

Abstract
Aims: Lifelong pain is a hallmark feature of sickle cell disease (SCD). How sickle pathobiology evokes pain remains unknown. We hypothesize that increased cell-free heme due to ongoing hemolysis activates toll-like receptor 4 (TLR4), leading to the formation of reactive oxygen species (ROS) and endoplasmic reticulum (ER) stress. Together, these processes lead to spinal microglial activation and neuroinflammation, culminating in acute and chronic pain.
Results: Spinal heme levels, TLR4 transcripts, oxidative stress, and ER stress were significantly higher in sickle mice than controls. In vitro, TLR4 inhibition in spinal cord microglial cells attenuated heme-induced ROS and ER stress. Heme treatment led to a time-dependent increase in the characteristic features of sickle pain (mechanical and thermal hyperalgesia) in both sickle and control mice; this effect was absent in TLR4-knockout sickle and control mice. TLR4 deletion in sickle mice attenuated chronic and hypoxia/reoxygenation (H/R)-evoked acute hyperalgesia. Sickle mice treated with the TLR4 inhibitor resatorvid; selective small-molecule inhibitor of TLR4 (TAK242) had significantly reduced chronic hyperalgesia and had less severe H/R-evoked acute pain with quicker recovery. Notably, reducing ER stress with salubrinal ameliorated chronic hyperalgesia in sickle mice.
Innovation: Our findings demonstrate the causal role of free heme in the genesis of acute and chronic sickle pain and suggest that TLR4 and/or ER stress are novel therapeutic targets for treating pain in SCD.
Conclusion: Heme-induced microglial activation via TLR4 in the central nervous system contributes to the initiation and maintenance of sickle pain via ER stress in SCD. Antioxid. Redox Signal. 34, 279–293.
Keywords: pain, sickle cell disease, heme, toll-like receptor 4, endoplasmic reticulum stress, glial cell
Introduction
Sickle cell disease (SCD), caused by a point mutation in the β globin gene, afflicts millions of people worldwide (43). SCD is characterized by chronic hemolysis, inflammation, organ damage, and pain (16, 29, 60). In the deoxygenated state, hemoglobin containing mutated β globin (HbS) can polymerize, causing sickling of red blood cells (RBCs); these sickle RBCs occlude blood vessels, resulting in ischemia-reperfusion injury and acute painful vaso-occlusive crises (VOCs) (16). In addition to recurrent acute VOCs, many adults with SCD suffer from chronic pain (52). Pain is the most common lifelong manifestation of SCD, can lead to hospitalization, and can begin in infancy and worsen throughout life (52). Morphine is the common analgesic treatment; however, chronic systemic administration of morphine can lead to many complications (19).
Therefore, understanding how sickle pathobiology evokes pain is critical to developing targeted therapies to prevent pain at its source.
Emerging evidence reveals that neurogenic inflammation (29), peripheral nerve sensitization (26), and central sensitization (10) are involved in chronic sickle pain. Thus, pain in SCD may be a manifestation of events that are activated by multiple factors (57). Patients with SCD have a higher plasma concentration of cell-free heme (iron with porphyrin) than healthy individuals (1). Chronic hemolysis, frequent blood transfusions, and shortened RBC survival cause elevated heme levels in the circulation and tissues (1, 59). Transcriptomic analysis of peripheral blood mononuclear cells from SCD patients showed a > 100-fold increase in toll-like receptor 4 (TLR4) and an iron-regulated gene signature (59).
Heme has been shown to activate TLR4 (15) and potentiate microglial activation (36). Moreover, TLR4 activation is involved in pathological pain induced by nerve injury, peripheral inflammation, cancer, and opioids (2, 25, 30, 33, 35, 39, 48, 57, 62). TLR4 activation in microglial cells facilitates neuropathic and inflammatory pain (61). TLR4 is also crucial to the development of endoplasmic reticulum (ER) stress (28, 44), and ER stress has been associated with the initiation and maintenance of inflammatory and neuropathic pain (24, 63). Whether and how heme drives TLR4 activation in sickle pain is unknown.
Innovation
Acute and chronic pain in sickle cell disease (SCD) can start in infancy and may continue even after curative therapies. Although analgesic therapies are being investigated, an unmet need is to prevent pain from being evoked in SCD. We demonstrate that cell-free heme from ongoing hemolysis in SCD stimulates endoplasmic reticulum (ER) stress via the activation of glial cell toll-like receptor 4 (TLR4) leading to pain. Targeting of TLR4 and ER stress prevents and/or reduces chronic as well as acute pain. Thus, TLR4 and ER stress are novel, treatable targets to prevent sickle pain with a translational potential.
In sickle mice, cell-free heme was shown to trigger endothelial activation, vaso-occlusion (3, 4), and acute lung injury (17) via TLR4. We previously demonstrated elevated TLR4 transcripts in spinal cords from sickle mice as well as increased glial activation in the spinal cords (29, 58, 60). In this study, we hypothesized that in the sickle microenvironment heme induces TLR4 activation, which exacerbates neuroinflammation, culminating in hyperalgesia.
We found that heme induced hyperalgesia in sickle and normal control mice via TLR4. Pharmacological inhibition or genetic deletion of TLR4 decreased chronic hyperalgesia and hypoxia/reoxygenation (H/R)-evoked acute hyperalgesia in sickle mice. The inhibition of TLR4 in sickle mice decreased spinal inflammation. TLR4 inhibition attenuated heme-evoked spinal microglial cell activation. Heme significantly enhanced the formation of reactive oxygen species (ROS) in microglial cells, which was abolished by TLR4 inhibition. TLR4 inhibition also reduced markers of ER stress. Further, reducing ER stress ameliorated chronic hyperalgesia in sickle mice. Our results reveal a novel mechanism whereby heme-activated TLR4 results in elevated ER stress, leading to spinal microglial activation, which, in turn, underlies sickle pain.
Results
Heme-induced chronic hyperalgesia in sickle and normal mice
Significantly elevated heme, which is found in the circulation and tissues of SCD patients, was present in the spinal cords of sickle (transgenic mouse model of SCD expressing exclusively human HbS developed at Lawrence Berkeley National Laboratory [HbSS-BERK]) mice compared with control (HbAA-BERK [transgenic mouse model expressing exclusively normal human HbA developed at Lawrence Berkeley National Laboratory]) mice (Supplementary Fig. S1A). We determined the extent to which heme might affect chronic hyperalgesia.
In HbAA-BERK control mice, heme significantly increased mechanical hyperalgesia from day 2 (p < 0.001 vs. baseline and vehicle, Fig. 1A), heat sensitivity from day 4 (p < 0.05 vs. baseline and vehicle, Fig. 1B), and cold sensitivity from day 2 (p < 0.001 vs. baseline and vehicle, Fig. 1C). In HbSS-BERK sickle mice, heme administration also increased mechanical and cold hyperalgesia from day 4; however, heat hyperalgesia was only affected on day 6 (vs. baseline and vehicle, p < 0.05 for mechanical; p < 0.001 for heat and cold; Fig. 1A–C). Increased hyperalgesia was reversed after discontinuation of heme in a time-dependent manner (Fig. 1A–C). Heme had no effect on musculoskeletal hyperalgesia in control or sickle mice (Supplementary Fig. S2A, C). Heme also had no effect on body weight (Supplementary Fig. S2E).
FIG. 1.

Heme administration induces hyperalgesia in sickle and control. Mice received hemin (Hem, 32 μmol/kg/day of body weight, i.p.) or Veh for 5 days. Pain testing was performed before any treatment (BL), 20 h after each injection during treatment, and daily after hemin treatment for 7 days as described in treatment schema (Supplementary Fig. S5A). (A) Mechanical hyperalgesia (increase = more pain). (B) Heat sensitivity (decrease = more pain). (C) Cold sensitivity (increase = more pain). Age in months ± SEM: HbAA-BERK: 4.81 ± 0.06 (Veh, n = 5); 4.77 ± 0.03 (hemin, n = 6) and HbSS-BERK: 4.79 ± 0.04 (Veh, n = 5); 4.75 ± 0.05 (hemin, n = 6). (D, E) Deep tissue/musculoskeletal hyperalgesia as measured by grip force. (F) Body weight. Age in months ± SEM. HbAA-BERK: 4.81 ± 0.06 (Veh, n = 5); 4.77 ± 0.03 (hemin, n = 6) and HbSS-BERK: 4.79 ± 0.04 (Veh, n = 5); 4.75 ± 0.05 (hemin, n = 6). All data are mean ± SEM. *p < 0.05, **p < 0.001 in HbAA-BERK, and #p < 0.05, ##p < 0.001 in HbSS-BERK for treatment versus corresponding vehicle at each time-point (two-way ANOVA, Bonferroni). †p < 0.05, ††p < 0.001 in HbAA-BERK, and &p < 0.05, &&p < 0.001 in HbSS-BERK for each time-point versus BL in each treatment group (one-way ANOVA, Bonferroni). With respect to deep tissue/musculoskeletal hyperalgesia and body weight, statistical analysis of hemin versus corresponding vehicle at each time-point (two-way ANOVA, Bonferroni) and each time-point versus BL in hemin groups (one-way ANOVA, Bonferroni) showed no significant differences. AA: HbAA-BERK; Hem: hemin; SS: HbSS-BERK. ANOVA, analysis of variance; BL, baseline; HbA, normal human hemoglobin A (human alpha and beta A globins); HbAA-BERK, transgenic mouse model expressing exclusively normal human HbA developed at Lawrence Berkeley National Laboratory; HbS, hemoglobin containing mutated β globin; HbSS-BERK, transgenic mouse model of SCD expressing exclusively human HbS developed at Lawrence Berkeley National Laboratory; i.p., intraperitoneal; PWF, paw withdrawal frequency; PWL, paw withdrawal latency; SEM, standard error of the mean; Tx, treatment; Veh, vehicle; VF, von Frey (Semmes-Weinstein) monofilament. Color images are available online.
These data demonstrate the ability of heme to evoke mechanical and thermal hyperalgesia. Importantly, although the parameters of chronic hyperalgesia were high at baseline in the sickle mice, the effects of heme administration on these parameters, with respect to extent of change (increase), were significantly greater in the control mice. This suggests that changes in the measured parameters for the sickle mice were less affected by heme administration compared with control mice as a consequence of already elevated heme levels and hyperalgesia at baseline.
Heme-induced chronic hyperalgesia requires TLR4
In TLR4 knockout HbSS-BERK sickle (TLR4 knockout, genetic deletion [TLR4-KO]-HbSS-BERK) and TLR4 knockout HbAA-BERK control (TLR4-KO-HbAA-BERK) mice, heme failed to induce significant alterations in mechanical, thermal, or musculoskeletal hyperalgesia (Fig. 1D–F and Supplementary Fig. S2B, D) compared with vehicle treatment or baseline in sickle or control TLR4-knockout mice. Notably, there was no difference in baseline pain for control- or sickle-TLR4-knockout mice. These findings suggest that heme-induced mechanical and thermal hyperalgesia in mice requires and is mediated by TLR4. Heme treatment did not cause a change in the body weight of mice (Supplementary Fig. S2F).
TLR4 contributes to chronic hyperalgesia in sickle mice
The SCD patients have significantly increased expression of TLR4 (59); TLR4 activation, specifically in microglial cells, facilitates neuropathic and inflammatory pain (61). Moreover, heme has the potential to activate TLR4 (15, 36). Transcriptional expression of TLR4 was significantly higher in the spinal cords of sickle mice compared with control mice (Supplementary Fig. S1B). We, therefore, examined the role of TLR4 in chronic sickle pain both pharmacologically and genetically. TAK 242 is a potent and selective TLR4 inhibitor, as determined by its ability to inhibit TLR4-mediated inflammatory cytokine (interleukin 6 [IL-6], tumor necrosis factor α [TNF-α]) production. Resatorvid, a selective small-molecule inhibitor of TLR4 (TAK242) administration, significantly inhibited the production of IL-6 in the spinal cords of HbSS-BERK sickle mice (Supplementary Fig. S1C).
In HbSS-BERK mice, the administration of TAK 242 after 4 days reduced mechanical hyperalgesia (p < 0.05 vs. baseline; p < 0.001 vs. vehicle, Fig. 2A), heat sensitivity (p < 0.05 vs. baseline; p < 0.001 vs. vehicle, Fig. 2B), and cold sensitivity (p < 0.01 vs. vehicle, Fig. 2C). Musculoskeletal hyperalgesia was reduced by day 5 (p < 0.05 vs. baseline and vehicle, Fig. 2D). The discontinuation of TAK242 administration led to a time-dependent restoration of mechanical, thermal, and musculoskeletal hyperalgesia. After withdrawal of TAK242, the effect of TAK242 was sustained for 3 days for cold sensitivity and musculoskeletal hyperalgesia, 5 days for heat sensitivity, and 7 days for mechanical hyperalgesia (Fig. 2A–D). TAK242 showed no significant effect on mechanical, thermal, or musculoskeletal hyperalgesia in HbAA-BERK control mice (Fig. 2A–D).
FIG. 2.

TLR4 inhibition (TAK242) and TLR4 deletion reduces hyperalgesia in sickle mice. Mice (A–D) received TAK242 (1 mg/kg/day, i.v.) or Veh for 5 days. Pain testing was performed before any treatment (BL), 20 h after each injection during treatment, and daily after TAK242 treatment for 9 days (Supplementary Fig. S5B). (E–H) Pain performance was measured in HbSS-BERK without TLR4 (SS-TLR4-KO), HbAA-BERK without TLR4 (AA-TLR4-KO), receptor WT HbSS-BERK sickle mice, and HbAA-BERK control mice. (A, E) Mechanical hyperalgesia. (B, F) Heat sensitivity. (C, G) Cold sensitivity. (D, H). Deep tissue/musculoskeletal hyperalgesia. Mean age of each group in months ± SEM: (A–D) HbAA-BERK: 6.04 ± 0.18 (Veh, n = 6); 5.92 ± 0.33 (TAK242, n = 6) and HbSS-BERK: 6.07 ± 0.05 (Veh, n = 8); 6.00 ± 0.33 (TAK242, n = 8). (E–H) HbAA-BERK (5.92 ± 0.72, n = 8); HbSS-BERK (6.00 ± 0.88, n = 8); AA-TLR4-KO (5.88 ± 0.96, n = 8); SS-TLR4-KO (6.02 ± 0.89, n = 5). (A–D) HbAA-BERK: 6.04 ± 0.18; Veh n = 6, TAK242 n = 6; HbSS-BERK: 6.0 ± 0.33; Veh, n = 8, TAK242 n = 8). (E–H) HbAA-BERK: 5.9 ± 0.7 (n = 8); HbSS-BERK: 6.0 ± 0.9 (n = 8); AA-TLR4-KO: 5.9 ± 1.0 (n = 8); and SS-TLR4-KO: 6.0 ± 0.9 (n = 5). All data are mean ± SEM. #p < 0.05, ##p < 0.001 in HbSS-BERK for treatment versus corresponding vehicle at each time-point (two-way ANOVA, Bonferroni). &p < 0.05, &&p < 0.001 in HbSS-BERK for each time-point versus BL in each treatment group (one-way ANOVA, Bonferroni). **p < 0.001 (Student's unpaired t test). AA: HbAA-BERK; AA-TLR4-KO: TLR4 knockout HbAA-BERK; SS: HbSS-BERK; SS-TLR4-KO: TLR4 knockout HbSS-BERK; WT: TLR4 wild type. i.v., intravenous; TAK242, resatorvid; selective small molecule inhibitor of TLR4; TLR4, toll-like receptor 4; TLR4-KO, TLR4 knockout (genetic deletion); WT, wild type. Color images are available online.
These findings demonstrate that TLR4 inhibition ameliorates features of chronic hyperalgesia in the context of SCD in which TLR4 expression/activation is high; TLR4 activation may, thus, underlie the pathobiology of sickle pain. No changes in the body weight of mice were observed with TAK242 treatment (Supplementary Fig. S3A).
To validate the contribution of TLR4 in chronic sickle pain, we examined baseline pain characteristics in TLR4-KO-HbSS-BERK and TLR4-KO-HbAA-BERK mice. Compared with HbSS-BERK sickle mice, TLR4-KO-HbSS-BERK mice had significantly reduced mechanical hyperalgesia as measured by paw withdrawal frequency (PWF, p < 0.001, Fig. 2E) or mechanical threshold (p < 0.001, Supplementary Fig. S3B). These mice also demonstrate significantly reduced heat hyperalgesia (p < 0.001, Fig. 2F), cold sensitivity (p < 0.001, Fig. 2G), and musculoskeletal hyperalgesia (p < 0.001, Fig. 2H). There was no significant difference in pain behaviors between HbAA-BERK mice and either TLR4-KO-HbSS-BERK or TLR4-KO-HbAA-BERK (Fig. 2E–H). These data further confirm that TLR4 activation contributes to chronic pain in sickle mice.
TLR4 contributes to acute hyperalgesia evoked by H/R in sickle mice
H/R results in ischemia/reperfusion injury and acute pain in sickle mice that mimics features of VOC in SCD (32, 34). To determine whether TLR4 inhibition ameliorates H/R-induced sickle pain, we pretreated HbSS-BERK mice with TAK242 or vehicle. Pretreatment with TAK242 significantly reduced hyperalgesia before H/R exposure (Fig. 3A–D). Subsequent H/R exposure significantly increased mechanical, thermal, and musculoskeletal hyperalgesia in sickle mice pretreated with vehicle or TAK242 compared with the respective pain behaviors before H/R treatment on day 6 (p < 0.01, Fig. 3A–D). H/R-evoked mechanical, thermal, and musculoskeletal hyperalgesia were significantly lower in mice treated for 5 days with TAK242 than in vehicle-treated mice both immediately after H/R and for a period of at least 4 days after H/R (Fig. 3A–D). TAK242 treatment did not completely prevent H/R-evoked hyperalgesia.
FIG. 3.

TLR4 mediates H/R-induced acute pain in sickle mice. (A–D) HbSS-BERK (SS) mice received intralipid Veh or TAK242 (1 mg/kg/day, i.v.) for 5 days. Twenty-four hours after the last injection, mice were subjected to H/R treatment. Pain testing was performed before any treatment (BL), under normoxia before H/R (Tx, pre-H/R), immediately after H/R (H/R), and daily post-H/R (Supplementary Fig. S5C). (E–G) SS-TLR4-KO and age-matched TLR4 WT HbSS-BERK (SS-WT) sickle mice were subjected to H/R treatment. Pain behaviors were assessed under normoxia before H/R (BL), immediately after H/R (H/R), and at the indicated time-points. (A, E) Mechanical hyperalgesia. (B, F) Heat sensitivity. (C) Cold sensitivity. (D, G) Deep tissue hyperalgesia. Mean age of each group in months ± SEM. (A–D) 7.51 ± 0.04 (n = 5) for SS-Veh and 7.39 ± 0.07 (n = 5) for SS-TAK242. (E–G) 6.25 ± 1.02 (n = 5) for SS-WT and 5.92 ± 0.93 (n = 5) for SS-TLR4-KO. Each value is the mean ± SEM. †p < 0.05, ††p < 0.01 for TAK242 versus corresponding vehicle at each time-point (two-way ANOVA, Bonferroni). *p < 0.05, **p < 0.01 versus BL of corresponding group (one-way ANOVA, Bonferroni). #p < 0.05, ##p < 0.01 versus day 6 before H/R of corresponding group (one-way ANOVA, Bonferroni). £p < 0.05, ££p < 0.01 versus day 6 post H/R of corresponding group. ¶p < 0.001 for SS-TLR4-KO versus SS-WT at the respective time-point (two-way ANOVA, Bonferroni). TLR4-KO-SS, TLR4 knockout HbSS-BERK. H/R, hypoxia/reoxygenation. Color images are available online.
These data show that TAK242 pretreatment reduced the severity of hyperalgesia induced by H/R in sickle mice. No changes in weight of mice were observed after H/R or TAK242 treatment (Supplementary Fig. S4).
Heme mediates spinal microglial activation via TLR4-induced ROS and ER stress
ROS and ER stress, the critical players in nociception, are associated with microglial activation (18, 34, 41, 64, 67), and TLR4 is involved in the ROS function and the development of ER stress (31, 44). Heme stimulation led to increased transcripts of TLR4 in primary spinal microglial cells from sickle mice in vitro (Fig. 4A), suggesting a possible association between heme, TLR4, and spinal microglial activation. Heme significantly enhanced the formation of ROS in both control and sickle microglial cells in culture (Fig. 4B). The heme-induced increase in ROS was abolished in control and sickle microglial cells by two mechanistically independent TLR4 inhibitors, TAK242 and Lipopolysaccharide from Rhodobacter sphaeroides (LPS-RS) (Fig. 4B).
FIG. 4.

Heme-induced ROS and ER stress in microglial cells requires TLR4. (A) TLR4 transcripts in spinal microglial cells from HbSS-BERK mice (6.31 ± 0.2 months old, n = 6) incubated with or without hemin (40 μM) for 1 h, normalized to GAPDH and relative to transcripts in unstimulated cells (n = 8/treatment). (B) Hemin-induced ROS requires TLR4. Hemin-induced ROS was attenuated in isolated microglial cells from adult HbSS-BERK and HbAA-BERK mice by pretreatment with LPS-RS (100 ng/mL; L), TAK-242 (400 nM; T), or salubrinal (5 μM; S) overnight before hemin (40 μM; H) for 1 h (n = 6 independent experiments). (C, D) Hemin-induced ER stress requires TLR4. Hemin-induced expression of sXBP1 and CHOP (markers of increased ER stress) was attenuated in isolated microglial cells from adult HbAA-BERK (C) and HbSS-BERK (D) mice by TLR4 inhibition. Microglial cells were incubated with Veh, hemin (40 μM; H), hemin with 100 ng/mL LPS-RS (H+L), or LPS-RS (L) for 60 min. At the end of the incubation period, sXBP1 and CHOP protein expression was evaluated by immunoblotting. Density of individual protein bands is normalized to GAPDH and is presented relative to vehicle. Relative density of vehicle was given an arbitrary value of 100. Data are mean ± SEM (n = 3 independent experiments). H: hemin; L: LPS-RS (TLR4 inhibitor); S: salubrinal; T: TAK-242 (TLR4 inhibitor). ER, endoplasmic reticulum; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; LPS-RS, lipopolysaccharide from Rhodobacter sphaeroides; ROS, reactive oxygen species; sXBP1, spliced XBP1. Color images are available online.
Further, the increase in ROS induced by heme in sickle microglial cells was also abolished by reducing ER stress with salubrinal, a selective inhibitor of eukaryotic initiation factor 2 α (eIF2α) dephosphorylation, which has been shown to protect cells against ER stress (7) (Fig. 4B). Heme treatment also increased the expression of key ER stress proteins spliced XBP1 (sXBP1) and CHOP in control (Fig. 4C) and sickle (Fig. 4D) primary microglial cells. Expression of both sXBP1 and CHOP proteins was attenuated by pretreatment of cells with the TLR4 inhibitor LPS-RS (Fig. 4C, D). Collectively, these data reveal that heme activates microglial cells in spinal cords via TLR4-induced ROS and ER stress.
ER stress contributes to chronic pain in sickle mice
Due to involvement of ROS-induced ER stress in microglial activation, we examined the role that ER stress plays in chronic sickle pain. We first examined whether increased ROS is associated with ER stress in the spinal cord. We observed increased ROS in the spinal cords of sickle mice compared with control mice (Fig. 5A, B). Electron micrographs of HbAA-BERK control spinal cords showed stacks of intact ER with well-organized lumen (Fig. 5C). In contrast, the spinal cords from HbSS-BERK sickle mice revealed disruption of the structural organization of the ER with swollen lumens of the ER cisternae (Fig. 5D). These data indicate that increased spinal neuroinflammation and oxidative stress are associated with ER stress in the sickle mice.
FIG. 5.
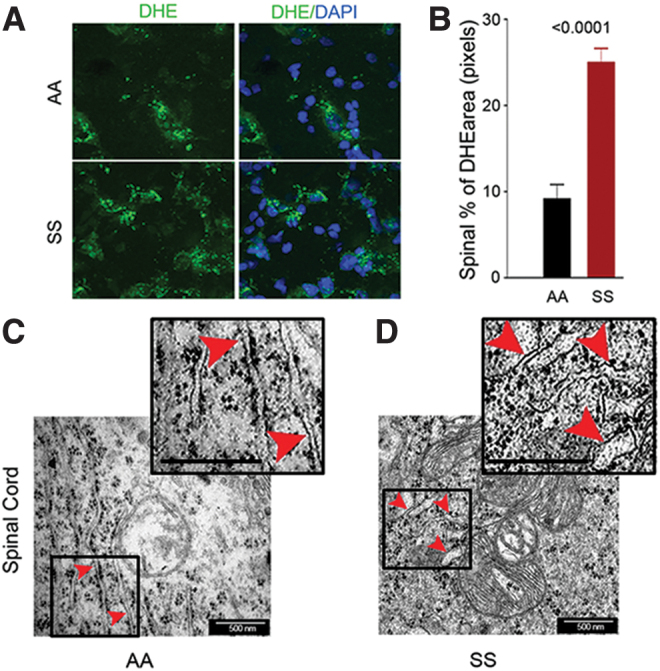
Spinal cords of sickle mice have increased ROS and ER stress. (A) Representative images of L4 and L5 spinal sections from HbSS-BERK and HbAA-BERK mice stained for ROS formation (DHE, green) and nuclei (DAPI, blue). Magnification 600 × ; bar = 10 μm. Each image is representative of images from 8 mice. (B) ROS expression as percentage of DHE-positive pixels was obtained from the right and left dorsal horn of each section (n = 8 per group). (C, D) Representative TEM images of ER (red arrowheads) in the spinal cords (magnification 60,000 × , bar = 500 nm) from the L4–L5 region. HbSS-BERK sickle mice exhibit pronounced swelling of ER cisternae (D) as compared with the normal, narrow ER cisternae in HbAA-BERK control normal mice (C). Insets (demarcated with boxes) show enhanced details of cisternae swelling. Each image is representative of images from three mice. DAPI, 4′,6-diamidino-2-phenylindole; DHE, dihydroethidium; TEM, transmission electron microscopy. Color images are available online.
We next determined the effect of reducing ER stress on chronic sickle pain. Treatment of HbSS-BERK sickle mice with the selective ER stress inhibitor, salubrinal, gradually attenuated mechanical hyperalgesia from treatment day 6 (p < 0.05, vs. vehicle; p < 0.0001 vs. baseline; Fig. 6A), heat sensitivity at 4 h (p < 0.05, vs. vehicle; p < 0.0001 vs. baseline; Fig. 6B), and cold sensitivity from treatment day 6 (p < 0.0001, vs. vehicle; and p < 0.0001 vs. baseline; Fig. 6C). These effects were maintained for 6 days after the last salubrinal injection. Cessation of salubrinal treatment led to a time-dependent restoration of mechanical, heat, and cold hyperalgesia in sickle mice. In addition, salubrinal significantly reduced musculoskeletal hyperalgesia (increased grip force) from day 2 post-treatment (p < 0.05, vs. vehicle and baseline, Fig. 6D) in sickle mice, and its effect was significantly sustained for 18 days (last period of observation) even after the discontinuation of salubrinal injection on day 10 (p < 0.05, vs. vehicle; p < 0.001 vs. baseline; Fig. 6D). Salubrinal did not affect the features of hyperalgesia of HbAA-BERK control mice (Fig. 6A–D).
FIG. 6.

ER stress contributes to chronic hyperalgesia in sickle mice. After BL pain determination, HbAA-BERK and HbSS-BERK mice were treated with i.p. injection of vehicle (0.2% DMSO in saline) or salubrinal (1 mg/kg/day) for 10 days. Pain measures were recorded 4 h after first injection and every other day during injection and postinjection (Supplementary Fig. S5D). (A) Mechanical hyperalgesia. (B) Heat sensitivity. (C) Cold sensitivity. (D) Deep tissue hyperalgesia. Data are mean ± SEM of eight mice. *p < 0.05, **p < 0.001, and ***p < 0.0001 for each time-point versus BL of corresponding group (one-way ANOVA, Bonferroni) in HbSS-BERK. †p < 0.05, ††p < 0.001 and †††p < 0.0001 for salubrinal versus corresponding vehicle at each time-point in HbSS-BERK (two-way ANOVA, Bonferroni). Mean age of mice (months ± SEM) was 5.3 ± 0.1 for HbAA-BERK vehicle, 5.3 ± 0.3 for HbAA-BERK salubrinal, 5.2 ± 0.5 for HbSS-BERK vehicle, and 5.4 ± 0.4 for HbSS-BERK salubrinal. Sal: salubrinal. DMSO, dimethyl sulfoxide. Color images are available online.
Together, these data suggest that targeting ER stress downstream from TLR4 activation may reduce/prevent chronic sickle pain. More importantly, inhibition of ER stress had a sustained effect on nonevoked musculoskeletal hyperalgesia in sickle mice.
Discussion
How sickle pathobiology evokes pain remains unknown. Chronic hemolysis, a characteristic feature of SCD, results in elevated tissue and circulating levels of free heme (59). We demonstrate for the first time the causal role of free heme in evoking pain in sickle mice via TLR4-mediated ER stress involving mechanisms in the central nervous system (CNS). In the spinal cord, TLR4 inhibition led to a significant reduction in heme-induced increases in oxidative stress, microglial activation, and ER stress in sickle mice, all of which are known contributors of neuropathic and inflammatory pain. Notably, both TLR4 inhibition and reduction of downstream ER stress ameliorated hyperalgesia in sickle mice.
The present findings complement our previous observations of increased spinal ROS, substance P, microglial activation, and phosphorylated signal transducer and activator of transcription 3 (phospho-STAT3) and -p38MAPK (P38 mitogen-activated protein kinase) and hyperexcitability of spinal neurons in sickle mice (10, 58). It is, therefore, likely that the increased TLR4 observed in the spinal cords of sickle mice in this study contributes to the hyperexcitability of dorsal horn neurons (10). Most importantly, our present study demonstrated that cell-free heme plays a causal role in the genesis of chronic and acute hyperalgesia via TLR4 and ER stress in sickle mice.
Pain processing in the CNS involves activation of spinal microglial cells. Activated spinal microglial cells release inflammatory cytokines and neuromodulators that sensitize spinal cord neurons (9), CNS inflammation (47), microglial cell activation (6), and increased oxidative stress (58), all of which likely contribute to pain in SCD. In this study, we found that heme stimulated an increase in TLR4 transcripts in spinal microglial cells from sickle mice. The inhibition of TLR4 attenuated activation of spinal microglial cells and decreased heme-induced microglial ROS. However, due to possible off-target/systemic effects of the TLR4 inhibitors, as well as the use of full-body TLR4 knockout mice in this study, it is possible that additional mechanism(s) may contribute to heme-induced hyperalgesia via TLR4 in addition to microglial activation.
TAK242 is a selective inhibitor of TLR4 and is able to cross the blood–brain barrier (22). Nevertheless, TAK242 failed to significantly decrease serum IL-6 levels and mortality in a clinical trial of 274 patients with severe sepsis (45). However, several features of this trial have been called into question, including the primary end point, patient selection, and TAK242 dose (46). The primary end point was serum IL-6 concentration within 96 h of TAK242 treatment. This end point may be too soon, as we observed a decrease in IL-6 after 5 days of TAK-242 treatment in sickle mice. In addition, TLR4 is a primary mediator of inflammation in the CNS (14, 54, 56), and its activity may not be reflected in short-term changes in serum IL-6.
In addition to our studies, TAK242 has been used successfully to attenuate chronic stress-induced visceral pain sensation in mice (62). Several novel antagonists and inhibitors of TLR4 activity have been described with analgesic ability (50). Although the inhibition of TLR4 with TAK242 is a promising approach to treat pain in SCD, it is also important to target downstream mechanisms orchestrated by TLR4 activation, such as ER stress. This is of particular consideration since TLR4 plays an integral role in immunity and defense. Therefore, long-term suppression of TLR4, as in the case of treating SCD pain, could potentially provoke unintentional consequences with respect to pathogen surveillance/clearance and infection severity/duration.
ER stress has recently been associated with microglial activation (18, 34). In addition, TLR4 is crucial to the development of ER stress in human aortic endothelial cells (28) and in liver, skeletal muscle, and adipose tissue after a high-fat diet in mice (44). Heme induces microglial activation and neuronal death via cofilin and mediates oxidative stress, ER stress, and microglial migration (49).
In this study, sickle mice showed ER stress in spinal cords. TLR4 inhibition attenuated heme-induced ROS formation; inhibition of ROS reduced ER stress in spinal microglial cells. These findings suggest that heme may evoke ER stress in microglial cells via TLR4. Heme-induced ROS formation in microglial cells was abolished by either the TLR4 inhibitor TAK242 or reducing ER stress with salubrinal, suggesting that there is a vicious cycle in which heme increases ROS formation to induce ER stress, which, in turn, generates more ROS, exaggerating oxidative stress in spinal cords.
Oxidative stress, inflammation, and hypoxia, the hallmarks of SCD, cause ER stress (11, 62). Recent reports advocate a role of ER stress in the initiation and maintenance of inflammatory and neuropathic pain (24, 53, 60, 65). It is plausible that in SCD, oxidative stress, inflammation, and hypoxia cause ER stress, which, in turn, augments oxidative stress and inflammation, thereby increasing chronic pain via central mechanisms of nociceptive processing.
TLR4 has been shown to increase ROS in microglial cells via activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) (40). It is, therefore, possible that increased TLR4 expression/activation in SCD (particularly in the context of elevated heme) results in increased ROS by the activation of one or more NOX members. This, in turn, might explain the ensuing ER stress, which leads to the microglial activation underlying the central mechanisms of sickle pain. It is noted, however, that a direct causal relationship between ER stress and ROS formation in microglial cells from the sickle mice needs further investigation.
In this study, salubrinal, which reduces ER stress via inhibition of eIF2α dephosphorylation, significantly attenuated hyperalgesia, indicative of the contribution of ER stress in the induction and maintenance of sickle pain. We also found consistent and sustained improvement in deep tissue hyperalgesia in sickle mice after salubrinal administration, which highlights that salubrinal could be a useful intervention. Salubrinal has been reported to induce resistance against glutamate toxicity-induced oxidative stress (32), protect against excitotoxic neuronal injury (56), accelerate bone healing after surgery in rodent models (66), and increase fetal hemoglobin (HbF) (20), all of which can mitigate the severity of SCD (12).
Therefore, salubrinal appears to be a promising novel pharmacological agent because of its beneficial effects on alleviating oxidative and ER stress, improving bone health, and inducing HbF in SCD, thus ameliorating hyperalgesia by attenuating sickle pathobiology. HbF has been suggested to be inversely associated with pain in patients with SCD (51) and sickle mice (27). BERK sickle mice used in this study do not express HbF, thus salubrinal may influence hyperalgesia in sickle mice independent of HbF.
In conclusion, our findings demonstrate that heme-induced TLR4 contributes to the initiation and maintenance of sickle pain. TLR4 also plays a critical role in oxidative stress and vascular and inflammatory pathobiology in sickle mice (4, 16). It is likely that targeting TLR4 (or ER stress) may ameliorate pain by mitigating the underlying pathobiology of SCD as well as central nociceptive mechanisms. Therefore, this study identifies the critical role of heme-driven TLR4 activation in central mechanisms associated with sickle pain and suggests that TLR4 and/or ER stress may be novel therapeutic targets for treating pain in SCD.
Materials and Methods
Animals
Sickle (HbSS-BERK) and control mice (HbAA-BERK)
HbSS-BERK demonstrate hematologic features of human sickle cell anemia, organ damage, pain, and shortened life span (3, 8, 42). HbSS-BERK are homozygous for knockout of both murine alpha and beta globins and carry linked transgenes for human alpha and beta-S globins. Control HbAA-BERK are from cross-breeding of HbSS-BERK, but they exclusively express normal human hemoglobin A (human alpha and beta A globins) and no murine alpha or beta globins (40). HbSS-BERK sickle mice express >99% human sickle hemoglobin, and littermate HbAA-BERK express normal human hemoglobin A (57). HbSS-BERK and HbAA-BERK mice were genotyped by using Transnetyx services and phenotyped by isoelectric focusing for homozygous human sickle and normal human hemoglobin as previously described (28).
TLR4 knockout (TLR4−/−) sickle (TLR4-KO HbSS-BERK) and control (TLR4-KO HbAA-BERK) mice
Tlr4lps-del mice on a C57BL/6 background with a 74,723 bp deletion that removes the TLR4 coding sequence were obtained from Jackson Laboratory (B6.B10ScN-Tlr4lps-del/JthJ, Stock No. 007227; MGI: 1860755; Bar Harbor, ME); they were backcrossed with HbSS-BERK and HbAA-BERK to obtain sickle and control homozygous TLR4 knockout mice (HbSS/TLR4R−/−; HbAA/TLR4R−/−), and “wildtype” TLR4 littermates (HbSS/TLR4+/+; HbAA/TLR4+/+). For each mouse in each backcross cycle, polymerase chain reaction (PCR) genotyping was conducted on three loci: α-globin, β-globin, and TLR4. Mice were phenotyped for the presence of homozygous human sickle hemoglobin by isoelectric focusing, as described earlier.
TLR4 gene deletion was analyzed by PCR using the PTC 100 thermal cycler (Bio-Rad) with the following primer sequences and strain genotyping protocol (Jackson Laboratory), which produced 390 bp (TLR4+/+) and 140 bp (TLR4−/−) products.
-
(i)
oIMR8365—GCA AGT TTC TAT ATG CAT TCT C Knockout Forward
-
(i
i) oIMR8366—CCT CCA TTT CCA ATA GGT AG Knockout Reverse
-
(i
ii) oIMR8367—ATA TGC ATG ATC AAC ACC ACA G Wild Type Forward
-
(i
v) oIMR8368—TTT CCA TTG CTG CCC TAT AG Wild Type Reverse.
All sickle and control mice with TLR4−/− or TLR4+/+ were also genotyped and phenotyped for hemoglobin as described earlier. Mice were bred and maintained under controlled environmental conditions (12 h light-to-dark cycle, at 23°C) in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-approved housing at the University of Minnesota as previously described (29). All experiments were performed following approved protocols from the University of Minnesota's Institutional Animal Care and Use Committee (protocol No. 1706–34904A) and conformed to the statutes of the Animal Welfare Act and the guidelines of the Public Health Service as issued in the Guide for the Care and Use of Laboratory Animals (revised 2011). An equal number of sex- and aged-matched male and female AA (control) and SS (sickle) mice were used in the studies.
Preparation of hemin, TAK242, LPS-RS, and salubrinal
Hemin (2.67 mM) was prepared immediately before use by mixing 10 mg hemin chloride (Catalog No. H651-9; Frontier Scientific), 10 mg d-sorbitol, and 6.9 mg sodium carbonate in 5.7 mL of sterile saline for 60 min in the dark. This solution was filtered with 0.22 μm filter (4). The solvent solution was used as vehicle.
TAK242 (resatorvid), a specific intracellular inhibitor of TLR4 via disruption of adaptor (TIRAP) binding (Catalog No. 614316; Sigma-Aldrich, St. Louis, MO), was dissolved in intralipid (20% of emulsion, Catalog No. I141; Sigma-Aldrich) or dimethyl sulfoxide (DMSO; in vitro studies) (38).
LPS-RS, a specific extracellular antagonist of TLR4 via inhibition of ligand binding (Catalog No. tlrl-prslps; InvivoGen, San Diego, CA), was dissolved in water.
Salubrinal (3-Phenyl-N-[2,2,2-trichloro-1-[[(8-quinolinylamino)thioxomethyl]amino]ethyl]-2-propenamide), a selective inhibitor of eIF2α dephosphorylation that protects against ROS-induced ER stress (catalog No. SML0951; Sigma-Aldrich), was dissolved in intralipid (20% of emulsion) or DMSO (in vitro studies).
Treatments
Mice were treated daily intraperitoneally with heme (32 μmol/kg/day, 5 days) prepared from hemin chloride. TAK242 (1 mg/kg/day, 5 days) and salubrinal (1 mg/kg/day, 10 days) were given intravenously and intraperitoneally, respectively. Corresponding solvents were used as vehicle (Supplementary Fig. S5). Dosages and routes of administration were based on previous studies showing inhibitor specificity and efficacy in the absence of toxicity or side-effects (51, 62).
Pain-related behaviors
Mice were tested daily for pain behaviors before the injection, during the period of injection, and every day postinjection. Pain behaviors were obtained on day 1 before drug administration (baseline) and after drug administration on days 2, 4, 6, 8, and 10. Pain measures were also recorded post-treatment for 8 days on days 12, 13, 16, and 18.
Mice were acclimatized to each test protocol in a quiet room at constant temperature and tested in the following order for each test: (i) mechanical, (ii) heat, (iii) grip force, and (iv) cold (29).
Mechanical hyperalgesia
PWF was determined by 10 consecutive applications of a 1.0 g (4.08 mN) von Frey (Semmes-Weinstein) monofilament (catalog No. 58028; Stoelting Co., Wood Dale, IL) to the plantar surface of each hind paw for 1–2 s with a force sufficient to bend the filament. An inter-stimulus interval of at least 5 s was observed. Only vigorous withdrawal responses were counted. Paw withdrawal threshold was determined by using the up–down method (20): A series of von Frey filaments, ranging from 0.4–8.0 g, were applied to the hind paws of mice. The resulting pattern of withdrawal responses was tabulated by using the convention: X = withdrawal; O = no withdrawal; the 50% response threshold was calculated by using the following formula (21):

Heat hyperalgesia
A radiant heat stimulus was applied to the plantar surface of the hind paw from below with a projector lamp bulb (CXL/CXR, 8 V, 50 W) by using the PAW Thermal Stimulator (University Anesthesia Research & Development Group, University of California, San Diego, CA). Paw withdrawal latency to the nearest 0.1 s was recorded when the mouse withdrew its paw from the stimulus.
Grip force
To assess deep tissue hyperalgesia, peak forepaw grip force was measured by using a computerized grip force meter (SA Maier Co., Milwaukee, WI). Mice held by the tail were made to pull on a wire-mesh gauge with their forepaws. As they were gradually pulled by the tail, the peak force (in g) exerted was recorded. Deep tissue hyperalgesia was defined as a decrease in the grip force.
Cold hyperalgesia
For cold sensitivity, the latency to initial lifting of either forepaw on cold plate (4°C; Ugo Basile Hot/Cold Plantar Test apparatus 35100; Stoelting Co.) and PWF over a period of 2 min was determined.
Quantification of heme in spinal cords
Whole spinal cords were lysed in hemin assay buffer, and protein concentration was assessed. Heme content in the spinal cords of control and sickle mice was measured with the hemin colorimetric assay kit (catalog No. K672; Biovision, Milpitas, CA) following the manufacturer's protocol. The optical density of each sample was analyzed at 570 nm, and hemin concentration was calculated with Gen5™ 1.0 data analysis software (Biotek), as previously described (23).
Microglial primary cell isolation, culture, and treatments
Spinal cords from 6–10 adult control and sickle mice (∼6 months old) were mechanically dissociated followed by treatment with 1.4 mg/mL of collagenase type A (catalog No. 10103578001; Sigma-Aldrich) in Dulbecco's modified Eagle medium (DMEM; Catalog No. 11995065; ThermoFisher Scientific, Waltham, MA) and penicillin/streptomycin (Catalog No. 15140122; ThermoFisher Scientific) for 30 min at 37°C. The pooled spinal single-cell suspension was washed in DMEM containing 10% fetal bovine serum (FBS; Hyclone Standard; Catalog No. SH3008803; ThermoFisher Scientific) and filtered through a 70-μm cell strainer. The spinal single-cell suspension was seeded in a T-25 flask coated with poly-d-lysine (1 μg/mL;catalog No. P1399; Sigma-Aldrich) in DMEM containing 10% FBS, penicillin/streptomycin, glutamate, and sodium pyruvate. After 7 days of culture, nonadherent cells were removed. The remaining adherent cells (microglial cells) were allowed to replicate further (2–3 weeks) in the T-25 flask (13).
Microglial cells were characterized by the expression of the surface marker, cluster of differentiation molecule 11b (CD11b; OX42, catalog No. CH23021; Neuromics, Edina, MN), and the absence of glial fibrillary acidic protein (GFAP; catalog No. RA22101; Neuromics). Microglial cells were cultured at >98% purity to 90% confluence. After dilution to 40 μM in medium, hemin was filtered with a 0.22 μm filter (4). The inhibitors of TLR4, LPS-RS in water (5 mg/mLl; Invivogen) and TAK242 in DMSO (1 mg/mL), were used at a concentration of 100 ng/mL and 400 nM, respectively, in cell culture medium. Salubrinal in DMSO (10 mg/mL; Sigma-Aldrich) was used at a concentration of 5 μM in serum-free cell culture medium.
Transmission electron microscopy
Spinal cord segments (L4 and L5) from the lumbar region were fixed with 2.5% glutaraldehyde overnight at 4°C, washed with 0.1 M sodium cacodylate, and postfixed with 1% OsO4 for 40–60 min. Samples were dehydrated in a grade series of ethanol and embedded in Epon 812 resin. Ultrathin sections (65 nm) were stained with uranyl acetate and lead citrate and examined at 60K with a JEOL 1200EX II electron microscope at 80 kV (Peabody, MA).
Quantification of ROS in spinal cord and microglial cells
L4 and L5 spinal cord segments were embedded in optimal cutting temperature embedding compound (Tissue Tek No. 4583, catalog No.62550-01; Electron Microscopy Sciences, Hatfield, PA). Cryosections (30 μm) were incubated with 5 μM dihydroethidium (DHE; catalog No. D23107; ThermoFisher Scientific) for 30 min at 37°C followed by staining for nuclei with 4′,6-diamidino-2-phenylindole (DAPI; catalog No. D1306; ThermoFisher Scientific) (55). Images were captured by using a confocal microscope (Olympus Fluo View 1000 BX2) with a 60 × objective (PLAPON 60X o), and the ratio of total number of fluorescent pixels to the total area was quantified with Adobe Photoshop (Adobe, San Jose, CA).
The ROS formation in microglial cells was detected by utilizing the cell-permeable reagent 2′,7′-dichlorofluorescein (DCFDA), which is oxidized by ROS to form a fluorescent compound, with excitation and emission spectra of 495 and 529 nm, respectively (catalog No. ab113851; Abcam, Cambridge, MA), according to the manufacturer's instructions.
Quantification of IL-6
Spinal cord proteins were extracted with 25 mM Hepes buffer (pH 7.6) containing 300 mM NaCl, 1.5 mM MgCl, 0.1% (vol/vol) Triton X-100, protease inhibitors (10 mM phenylmethylsulfonyl fluoride [PMSF], 20 μg/mL leupeptin), and phosphatases inhibitors (0.1 mM sodium orthovanadate, 20 mM β-glycerophosphate). Whole spinal cord lysates (250 μg of protein) were analyzed by an enzyme-linked immunosorbent assay for IL-6 (catalog No. M6000B; R&D Systems, Minneapolis, MN). Assays were performed according to the manufacturer's protocols by using a microplate reader (Synergy HT; Biotek) and calculated with the plate reader Gen5 1.0 data analysis software (Biotek). All analyses and calibrations were performed in duplicate, in multiple samples per condition with appropriate controls (60).
Real-time quantitative PCR
RNA isolation was performed by using TRIzol (catalog No. 15596026; ThermoFisher Scientific) followed by purification with RNeasy mini kit (catalog No. 74104; Qiagen, Valencia, CA). Thereafter, up to 2 μg of RNA was converted into complementary DNA (cDNA) by using a high-capacity RNA to cDNA kit (catalog No. 4387406; ThermoFisher Scientific). Real-time PCR reactions were performed in a total volume of 20 μL, containing 10 μL of TaqMan gene expression master mix (catalog No. 4369016; ThermoFisher Scientific), 2 μL of the diluted cDNA, 1 μL of predesigned FAM (6-carboxyfluorescein) labeled TaqMan probe (catalog No. 4331182; ThermoFisher Scientific) for TLR4 (Mm00445274_m1), and 7 μL of nuclease-free water.
Quantification of RNA expression levels was performed in an Applied Biosystems 7000 RT-PCR system by using the following PCR conditions: hold at 50°C and 95°C for 2 and 10 min, respectively, followed by 40 cycles of denaturation at 95°C for 15 s and annealing/extension at 60°C for 1 min. TaqMan FAM probe (catalog No. 4331182; ThermoFisher Scientific) for glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Mm99999915_g1) was used for normalization. Calculations of relative gene expression were done by using the 2(-Delta C(T)) method (37).
Statistical analysis
Data were analyzed by using Prism software (v 6.0; GraphPad Prism, Inc.). One- or two-way analysis of variance with Bonferroni's or unpaired Student's t test was used to compare treatments. A p value of <0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors thank Michael J. Franklin for editorial assistance. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations Used
- ANOVA
analysis of variance
- BL
baseline
- BW
body weight
- cDNA
complementary DNA
- CNS
central nervous system
- DAPI
4′,6-diamidino-2-phenylindole
- DHE
dihydroethidium
- DMEM
Dulbecco's modified Eagle medium
- DMSO
dimethyl sulfoxide
- eIF2α
eukaryotic initiation factor 2 α
- ELISA
enzyme-linked immunosorbent assay
- ER
endoplasmic reticulum
- FAM
6-carboxyfluorescein
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HbA
normal human hemoglobin A (human alpha and beta A globins)
- HbAA-BERK
transgenic mouse model expressing exclusively normal human HbA developed at Lawrence Berkeley National Laboratory
- HbF
fetal hemoglobin
- HbS
hemoglobin containing mutated β globin
- HbSS-BERK
transgenic mouse model of SCD expressing exclusively human HbS developed at Lawrence Berkeley National Laboratory
- H/R
hypoxia/reoxygenation
- IL-6
interleukin 6
- i.v.
intravenous
- LPS-RS
Lipopolysaccharide from Rhodobacter sphaeroides
- NOX
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase
- PCR
polymerase chain reaction
- PWF
paw withdrawal frequency
- PWL
paw withdrawal latency
- RBCs
red blood cells
- ROS
reactive oxygen species
- SCD
sickle cell disease
- SEM
standard error of the mean
- sXBP1
spliced XBP1
- TAK242
resatorvid; selective small molecule inhibitor of TLR4
- TEM
transmission electron microscopy
- TLR4
toll-like receptor 4
- TLR4-KO
TLR4 knockout (genetic deletion)
- Tx
treatment
- Veh
vehicle
- VF
von Frey (Semmes-Weinstein) monofilament
- VOCs
vaso-occlusive crises
- WT
wild type
Authors' Contributions
K.G. developed the concept, supervised the study, and edited the article; K. G., M.G., and J.L. designed the experiments, and analyzed and interpreted the data; J.L., J.P., D.V., J. N., Y.V., and Y.L. performed research; S.T. and R.J. conducted the breeding, pheno- and geno-typing, and maintenance of sickle, control, and different cross-bred TLR4/sickle/control mice; J.L., J.P., Y.W., and D.V. analyzed data; and J.L., M.G., and M.K.J. wrote the article.
Author Disclosure Statement
K.G. is a consultant for Tautona Group and received honoraria from Novartis and CSL Behring; received research funding from 1910 Genetics and Cyclerion; and received a research drug from Grifols for another research project, but it has no conflict with the work presented in this article. All other authors state that no competing financial interests exist.
Funding Information
This work is supported by the National Institutes of Health grants R01HL103773, U01 HL117664, and HL147562, and grants from the Institute for Engineering in Medicine to K.G.
Supplementary Material
References
- 1. Adisa OA, Hu Y, Ghosh S, Aryee D, Osunkwo I, and Ofori-Acquah SF. Association between plasma free haem and incidensce of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. Br J Haematol 162: 702–705, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, Karanjia R, Barajas-Lopez C, Vanner S, Vergnolle N, and Bunnett NW. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol 575(Pt 2): 555–571, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Belcher JD, Bryant CJ, Nguyen J, Bowlin PR, Kielbik MC, Bischof JC, Hebbel RP, and Vercellotti GM. Transgenic sickle mice have vascular inflammation. Blood 101: 3953–3959, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, and Vercellotti GM. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123: 377–390, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. This reference has been deleted
- 6. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, and Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307: 935–939, 2005 [DOI] [PubMed] [Google Scholar]
- 7. Brandow AM, Wandersee NJ, Dasgupta M, Hoffmann RG, Hillery CA, Stucky CL, and Panepinto JA. Substance P is increased in patients with sickle cell disease and associated with haemolysis and hydroxycarbamide use. Br J Haematol 175: 237–245, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cain DM, Vang D, Simone DA, Hebbel RP, and Gupta K. Mouse models for studying pain in sickle disease: effects of strain, age, and acuteness. Br J Haematol 156: 535–544, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cao H and Zhang YQ. Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev 32: 972–983, 2008 [DOI] [PubMed] [Google Scholar]
- 10. Cataldo G, Rajput S, Gupta K, and Simone DA. Sensitization of nociceptive spinal neurons contributes to pain in a transgenic model of sickle cell disease. Pain 156: 722–730, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaudhari N, Talwar P, Parimisetty A, Lefebvre d'Hellencourt C, and Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci 8: 213, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darbari DS, Hampson JP, Ichesco E, Kadom N, Vezina G, Evangelou I, Clauw DJ, Taylor Vi JG, and Harris RE. Frequency of hospitalizations for pain and association with altered brain network connectivity in sickle cell disease. J Pain 16: 1077–1086, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Devarajan G, Chen M, Muckersie E, and Xu H. Culture and characterization of microglia from the adult murine retina. Sci World J 2014: 894368, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Falip M, Salas-Puig X, and Cara C. Causes of CNS inflammation and potential targets for anticonvulsants. CNS Drugs 27: 611–623, 2013 [DOI] [PubMed] [Google Scholar]
- 15. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, and Bozza MT. Characterization of heme as activator of toll-like receptor 4. J Biol Chem 282: 20221–20229, 2007 [DOI] [PubMed] [Google Scholar]
- 16. Frenette PS and Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest 117: 850–858, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, and Ofori-Acquah SF. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest 123: 4809–4820, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo ML, Liao K, Periyasamy P, Yang L, Cai Y, Callen SE, and Buch S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 11: 995–1009, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupta M and Msambichaka L. Morphine for the treatment of pain in sickle cell disease. Sci World J 2015: 540154, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hahn CK and Lowrey CH. Induction of fetal hemoglobin through enhanced translation efficiency of gamma-globin mRNA. Blood 124: 2730–2734, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamamoto DT, Giridharagopalan S, and Simone DA. Acute and chronic administration of the cannabinoid receptor agonist CP 55,940 attenuates tumor-evoked hyperalgesia. Eur J Pharmacol 558: 73–87, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hua F, Tang H, Wang J, Prunty MC, Hua X, Sayeed I, and Stein DG. TAK-242, an antagonist for toll-like receptor 4, protects against acute cerebral ischemia/reperfusion injury in mice. J Cereb Blood Flow Metab 35: 536–542, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang Z, Chen K, Xu T, Zhang J, Li Y, Li W, Agarwal AK, Clark AM, Phillips JD, and Pan X. Sampangine inhibits heme biosynthesis in both yeast and human. Eukaryot Cell 10: 1536–1544, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inceoglu B, Bettaieb A, Trindade da Silva CA, Lee KS, Haj FG, and Hammock BD. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc Natl Acad Sci U S A 112: 9082–9087, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kato J and Svensson CI. Role of extracellular damage-associated molecular pattern molecules (DAMPs) as mediators of persistent pain. Prog Mol Biol Transl Sci 131: 251–279, 2015 [DOI] [PubMed] [Google Scholar]
- 26. Kenyon N, Wang L, Spornick N, Khaibullina A, Almeida LE, Cheng Y, Wang J, Guptill V, Finkel JC, and Quezado ZM. Sickle cell disease in mice is associated with sensitization of sensory nerve fibers. Exp Biol Med 240: 87–98, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khaibullina A, Almeida LE, Wang L, Kamimura S, Wong EC, Nouraie M, Maric I, Albani S, Finkel J, and Quezado ZM. Rapamycin increases fetal hemoglobin and ameliorates the nociception phenotype in sickle cell mice. Blood Cells Mol Dis 55: 363–372, 2015 [DOI] [PubMed] [Google Scholar]
- 28. Kim JA, Jang HJ, and Hwang DH. Toll-like receptor 4-induced endoplasmic reticulum stress contributes to impairment of vasodilator action of insulin. Am J Physiol Endocrinol Metab 309: E767–E776, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kohli DR, Li Y, Khasabov SG, Gupta P, Kehl LJ, Ericson ME, Nguyen J, Gupta V, Hebbel RP, Simone DA, and Gupta K. Pain-related behaviors and neurochemical alterations in mice expressing sickle hemoglobin: modulation by cannabinoids. Blood 116: 456–465, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lan LS, Ping YJ, Na WL, Miao J, Cheng QQ, Ni MZ, Lei L, Fang LC, Guang RC, Jin Z, and Wei L. Down-regulation of Toll-like receptor 4 gene expression by short interfering RNA attenuates bone cancer pain in a rat model. Mol Pain 6: 2, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee CW, Wu CH, Chiang YC, Chen YL, Chang KT, Chuang CC, and Lee IT. Carbon monoxide releasing molecule-2 attenuates Pseudomonas aeruginosa-induced ROS-dependent ICAM-1 expression in human pulmonary alveolar epithelial cells. Redox Biol 18: 93–103, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lewerenz J and Maher P. Basal levels of eIF2alpha phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem 284: 1106–1115, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li XQ, Wang J, Fang B, Tan WF, and Ma H. Intrathecal antagonism of microglial TLR4 reduces inflammatory damage to blood-spinal cord barrier following ischemia/reperfusion injury in rats. Mol Brain 7: 28, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liao K, Guo M, Niu F, Yang L, Callen SE, and Buch S. Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J Neuroinflammation 13: 33, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin JJ, Du Y, Cai WK, Kuang R, Chang T, Zhang Z, Yang YX, Sun C, Li ZY, and Kuang F. Toll-like receptor 4 signaling in neurons of trigeminal ganglion contributes to nociception induced by acute pulpitis in rats. Sci Rep 5: 12549, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY, and Yang QW. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation 9: 46, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 38. Matsunaga N, Tsuchimori N, Matsumoto T, and Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol 79: 34–41, 2011 [DOI] [PubMed] [Google Scholar]
- 39. Pan R, Di H, Zhang J, Huang Z, Sun Y, Yu W, and Wu F. Inducible lentivirus-mediated siRNA against TLR4 reduces nociception in a rat model of bone cancer pain. Mediators Inflamm 2015: 523896, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park HS, Jung HY, Park EY, Kim J, Lee WJ, and Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol 173: 3589–3593, 2004 [DOI] [PubMed] [Google Scholar]
- 41. Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, Kong IK, Chang KT, and Lee DS. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci Lett 584: 191–196, 2015 [DOI] [PubMed] [Google Scholar]
- 42. Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, and Rubin EM. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science 278: 876–878, 1997 [DOI] [PubMed] [Google Scholar]
- 43. Piel FB, Hay SI, Gupta S, Weatherall DJ, and Williams TN. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med 10: e1001484, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pierre N, Deldicque L, Barbe C, Naslain D, Cani PD, and Francaux M. Toll-like receptor 4 knockout mice are protected against endoplasmic reticulum stress induced by a high-fat diet. PLoS One 8: e65061, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, and Cohen J. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med 38: 1685–1694, 2010 [DOI] [PubMed] [Google Scholar]
- 46. Salluh JI and Povoa P. Biomarkers as end points in clinical trials of severe sepsis: a garden of forking paths. Crit Care Med 38: 1749–1751, 2010 [DOI] [PubMed] [Google Scholar]
- 47. Salter MW and Stevens B. Microglia emerge as central players in brain disease. Nat Med 23: 1018–1027, 2017 [DOI] [PubMed] [Google Scholar]
- 48. Sauer RS, Hackel D, Morschel L, Sahlbach H, Wang Y, Mousa SA, Roewer N, Brack A, and Rittner HL. Toll like receptor (TLR)-4 as a regulator of peripheral endogenous opioid-mediated analgesia in inflammation. Mol Pain 10: 10, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sayeed MSB, Alhadidi Q, and Shah ZA. Cofilin signaling in hemin-induced microglial activation and inflammation. J Neuroimmunol 313: 46–55, 2017 [DOI] [PubMed] [Google Scholar]
- 50. Selfridge BR, Wang X, Zhang Y, Yin H, Grace PM, Watkins LR, Jacobson AE, and Rice KC. Structure-activity relationships of (+)-naltrexone-Inspired Toll-like Receptor 4 (TLR4) antagonists. J Med Chem 58: 5038–5052, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smith WR, Ballas SK, McCarthy WF, Bauserman RL, Swerdlow PS, Steinberg MH, and Waclawiw MA; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. The association between hydroxyurea treatment and pain intensity, analgesic use, and utilization in ambulatory sickle cell anemia patients. Pain Med 12: 697–705, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith WR and Scherer M. Sickle-cell pain: advances in epidemiology and etiology. Hematology Am Soc Hematol Educ Program 2010: 409–415, 2010 [DOI] [PubMed] [Google Scholar]
- 53. Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, and Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci 27: 901–908, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun Y, Yang M, Tang H, Ma Z, Liang Y, and Li Z. The over-production of TNF-alpha via Toll-like receptor 4 in spinal dorsal horn contributes to the chronic postsurgical pain in rat. J Anesth 29: 734–740, 2015 [DOI] [PubMed] [Google Scholar]
- 55. Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y, and Ii M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol 157: 1250–1262, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tanga FY, Nutile-McMenemy N, and DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A 102: 5856–5861, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tran H, Gupta M, and Gupta K. Targeting novel mechanisms of pain in sickle cell disease. Blood 130: 2377–2385, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Valverde Y, Benson B, Gupta M, and Gupta K. Spinal glial activation and oxidative stress are alleviated by treatment with curcumin or coenzyme Q in sickle mice. Haematologica 101: e44–e47, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Beers EJ, Yang Y, Raghavachari N, Tian X, Allen DT, Nichols JS, Mendelsohn L, Nekhai S, Gordeuk VR, Taylor JG 6th, and Kato GJ. Iron, inflammation, and early death in adults with sickle cell disease. Circ Res 116: 298–306, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vincent L, Vang D, Nguyen J, Gupta M, Luk K, Ericson ME, Simone DA, and Gupta K. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood 122: 1853–1862, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang X, Grace PM, Pham MN, Cheng K, Strand KA, Smith C, Li J, Watkins LR, and Yin H. Rifampin inhibits Toll-like receptor 4 signaling by targeting myeloid differentiation protein 2 and attenuates neuropathic pain. FASEB J 27: 2713–2722, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Woller SA, Ravula SB, Tucci FC, Beaton G, Corr M, Isseroff RR, Soulika AM, Chigbrow M, Eddinger KA, and Yaksh TL. Systemic TAK-242 prevents intrathecal LPS evoked hyperalgesia in male, but not female mice and prevents delayed allodynia following intraplantar formalin in both male and female mice: the role of TLR4 in the evolution of a persistent pain state. Brain Behav Immun 56: 271–280, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang ES, Bae JY, Kim TH, Kim YS, Suk K, and Bae YC. Involvement of endoplasmic reticulum stress response in orofacial inflammatory pain. Exp Neurobiol 23: 372–380, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuksel E, Naziroglu M, Sahin M, and Cig B. Involvement of TRPM2 and TRPV1 channels on hyperalgesia, apoptosis and oxidative stress in rat fibromyalgia model: protective role of selenium. Sci Rep 7: 17543, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int J Clin Exp Med 3: 33–40, 2010 [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang P, Hamamura K, Jiang C, Zhao L, and Yokota H. Salubrinal promotes healing of surgical wounds in rat femurs. J Bone Miner Metab 30: 568–579, 2012 [DOI] [PubMed] [Google Scholar]
- 67. Zhou YQ, Liu DQ, Chen SP, Sun J, Zhou XR, Rittner H, Mei W, Tian YK, Zhang HX, Chen F, and Ye DW. Reactive oxygen species scavengers ameliorate mechanical allodynia in a rat model of cancer-induced bone pain. Redox Biol 14: 391–397, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.