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. von Arx T., Szentkuti A., Zehnder T. N., Blacque O., Venkatesan K., J. Mater. Chem. C 2017, 5, 3765–3769. [Google Scholar]
- 30. Crespo O., Gimeno M. C., Laguna A., Montanel-Pérez S., Villacampa M. D., Organometallics 2012, 31, 5520–5526. [Google Scholar]
- 31. Holzer C., Klopper W., J. Chem. Phys. 2018, 149, 101101. [DOI] [PubMed] [Google Scholar]
- 32. Usón R., Vicente J., Cirac J. A., Chicote M. T., J. Organomet. Chem. 1980, 198, 105–112. [Google Scholar]
- 33. Uson R., Laguna A., Vicente J., J. Organomet. Chem. 1977, 131, 471–475. [Google Scholar]
- 34. David B., Monkowius U., Rust J., Lehmann C. W., Hyzak L., Mohr F., Dalton Trans. 2014, 43, 11059–11066. [DOI] [PubMed] [Google Scholar]
- 35. Wu C. Y., Horibe T., Jacobsen C. B., Toste F. D., Nature 2015, 517, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Joost M., Estevez L., Miqueu K., Amgoune A., Bourissou D., Angew. Chem. Int. Ed. 2015, 54, 5236–5240; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5325–5329. [Google Scholar]
- 37. Chu J., Munz D., Jazzar R., Melaimi M., Bertrand G., J. Am. Chem. Soc. 2016, 138, 7884–7887. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Nilakantan L., McMillin D. R., Sharp P. R., Organometallics 2016, 35, 2339–2347; [Google Scholar]
- 38b. Chan K. T., Tong G. S. M., Wan Q., Cheng G., Yang C., Che C. M., Chem. Asian J. 2017, 12, 2104–2120. [DOI] [PubMed] [Google Scholar]
- 39. Neugebauer W., Kos A. J., von Ragué Schleyer P., J. Organomet. Chem. 1982, 228, 107–118. [Google Scholar]
- 40. Kürti L., Czakó B., Strategic Applications of Named Reactions in Organic Synthesis, Cambridge Academic Press, Cambridge, 2005. [Google Scholar]
- 41.
- 41a. Espinet P., Echavarren A. M., Angew. Chem. Int. Ed. 2004, 43, 4704–4734; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4808–4839; [Google Scholar]
- 41b. Renaldo A. F., Labadie J. W., Stille J. K., Org. Synth. 1989, 67, 86–97. [Google Scholar]
- 42.
- 42a. Düfert M. A., Billingsley K. L., Buchwald S. L., J. Am. Chem. Soc. 2013, 135, 12877–12885; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42b. Bruno N. C., Tudge M. T., Buchwald S. L., Chem. Sci. 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- 44. Sun H., Tang S., Li D., Zhou Y., Huang J., Zhu Q., Org. Biomol. Chem. 2018, 16, 3893–3896. [DOI] [PubMed] [Google Scholar]
- 45. Baik W., Luan W., Lee H. J., Yoon C. H., Koo S., Kim B. H., Can. J. Chem. 2005, 83, 213–219. [Google Scholar]
- 46. Doyle M. P. S., Siegfried B., F. F. Dellaria, Jr. , J. Org. Chem. 1977, 42, 2426–2431. [Google Scholar]
- 47. Qiu D., Meng H., Jin L., Wang S., Tang S., Wang X., Mo F., Zhang Y., Wang J., Angew. Chem. Int. Ed. 2013, 52, 11581–11584; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11795–11798. [Google Scholar]
- 48.
- 48a. Cornforth J., Huguenin L. M., Wilson J. R. H., J. Chem. Soc. Perkin Trans. 1 1987, 871–875; [Google Scholar]
- 48b. Ross S. D., Kuntz I., J. Am. Chem. Soc. 1951, 73, 1297–1302; [Google Scholar]
- 48c. Lee D. S., Chatterjee T., Ban J., Rhee H., Cho E. J., ChemistrySelect 2018, 3, 2092–2095. [Google Scholar]
- 49. Liu C. Y., Knochel P., Org. Lett. 2005, 7, 2543–2546. [DOI] [PubMed] [Google Scholar]
- 50. Krasovskiy A., Knochel P., Angew. Chem. Int. Ed. 2004, 43, 3333–3336; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3396–3399. [Google Scholar]
- 51. King A. O., Okukado N., Negishi E.-i., J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar]
- 52.
- 52a. Rieke R. D., Science 1989, 246, 1260–1264; [DOI] [PubMed] [Google Scholar]
- 52b. Zhu L., Wehmeyer R. M., Rieke R. D., J. Org. Chem. 1991, 56, 1445–1453; [Google Scholar]
- 52c. Kudret S., Haen J. D., Lutsen L., Vanderzande D., Maes W., Adv. Synth. Catal. 2013, 355, 614. [Google Scholar]
- 53.
- 53a. Liu C.-Y., Knochel P., Synlett 2007, 2007, 2081–2085; [Google Scholar]
- 53b. Krasovskiy A., Malakhov V., Gavryushin A., Knochel P., Angew. Chem. Int. Ed. 2006, 45, 6040–6044; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6186–6190; [Google Scholar]
- 53c. Piller F. M., Metzger A., Schade M. A., Haag B. A., Gavryushin A., Knochel P., Chem. Eur. J. 2009, 15, 7192–7202. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Nishiwaki K., Ogawa T., Shigeta K., Takahashi K., Matsuo K., Tetrahedron 2006, 62, 7034–7042; [Google Scholar]
- 54b. Nishiwaki K., Ogawa T., Tagami K.-i., Tanabe G., Muraoka O., Matsuo K., Tetrahedron 2006, 62, 10854–10858; [Google Scholar]
- 54c. Reingruber R., Vanderheiden S., Wagner A., Nieger M., Muller T., Es-Sayed M., Bräse S., Eur. J. Org. Chem. 2008, 3314–3327. [Google Scholar]
- 55. Barbero M., Degani I., Diulgheroff N., Dughera S., Fochi R., Synthesis 2001, 2001, 2180–2190. [Google Scholar]
- 56. Satyamurthy N., Barrio J. R., J. Org. Chem. 1983, 48, 4394–4396. [Google Scholar]
- 57.
- 57a. Ku H., Barrio J. R., J. Org. Chem. 1981, 46, 5239–5241; [Google Scholar]
- 57b. Wu Z., Moore J. S., Tetrahedron Lett. 1994, 35, 5539–5542. [Google Scholar]
- 58. Brückner R., Reaktionsmechanismen: organische Reaktionen, Stereochemie, moderne Synthesemethoden, Springer, 2014. [Google Scholar]
- 59. Miller W. T., Sun K. K., J. Am. Chem. Soc. 1970, 92, 6985–6987. [Google Scholar]
- 60. Unger Y., Meyer D., Molt O., Schildknecht C., Munster I., Wagenblast G., Strassner T., Angew. Chem. Int. Ed. 2010, 49, 10214–10216; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10412–10414. [Google Scholar]
- 61. Fleming I., Molecular Orbitals and Organic Chemical Reactions, Wiley, Chichester, 2010. [Google Scholar]
- 62. Turro N. J. T., Ramamurthy V., Scaiano J. C., Modern Molecular Photochemistry of Organic Molecules, Vol. 10, University Science Books, Sausalito, 2010. [Google Scholar]
- 63. Garg J. A., Blacque O., Fox T., Venkatesan K., Inorg. Chem. 2010, 49, 11463–11472. [DOI] [PubMed] [Google Scholar]
- 64. Bachmann M., Fessler R., Blacque O., Venkatesan K., Dalton Trans. 2019, 48, 7320–7330. [DOI] [PubMed] [Google Scholar]
- 65.The pentafluorophenyl derivative shows luminescence with a quantum yield of about 1 %, however, this is rooted in a fluorescence as clearly seen from a small Stokes shift, emission lifetimes in the nanosecond region and different vibronic bands.
- 66.The stronger donor properties of phenylethinyl compared to pentafluorophenyl is also reflected in a blue shifted absorption of the [(C^N^C)AuIII] phenylethinyl complex compared to its C6F5 pendant.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary