

Abstract
Neuroligins (NLGNs) are a class of postsynaptic cell adhesion molecules that interact with presynaptic neurexins (NRXNs) and regulate synapse function. NLGN4 is a member of the NLGN family and consists of a unique amino acid sequence in humans that is not evolutionarily well conserved in rodents. The human-specific NLGN4 gene has been reported to be mutated in many patients with autism and other neurodevelopmental disorders. However, it remained unclear how these mutations might alter the molecular properties of NLGN4 and affect synaptic transmission in human neurons. Here, we describe a severely autistic male patient carrying a single amino acid substitution (R101Q) in the NLGN4 gene. When expressed in HEK293 cells, the R101Q mutation in NLGN4 did not affect its binding affinity for NRXNs or its capacity to form homodimers. This mutation, however, impaired the maturation of NLGN4 protein by inhibiting N-linked glycosylation at an adjacent residue (N102), which is conserved in all NLGNs. As a result, the R101Q substitution significantly decreased the surface trafficking of NLGN4 and increased its retention in the endoplasmic reticulum and Golgi apparatus. In human neurons derived from male stem cell lines, the R101Q mutation also similarly reduced the synaptic localization of NLGN4, resulting in a loss-of-function phenotype. This mutation-induced trafficking defect substantially diminished the ability of NLGN4 to form excitatory synapses and modulate their functional properties. Viewed together, our findings suggest that the R101Q mutation is pathogenic for NLGN4 and can lead to synaptic dysfunction in autism.
Keywords: AMPA receptor, autism, glycosylation, neuroligin-4, synaptic transmission, trafficking
Significance Statement
Several single amino-acid substitutions in the X-linked neuroligin 4 (NLGN4) gene (NLGN4X) have been identified in autistic patients. However, it remained unclear how these point mutations might affect NLGN4 properties and influence synapse function in humans. Here, we analyzed the mechanisms of an autism-associated R101Q variant in NLGN4. We demonstrate that this mutation can directly impair the post-translational modification of NLGN4 by inhibiting N-linked glycosylation. These immaturely glycosylated products exhibit categorical deficits in surface trafficking and synaptic localization, and thus represent a loss-of-function phenotype. In reprogrammed human neurons, the loss of NLGN4 function by R101Q mutation resulted in enhanced excitatory synaptic transmission. Our results highlight the pathologic reactions of a NLGN4 point mutation, which may lead to future mechanism-based therapy.
Introduction
Autism spectrum disorder (ASD) is an early neurodevelopmental syndrome associated with impaired social interaction, delayed speech and nonverbal communication, and repetitive and restrictive behavior (American Psychiatric Association, 2013; Volkmar, 2013). Genetic studies suggest that a large cohort of autistic probands carry either familial or de novo mutations in various synaptic proteins (Jacquemont et al., 2006; Sebat et al., 2007). One of these genes that exhibit high penetrance for ASD is the X chromosome-linked neuroligin 4 (NLGN4), a postsynaptic cell adhesion molecule (CAM; for review, see Südhof, 2008). More than 50 distinct mutations in NLGN4 have been reported in ASD patients with a nearly 100% penetrant phenotype (Jamain et al., 2003; Laumonnier et al., 2004; Yan et al., 2005; Chocholska et al., 2006; Talebizadeh et al., 2006; Macarov et al., 2007; Lawson-Yuen et al., 2008; Pampanos et al., 2009).
NLGN4 belongs to the family of NLGN genes (NLGN1–4) that participate in synapse organization and maintenance, and assign synaptic properties (Südhof, 2008). NLGNs generally form a trans-synaptic complex with presynaptic neurexins (NRXNs; Ichtchenko et al., 1995; Varoqueaux et al., 2006; Arac et al., 2007; Fabrichny et al., 2007; Koehnke et al., 2008; Ko et al., 2009) and establish cis-interaction with postsynaptic scaffolding molecules (e.g., PSD-95, SHANK3, and Gephyrin; Irie et al., 1997; Meyer et al., 2004; Varoqueaux et al., 2004; Nam and Chen, 2005; Mondin et al., 2011). Different NLGN genes differentially regulate postsynaptic clustering of neurotransmitter receptors at excitatory versus inhibitory synapses (Song et al., 1999; Varoqueaux et al., 2004; Budreck and Scheiffele, 2007; Heine et al., 2008; Chanda et al., 2017; Jiang et al., 2017).
Although NLGN1/2/3 are evolutionarily conserved, NLGN4 exhibits low abundance and poor sequence conservation in rodents (Bolliger et al., 2008; Maxeiner et al., 2020). No NLGN4 homolog was identified in Rattus norvegicus. Moreover, the mouse ortholog (termed NLGN4-like in Mus musculus) shows considerable variability across different strains and contains only ∼51% sequence similarity with human NLGN4 (Bolliger et al., 2008; Maxeiner et al., 2020). Therefore, to understand the function of human NLGN4, we have previously used autopsied human brain samples and cultured human neurons differentiated from embryonic stem (ES) cells. We demonstrated that human NLGN4 is expressed in the cerebral cortex, preferentially localizes at glutamatergic postsynapses, and specifically modulates excitatory synaptic transmission (Marro et al., 2019).
Given this regulatory role of NLGN4 at human synapses, a definitive loss of NLGN4 function by deleterious mutations suggest a causative connection between synaptic dysfunction and ASD. In fact, several internal deletions and insertions, frameshift mutations, premature nonsense codons, and copy number variations (CNVs) constitute the majority of NLGN4 mutations identified in autistic probands (Marshall et al., 2008).
However, a number of ASD patients also carry NLGN4 missense mutations with relatively unknown pathologic consequences (Fabrichny et al., 2007; Schepici et al., 2019). Although some of these amino acid substitutions have been shown to disrupt the structural integrity of NLGN4 (e.g., R87W; Zhang et al., 2009) or enhance its binding affinity for synaptic receptors (e.g., R704; Chanda et al., 2016; Marro et al., 2019), other variants might simply represent genetic polymorphisms without any functional significance (e.g., G84R, G99S, Q162K, A283T; Fabrichny et al., 2007; Xu et al., 2017). Therefore, it remained unclear whether there are other pathogenic mechanisms by which point mutations can affect NLGN4 properties and impair synapse function in ASD.
In this current study, we analyze the potential role of a NLGN4 missense variant in ASD by investigating the case of an autistic boy carrying R101Q substitution in the NLGN4 gene. We show that this mutation impairs NLGN4 maturation, decreases its surface trafficking, and inhibits NLGN4-mediated silencing of excitatory synapses in reprogrammed human neurons. Our report draws a direct correlation between the deleterious effects of a NLGN4 point mutation and clinical symptoms observed in a human patient.
Materials and Methods
Clinical evaluation
The proband is a boy with autism and intellectual disability. He exhibits substantially delayed language skills, executive functions, and aberrant behavior. His social skills are substantially delayed in all domains (cognitive, behavior, and adaptive) compared with his peers. However, he is fully aware of his functional abilities as well as social limitations; he can express himself and answer prompted questions through independent (i.e., unassisted) use of a letterboard and/or computer keyboard. His hearing is normal, but vision is impaired by a lazy eye and a congenital cataract for which he wears corrective glasses. The patient is not dysmorphic; he has difficulty falling asleep and is a restless sleeper.
This individual has never had witnessed clinical seizures, but persistent abnormalities were observed in his electroencephalogram (EEG) recordings dating back to 2013, including multifocal independent epileptiform discharges (bilateral frontal, temporal, central, parietal, and occipital regions), occasional generalized epileptiform discharges, and rare bursts of generalized paroxysmal fast activity. These EEG patterns are indicative of a predisposition toward focal onset and generalized onset seizures. Between 2013 and 2017, discharges were seen frequently during both wakefulness and sleep. However, no electrographic seizures or continuous discharges (e.g., continuous spike-wave discharge during slow-wave sleep) were ever recorded. Epileptiform discharges decreased in frequency from 2018 and now only occur rarely. Formerly (2014–2016), the patient was prescribed Trileptal (oxcarbazepine) daily for seizure prevention. In addition, he also took l-carnitine, leucovorin (folinic acid), and Namenda (memantine). He currently (2016 to present) takes Lamictal (lamotrigine) and l-carnitine, as recommended.
Neuropsychological tests
All interviews for neuropsychological assessments were administered with full permission of the patient family.
Social responsiveness scales
The Social Responsiveness Scale (SRS; Constantino, 2005) test and its revised second edition (SRS-2; Constantino and Gruber, 2012) are self-reported questionnaires, and were filled out by parents of the affected individual when he was 4 and 14 years old. The SRS/SRS-2 scale is designed to identify the presence and severity of social impairments associated with ASD and to differentiate them from clinical groups with other neurologic disorders. The test scores (T-scores) are normalized to healthy children matching the patient's age, sex, ethnicity, and education. T-scores >60 are generally considered as moderate autism symptoms, while T-scores >75 indicate severe ASD symptoms. Nonautistic children typically score between 0 and 35 (Bölte et al., 2011).
Aberrant behavior checklists
The Aberrant Behavior Checklist (ABC-2; Aman and Singh, 1994, 2017) test is a parental questionnaire that measures a list of problematic secondary behaviors for multiple intellectual and learning disorders. These subdomain scores (range, 0–3) are categorized into five subscales that are empirically derived by principal component analysis. Final scores rate how the tested person compares to age-matched and gender-matched healthy people, with greater scores representing more severity overall.
Vineland adaptive behavior scales
The Vineland Adaptive Behavior Scale (VABS; Sparrow and Cicchetti, 1985; Sparrow et al., 2016) test measures the ability of an individual to live independently and follow social rules. The Vineland-3 test is typically used to track functional, day-to-day abilities of an autistic patient and progress along planned intervention programs. The three major domains of the Vineland-3 test include communication (one's capacity to both understand and effectively communicate), daily living skills (self-reliability in a community), and socialization (ability to form interpersonal relationships). The composite score of a human subject is compared with those of healthy, age-matched individuals (normalized to 100). A low VABS score indicates severe deficits.
Childhood autism rating scales
The Childhood Autism Rating Scale (CARS-2; Schopler and Van Bourgondien, 2010) test provides a diagnostic and severity measurement for ASD. A professional observer rates the subject's behavior in 14 different categories that described emotional, visual, and listening responses, as well as verbal and nonverbal communications. Scores from each category ranged from 0 to 4, with higher scores representing more severe maladaptation and likelihood of ASD diagnosis. Recommended final cutoff scores vary by age groups and intellectual development, and was set at 28 in this study.
Whole exome sequencing
No human tissues or bodily fluids were directly handled in this study. The patient family privately conducted genetic analysis of patient samples. This article only reports this genetic information along with a brief description of the analysis methods (see below), with full knowledge of the Colorado State University Institutional Review Board and permission from the parents of this child.
The blueprint genetics (BpG) whole exome family plus test (version 2) was performed, which included whole exome sequencing (WES) analysis coupled with whole exome deletion/duplication (CNV) analysis for the proband and unaffected family members. The test targeted all protein coding exons, exon–intron boundaries (±20 bp), and clinically relevant noncoding variants, and also detected single nucleotide substitutions and small insertions and deletions. The analysis of the WES test was primarily focused on established genes (total, >3750) that have been previously associated with genetic disorders, curated from BpG diagnostic panels, clinical genomics database, and the developmental disorders genotype–phenotype database. Variants classified as potentially deleterious were further confirmed by bidirectional Sanger sequencing.
Structural prediction of NLGN4
The amino acid sequence of human NLGN4 was uploaded to the Phyre2 web portal and fit against the previously resolved rat NLGN1 crystal structure with 100% confidence. The resulting PDB file was modeled and colored using Chimera (University of California, San Francisco). A 40% transparency was applied to the surface model to visualize its secondary structures. The α-helix bundle involved in NLGN4 dimerization and the residues creating NRXN1 contact sites were highlighted with purple and green, respectively (Leone et al., 2010). The location of the R101Q mutation and adjacent N-linked glycosylation site (N102) were colored coded blue and red, respectively.
General experimental design
All experiments were conducted with the approval of the Colorado State University Institutional Biosafety Committee (protocol 19-059B). Experiments (except for the Western blots and induced neurogenesis) were conducted in “blinded” fashion (i.e., the experimenters were unaware of the sample types being analyzed).
Vector design
Reliable antibodies for NLGN4 do not exist. Therefore, we introduced a hemagglutinin (HA) or FLAG epitope-tag in wild-type (WT) human NLGN4X coding sequence (CDS) after its N-terminal signal peptide (1–41 aa) cleavage site, which does not interfere with the surface trafficking of NLGN4 (Chanda et al., 2016; Marro et al., 2019). The R101Q and N102A mutations were introduced in HA-NLGN4X CDS using site-directed mutagenesis. Similar strategies were implemented for generating HA-tagged rat NLGN1/2 and mouse NLGN3 expression cassettes carrying R101Q-equivalent mutations. These constructs were then inserted into a vector preceded by cytomegalovirus promoter, a Kozak sequence, and followed by an internal ribosome entry site (IRES)-driven fluorescent protein, either enhanced green fluorescent protein (EGFP) or mOrange (see Fig. 2A). The HA-tagged rat NRXN1β without splice-site #4 (NRXN1β-SS4) was cloned and expressed using a similar approach (see Fig. 2G).
Figure 2.

R101Q mutation prevents maturation and surface localization of NLGN4. A, Design of NLGN4 (WT/R101Q) expression vectors (for details, see Materials and Methods). B, Representative immunoblot of HA-tagged NLGN4 (top) collected from HEK293 cells expressing NLGN4 WT or R101Q variant; arrowheads indicate mature (green) versus immature (purple) NLGN4 products. GAPDH was used as a loading control (middle), and EGFP was used as a transfection control (bottom). C, Bar graphs summarize relative expression of total (mature + immature; left), mature (middle), or immature (right) products in WT versus R101Q conditions, when normalized to corresponding EGFP levels. D, Sample images (left) of HEK293 cells transfected with NLGN4 WT (top) and R101Q (bottom) constructs coexpressing mOrange (red), immunolabeled with HA antibody under nonpermeabilized condition (green), and counterstained for DAPI (blue). Yellow versus white arrowheads point at transfected versus nontransfected cells, respectively. Summary graph (right) of HA-NLGN4 surface expression normalized to mOrange signal intensity. Ei, Immunoprecipitation experiment from HEK293 cells in untransfected control (Ctrl) or those expressing HA-tagged NLGN4 (WT vs R101Q) constructs: sample immunoblots (IBs) with anti-HA antibody for 10% input (left; with GAPDH as a loading control), or when immunoprecipitated (IP) with anti-HA antibody versus IgG control (right); arrowheads point at mature (green) versus immature (purple) NLGN4 products, IgG heavy chains (blue) or IgG light chains (red), and asterisk indicates a nonspecific (ns) “sticky” band. Eii, To assess NLGN4 dimerization, both HA- and FLAG-tagged versions of NLGN4 were coexpressed in HEK cells, immunoprecipitated with anti-HA antibody (right), and the same lanes were immunolabeled with mouse anti-HA versus rabbit anti-FLAG antibodies. F, Summary graph indicates the relative intensity of immunoprecipitated FLAG-NLGN4 compared with HA-NLGN4, for mature versus immature products in WT versus R101Q conditions. G, Experimental strategy for probing NLGN4–NRXN interaction (for details, see Materials and Methods). H, I, Average bar graphs (H) and example images (I) of aggregates (white arrowheads) formed by HEK cells expressing NRXN with mOrange reporter, and either control vector or NLGN4 (WT/R101Q) with EGFP reporter. Quantification of cell aggregates were performed using Mander's coefficient of colocalization, and normalized to control condition. All numerical data are presented as the mean ± SEM, with total number of batches analyzed (for Western blots) or field of views analyzed/independent batches (for imaging). Statistical significance was evaluated by two-tailed, unpaired (paired for batchwise comparisons, C, F), Student's t test or one-way ANOVA (across all conditions, F). ***p < 0.005; *p < 0.05; ns, not significant (p > 0.05).
NLGN immunoblotting
Human embryonic kidney 293 (HEK293) cells were transfected with respective vectors using polyethylenimine (PEI) and were allowed to express the constructs for 48 h with media exchanges at 8 and 24 h. Cells were collected by scraping and lysed in RIPA buffer (150 mm NaCl, 5 mm EDTA, 25 mm Tris pH 7.4, 1% Nonidet P-40 substitute, 0.5% sodium deoxycholate) supplemented with Halt protease inhibitor cocktail (PIC; catalog #78429, Thermo Fisher Scientific). Lysates were mixed with 2.5–5% SDS loading buffer, run on a 7.5% PAGE, and transferred to a nitrocellulose membrane. Membranes were blocked with 1–3% bovine serum albumin in Tris-buffered saline and then immunostained overnight at 4°C with mouse anti-HA (1:1000; catalog #h3663, Sigma-Aldrich) or rabbit anti-HA (1:3000; catalog #51 064–2-AP, Proteintech) antibodies. Membranes were stained with fluorescent secondary antibodies and imaged using the LI-COR Odyssey CLx imaging system. Mouse anti-GAPDH primary antibodies (1:60,000; catalog #60 004–1-Ig, Proteintech; or 1:10,000; catalog #MAB374, Sigma-Aldrich) were used as well loading control, whereas a mouse anti-GFP antibody (1:1000; catalog #AE012, ABClonal) was used as transfection control for quantifying relative protein expressions (NLGNs/EGFP intensity). Normalized ratios of mature versus immature NLGNs (see Fig. 4C–E) were calculated as corresponding fractions of total (mature + immature) NLGN1/2/3 levels.
Figure 4.
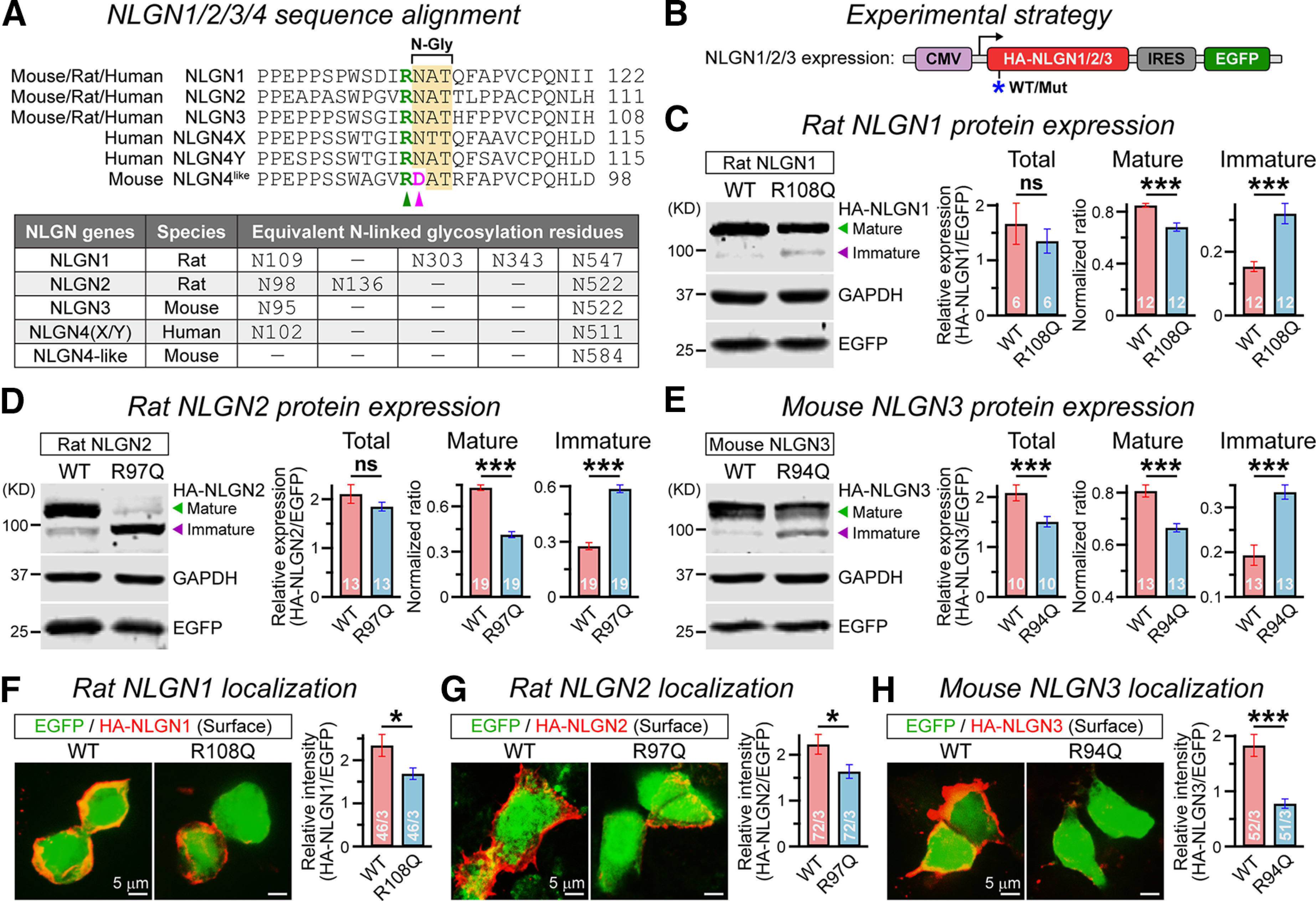
R101Q-equivalent mutations similarly affect maturation and surface trafficking of all NLGNs. A, Sequence alignment (top) of mouse/rat/human versions of NLGN1/2/3/4×/4Y indicate a conserved arginine residue (green arrowhead) followed by an asparagine glycosylation site (N-Gly) in all NLGNs, except in mouse NLGN4 that contains an aspartate residue instead (pink arrowhead). No NLGN4 homolog was identified in the rat. The table (bottom) lists all comparable N-linked glycosylation sites of NLGN1/2/3/4 from different species. B, Strategy for expressing HA-tagged NLGN1/2/3 with or without the R101Q-equivalent mutations (for details, see Materials and Methods). C, Representative immunoblot (left) and normalized ratios (right) of HA-tagged mature (green arrowhead) versus immature (purple arrowhead) NLGN1 products extracted from HEK293 cells expressing rat NLGN1 WT or R108Q mutant proteins. EGFP and GAPDH were respectively used as transfection control and loading control. D, E, Same as C, except for rat NLGN2 containing R97Q mutation (D), and mouse NLGN3 containing R94Q mutation (E). F–H, Sample images (left) represent immunostaining of HA-tagged NLGNs under nonpermeabilized conditions in HEK293 cells coexpressing IRES-driven EGFP, and summary graphs (right) of HA-NLGN signal intensity relative to EGFP, for rat NLGN1 WT versus R108Q mutant (F), rat NLGN2 WT versus R97Q mutant (G), and mouse NLGN3 WT versus R94Q mutant (H). All summary data are presented as the mean ± SEM, with the total number of independent replicates (C–E) or fields of view (F–H)/number of experimental batches. Statistical significance was assessed by two-tailed, paired (C–E) or unpaired (F–H), Student's t test. ***p < 0.005; *p < 0.05; ns, not significant (p > 0.05).
Cell-aggregation assay
HEK293 cells (60–70% confluent) were separately transfected with either EGFP-only control or NLGN4(WT/R101Q)-IRES-EGFP and NRXN1β-IRES-mOrange constructs. After 48 h, cells were washed once with PBS and dissociated with EDTA (1 mm in PBS). Cells were triturated and resuspended at a concentration of 1 × 106 cells/ml in DMEM media (Genesee Scientific) supplemented with 10 mm CaCl2. NRXN-expressing cells (300 µl) were mixed with appropriate combinations of control versus NLGN4 WT versus R101Q mutant-expressing cells (300 µl) in a tube rotator for 2 h at room temperature. Cell mixtures were imaged using a Keyence BZ-X710 microscope and analyzed using Mander's coefficient of colocalization.
Immunoprecipitation
To monitor NLGN4 dimerization, lysates from HEK293 cells expressing both HA-tagged and FLAG-tagged NLGN4 constructs (WT/R101Q) were collected in RIPA buffer and incubated with mouse anti-HA antibody (1:250; catalog #h3663, Sigma-Aldrich) or nonspecific mouse IgG antibody as an isotype control (1:250; catalog #PP100, Sigma-Aldrich) in a tube rotator overnight at 4°C. The lysate and antibody mix were incubated with protein G-Sepharose beads (catalog #6511–1, BioVision) while rotating for 4 h at 4°C. These mixes were then washed three times with RIPA buffer supplemented with PIC. Next, the beads were resuspended in 25 µl of SDS loading buffer and placed in boiling water for 20 min. The mixture was vortexed, spun (13,000 rpm for 5 min), and the eluate was collected to run on an SDS-PAGE. The gels were transferred to a nitrocellulose membrane and blotted with mouse anti-HA (1:1000; catalog #h3663, Sigma-Aldrich) and rabbit anti-FLAG (1:2000; catalog #F7425, Sigma-Aldrich) antibodies.
Glycosidase treatment
For deglycosylation experiments, HA-tagged NLGN4 WT versus R101Q mutant proteins were immunoprecipitated using anti-HA antibody and protein G-Sepharose beads, as described above. The beads were then spun down at 13,000 rpm for 2 min, resuspended in 20 µl RIPA + PIC, and treated with either Endoglycosidase H (Endo H; catalog #P0702S, New England Biolabs) or PNGase F (catalog #P0704S, New England Biolabs) per manufacturer specifications for 1 h at 37°C in a tube rotator. Nonidet P-40 substitute was omitted from PNGase F treatments because of its presence in RIPA buffer. These deglycosylated products were collected by centrifugation, the supernatants were removed, and 15 µl of SDS loading buffer was added. Tubes were boiled for 10 min, spun again to remove any aggregates, and the eluates were collected for Western blot analysis.
Lentivirus production
For neuronal expression, the EGFP-only control or NLGN4(WT/R101Q)-IRES-EGFP cassettes were cloned into a lentiviral vector under human Synapsin (hSyn1) promoter, followed by a Woodchuck regulatory element, and flanked by 5′ and 3′ long terminal repeats. A similar policy was adopted to generate lentiviruses for NLGN4(WT/R101Q)-IRES-mOrange and SEP-GluA1 [AMPA receptor (AMPAR) subunit GluA1 fused to super ecliptic pHluorin (SEP), a pH-sensitive variant of EGFP] constructs. The viral vectors used for Neurogenin-2 (Ngn2)-induced neuronal reprogramming of ES cells included Ngn2-T2A-PuromycinResistance and rtTA (driven by Tet-on and Ubiquitin promoters, respectively).
Three helper plasmids (pRSV-REV, pMDLg/pRRE, and VSV-G; 7 µg each) and corresponding expression vectors (20 µg) were cotransfected with PEI into 70–80% confluent HEK293T (containing SV40 T-antigen to facilitate virus production) cells plated on 10 cm dishes. At 8–9 h post-transfection, the culture medium was exchanged completely, and the supernatant containing viral particles was collected after 36 and 60 h. The supernatant was then pooled and spun at 1200 rpm for 6 min to remove any HEK293 cell debris. The supernatant was then spun at 30,000 rpm for 2 h at 4°C (Beckman L8-70 M ultracentrifuge equipped with SW41Ti rotor). The viral pellet was resuspended overnight in ∼100 µl DMEM media, subsequently aliquoted, and frozen at −80°C before use.
Generation of human neurons
Human ES cells (male H1-line, WiCell WA01) were maintained under feeder-free conditions in mTeSR Plus media (StemCell Technologies). Media were changed every day. When cell density reached ∼70%, they were dissociated with PBS-EDTA (0.5 mm) and plated at a 1:6 dilution onto Matrigel (BD Biosciences)-coated wells. During passaging, the cultures were additionally supplemented with ROCK-inhibitor Y-27 632 (2.5 μm; MedChemExpress) overnight.
Differentiation of human ES cells into neural stem cell (NSC) monolayer was achieved by dual SMAD inhibition (Chambers et al., 2009) with minor modifications as described previously (Chanda et al., 2019). In brief, Oct3/4-positive H1-ES cells were treated with LDN193189 (100 nm; STEMCELL Technologies) and SB431542 (10 μm; STEMCELL Technologies) for 6 d to induce them into Nestin-positive NSCs (see Fig. 5C). The NSCs were then passaged and expanded by a 1:6 split, and subsequently treated with Cytarabine (Ara-C; 2 μm) to prevent further proliferation. During this period, the NSCs spontaneously differentiated into doublecortin (Dcx)-positive neurons. The cells were finally dissociated with Accutase (Innovative Cell Technologies) and replated with primary mouse glia (passages 1–2, derived from C57BL/6 strain) on Matrigel-coated coverslips at a 1:50 ratio. These cells were cultured in N3 media [DMEM/F12 (Thermo Fisher Scientific), N2 (Thermo Fisher Scientific), and B27 (Thermo Fisher Scientific), supplemented with insulin (20 μg/ml, Sigma-Aldrich) and penicillin/streptomycin (Thermo Fisher Scientific)].
Figure 5.

R101Q mutation inhibits NLGN4-induced excitatory synapse formation in NSC-derived human neurons. A, NSCs were induced from H1-ES cells by dual-SMAD inhibition. The NSC-derived neurons were cocultured with glia, infected with lentivirus-expressing transgenes, and analyzed at indicated time points (arrowheads). B, Design of lentiviral vectors expressing NLGN4 WT or R101Q variant under hSyn1 promoter followed by an IRES-EGFP construct. An EGFP-only vector was used as infection control (Ctrl). C, Images illustrate cellular identities, when immunolabeled for ES cell marker (Oct3/4), NSC marker (Nestin), or neuronal marker (Dcx), at different stages of the differentiation protocol. DAPI was used for nuclear stain. At day 60, elaborate neuronal morphology was confirmed by EGFP signal. Arrowheads point at neurons (yellow) and glial cells (white) in the culture. D, Sample images (top) depict NSC-derived neurons expressing NLGN4 WT versus R101Q variant, when immunostained for cytoskeletal marker Tuj1 and synaptic marker Synapsin. Bar graphs (below) represent normalized counts of cell bodies per field of view (left), total neurite volume (middle), and integrated signal intensity of Synapsin (right), for WT versus R101Q conditions. E, Representative images (top) of MAP2-positive dendritic segments counterstained for Synapsin, and summary graphs (bottom) of the density and size of Synapsin puncta, as quantified from cells expressing NLGN4 WT versus R101Q mutant. F, Example images (left) of MAP2-positive dendritic branches from cells in control, NLGN4 WT, and R101Q mutant conditions, as counterstained for an inhibitory synapse marker vGAT. Summary graphs (right) of the density and size of vGAT puncta. G, Same as F, except the neurons were immunolabeled for VGluT, an excitatory synapse marker. Averages indicate the mean ± SEM, with the total number of fields of view analyzed/number of batches. Statistical significance was tested by two-tailed, unpaired, Student's t test. ***p < 0.005; **p < 0.01; *p < 0.05; ns, not significant (p > 0.05).
Transcription factor (TF)-mediated direct neuronal reprogramming was achieved as described previously (Zhang et al., 2013; Chanda et al., 2019). In brief, the H1-ES cells were coinfected with lentiviruses expressing rtTA and Ngn2-t2A-PuromycinResistance, induced with doxycycline (2 μg/ml), selected using puromycin (1 μg/ml), and cultured together with mouse glia in N3 media (see Fig. 7A). The NSC-derived or Ngn2-induced human neurons were additionally infected with control or NLGN4(WT/R101Q)-expressing lentiviruses, as needed.
Figure 7.

NLGN4 WT but not the R101Q variant suppresses AMPAR-mediated synaptic transmission in Ngn2-induced human neurons. A, Neurogenesis was induced in H1-ES cells by ectopic expression of Ngn2 using high-titer lentivirus, and cells were selected using puromycin and cocultured with mouse glia for an extended period (timeline; top). In addition, cells were also sparsely infected with lower-titer lentiviruses expressing either a control vector or an HA-tagged NLGN4 WT versus R101Q variant under hSyn1 promoter (abbreviated construct maps; bottom), each followed by an IRES-EGFP or IRES-mOrange reporter (R). B, Representative images (left) depict Ngn2-induced neurons coinfected with lentivirus expressing a control vector or NLGN4 WT versus R101Q variant, when immunostained for dendritic marker MAP2; bar graph (right) represents normalized counts of cell bodies per field of view. Arrowheads point at neurons (yellow) and glial cells (white) in these cocultures. C, Sample image of an Ngn2-induced human neuron expressing NLGN4 transgene, as visualized by EGFP (yellow arrowhead) and patched using a recording pipette (Rec). White arrowhead points at an uninfected cell. D, Representative traces (left) of AMPAR-mediated evoked EPSCs with arrowheads indicating stimulation artifacts. Summary graph (right) of EPSC amplitude, as measured from Ngn2-induced neurons expressing either a control vector or NLGN4 WT versus R101Q mutant constructs, while the recording quality was monitored by Rs measurements. E, F, Example traces of AMPAR-mediated mEPSCs (E) and cumulative probability plots with average bar graphs (F) for mEPSC event frequency and amplitude, as recorded from control, NLGN4 WT, and NLGN4 R101Q conditions. Data are presented as the mean ± SEM, with corresponding number of cells patched (for electrophysiology) or frames analyzed (for imaging)/number of experiments. Statistical significance was assessed by two-tailed, unpaired, Student's t test (all bar graphs), or KS test (probability plots; F). ***p < 0.005; **p < 0.01; ns, not significant (p > 0.05).
Immunostaining
After 48 h of transfection, the HEK293 cells were washed once with PBS and immediately fixed with 4% paraformaldehyde (PFA) for 20–30 min at room temperature, followed by an additional wash with PBS. Human neurons expressing NLGN constructs were similarly fixed with 4% PFA. Cells were blocked in 10% cosmic calf serum for 1 h at 37°C, incubated with primary antibodies for 1–2 h at 37°C, washed four times with blocking buffer, followed by incubation with fluorophore-conjugated secondary antibodies for 1 h at 37°C. Cells were then washed four times with PBS and mounted on glass slides using Fluoromount-G (Southern Biotech). For immunostaining experiments where permeabilization was required, 0.1% Triton X-100 was added to the blocking buffer and for all subsequent steps including washes or antibody dilution. Cells were also nuclear stained with DAPI (1:50,000; catalog #D1306, Thermo Fisher Scientific) for 10 min when applicable. Images were acquired using an inverted microscope (model IX83, Olympus) equipped with LaserStack units (model 3iL33, Intelligent Imaging Innovations) and spinning disk confocal scanner (model CSU22, Yokogawa), or a STELLARIS 5 (Leica Microsystems) laser-scanning microscope. A series of z-projections were obtained with 0.5–1 µm optical thickness using either a 20× dry objective or 40–63× oil-immersion objectives.
Primary antibodies for immunolabeling included mouse anti-HA (1:1000; catalog #h3663, Sigma-Aldrich), rabbit anti-calnexin (1:500; catalog #PA5-34 754, Thermo Fisher Scientific), rabbit anti-GM130 (1:300; catalog #A5344, ABClonal), chicken anti-GFP (1:500; catalog #GFP-1020, Aves Labs), mouse anti-Oct3/4 (1:200; catalog #sc-5279, Santa Cruz Biotechnology), mouse anti-Nestin (1:500; catalog #MAB1259, R&D Systems), goat anti-Dcx (1:500; mixture of catalog #sc-8066 and #8067, Santa Cruz Biotechnology), mouse anti-Tuj1 (1:1000; catalog # 801202, BioLegend), chicken anti-MAP2 (1:400; catalog #ab5392, Abcam), mouse anti-MAP2 (1:500; catalog #M1406, Sigma-Aldrich), rabbit anti-Synapsin1/2 (1:500; catalog #106002, Synaptic Systems), rabbit anti-vGAT (1:200; catalog #131003, Synaptic Systems), rabbit anti-VGluT1/2 (1:500; YENZYM ANTIBODIES, mixture of catalog #YZ6089, #6093, #6102), and Alexa Fluor 488/555/647-conjugated secondary antibodies (Thermo Fisher Scientific). A mouse anti-GFP antibody (1:500; catalog #AE012, ABclonal) was used to further amplify surface SEP–GluA1 signals under nonpermeabilized condition.
Image analysis
All images were analyzed using FIJI-ImageJ (NIH). Images of HA-NLGN surface localization in HEK293 cells were converted into maximum-intensity projections (two to three optical slices) and the mean signal intensity per cell was normalized to corresponding fluorescent reporter protein intensity. Images of NLGN4 colocalization at endoplasmic reticulum (ER) and Golgi were appropriately thresholded and analyzed using Mander's coefficient within the JACoP plugin. To quantify synaptic puncta along neuronal processes, images were obtained with maximum intensity projection, and channels containing neurite arbors were converted into ROIs to specify analysis areas for synaptic markers. The ROI masks were then skeletonized, and the total pixel length of these skeletons was measured for normalization.
Electrophysiology
Whole-cell patch-clamp recordings were performed similarly to those described previously (Chanda et al., 2013). In brief, reprogrammed human neurons were patched using an internal solution containing (for current clamp; in mm) 130 KMeSO3, 10 NaCl, 2 MgCl2, 0.5 EGTA, 0.16 CaCl2, 4 Na2ATP, 0.4 NaGTP, 14 Tris-creatine phosphate, and 10 HEPES-KOH, pH 7.3, 310 mOsm; or (for voltage clamp; in mm) 135 CsCl2, 1 EGTA, 1 NaGTP, 2 QX-314, and 10 HEPES-CsOH, pH 7.4, 310 mOsm. The extracellular solution contained the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES-NaOH, pH 7.4. Recordings were conducted using an integrated patch-clamp amplifier (Sutter Instrument) with a customized Igor Pro (WaveMetrics) data acquisition system. Current-induced action potential (AP) recordings were performed at approximately −60 mV, using a small holding current to adjust the membrane potential accordingly. Voltage-clamp recordings for AMPAR-mediated EPSCs and GABAA receptor (GABAAR)-mediated IPSCs were conducted at a holding potential (Vhold) of −70 mV. The evoked synaptic currents were triggered by field stimulation using a matrix electrode (catalog #MX21AEW-RT2, FHC) connected to an A365RC isolated pulse stimulator (World Precision Instruments). AMPAR-mediated or GABAAR-mediated synaptic currents were isolated using, respectively, picrotoxin (100 μm; GABAAR/GlycineR Blocker, Tocris Bioscience) or CNQX (AMPAR blocker; 25 μm; Tocris Bioscience). Tetrodotoxin (2 μm; Ascent Scientific) was additionally added to the external solution during all miniature EPSC (mEPSC) and miniature IPSC (mIPSC) recordings, to avoid presynaptic release induced by spontaneous APs.
Quantitative RT-PCR
Ngn2-induced human neurons from control, NLGN4 WT, and NLGN4 R101Q conditions were washed with ice-cold PBS and collected in 500 µl TRIzol reagent. Immediately, 250 μl chloroform was added to the cell lysate, vortexed vigorously, centrifuged at 12,000 × g for 15 min, aqueous phase collected, and RNA precipitated by adding 250 µl of isopropanol and centrifuging at 12,000 × g for 10 min. The RNA pellets were washed with 70% ethanol, air dried, and dissolved in nanopure water.
cDNA was generated from 200 to 400 ng to total RNA using Invitrogen SuperScript III First-Strand Synthesis SuperMix (catalog #11752–050, Thermo Fisher Scientific) following the manufacturer protocol. Quantitative PCR (qPCR) was performed on a CFX-96 (Bio-Rad) machine using SYBR Green Master Mix (catalog #M-915, Gold Biotechnology). Two independent primer sets were designed that spanned two NLGN4 exons (set 1: forward, 5′-GAAGCCCGTCATGGTCTATATC-3′; reverse, 5′-AGTATTCCCAGACGGTAGTTAATG-3′; set 2: forward, 5′-ACCTGGATGAGAGATCCTTACT-3′; reverse, 5′-CGTGGGCACGTAGATGTTTA-3′). A human GAPDH primer set (forward, 5′-TTGAGGTCAATGAAGGGGTC-3′; reverse, 5′-GAAGGTGAAGGTCGGAGTCA-3′) was used as an internal control to normalize the NLGN4 expression levels per sample.
Data presentation
For batchwise comparisons (i.e., immunoblots and qPCR), the total number of experimental replicates are indicated with corresponding average values. For all other experiments (i.e., electrophysiology and imaging), the average values are presented with X/Y, where “X” represents the total number of neurons recorded (electrophysiology) or field of views analyzed (imaging) from “Y” number of independent batches. All average data indicate the mean ± SEM (SD of parameter tested divided by the square root of number of samples). In most cases, statistical comparisons between conditions were made using unpaired (paired for batchwise comparisons), two-tailed, Student's t test [***p < 0.005; **p < 0.01; *p < 0.05; ns, not significant (p > 0.05)]. In some cases, one-way ANOVA (for group assessments) or Kolmogorov–Smirnov (KS; for cumulative distribution) tests were performed, as mentioned in the figure legends.
Results
Autistic individual carries a missense mutation in NLGN4
The patient we describe in this study is a male child who began to manifest signs of pervasive developmental abnormality at ∼1 year of age and was diagnosed with autism when he was 2 years old. He was first seen in a neurology clinic for the evaluation of recurring unresponsive staring spells. At 8 years of age, EEG recordings identified abnormal activity with independent epileptiform discharges in the cerebral cortex. He was since treated with Trileptal and Lamictal, and subsequently showed subjectively enhanced social response including elevated nonverbal interactions and spontaneous greetings. He is partially capable of communicating with others using rapid prompting method aids and often demonstrates emotional intelligence and the ability to comprehend social cues by responding accordingly. However, despite these improvements, the subject continues to lack verbal expression, and struggles with repetitive behavior and social detachment.
At the age of 4 years, his social responsiveness was evaluated using SRS scales, and his overall test score met the rating criteria for severe autism (Fig. 1Ai). By 14 years of age, he showed moderate improvement in social awareness and cognition but remained equally impaired for social communication, placing him into the moderate autism category (Fig. 1Ai). The patient was also assessed for his aberrant behavior in residential-, educational-, and community-based facilities. ABC-2 test subdomains ranked him as severely autistic, as he continued to struggle with hyperactivity, irritability, and repetitive interest or stereotyped behavior (Fig. 1Aii). The Vineland-3 test was performed to evaluate his adaptive behavior skills. The composite score suggested that his communication, socialization, and daily living skills range well below the expected age classification (Fig. 1Aiii). In addition, when assessed by the CARS-2 test, he scored beyond the cutoff value to be characterized as severely autistic (Fig. 1Aiv).
Figure 1.
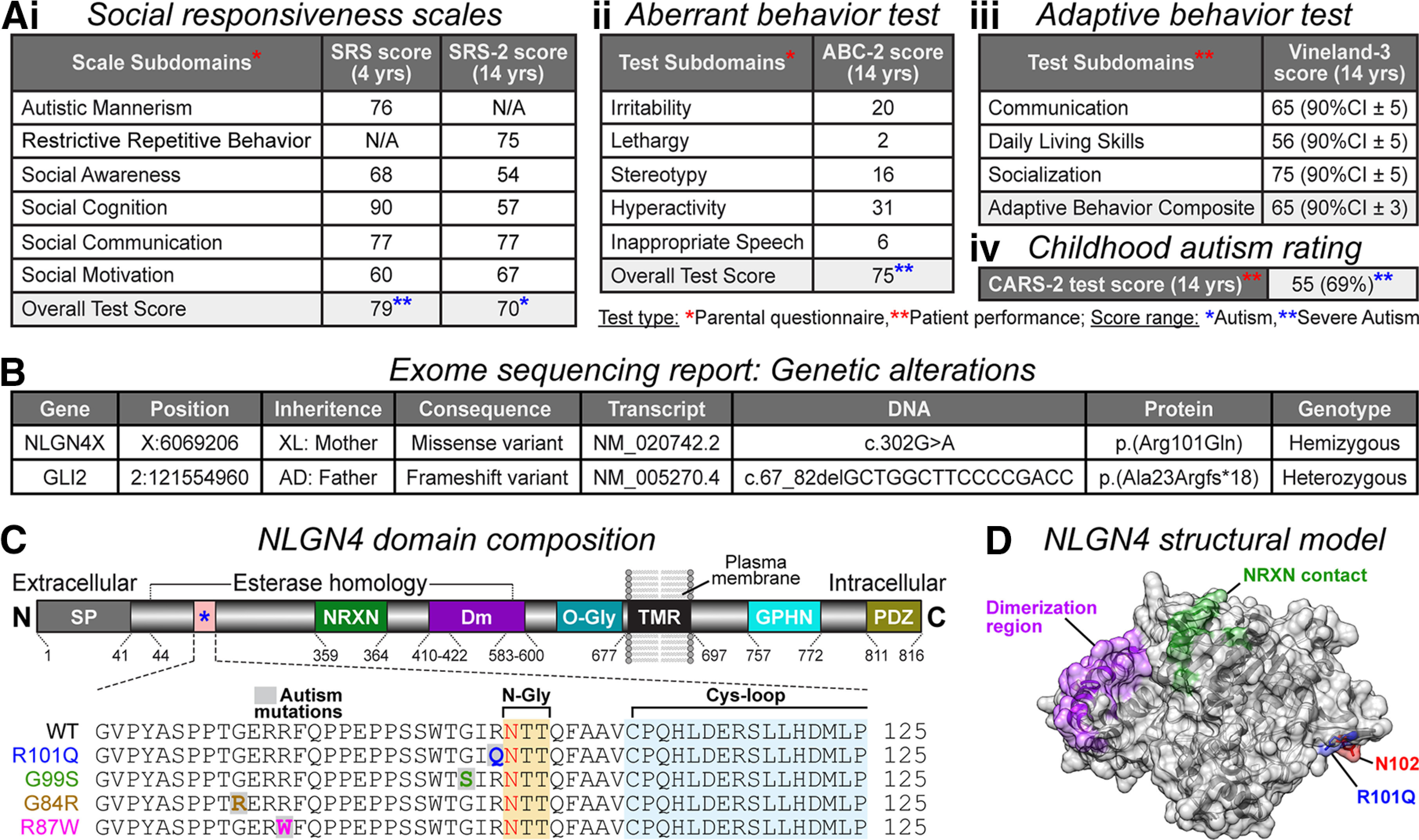
Identification of NLGN4 missense mutation in an autistic patient. Ai–iii, Behavior assessments leading to ASD diagnosis in the proband. Subdomain scores for social responsiveness tests (SRS and SRS-2, respectively conducted at ages 4 and 14 years using parental questionnaire; severe autism score, ≥76, moderate autism score, 66–75; mild deficit score, 60–65; Constantino, 2005; Constantino and Gruber, 2012; i), aberrant behavior checklist (ABC-2 test performed at age 14 years using the parental questionnaire; severe autism score, 75; Aman and Singh, 2017; ii), adaptive behavior scales (Vineland-3 assessment at age 14 years via professional observation; results are represented as 90% confidence interval ± SD; below-average score, <100; Sparrow et al., 2016; iii), and childhood autism rating (CARS-2 test at age 14 years via professional observation; severe autism score, ≥35; Schopler and Van Bourgondien, 2010; iv). B, WES report listing genetic alterations in NLGN4X and GLI2. Sequencing shows that the patient is hemizygous for a maternally inherited missense mutation in the NLGN4X gene, producing the Arg101Gln (R101Q) protein variant. The GLI2 heterozygous mutation originated from an asymptomatic father. C, Annotated map of NLGN4 with domain composition and corresponding amino acid numbers (top, not to scale): signal peptide (SP; gray), NRXN-binding domain (NRXN; green), dimerization residues (Dm; purple), O-glycosylation site (O-Gly; teal), transmembrane region (TMR; black), Gephyrin-binding domain (GPHN; cyan), and PDZ-binding sequence (PDZ; gold). The R101Q mutation is located within the esterase homology domain of NLGN4 (bottom, magnified view), neighboring a consensus site for N-linked glycosylation (N-Gly; yellow), and proximal to a cysteine-loop structure (Cys-loop; blue). The R101Q variant is also adjacent to previously reported ASD mutations (e.g., G99S, G84R, and R87W). D, Model of NLGN4 extracellular domain indicating spatial orientation of the R101Q mutation (blue), proximal to N-glycosylation (red) residue, and distal from dimerization (purple) and NRXN-contacting (green) regions.
To determine whether any underlying genetic mutations could help explain the clinical symptoms of this child, a high-throughput WES test was conducted privately in a clinic. This genomic analysis followed by bidirectional Sanger sequencing identified the presence of two mutations in the patient exome; a maternally inherited missense variant 302G>A creating a hemizygous R101Q substitution in NLGN4 CDS, and a paternally inherited frameshift variant causing heterozygous deletion in GLI2 (Fig. 1B). Both parents are heterozygous for their corresponding mutations and are asymptomatic, suggesting compensatory effects from their second alleles. Although the male patient is also heterozygous for his autosomal GLI2 deletion, he lacks a second allele to compensate for the X-linked NLGN4 mutation. Therefore, we hypothesized that the NLGN4X missense mutation could manifest pathogenic potential. Moreover, several mutations in NLGN4X gene have been previously reported in autistic probands, making the R101Q substitution a likely candidate leading to ASD diagnosis in this individual.
The R101 residue is located within the esterase homology domain of NLGN4, adjacent to a consensus N-linked glycosylation sequon (N102), and proximal to a downstream cysteine-loop (Fig. 1C). Interestingly, the R101Q mutation is also situated next to a region with multiple previously identified ASD mutations (e.g., G84R, R87W, and G99S), some with mostly unknown mechanistic consequences (Fig. 1C; Fabrichny et al., 2007; Zhang et al., 2009; Xu et al., 2017; Schepici et al., 2019; Nguyen et al., 2020). A structural model of the extracellular domain of NLGN4 predicted that the R101Q mutation is spatially oriented opposite to its dimerization domain and away from its NRXN contact site (Fig. 1D; Fabrichny et al., 2007; Leone et al., 2010).
R101Q mutation impairs NLGN4 maturation and its trafficking to cell surface
We first inquired whether R101Q substitution has any impact on the maturation of NLGN4 protein. We transfected HEK293 cells with constructs expressing HA-tagged NLGN4 WT versus R101Q variant followed by either an IRES-EGFP or IRES-mOrange reporter (Fig. 2A). We extracted total cell lysate proteins after 48 h and performed quantitative immunoblotting with HA antibody. The molecular weight (MW) of WT NLGN4 is ∼92 kDa, but it generated a predominantly high MW product (>100 kDa) which likely reflected a maturely glycosylated version, as described for other NLGNs (Fig. 2B; also see Boucard et al., 2005; Budreck and Scheiffele, 2007; Zhang et al., 2009; Poulopoulos et al., 2012). The WT NLGN4 also generated a minor, low MW (<100 kDa) version, which indicated an immaturely glycosylated product that was possibly undergoing post-translational modifications (PTMs) in intracellular organelles (Fig. 2B; also see Zhang et al., 2009; Poulopoulos et al., 2012). We noticed that both products were similarly present in R101Q condition, however, their relative abundance was significantly altered (Fig. 2B). R101Q substitution reduced the total NLGN4 protein content, considerably decreased its mature version, and increased the immature form, when normalized to coexpressed EGFP levels (Fig. 2C).
Because the R101Q mutation partially inhibited NLGN4 maturation, we next asked whether it influenced the surface trafficking of NLGN4, an important feature of all synaptic CAMs. We immunostained transfected HEK293 cells after 48 h with HA antibody under nonpermeabilized conditions and normalized the signal to a coexpressed intracellular mOrange intensity. We noticed that R101Q substitution considerably diminished the localization of NLGN4 at the cell surface, suggestive of a major trafficking defect (Fig. 2D).
We next asked whether R101Q mutation alters other critical properties of NLGN4, e.g., dimerization or NRXN-binding. We first confirmed the specificity of HA antibody as it failed to probe any similar MW product in cell lysates from untransfected control; moreover, both NLGN4 WT and R101Q variants did not exhibit any nonspecific binding when immunoprecipitated with the HA antibody versus a control IgG (Fig. 2Ei). To monitor NLGN4 dimerization, we next coexpressed FLAG-tagged and HA-tagged NLGN4 in HEK293 cells, lysed them after 48 h, immunoprecipitated NLGN4 (WT vs R101Q mutant) using HA antibody, and immunolabeled the products with HA and FLAG antibodies of two different species (Fig. 2Eii). We observed that both mature and immature versions of NLGN4 WT and R101Q mutant were able to form homodimers in similar proportions, as evidenced by HA and FLAG colabeling (Fig. 2Eii,F).
To examine whether R101Q substitution affects the ability of NLGN4 to bind NRXN, we transfected HEK293 cells with a NRXN1β-SS4 construct followed by IRES-mOrange reporter, and EGFP-only control, NLGN4 WT, or R101Q mutant constructs followed by IRES-EGFP reporter (Fig. 2G). At 48 h post-transfection, we dissociated the NRXN-expressing and NLGN4-expressing cells, mixed them together, and evaluated their capacity to form aggregates via molecular interaction. We found that, with respect to control condition, expression of both NLGN4 WT and R101Q mutant substantially increased the formation of multicellular clusters, as identified by colocalization of mOrange and EGFP-positive cells (Fig. 2H,I). This result suggests that the NLGN4 R101Q variant can interact with NRXN. However, when compared with WT, the R101Q condition showed a small but significant reduction in cell aggregation, likely because of the lower surface export of mutant protein (Fig. 2H,I).
In summary, our data revealed that the R101Q mutation is pathogenic, inhibits maturation of NLGN4 protein and its transport to the cell surface, but does not severely alter its ability to dimerize or its affinity for NRXN.
R101Q mutation causes glycosylation defects in NLGN4 and increases its retention at endoplasmic reticulum and Golgi
The vicinity of the R101 residue of NLGN4 to a potential N-glycosylation sequon (N102; Fig. 1C,D) and the appearance of a lower MW product in R101Q conditions (Fig. 2B) made us hypothesize that the R101Q mutation could indirectly impair glycosylation at the adjacent N102 residue. To probe this, we first substituted the N102 residue for an alanine (N102A) to directly eliminate its glycosylation (Fig. 3A). We expressed this HA-tagged mutant construct in HEK293 cells for 48 h and performed Western blot with HA antibody under a denaturing condition. We found that the N102A mutation similarly generated a prominent immature product, and phenocopied the R101Q variant by substantially decreasing mature NLGN4 level (Fig. 3B).
Figure 3.
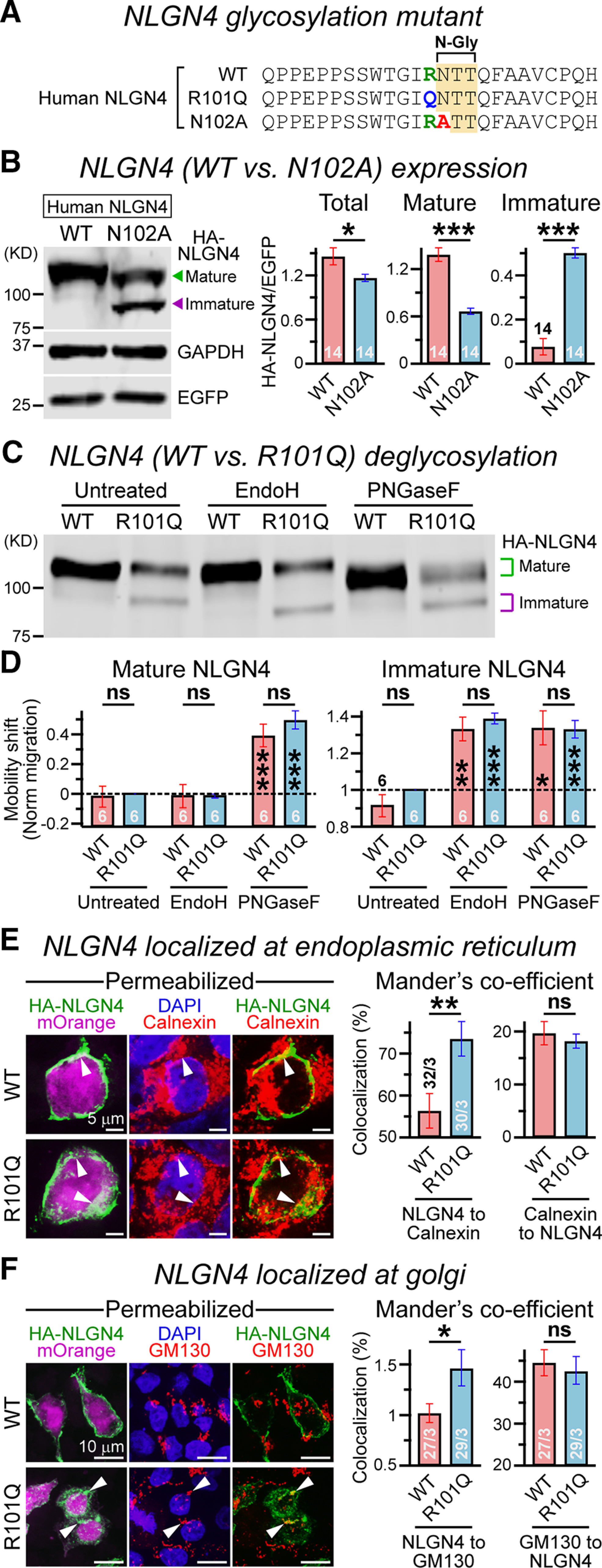
R101Q mutation blocks NLGN4 glycosylation and retains it at intracellular compartments. A, Alanine substitution of NLGN4 N102 residue to prevent N-linked glycosylation (N-Gly). B, Western blot (left) illustrating mature (green arrowhead) versus immature (purple arrowhead) HA-NLGN4 products harvested from HEK293 cells expressing NLGN4 WT versus N102A mutant, with EGFP as a transfection control and GAPDH as a loading control. Summary graphs (right) of total, mature, and immature NLGN4 normalized to coexpressed EGFP levels. C, Example immunoblot (C) for NLGN4 WT and R101Q variant, before (untreated) and after enzymatic deglycosylation with glycosidases (i.e., Endo H and PNGase F). D, Bar graphs represent the relative positions of mature (left) and immature (right) NLGN4 products. To compare across multiple experimental batches with slightly variable mobility shifts, the position of all bands was normalized (dotted lines) to the corresponding distance between the mature and immature bands in the untreated R101Q condition. E, Representative images (left) of HEK293 cells expressing NLGN4 WT (top) or R101Q mutant (bottom), as visualized by an IRES-driven mOrange reporter, and stained with DAPI, HA antibody, and calnexin antibody (respective channels) under permeabilized conditions. Arrowheads point at intracellular HA-NLGN4 colocalized with soluble mOrange and ER marker calnexin. Summary graphs (right) quantify the correlation between HA-NLGN4 and calnexin signals, measured via Mander's coefficients. F, Same as E, except the cells were counterstained with GM130 antibody. The values on bar graphs represent the mean ± SEM for number of experimental batches (immunoblots) or fields of view analyzed (imaging)/number of independent batches. Statistical significance was weighed by two-tailed, paired (for immunoblots) or unpaired (for imaging), Student's t test. ***p < 0.005; **p < 0.01; *p < 0.05; ns, not significant (p > 0.05).
To further characterize the glycosylation profile of mature versus immature NLGN4 products, we next treated the WT and R101Q variant with glycosidases: Endo H (which cleaves only the ER-associated, immature, high-mannose oligosaccharides from N-linked glycoproteins) or PNGase F (which removes all N-linked oligosaccharides from glycoproteins, including both complex and hybrid forms as well as immature high-mannose oligosaccharides). We noticed that Endo H treatment specifically downshifted the immature NLGN4 bands of lower MW without affecting the position of mature NLGN4 bands of higher MW; whereas PNGase F treatment displaced both high- and low-MW NLGN4 products for both the WT and R101Q conditions (Fig. 3C,D). Therefore, the low- and high-MW bands, respectively, correspond with immaturely and maturely glycosylated versions of NLGN4, and the ratio between these two was significantly altered by R101Q substitution.
Because R101Q mutation impaired the maturation of NLGN4 and prevented its transport to the cell surface (Fig. 2), we postulated that the mutant protein might accumulate in intracellular organelles (e.g., ER and Golgi, which play major roles in PTM pathways including glycosylation). We transfected HEK293 cells with HA-tagged NLGN4 WT versus R101Q followed by an IRES-mOrange reporter, and coimmunostained them for ER-specific marker calnexin and HA antibody under permeabilized conditions. We noticed that the R101Q variant exhibited higher localization at ER, when compared with WT NLGN4 (Fig. 3E). We also observed a similar increase in colocalization between the NLGN4 R101Q variant and the Golgi-specific marker GM130 (Fig. 3F). These data indicate that the R101Q substitution inhibits the glycosylation of NLGN4 and increases its retention at intracellular compartments.
R101Q-equivalent mutations trigger trafficking defects in all NLGNs with conserved glycosylation sites
The R101 residue and its adjacent glycosylation site are highly conserved among all NLGN genes from different species, except mouse NLGN4 (also referred to as NLGN4-like), which contains an aspartic acid residue (D85) instead of asparagine in the equivalent location, eliminating the consensus sequence for N-linked glycosylation (Fig. 4A). Therefore, we next introduced R101Q-equivalent mutations in HA-tagged versions of rat NLGN1, rat NLGN2, and mouse NLGN3, and asked whether that can similarly affect their molecular properties (Fig. 4B). Our immunoblotting experiments from HEK293 cells transfected with NLGN1 R108Q mutant indicated a definitive increase in immature product and a decrease in mature product (Fig. 4C). This effect was similarly reproduced by R97Q mutation in NLGN2 and R94Q mutation in NLGN3 (Fig. 4D,E). Of note, R101Q-equivalent mutations also decreased the total protein content of NLGN3 (similar to NLGN4; Fig. 2B), but not that of NLGN1 or NLGN2 (Fig. 4C–E), which contained a higher number of N-glycosylation sites (Fig. 4A). Thus, R101Q-equivalent mutations similarly affected the maturation of all NLGNs and also impaired the stability of different NLGNs depending on their potential glycosylation profiles.
To further assess whether these mutation-induced deficiencies in NLGN1/2/3 maturation can also affect their ability to localize at the cell surface, we conducted immunostaining of NLGN-expressing HEK293 cells under nonpermeabilized conditions. We found that NLGN1 R108Q, NLGN2 R97Q, and NLGN3 R94Q substitutions displayed significant reduction in surface intensity, with respect to their corresponding WT versions (Fig. 4F–H). These results demonstrate that the N102-equivalent glycosylation sites play a critical role in the maturation and surface trafficking of all NLGNs, and that the R101-equivalent upstream residues can strongly influence this process.
R101Q mutation blocks the ability of NLGN4 to induce excitatory synaptogenesis
After we used HEK293 cells as a reduced system to establish that R101Q substitution is not benign and directly impairs the molecular properties of NLGN4, we next inquired whether this pathogenic mutation affects the morphologic and/or functional properties of human neurons. To this end, we first performed dual-SMAD inhibition in human ES cells to generate NSCs, then differentiated the NSCs into neurons, and subsequently cocultured them with mouse primary astrocytes, as described previously (Fig. 5A; Chambers et al., 2009; Chanda et al., 2019). During this period, the cells gradually changed their identity in terms of relevant marker expression, from Oct3/4-positive ES cells to Nestin-positive NSCs to Dcx-positive neurons (Fig. 5C). We infected the neurons with lentivirus expressing either a control EGFP or NLGN4 WT versus R101Q mutant constructs under hSyn1 promoter and followed by IRES-driven EGFP reporter (Fig. 5B). After an additional 30 d, i.e. at 60 d postinfection, we observed robust EGFP fluorescence in these neurons, confirming successful transgene expression (Fig. 5C).
To monitor synapse formation in NSC-derived neurons, we immunostained them for cytoskeletal marker Tuj1 (class III β-tubulin) and synaptic marker Synapsin. We found that the NLGN4 R101Q mutation did not affect the cell number or total neurite volume, but substantially diminished Synapsin puncta intensity (Fig. 5D). Further analysis confirmed that, compared with WT NLGN4, the R101Q substitution considerably reduced both the density and size of synapses formed along Map2-positive dendrites (Fig. 5E).
To gain insight into the type of synapses affected by NLGN4 R101Q mutation, we immunolabeled the neurons with GABAergic or glutamatergic synapse markers (i.e., vGAT or VGluT, respectively). Compared to neurons expressing a control vector, the NLGN4 WT and R101Q conditions displayed a minor increase in GABAergic synapse density without significantly affecting synapse size (Fig. 5F). However, for GABAergic synapses, there was no apparent change in either synaptic parameters between the WT and R101Q conditions (Fig. 5F). For glutamatergic synapses, the expression of WT NLGN4 substantially elevated both the number and size of synaptic puncta, presumably by recruiting excitatory presynaptic terminals via the NRXN interaction, whereas R101Q mutant failed to demonstrate any sizable impact compared with the control condition (Fig. 5G). These data indicate that R101Q substitution substantially impairs the ability of human NLGN4 to induce excitatory synapses when overexpressed, again suggesting a loss-of-function phenotype.
R101Q mutation inhibits NLGN4-mediated suppression of excitatory synaptic transmission
To investigate the effects of NLGN4 R101Q mutation on the functional properties of human neurons, we made electrophysiological recordings from NSC-derived neurons expressing either a control vector or NLGN4 WT versus R101Q constructs (Fig. 5B). Current-clamp recordings detected no noticeable differences among the control versus NLGN4 WT versus R101Q conditions in terms of their ability to fire repetitive APs (Fig. 6A). Furthermore, the membrane capacitance (Cm), membrane resistance (Rm), and resting membrane potential (Vm) did not significantly differ across conditions (Fig. 6B). Voltage-clamp recordings revealed no effect on the amplitude or frequency of GABAergic mIPSCs by either NLGN4 WT or R101Q mutant (Fig. 6C,D). However, the expression of NLGN4 WT but not the R101Q mutant attenuated the frequency of AMPAR-mediated mEPSCs and augmented their interevent interval without affecting mEPSC amplitude (Fig. 6E,F).
Figure 6.
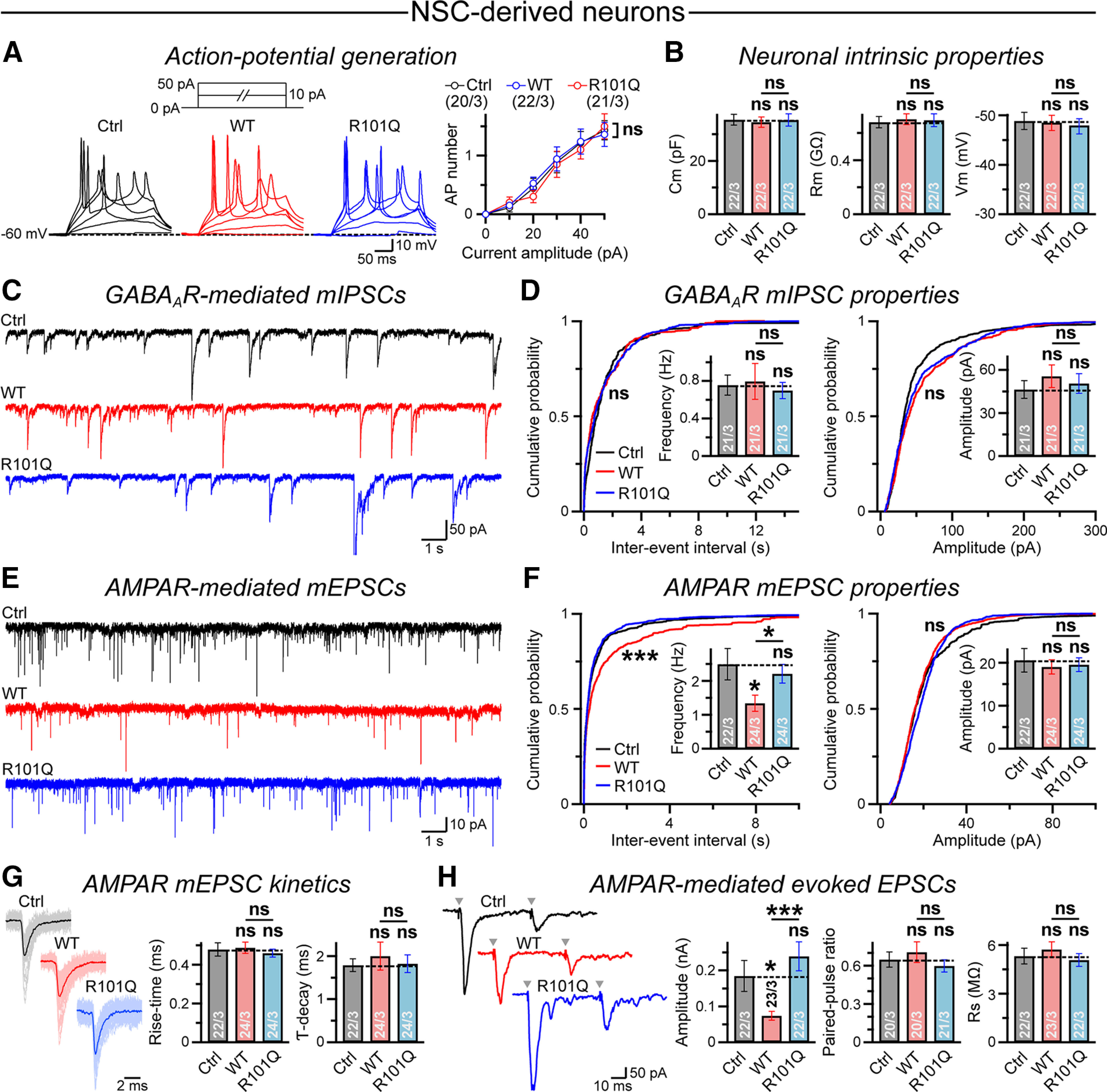
NLGN4 WT but not the R101Q variant suppresses AMPAR-mediated synaptic transmission in NSC-derived human neurons. A, Recordings were conducted from NSC-derived neurons expressing either a control vector (Ctrl; EGFP only) or NLGN4 WT versus R101Q mutant constructs followed by an IRES-driven EGFP reporter. Sample traces (left) of APs induced by step current injections (protocol is shown on top, Vhold = −60 mV), and summary plot (right) of AP numbers as a function of injected current–amplitude. B, Average values of Cm (left), Rm (middle), and Vm (right), as measured from control, NLGN4 WT, and R101Q conditions. C, D, Example traces of GABAAR-mediated mIPSCs (C) and cumulative plots with average bar graphs (D) for mIPSC event frequency and amplitude, as recorded from control, NLGN4 WT, and R101Q conditions. E, F, Same as C and D, except for AMPAR-mediated mEPSC events. G, Superimposed mEPSC waveforms (left) from control, NLGN4 WT, and R101Q conditions. Summary graphs (right) of mEPSC rise time and decay kinetics. H, Representative traces (left) of AMPAR-mediated evoked EPSCs stimulated in pairs (Δt = 50 ms); arrowheads indicate stimulation artifacts. Summary graphs (right) of EPSC amplitude, PPR (EPSC2/EPSC1) as an indirect measure of presynaptic release probability, and Rs as a measure of recording quality. All quantifications are the mean ± SEM. All summary data include the number of cells patched/experimental batches. Statistical significance was calculated by two-tailed, unpaired, Student's t test (all bar graphs), one-way ANOVA (summary plot; A), or KS test (WT vs R101Q probability plots; D–F). ***p < 0.005; *p < 0.05; ns, not significant (p > 0.05).
The kinetic parameters of mEPSC events were not considerably altered by NLGN4 WT/R101Q expression, suggesting no apparent impact on AMPAR composition (Fig. 6G). Finally, the overexpression of NLGN4 WT but not the R101Q mutant also suppressed the amplitude of AMPAR-evoked EPSCs without affecting presynaptic release probability [as estimated by paired-pulse ratios (PPRs)], when measured under similar recording conditions [as assessed by series resistance (Rs) parameters; Fig. 6H]. These results suggest that NLGN4 overexpression can negatively modulate the strength of excitatory synapses in human neurons by a potentially postsynaptic mechanism, and the R101Q substitution causes a major loss of this NLGN4-induced synaptic phenotype.
To test whether our conclusions apply to neurons generated by a different protocol, we transdifferentiated human ES cells into cortical glutamatergic neurons by ectopic expression of a single TF, Ngn2 (Fig. 7A; Zhang et al., 2013). We coinfected the neurons with lower-titer lentivirus encoding either a control vector or HA-tagged NLGN4 WT versus R101Q mutant, and detected no noticeable difference in their cell viability (Fig. 7A,B). We next identified infected neurons by coexpressed fluorescent reporters and made patch-clamp recordings to evaluate their functional properties (Fig. 7A,C). Similar to NSC-derived neurons, the Ngn2-induced human neurons expressing WT NLGN4 showed a significantly lower amplitude of AMPAR-mediated evoked EPSCs; whereas the R101Q variant largely reversed this phenotype (Fig. 7D). Furthermore, R101Q substitution in NLGN4 also elevated the AMPAR mEPSC frequency compared with WT version, with a relatively minor effect on mEPSC amplitude (Fig. 7E,F). Thus, both NLGN4 WT and R101Q variant had highly consistent and reproducible effects on the excitatory synaptic transmission in human neurons irrespective of their differentiation or reprogramming methods.
R101Q mutation prevents NLGN4-mediated silencing of excitatory postsynapses
We aimed to understand how loss of NLGN4 function by R101Q mutation can contribute to aberrant synaptic transmission. We first used IRES-driven EGFP fluorescence to visualize the morphology of Ngn2-induced neurons from control, WT, and R101Q conditions, but failed to observe any difference in their soma size, primary dendrite number, or total neurite length (Fig. 8A). To determine NLGN4 levels in these neurons, we performed qPCR analysis, and found that cells overexpressing WT versus R101Q versions displayed an equivalent increase in mRNA levels relative to control condition (Fig. 8B). We next tracked the distribution of NLGN4 protein along dendrites by immunostaining the cells with HA antibody under nonpermeabilized conditions, and then permeabilized them to counterstain for Synapsin. We noticed that R101Q mutation caused a substantial reduction in the density of NLGN4 clusters along the surface of dendritic branches (Fig. 8C) and thus decreased the colocalization of NLGN4 with the Synapsin signal (Fig. 8D). Therefore, R101Q substitution inhibited the surface trafficking of NLGN4 in human neurons, even to a greater extent than our observations in HEK293 cells (compare with Fig. 2D) and simultaneously diminished its targeting at synapses.
Figure 8.
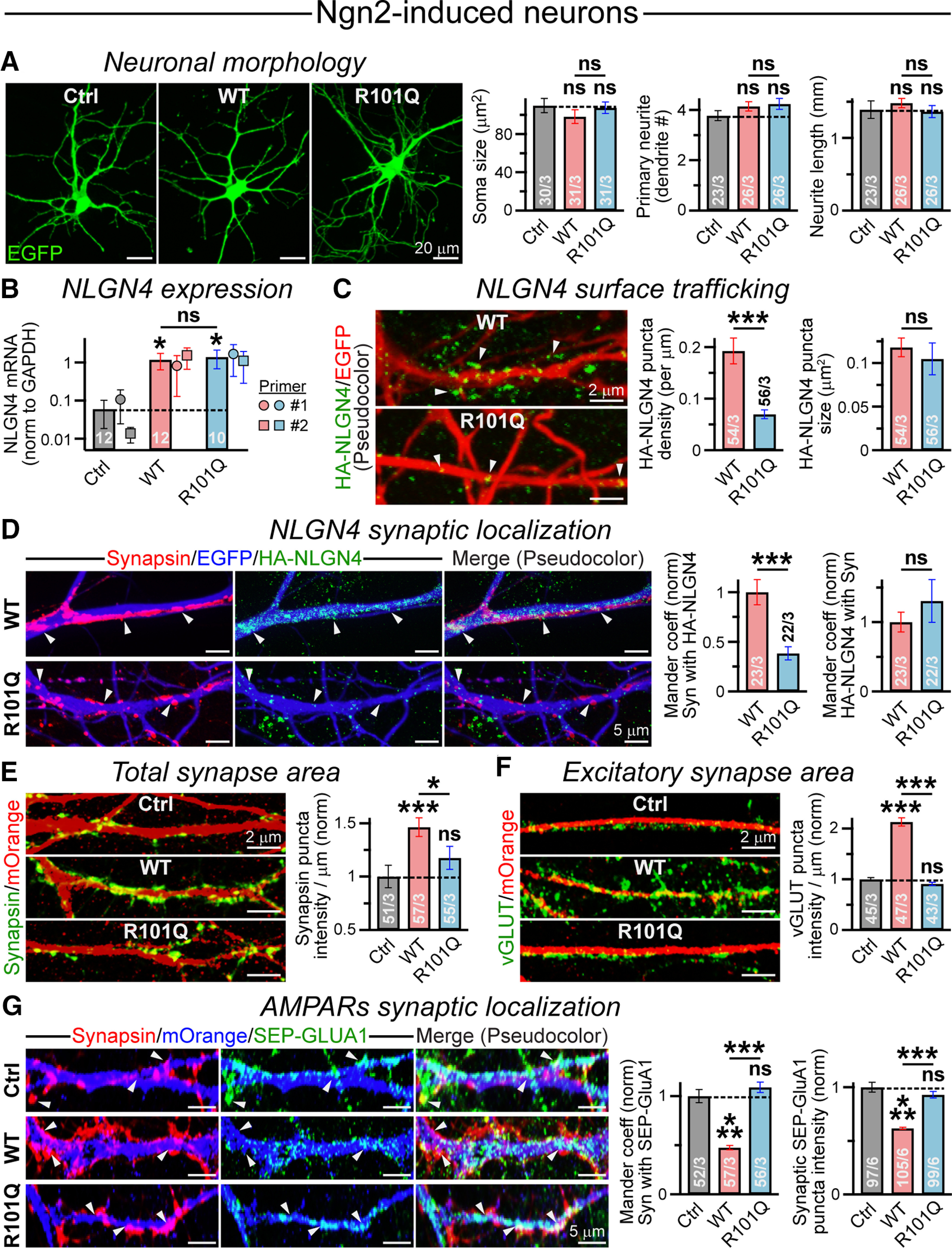
R101Q mutation impairs NLGN4 synaptic localization and inhibits NLGN4-mediated reduction in synaptic AMPAR clusters. A, Representative images (left) of Ngn2-induced human neurons expressing either a control (Ctrl) vector or NLGN4 WT versus R101Q variant, as their morphologic parameters were assessed using an EGFP reporter. Summary graphs (right) of soma size, primary dendrite number, and average neurite length per cell. B, NLGN4 mRNA levels were estimated by quantitative real-time PCR and normalized to those of GAPDH (internal control) for Ngn2-induced neurons expressing either a control vector, or NLGN4 WT versus R101Q mutant constructs. The average bar graphs combine values from two independent primer sets (set 1: circles; n = 6 Ctrl, 6 WT, and 5 for R101Q; and set 2: squares; n = 6 Ctrl, 6 WT, and 5 for R101Q). C, Sample images (left) of EGFP-expressing dendritic segments (pseudocolored for better visibility) from NLGN4 WT (top) versus R101Q (bottom) conditions, when immunostained with anti-HA antibody (arrowheads) under nonpermeabilized state. Summary graphs (right) of surface HA-NLGN4 puncta density and size. D, Representative images (left) of EGFP-labeled dendritic segments from WT (top) versus R101Q (bottom) conditions, when incubated with anti-HA antibody under nonpermeabilized state, then permeabilized and coimmunostained with Synapsin antibody. The channel colors were artificially reassigned to provide better visibility for synaptic signals; arrowheads point at synaptic puncta. Summary graphs (right) indicate synaptic distribution of NLGN4, characterized using normalized Mander's colocalization coefficients between HA-NLGN4 and Synapsin signals. E, F, Dendritic sections of mOrange-expressing human neurons from control, NLGN4 WT, and NLGN4 R101Q conditions immunostained with either Synapsin (E, left) or VGluT (F, left) antibody, and integrated intensity of Synapsin (E, right) or VGluT (F, right) puncta normalized with respect to the dendrite length. G, Neurons expressed either a control vector or NLGN4 WT versus R101Q mutant followed by mOrange reporter. Sample images (left; pseudocolor) of dendritic branches immunolabeled for surface AMPARs (SEP-GluA1) using anti-GFP antibody under nonpermeabilized condition, subsequently permeabilized and stained with anti-Synapsin antibody. Arrowheads indicate synaptic clusters; note that many lack copresence of SEP-GluA1 in WT condition. Summary graphs (right) of normalized Mander's coefficient indicating a Synapsin (Syn) puncta fraction containing SEP-GluA1 signal and the intensity of SEP-GluA1 puncta colocalized with Synapsin. Average bar graphs represent the mean ± SEM, with the number of frames analyzed/number of experiments, or only the number of independent batches (for B). Statistical significance was assessed by two-tailed, unpaired (one-tailed and paired for B), Student's t test. ***p < 0.005; *p < 0.05; ns, not significant (p > 0.05).
To further evaluate the consequences of reduced NLGN4 content at excitatory synapses, we immunostained Ngn2-induced neurons with Synapsin and VGluT antibodies. We found that, compared with control condition, the overexpression of WT NLGN4 but not the R101Q version caused a considerable increase in both Synapsin and VGluT intensities at dendritic branches (Fig. 8E,F), mimicking our observations in NSC-derived neurons (Fig. 5G). Although these results suggest that overexpression of NLGN4 WT but not the R101Q mutant can induce the formation of glutamatergic synapses, their function depends on the availability of postsynaptic AMPARs. To directly assess that, we infected Ngn2-induced neurons with lentiviruses expressing SEP-GluA1, and either control or NLGN4 WT versus R101Q constructs followed by an IRES-mOrange reporter. We noticed that, compared with control condition, WT NLGN4 significantly reduced the localization and signal intensity of SEP-GluA1 at Synapsin-positive synaptic clusters, whereas the R101Q variant alleviated these phenotypes (Fig. 8G). Together, these results indicate that R101Q substitution increases excitatory synaptic transmission (Fig. 7C–F) by preventing NLGN4-induced loss of postsynaptic AMPARs.
Discussion
The X chromosome-linked human NLGN4 gene has been found to be mutated in several patients with autism and other neuropsychiatric disorders (Jamain et al., 2003; Laumonnier et al., 2004; Yan et al., 2005; Chocholska et al., 2006; Talebizadeh et al., 2006; Macarov et al., 2007; Lawson-Yuen et al., 2008; Pampanos et al., 2009). A majority of these mutations represent CNVs, truncations, frameshifts, and premature stop codons, suggesting an ultimate loss of NLGN4 expression. However, a fraction of NLGN4 mutations also include single nucleotide polymorphisms creating missense variants of relatively unknown physiological consequence. Earlier studies indicate that while some of these missense mutations do exhibit pathogenic phenotypes, other variants may not lead to significant functional inactivation (Xu et al., 2017). In this study, we describe a severely autistic patient carrying a single amino acid substitution (i.e., R101Q) in NLGN4 (Figs. 1, 9A). We demonstrate that the R101Q mutation prevents NLGN4 maturation by causing incomplete glycosylation (Figs. 2, 3, 9B). As a result, this mutation blocks the surface trafficking of NLGN4 by enhancing its retention at intracellular compartments (Figs. 3, 9B). R101Q-equivalent mutations also affect the maturation of other NLGNs with conserved glycosylation sites (Fig. 4). This mutation-induced trafficking deficiency was already visible in reduced systems (e.g., HEK293 cells, Figs. 2–4) and inhibited the synaptic properties of NLGN4 even more prominently in human neurons when overexpressed for a prolonged time period (Figs. 5–8). R101Q substitution causes a major reduction of NLGN4 localization at human synapses, and largely inhibits NLGN4-dependent effects on synapse formation and synaptic transmission (Figs. 5, 6, 9C). These phenotypes were highly reproducible and mediated by altered synaptic density of AMPARs (Figs. 7, 8, 9C). These results confirm that the R101Q mutation is not benign, as it impairs the synaptic properties of NLGN4 by a partial loss-of-function mechanism.
Figure 9.

Schematic diagram of NLGN4 R101Q mutation-induced defects in cellular models. A, Summary of clinical evaluations performed on an autistic patient carrying R101Q substitution in NLGN4. B, In HEK cells, R101Q mutation directly impairs N-linked glycosylation of NLGN4, increases its accumulation in ER and Golgi, and, as a result, indirectly decreases its surface trafficking to the cell membrane. C, In stem cell -derived human neurons, R101Q mutation-mediated reduction in surface trafficking relieves NLGN4-induced inhibition of AMPAR clustering at the excitatory postsynapses, and thus enhances neuronal excitability.
The R101Q mutation is located in close proximity to a number of previously identified NLGN4 mutations. Of these, an ASD-associated R87W substitution has been shown to disrupt the nucleation site for the overall folding of NLGN4, leading to its degradation and substantial loss in total protein content (Zhang et al., 2009). Unlike R87W mutation, the R101Q variant only moderately reduced NLGN4 protein level, but interrupted N-linked glycosylation at an adjacent N102 residue (Figs. 2B, 3A–D). This mutant protein, although immaturely glycosylated, was fully capable of forming homodimers and interacting with presynaptic NRXNs, indicating that its secondary structure likely remained intact (Fig. 2E–I). These findings are in accordance with previous studies with recombinant proteins suggesting that deglycosylated versions of soluble NLGN1 fragments can bind to NRXNs, often with a higher affinity (Comoletti et al., 2003). However, in a cellular environment, mutation-induced lack of glycosylation also modulated the availability of NLGN4 at the cell surface and indirectly reduced its ability to bind NRXNs when compared with an adequately glycosylated WT product (Fig. 2D,G–I). Thus, the pathogenic effects of R101Q differed significantly from R87W in that the former mutation affected NLGN4 properties primarily by impairing post-translational modification with a relatively minor effect on the stability of this protein. These results highlight divergent mechanisms of various missense mutations that can directly or indirectly trigger the loss of NLGN4 function. Moreover, although the pathologic mechanisms of other missense mutations (e.g., G99S, G84R) remain uncertain or even controversial, at least some variants may also alter NLGN4 properties by gain-in-function mechanisms (e.g., R704C; Marro et al., 2019).
All NLGNs are heavily modified by N-glycosylation at multiple putative residues located in their corresponding extracellular domains (Comoletti et al., 2003; Bemben et al., 2015; Jeong et al., 2017). While some of these glycosylation sites are specific for individual NLGNs, the N102 site of human NLGN4 is highly conserved among all other NLGN genes (NLGN1/2/3) regardless of species, and is positioned next to an equally conserved R101 residue (Fig. 4A). We found that R101Q-equivalent substitutions also affected the protein processing and surface trafficking of NLGN1/2/3 to variable degrees (Fig. 4B–H). This is in contrast to the N303 glycosylation site of NLGN1, which actively modulates its NRXN binding, but is not conserved in NLGN2/3/4 to affect their properties (Fig. 4A; Comoletti et al., 2003; Lee et al., 2010). Although our data suggest that the N102 glycosylation site could be necessary for NLGN function in general, further investigation is required to comprehend its mechanistic contribution relative to other specific and/or conserved glycosylation sites. Interestingly, the evolutionarily divergent mouse NLGN4 completely lacks this additional N102 glycosylation site. This contrast in glycosylation profile further highlights the differences between rodent and human versions of NLGN4, which might also contribute to their differential effects on synapse regulation. Moreover, it was recently shown that the Y chromosome linked human NLGN4 (NLGN4Y) only produces an immature and low-MW product (Nguyen et al., 2020). Although both N102 and N511 glycosylation sites are preserved in NLGN4Y (Fig. 4A), subtle but inherent differences in NLGN4X versus NLGN4Y amino acid residues might indirectly influence their accessibility for PTMs, differentially affecting their maturation and surface localization.
We noticed that the overexpression of WT NLGN4X enhanced synaptogenesis in NSC-derived human neurons (Fig. 5). However, this increase in synapse number and size was mainly restricted to excitatory synapses, with only minor changes observed at inhibitory synapses (Fig. 5F,G). Furthermore, the overexpression of WT NLGN4 also unambiguously affected the functional properties of excitatory synapses (Fig. 6E–H). The NLGN4-mediated induction in synapse formation and modulation of synaptic transmission was substantially impaired by the R101Q mutation, supporting a primarily loss-of-function hypothesis (Figs. 5, 6). These phenotypes, in conjunction with previous findings, support the notion that the role of human NLGN4 could be limited to excitatory synapses (Chanda et al., 2016; Marro et al., 2019). These characteristics of human NLGN4 were surprisingly different from its mouse counterpart, because earlier elegant studies demonstrated that mouse NLGN4 preferentially localizes at glycinergic and/or GABAergic synapses and regulates inhibitory synaptic transmission (Hoon et al., 2011; Unichenko et al., 2018). Given the striking disparity between the amino acid compositions of human versus mouse NLGN4, it is possible that they diverge in synaptic localization and/or function. Indeed, in differentiated human neurons, an epitope-tagged version of endogenous NLGN4 preferably localized at excitatory synapses and only a subset of inhibitory synapses (Marro et al., 2019).
Similar to earlier findings, the overexpression of WT NLGN4 in human neurons significantly reduced the frequency of AMPAR-mediated mEPSCs without affecting their amplitude (Fig. 6E,F; also see Marro et al., 2019). Although the mechanism of this effect remains unclear, molecular overcrowding by overexpressed NLGN4 might outcompete endogenous NLGN1/3 homodimers and heterodimers for NRXN or PSD-95 binding, which play critical role in recruiting AMPARs (NMDA receptor dependent or independent) at postsynaptic density (Fig. 9C). It is possible that, compared to NLGN1/3, human NLGN4 could be a relatively weak inducer of postsynaptic AMPAR clustering. However, in the absence of other NLGNs (e.g. in NLGN1/2/3 knockdown cells with severely impaired basal excitatory synaptic transmission), the overexpressed NLGN4 homodimers might enhance EPSC strength by partially substituting for NLGN1/3 (Nguyen et al., 2020).
Interestingly, overexpression of the R101Q variant inhibited NLGN4-induced suppression of synaptic strength, again supporting our hypothesis that this mutation operates via an obligatory loss-of-function mechanism. These phenotypes were independent of differentiation protocol and equally observed in neurons reprogrammed by different procedures (Fig. 7D–F). The NLGN4-mediated suppression of mEPSC frequency could result from (1) the decrease in presynaptic release probability, (2) the reduction in total synapse numbers, or (3) the silencing of excitatory synapses. Analysis of AMPAR-mediated evoked EPSCs indicated that the overexpression of WT NLGN4 substantially attenuated basal synaptic strength without changing PPRs, arguing against any effect on release probability (Fig. 6H). In addition, overexpression of WT NLGN4 increased the number of excitatory synapses instead of decreasing it (Figs. 5E–G, 8E,F). Therefore, an alternative hypothesis suggests that overexpressed WT NLGN4 might induce silencing of excitatory synapses, and R101Q mutation could prevent this process by eliminating NLGN4 localization from dendritic surfaces and synapses. In support of this prediction, we found that R101Q mutation decreased dendritic clusters of NLGN4, apparently reduced its synaptic localization (Fig. 8D), and concurrently facilitated the synaptic localization of AMPARs as monitored by SEP-GluA1 (Fig. 8G). Based on these results, we propose that the autism-associated R101Q mutation interrupts NLGN4-mediated silencing of excitatory synapses, and thus increases cellular excitability in human neurons.
Footnotes
This study was supported by a start-up fund from Colorado State University to S.C. We thank the affected individual and family for participating in this study. We also thank the laboratory members of Dr. James Bamburg for helpful discussion and some experimental reagents, including cryopreserved mouse glia.
The authors declare no competing financial interests.
References
- Aman MG, Singh NN (1994) Aberrant behavior checklist. New York: Slosson Educational Publications. [Google Scholar]
- Aman MG, Singh NN (2017) Aberrant behavior checklist, Ed 2. East Aurora, NY: Slosson Educational Publications. [Google Scholar]
- Arac D, Boucard AA, Ozkan E, Strop P, Newell E, Südhof TC, Brunger AT (2007) Structures of neuroligin-1 and the neuroligin-1/neurexin-1 beta complex reveal specific protein-protein and protein-Ca2+ interactions. Neuron 56:992–1003. 10.1016/j.neuron.2007.12.002 [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (2013) DSM-5. Diagnostic and statistical manual of mental disorders, Ed 5 Washington, DC: American Psychiatric Association. [Google Scholar]
- Bemben MA, Shipman SL, Nicoll RA, Roche KW (2015) The cellular and molecular landscape of neuroligins. Trends Neurosci 38:496–505. 10.1016/j.tins.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolliger MF, Pei J, Maxeiner S, Boucard AA, Grishin NV, Südhof TC (2008) Unusually rapid evolution of Neuroligin-4 in mice. Proc Natl Acad Sci U S A 105:6421–6426. 10.1073/pnas.0801383105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Südhof TC (2005) A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48:229–236. 10.1016/j.neuron.2005.08.026 [DOI] [PubMed] [Google Scholar]
- Budreck EC, Scheiffele P (2007) Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur J Neurosci 26:1738–1748. 10.1111/j.1460-9568.2007.05842.x [DOI] [PubMed] [Google Scholar]
- Bölte S, Westerwald E, Holtmann M, Freitag C, Poustka F (2011) Autistic traits and autism spectrum disorders: the clinical validity of two measures presuming a continuum of social communication skills. J Autism Dev Disord 41:66–72. 10.1007/s10803-010-1024-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280. 10.1038/nbt.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Marro S, Wernig M, Südhof TC (2013) Neurons generated by direct conversion of fibroblasts reproduce synaptic phenotype caused by autism-associated neuroligin-3 mutation. Proc Natl Acad Sci U S A 110:16622–16627. 10.1073/pnas.1316240110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Aoto J, Lee SJ, Wernig M, Südhof TC (2016) Pathogenic mechanism of an autism-associated neuroligin mutation involves altered AMPA-receptor trafficking. Mol Psychiatry 21:169–177. 10.1038/mp.2015.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Hale WD, Zhang B, Wernig M, Südhof TC (2017) Unique versus redundant functions of neuroligin genes in shaping excitatory and inhibitory synapse properties. J Neurosci 37:6816–6836. 10.1523/JNEUROSCI.0125-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Ang CE, Lee QY, Ghebrial M, Haag D, Shibuya Y, Wernig M, Südhof TC (2019) Direct reprogramming of human neurons identifies MARCKSL1 as a pathogenic mediator of valproic acid-induced teratogenicity. Cell Stem Cell 25:103–119.e6. 10.1016/j.stem.2019.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chocholska S, Rossier E, Barbi G, Kehrer-Sawatzki H (2006) Molecular cytogenetic analysis of a familial interstitial deletion Xp22.2-22.3 with a highly variable phenotype in female carriers. Am J Med Genet A 140:604–610. 10.1002/ajmg.a.31145 [DOI] [PubMed] [Google Scholar]
- Comoletti D, Flynn R, Jennings LL, Chubykin A, Matsumura T, Hasegawa H, Südhof TC, Taylor P (2003) Characterization of the interaction of a recombinant soluble neuroligin-1 with neurexin-1beta. J Biol Chem 278:50497–50505. 10.1074/jbc.M306803200 [DOI] [PubMed] [Google Scholar]
- Constantino JN. (2005) Social responsiveness scale. Torrance, CA: Western Psychological Services. [Google Scholar]
- Constantino JN, Gruber CP (2012) Social responsiveness scale: SRS-2, Ed 2. Torrance, CA: Western Psychological Services. [Google Scholar]
- Fabrichny IP, Leone P, Sulzenbacher G, Comoletti D, Miller MT, Taylor P, Bourne Y, Marchot P (2007) Structural analysis of the synaptic protein neuroligin and its beta-neurexin complex: determinants for folding and cell adhesion. Neuron 56:979–991. 10.1016/j.neuron.2007.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D (2008) Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci U S A 105:20947–20952. 10.1073/pnas.0804007106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon M, Soykan T, Falkenburger B, Hammer M, Patrizi A, Schmidt KF, Sassoè-Pognetto M, Löwel S, Moser T, Taschenberger H, Brose N, Varoqueaux F (2011) Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc Natl Acad Sci U S A 108:3053–3058. 10.1073/pnas.1006946108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Südhof TC (1995) Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81:435–443. 10.1016/0092-8674(95)90396-8 [DOI] [PubMed] [Google Scholar]
- Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Südhof TC (1997) Binding of neuroligins to PSD-95. Science 277:1511–1515. 10.1126/science.277.5331.1511 [DOI] [PubMed] [Google Scholar]
- Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, Amiel J, Le Merrer M, Heron D, de Blois MC, Prieur M, Vekemans M, Carter NP, Munnich A, Colleaux L, Philippe A (2006) Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet 43:843–849. 10.1136/jmg.2006.043166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T (2003) Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34:27–29. 10.1038/ng1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Paskus JD, Roche KW (2017) Posttranslational modifications of neuroligins regulate neuronal and glial signaling. Curr Opin Neurobiol 45:130–138. 10.1016/j.conb.2017.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Polepalli J, Chen LY, Zhang B, Südhof TC, Malenka RC (2017) Conditional ablation of neuroligin-1 in CA1 pyramidal neurons blocks LTP by a cell-autonomous NMDA receptor-independent mechanism. Mol Psychiatry 22:375–383. 10.1038/mp.2016.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Zhang C, Arac D, Boucard AA, Brunger AT, Südhof TC (2009) Neuroligin-1 performs neurexin-dependent and neurexin-independent functions in synapse validation. EMBO J 28:3244–3255. 10.1038/emboj.2009.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J, Jin X, Budreck EC, Posy S, Scheiffele P, Honig B, Shapiro L (2008) Crystal structure of the extracellular cholinesterase-like domain from neuroligin-2. Proc Natl Acad Sci U S A 105:1873–1878. 10.1073/pnas.0711701105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthélémy C, Moraine C, Briault S (2004) X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet 74:552–557. 10.1086/382137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson-Yuen A, Saldivar JS, Sommer S, Picker J (2008) Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet 16:614–618. 10.1038/sj.ejhg.5202006 [DOI] [PubMed] [Google Scholar]
- Lee H, Dean C, Isacoff E (2010) Alternative splicing of neuroligin regulates the rate of presynaptic differentiation. J Neurosci 30:11435–11446. 10.1523/JNEUROSCI.2946-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone P, Comoletti D, Ferracci G, Conrod S, Garcia SU, Taylor P, Bourne Y, Marchot P (2010) Structural insights into the exquisite selectivity of neurexin/neuroligin synaptic interactions. EMBO J 29:2461–2471. 10.1038/emboj.2010.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarov M, Zeigler M, Newman JP, Strich D, Sury V, Tennenbaum A, Meiner V (2007) Deletions of VCX-A and NLGN4: a variable phenotype including normal intellect. J Intellect Disabil Res 51:329–333. 10.1111/j.1365-2788.2006.00880.x [DOI] [PubMed] [Google Scholar]
- Marro SG, Chanda S, Yang N, Janas JA, Valperga G, Trotter J, Zhou B, Merrill S, Yousif I, Shelby H, Vogel H, Kalani MYS, Südhof TC, Wernig M (2019) Neuroligin-4 regulates excitatory synaptic transmission in human neurons. Neuron 103:617–626.e6. 10.1016/j.neuron.2019.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CEJ, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, et al. (2008) Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 82:477–488. 10.1016/j.ajhg.2007.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxeiner S, Benseler F, Krasteva-Christ G, Brose N, Sudhof TC (2020) Evolution of the autism-associated neuroligin-4 gene reveals broad erosion of pseudoautosomal regions in rodents. Mol Biol Evol 37:1243–1258. 10.1093/molbev/msaa014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N (2004) The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology 47:724–733. 10.1016/j.neuropharm.2004.06.023 [DOI] [PubMed] [Google Scholar]
- Mondin M, Labrousse V, Hosy E, Heine M, Tessier B, Levet F, Poujol C, Blanchet C, Choquet D, Thoumine O (2011) Neurexin-neuroligin adhesions capture surface-diffusing AMPA receptors through PSD-95 scaffolds. J Neurosci 31:13500–13515. 10.1523/JNEUROSCI.6439-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam CI, Chen L (2005) Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci U S A 102:6137–6142. 10.1073/pnas.0502038102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TA, Wu K, Pandey S, Lehr AW, Li Y, Bemben MA, Badger JD 2nd, Lauzon JL, Wang T, Zaghloul KA, Thurm A, Jain M, Lu W, Roche KW (2020) A cluster of autism-associated variants on X-linked NLGN4X functionally resemble NLGN4Y. Neuron 106:759–768. 10.1016/j.neuron.2020.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampanos A, Volaki K, Kanavakis E, Papandreou O, Youroukos S, Thomaidis L, Karkelis S, Tzetis M, Kitsiou-Tzeli S (2009) A substitution involving the NLGN4 gene associated with autistic behavior in the Greek population. Genet Test Mol Biomarkers 13:611–615. 10.1089/gtmb.2009.0005 [DOI] [PubMed] [Google Scholar]
- Poulopoulos A, Soykan T, Tuffy LP, Hammer M, Varoqueaux F, Brose N (2012) Homodimerization and isoform-specific heterodimerization of neuroligins. Biochem J 446:321–330. 10.1042/BJ20120808 [DOI] [PubMed] [Google Scholar]
- Schepici G, Cavalli E, Bramanti P, Mazzon E (2019) Autism spectrum disorder and miRNA: an overview of experimental models. Brain Sci 9:265 10.3390/brainsci9100265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopler E, Van Bourgondien ME (2010) CARS2: the childhood autism rating scale, Ed 2. Los Angeles: Western Psychological Services. [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee Y-H, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, et al. (2007) Strong association of de novo copy number mutations with autism. Science 316:445–449. 10.1126/science.1138659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Südhof TC, Brose N (1999) Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci U S A 96:1100–1105. 10.1073/pnas.96.3.1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow S, Cicchetti D, Saulnier C, Doll EA (2016) Vineland adaptive behavior scales, Ed 3. Minneapolis, MN: Pearson Assessments. [Google Scholar]
- Sparrow SS, Cicchetti DV (1985) Diagnostic uses of the Vineland Adaptive Behavior Scales. J Pediatr Psychol 10:215–225. 10.1093/jpepsy/10.2.215 [DOI] [PubMed] [Google Scholar]
- Südhof TC. (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455:903–911. 10.1038/nature07456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talebizadeh Z, Lam DY, Theodoro MF, Bittel DC, Lushington GH, Butler MG (2006) Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J Med Genet 43:e21. 10.1136/jmg.2005.036897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unichenko P, Yang JW, Kirischuk S, Kolbaev S, Kilb W, Hammer M, Krueger-Burg D, Brose N, Luhmann HJ (2018) Autism related Neuroligin-4 knockout impairs intracortical processing but not sensory inputs in mouse barrel cortex. Cereb Cortex 28:2873–2886. 10.1093/cercor/bhx165 [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, Brose N (2004) Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol 83:449–456. 10.1078/0171-9335-00410 [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N (2006) Neuroligins determine synapse maturation and function. Neuron 51:741–754. 10.1016/j.neuron.2006.09.003 [DOI] [PubMed] [Google Scholar]
- Volkmar FR. (2013) Encyclopedia of autism spectrum disorders, Ed 1 New York: Springer. [Google Scholar]
- Xu X, Hu Z, Zhang L, Liu H, Cheng Y, Xia K, Zhang X (2017) Not all neuroligin 3 and 4X missense variants lead to significant functional inactivation. Brain Behav 7:e00793. 10.1002/brb3.793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Oliveira G, Coutinho A, Yang C, Feng J, Katz C, Sram J, Bockholt A, Jones IR, Craddock N, Cook EH Jr, Vicente A, Sommer SS (2005) Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry 10:329–332. 10.1038/sj.mp.4001629 [DOI] [PubMed] [Google Scholar]
- Zhang C, Milunsky JM, Newton S, Ko J, Zhao G, Maher TA, Tager-Flusberg H, Bolliger MF, Carter AS, Boucard AA, Powell CM, Südhof TC (2009) A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci 29:10843–10854. 10.1523/JNEUROSCI.1248-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, Xu W, Yang N, Danko T, Chen L, Wernig M, Südhof TC (2013) Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78:785–798. 10.1016/j.neuron.2013.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]