

Preconditioning peripheral nerve injury enhances the intrinsic growth capacity of DRGs sensory axons by inducing transcriptional upregulation of the regeneration-associated genes (RAGs). However, it is still unclear how preconditioning injury leads to the orchestrated induction of many RAGs. The present study identified Myc proto-oncogene as a transcriptional hub gene to regulate the expression of a distinct subset of RAGs in DRGs following the preconditioning injury.
Keywords: axon regeneration, axon retraction, hub transcription factor, Myc, preconditioning injury, regeneration-associated genes
Abstract
Preconditioning peripheral nerve injury enhances the intrinsic growth capacity of DRGs sensory axons by inducing transcriptional upregulation of the regeneration-associated genes (RAGs). However, it is still unclear how preconditioning injury leads to the orchestrated induction of many RAGs. The present study identified Myc proto-oncogene as a transcriptional hub gene to regulate the expression of a distinct subset of RAGs in DRGs following the preconditioning injury. We demonstrated that c-MYC bound to the promoters of certain RAGs, such as Jun, Atf3, and Sprr1a, and that Myc upregulation following SNI preceded that of the RAGs bound by c-MYC. Marked DNA methylation of the Myc exon 3 sequences was implicated in the early transcriptional activation and accompanied by open histone marks. Myc deletion led to a decrease in the injury-induced expression of a distinct subset of RAGs, which were highly overlapped with the list of RAGs that were upregulated by Myc overexpression. Following dorsal hemisection spinal cord injury in female rats, Myc overexpression in DRGs significantly prevented the retraction of the sensory axons in a manner dependent on its downstream RAG, June. Our results suggest that Myc plays a critical role in axon regeneration via its transcriptional activity to regulate the expression of a spectrum of downstream RAGs and subsequent effector molecules. Identification of more upstream hub transcription factors and the epigenetic mechanisms specific for individual hub transcription factors would advance our understanding of how the preconditioning injury induces orchestrated upregulation of RAGs.
Significance Statement
Identification of the transcriptional network regulating regeneration-associated gene (RAG) expression would enable us to understand how preconditioning injury enhances intrinsic regeneration capacity. It has been suggested that a handful of hub transcription factors may play an important role in regulating the expression of a large set of RAGs. However, the precise roles of the hub genes in the transcriptional activation of RAGs have not been verified. This study provides experimental evidence that Myc proto-oncogen functions as a transcriptional hub gene and that the DNA methylation may lead to early transcriptional activation of Myc. Dissecting out the hub transcription factors and their hierarchical relationship should provide important clues on the combinatorial genetic approaches to achieve more meaningful and robust axon regeneration.
Introduction
Regeneration failure of CNS axons following traumatic or vascular injuries is in part attributed to the limited intrinsic growth capability of CNS neurons (Curcio and Bradke, 2018; Mahar and Cavalli, 2018). Yet, experimental evidence of specific molecular readouts that are directly as well as critically linked to the intrinsic growth capacity is still lacking. It has been generally assumed that induction of a group of so-called regeneration-associated genes (RAGs) is correlated with successful axon regeneration (Hoffman, 2010; T. C. Ma and Willis, 2015; Senger et al., 2018). Expression of RAGs along with their functional consequences has been frequently studied in preconditioning injury model, where a preceding injury to the peripheral branches of DRG neurons allows successful regeneration of their central branches over the caudal lesion border (Richardson et al., 2009; Blesch et al., 2012; Kwon et al., 2013). However, it is still not known how the preconditioning injury leads to orchestrated induction of RAGs.
Multilevel bioinformatics analysis identified a core set of hub transcription factors (TFs) that are enriched in the promoter regions of RAG module genes upregulated following peripheral nerve injury (Chandran et al., 2016), suggesting a possibility that upstream TFs functioning as hub genes in the transcriptional network may play a role of master regulators in injury-induced RAG induction. Understanding precise roles of individual hub TFs would provide valuable information for designing combinatorial genetic approaches to render injured neurons capable of mounting regenerative responses (Lerch et al., 2014; Fagoe et al., 2015). However, it remains to be studied which hub TFs indeed influence transcription of specific RAGs or which RAGs are directly regulated by individual hub TFs. Furthermore, it is not clearly understood how hub TFs are induced or activated in the first place following preconditioning peripheral nerve injury.
Epigenetic mechanisms also play a crucial role in the transcriptional activation of RAGs in the preconditioning model (Cho et al., 2013; Puttagunta et al., 2014; Weng et al., 2017). Our previous study examined changes in DNA methylome in the preconditioning injury model and found that global DNA methylation does not correlate with injury-induced RAG upregulation (Shin et al., 2020). We reasoned that DNA methylation might be involved in the activation of upstream TF genes that in turn regulate transcription of many RAGs as a gene expression hub. The current study identified Myc proto-oncogene, which undergoes marked demethylation following peripheral nerve injury, as a hub TF gene that regulates transcription of a subset of RAGs and influences injury-induced intrinsic growth capacity of DRG sensory neuron. We also found that Myc overexpression in DRG sensory neurons partially mimics the preconditioning effects and distinctively prevents axonal retraction following an injury in the spinal cord.
Materials and Methods
Animals
All animal protocols were approved by the Institutional Animal Care and Use Committee of Ajou University School of Medicine. Adult female Sprague Dawley rats weighing 250-300 g were purchased from Orient Bio. Myc floxed mouse line (B6.129S6-Myctm2Fwa/Mmjax, stock #032046-JAX) was cryorecovered at Mutant Mouse Regional Resource Center and purchased from The Jackson Laboratory. Nestin-Cre line (B6.Cg-Tg(Nes-cre)1Kln/J, stock #003771) was also purchased from The Jackson Laboratory.
Surgical procedures
Rats were anesthetized with intraperitoneal injection of the ketamine and xylazine mixture (90 mg/kg and 8 mg/kg, respectively). For sciatic nerve injury (SNI) in rats, muscles were displaced to expose the right sciatic nerve, and the nerve was ligated proximal to its trifurcation. The sciatic nerve was completely transected below the ligation site with fine surgical scissors. For dorsal hemisection injury in the rat spinal cord, a dorsal laminectomy was performed at the T9 level to expose the thoracic spinal cord. After the dura was opened with a midline incision, bilateral dorsal columns with adjacent lateral columns were cut out with iridectomy scissors inserted at a depth of ∼1.5 mm. A small piece of Gelfoam was temporarily placed over the lesion site to prevent bleeding. The overlying muscles and fascia were sutured in layer, and the skin was stapled. To visualize regenerating axons, cholera toxin subunit B (CTB; List Biological Laboratories) was injected using a protocol modified slightly from that in the previous report (Kwon et al., 2013). After the sciatic nerve between the thigh muscles was exposed, a small incision was made on the perineurium just proximal to the trifurcation site; 4 μl of unconjugated CTB solution (0.1% in PBS) was slowly injected using the Hamilton syringe through the perineural incision, and animals were killed 5 d after the injection. Mice were anesthetized with intraperitoneal injection of the ketamine and xylazine mixture (100 mg/kg and 10 mg/kg, respectively). SNI in mice was performed following the same procedures as for rats.
Preparation and injection of recombinant adeno-associated virus (AAV)
Recombinant AAV vector expressing Myc was constructed by inserting the full-length cDNA for human Myc into the pAAV-CAG-GFP (Addgene). Then AAV serotype 5 (AAV5) viral particles were produced by the custom AAV production service at the University of North Carolina at Chapel Hill vector core. Premade control AAV5-GFP viral particles were purchased from the same facility. The titer of both viral particles was ∼1 × 1013 genome copies/ml. For intraganglionic injection, the L5 DRG was exposed after removal of the lateral process of the L5 vertebral bone. A Hamilton syringe configured with a glass pipette was controlled using a micromanipulator, and the tip of the glass pipette was slowly advanced into the L5 DRG under a surgical microscope; 2 μl of AAV5-Myc or AAV5-GFP was injected into the L5 DRG at a rate of 0.5 µl/min using a nanoinjector.
In vivo SP600125 administration through osmotic minipump
A c-Jun N-terminal kinase (JNK) inhibitor, SP600125 was administered to DRGs through Alzet osmotic minipumps (Durect) installed 3 weeks after intraganglionic injection of AAV5-Myc and 1 h before dorsal hemisection spinal cord injury. The polyethylene tubing (PE-10) was advanced below the L6/S1 vertebral bone on the right side. The catheter was slightly heated so that it could be bent to position the tubing tip exactly at the L5 DRG. The catheter position was secured with sutures tied to L6/S1 bone. The osmotic minipump was also sutured to the paravertebral muscle. The exact positioning of the tubing tip was confirmed at the time of death. The pumps delivered SP600125 (1 mm; Sigma Millipore) or 1% DMSO as a vehicle control at a rate of 1 μl/h continuously for 7 d.
Primary culture of dissociated adult DRG neurons and neurite outgrowth assay
Adult L4-6 DRGs in rats or mice were freshly dissected and treated with 125 U/ml Type XI collagenase (Sigma Millipore) dissolved in DMEM (Hyclone) for 90 min at 37°C with gentle rotation. For the experiment of intraganglionic injection of AAV5-Myc, only L5 DRG was dissected and subjected to dissociation. Dissociation of DRGs and plating of dissociated DRG neurons were done following the protocols previously reported in detail (Kwon et al., 2013). The culture duration in our neurite outgrowth assay was strictly confined to 15 h to minimize neurite outgrowth in control conditions (without conditioning injury). Neurite outgrowth was visualized by immunostaining with mouse anti-β III tubulin (1:1000; Promega) followed by Alexa-594-conjugated anti-mouse secondary antibody.
PC12 cell culture
PC12 cells were cultured in DMEM containing 10% FBS and 1% penicillin-streptomycin. PC12 cells (2 × 105 cells) were seeded on poly-D-lysine (PDL)-coated 6 well plates and incubated for 24 h. To induce neuronal differentiation, the medium was changed to DMEM containing 1% horse serum, 1% penicillin-streptomycin, and 100 ng/ml NGF. The cells were further cultured for 6 d, and the differentiation medium was replaced every 2 d. PC12 cells were treated with dibutyryl-cAMP (db-cAMP; 0.1 mm; Calbiochem) for the designated duration and then harvested for mRNA extraction. For S-adenosylmethionine (SAM) pretreatment, SAM was added at a concentration of 500 μm to medium overnight the day before db-cAMP treatment.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using the EZ-ChIP assay kit (#17-295; Millipore) according to the manufacturer's instructions. Briefly, freshly dissected DRGs were incubated for 15 min with gentle rotation in 1% formaldehyde solution. The cross-linking reaction was stopped by addition of glycine to a final concentration of 0.25 M. Cross-linked chromatin was sheared by enzymatic digestion following the manufacturer's recommendations to yield an average fragment size of 300-600 bp. Samples were incubated with 5-10 μg of antibodies against c-MYC (10 μg; rabbit polyclonal; Cell Signaling), H3K9ac (5 μg; rabbit polyclonal; Abcam), H3K9me2 (5 μg; mouse monoclonal; Abcam), H3K9me3 (5 μg; mouse monoclonal; Millipore), and H3K27me3 (5 μg, mouse monoclonal; Abcam) at 4°C overnight. Rabbit or mouse IgGs were added to each sample to control for nonspecific antibody binding. Then, the samples were incubated with protein A agarose/salmon sperm DNA for 1 h at 4°C. The immunoprecipitated complexes were eluted, subjected to reversal of crosslinking by heat, and incubated with proteinase K. The recovered DNA was purified using PCI solution and processed to qRT-PCR. Cross-linked chromatin samples diluted 10 times were used as input controls that were not subjected to immunoprecipitation.
Quantitative measurement of mRNA by real-time PCR
Total RNAs were extracted from rat DRGs using Trizol (Invitrogen) according to the manufacturer's protocol. The amount of RNA was determined using NanoDrop Lite Spectrophotometer (Thermo Fisher Scientific) at 260 nm; 2 μg of RNA was reverse-transcribed to cDNA using a standard RT protocol; 1 μl of cDNA was added to the PCR-reaction premix (Takara Bio) with 10 pm corresponding primer pairs. The following primers were used for PCR: 18S ribosomal RNA (rRNA), 5′-CGGCTACCACATCCAAGGAA-3′ (forward), 5′-TGCTGGCACCAGACTTGCCCTC-3′ (reverse); c-Myc, 5′-CGAGCTGAAGCGTAGCTTTT-3′ (forward), 5′-CTCGCCGTTTCCTCAGTAAG-3′ (reverse); c-Jun, 5′-ACCCCCACTCAGTTCTTGTG-3′ (forward), 5′-AGTTGCTGAGGTTGGCGTAG-3′ (reverse); Atf3, 5′-CCATCCAGAACAAGCACCTT-3′ (forward), 5′-TCAGTTCGGCATTCACACTC-3′ (reverse). qRT-PCR was performed according to the protocol supplied by the SYBR Green PCR kit using the 7500 Real-Time PCR System (Applied Biosystems). Cycling conditions were 94°C for 30 s, 55°C ∼ 64°C for 35 s, and 72°C for 60 s, with a total of 40 cycles. Melting curves were generated after the last extension step, and the CT values were quantified by the Applied Biosystems 7500 software. Target gene expression was normalized with the expression of 18S rRNA as an internal control.
Bisulfite sequencing
Genomic DNA was extracted from rat L4-6 DRGs and purified using the DNeasy Blood & Tissue kit (QIAGEN). Genomic DNA was treated with sodium bisulfite (Zymo Research) to convert unmethylated cytosines to uracils; 100 ng of the converted DNA was used as the template to amplify the ROIs using specific primers designed by Methprimer (Li and Dahiya, 2002). Primers covering the exon 3 of Myc gene were as follows: 5′- GGTGTTTGTGGAAAAGAGGTAATT-3′ (forward) and 5′- AAAACCCCAACCAAAATTATAAAAT-3′ (reverse). Cycling conditions were 95°C for 30 s, 55°C ∼56°C for 30 s, and 72°C for 3 min, with a total of 40 cycles. PCR products were purified and cloned by the TOPO-TA cloning kit (Invitrogen). Sequencing was done with 10 colonies randomly picked. The efficiency of bisulfate conversion is confirmed by the complete conversion of non-CG cytosines to thymidines in PCR-amplified sequences. Clones with <95% C-to-T conversion of non-CpG cytosine were excluded from the analysis. The sequencing data were compiled and compared using QUMA (QUantification tool for Methylation Analysis), a web-based bisulfite sequencing analysis tool (Kumaki et al., 2008; Petkova et al., 2011; Saied et al., 2012). The number of clones exhibiting methylated cytosine was expressed as the percent methylation at the designated CpG site.
PCR array
To select RAGs to be profiled for PCR array kits, we carefully reviewed previous studies reporting lists of upregulated genes in DRGs following peripheral nerve injury (Bonilla et al., 2002; Costigan et al., 2002; Xiao et al., 2002; Nilsson et al., 2005; Stam et al., 2007), and 44 genes were finally chosen based on relevance to axonal regeneration and/or growth. The RT2 Profiler PCR Array (QIAGEN, #330171) was customized to profile two sets of the selected 44 RAGs of rat or mouse origin in a 96-well format. For the rat experiment, the L5 DRGs injected with AAV5-Myc or AAV5-GFP were quickly dissected 28 d after injection. Two L5 DRGs obtained from 2 rats were pooled together to comprise one biological replicate. For mouse experiments, bilateral L4-6 DRGs were quickly dissected 7 d following bilateral SNI or sham operation. A total of 12 DRGs from 2 mice were pooled together to comprise one biological replicate. Dissected DRGs were rapidly frozen and stored at −70˚C until use. Total RNA was purified using the RNeasy mini kit (QIAGEN, #74104) following the manufacturer's instructions. cDNA was obtained from purified RNA using the RT2 First Strand Kit (QIAGEN, #330401) provided within the PCR array kit; 1 μg of cDNA template mixed with PCR master mix was dispensed in each well, and qRT-PCR was performed using the Applied Biosystems 7500 system. Three independent arrays (three biological replicates) were performed for each condition, and the average array value compared with the control condition was obtained.
Tissue processing and immunohistochemistry
Rats or mice were anesthetized with ketamine/xylazine mixture as described in Surgical procedures and perfused with heparinized PBS followed by 4% PFA in 0.2 m PB. DRGs or spinal cord tissues containing the lesion site were dissected and postfixed in 4% PFA for 2 h, followed by cryoprotection in a graded series of sucrose solutions. DRGs were cryosectioned at 20 μm thickness. For spinal cord tissue, parasagittal cryosections (at 50 µm thickness) were made in a 1:4 series. Tissue sections were mounted onto Super Frost plus slides (Thermo Fisher Scientific) and stored at −20°C until use. DRG sections underwent antigen retrieval with 0.1 m EDTA-Tris buffer, pH 9.0, at 98˚C and were treated with 10% normal goat serum and 0.3% Triton X-100 for 1 h, and then the primary antibodies, dissolved in the same blocking solution, were applied at 4°C overnight. The primary antibodies were rabbit anti-GFAP (1:500; Dako), rabbit anti-GAP43 (1:500; Millipore), chicken anti-GFP (1:300; Millipore), goat anti-CTB antibodies (1:1000; List Biological Laboratories), rabbit anti-c-Myc (1:750; Abcam), mouse anti-NeuN (1:300; Millipore), rabbit anti-ATF3 (1:300; Santa Cruz Biotechnology), rabbit anti-Gadd45α (1:100; Santa Cruz Biotechnology), and rabbit anti-c-Jun (1:100; Millipore). Tissue sections were washed thoroughly and then incubated with appropriate secondary antibodies tagged with AlexaFluor-488 or -594 (1:500; Invitrogen) for 1 h at room temperature. The coverslips were mounted onto slides with glycerol-based mounting medium (Biomeda). The images were taken using an FV 300 confocal microscope (Olympus).
Quantitative image analysis
To quantify the extent of dorsal column axon growth after injury, two consecutive parasagittal sections collected at a distance of 200 µm containing CTB-traced axons were used for analysis. After the caudal lesion border was identified using GFAP staining, lines perpendicular to the longitudinal axis were drawn at 200 µm intervals from the caudal lesion border to delineate counting blocks. The number of CTB-positive axons between successive lines was counted and recorded as the number of axons in the block. The values from the two sections were added to generate the value of each animal. In addition, the longest distance of regenerating axons beyond the caudal lesion border was recorded for each animal. The percentage of CTB-traced regenerating axons presenting retraction bulbs was analyzed in the parasagittal spinal cord sections containing CTB-traced axonal tips. A square ROI with 0.1 mm2 area covering the front-end of traced axons was drawn, and the second square ROI was determined 200 µm caudal to the first ROI to include the axonal tips located caudal to the front-end. The maximum diameter of the axonal tips within the designated squares was measured and divided by the cylindrical diameter of the corresponding axon shafts to calculate the tip/axon ratio. A previous study reported that the retraction bulbs of the dorsal column sensory axons exhibited the mean tip/axonal shaft ratio that was >4.0 at 1 d after injury and even greater at later time points (Erturk et al., 2007). Therefore, the retraction bulbs were defined here as the axonal tips with a measured tip/axonal shaft ratio >4.0. The percentage of the retraction bulbs was obtained by dividing the number of the retraction bulbs by that of the total axonal tips contained within an ROI and averaged with the values from different ROIs in the same animal. The number of axonal tips analyzed in each animal ranged from 101 to 284.
For quantification of GFAP-immunoreactive signal intensity, three ROIs (each covering an area of 250 mm2) were placed along the caudal lesion border at an equal distance. To quantify intensity of IBA-1 immunoreactivities, two ROIs each covering 150 mm2 were placed at the epicenter. All images were obtained using the same detector setting. After images were adjusted using the predetermined threshold setting using ImageJ software (available at: http://imagej.nih.gov/ij/), integrated intensity of the immunoreactivity per unit area was obtained.
Experimental design and statistical methods
For gene expression analysis of DRGs following SNI, L4-6 DRGs were collected ipsilateral to the injured sciatic nerve and pooled to extract mRNA for one biological replicate. Each time point group included 3 or 4 animals. For the ChIP-PCR experiment, L4-6 DRGs were collected ipsilateral to the injured sciatic nerve from 3 animals and pooled as one biological replicate. Thus, each biological replicate consisted of 9 DRGs from 3 animals. Each time point group included 3 animals. For in vitro experiments using PC12 cells, each biological replicate was from an independent culture, and each group consisted of 3-5 independent cultures. For neurite outgrowth assay of Mycfl/fl or Mycnes-cre mice, L4-6 DRGs ipsilateral to the injured sciatic nerve were collected and dissociated together as one biological replicate. Thus, each group consisted of 3 animals with three DRGs from each animal. For neurite outgrowth assay of rats, only L5 DRG that was injected with either AAV5-GFP or AAV5-Myc was dissected and dissociated as one biological replicate. Thus, each group consisted of 3 animals with one DRG from each animal. For the PCR array experiment, detailed experimental design was described in the corresponding Materials and Methods section. For in vivo regeneration assay in rats, three experimental groups were generated: (1) AAV5-GFP group (N = 8), (2) AAV5-Myc group (N = 9), and (3) preconditioning SNI group (N = 5). For another experiment to use JNK inhibitor, two experimental groups were generated: rats injected with AAV5-Myc and subjected to an osmotic minipump containing (1) DMSO vehicle (N = 3) or (2) SP600125 (N = 3).
Statistical comparison of mean values between two groups was performed using unpaired Student's t test. Multiple comparisons between more than two groups were conducted using one-way ANOVA followed by Tukey's post hoc test. Quantification graphs were generated using GraphPad Prism software version 5.0.
Results
Identification of myc as an upstream TF gene for RAGs
In our previous study to examine a potential role of DNA methylation in the orchestrated transcription of RAGs in the preconditioning injury model (Shin et al., 2020), we profiled the DNA methylome using methylated DNA immunoprecipitation sequencing (MeDIP-seq) in DRGs following SNI and found that the changes in DNA methylation were not significantly correlated with the transcription levels of the RAGs. As a next approach, we sought to examine whether upstream transcription TF genes that can bind to the RAG promoters are affected by DNA methylation. We curated 81 RAGs based on the literature review that were consistently reported to increase in DRGs following preconditioning SNI (Shin et al., 2020), and then listed TF genes that are known to interact with the curated RAGs based on the Open Regulatory Annotation (ORegAnno) database (Griffith et al., 2008). We selected TF genes of which methylation level was changed by more than twofold and the expression level was increased following SNI (Table 1). There were no TF genes that met the criteria and of which methylation changes occurred in the promoter regions. The majority of DNA methylation in the listed TF genes occurred in intron regions. Compared with intron DNA methylation, DNA methylation in exon regions is more closely associated with histone modification (Dhami et al., 2010; Singer et al., 2015), which is another important epigenetic mechanism regulating gene expression. Myc and Creb1 exhibited demethylation in exon sequences (Table 1). In the present study, we paid our attention to Myc because its expression level was quite robust compared with Creb1 at both 1 and 7 d time points.
Table 1.
List of TF genes that are expected to interact with RAG promoters and of which DNA methylation is significantly changeda
| Time point | Predicted TF gene | Chromosome # | MeDIP peak (start) | MeDIP peak (end) | DNA methylation | Location | Log2(FC) | Interacting RAGs |
|---|---|---|---|---|---|---|---|---|
| 1 d | Meox2 | 6 | 55931995 | 55932480 | Hyper | Intron (1/2) | 0.4 | Cdkn1a |
| 1 d | Meox2 | 6 | 55951319 | 55951733 | Hypo | Intron (1/2) | 0.4 | Cdkn1a |
| 1 d | Tp53 | 10 | 56408640 | 56409942 | Hyper | Intron (8/9) | 0.27 | Atf3, Cdkn1a |
| 1 d | Creb1 | 9 | 63202312 | 63203771 | Hypo | Exon (3/7) | 0.23 | Serpine1 |
| 1 d | Foxa1 | 6 | 77985644 | 77986679 | Hyper | Intron (7/8) | 2.08 | Gadd45a, Gadd45g, Pdlim1 |
| 1 d | Foxa1 | 6 | 77994969 | 77995631 | Hyper | Intron (5/8) | 2.08 | Gadd45a, Gadd45g, Pdlim1 |
| 1 d | Gata3 | 17 | 79929424 | 79930365 | Hypo | Intron (4/7) | 1.58 | Edn1, Gadd45a, Gadd45g, Pdlim1 |
| 1 d | Hif1a | 6 | 96435222 | 96435734 | Hypo | Intron (1/15) | 0.43 | Arg1, Jun, Serpine1 |
| 1 d | Hif1a | 6 | 96451192 | 96451950 | Hypo | Intron (6/15) | 0.43 | Arg1, Jun, Serpine1 |
| 1 d | Hif1a | 6 | 96456609 | 96457122 | Hypo | Intron (9/15) | 0.43 | Arg1, Jun, Serpine1 |
| 1 d | Myc | 7 | 98956881 | 98958075 | Hypo | Exon (3/4) | 1.58 | Cdkn1a, Gadd45a, Gal, Jun, Serpine1, Vgf |
| 1 d | Max | 6 | 99496672 | 99497343 | Hypo | Intron (9/11) | 0.12 | Gadd45a |
| 7 d | Gata1 | X | 26570505 | 26571477 | Hypo | Intron (5/5) | 1.19 | Gadd45a, Gadd45g, Il6, Jun, Pdlim1, Socs3 |
| 7 d | Meox2 | 6 | 55931995 | 55932480 | Hyper | Intron (1/2) | 0.83 | Cdkn1a |
| 7 d | Tp53 | 10 | 56408640 | 56409942 | Hyper | Intron (8/9) | 0.14 | Atf3, Cdkn1a |
| 7 d | Creb1 | 9 | 63202312 | 63203771 | Hypo | Exon (3/7) | 0.19 | Serpine1 |
| 7 d | Klf4 | 5 | 73446241 | 73447295 | Hyper | TTS | 0.43 | Cdkn1a |
| 7 d | Tp63 | 11 | 76989128 | 76990082 | Hyper | Intron (3/13) | 1.49 | Cdkn1a |
| 7 d | Tp63 | 11 | 77017983 | 77018713 | Hyper | Intron (3/13) | 1.49 | Cdkn1a |
| 7 d | Tp63 | 11 | 77043875 | 77045468 | Hyper | Intron (3/13) | 1.49 | Cdkn1a |
| 7 d | Tp63 | 11 | 77055573 | 77056745 | Hyper | Intron (3/13) | 1.49 | Cdkn1a |
| 7 d | Tp63 | 11 | 77066199 | 77066711 | Hyper | Intron (3/13) | 1.49 | Cdkn1a |
| 7 d | Tp63 | 11 | 77095731 | 77096589 | Hyper | Intron (3/13) | 1.49 | Cdkn1a |
| 7 d | Tp63 | 11 | 77099805 | 77100411 | Hyper | Intron (3/13) | 1.49 | Cdkn1a |
| 7 d | Tp63 | 11 | 77124033 | 77125944 | Hyper | Intron (1/13) | 1.49 | Cdkn1a |
| 7 d | Gata3 | 17 | 79929424 | 79930365 | Hypo | Intron (4/7) | 1.54 | Edn1, Gadd45a, Gadd45g, Pdlim1 |
| 7 d | Hif1a | 6 | 96435222 | 96435734 | Hypo | Intron (1/15) | 0.25 | Arg1, Jun, Serpine1 |
| 7 d | Hif1a | 6 | 96451192 | 96451950 | Hypo | Intron (6/15) | 0.25 | Arg1, Jun, Serpine1 |
| 7 d | Myc | 7 | 98956881 | 98958075 | Hypo | Exon (3/4) | 0.94 | Cdkn1a, Gadd45a, Gal, Jun, Serpine1, Vgf |
| 7 d | Max | 6 | 99496672 | 99497343 | Hypo | Intron (9/11) | 0.83 | Gadd45a |
| 7 d | Gata2 | 4 | 122282649 | 122283285 | Hypo | Intron (4/6) | 0.52 |
Adam8, Ankrd1, Apobec1, Atf3, Cdkn1a, Edn1, Gadd45a, Gadd45g, Gch1, Il6, Jun, Nos1, Pdlim1, Rhoq, Socs3 |
aHyper, Hypermethylation; Hypo, hypomethylation; Log2(FC), log2 value of the fold change in the gene expression level compared with the uninjured control condition; TTS, transcription termination site.
DNA hypomethylation of Myc within the third exon sequences was validated by bisulfite sequencing (Fig. 1A). In the uninjured control condition, the majority of CpG sites within the MeDIP peak sequences located in exon 3 were almost fully methylated, except a couple of CpG sites at the most 3′ position. Preconditioning SNI resulted in a decrease in the extent of methylation at many CpG sites across the entire exon 3 sequences similarly at both time points (Fig. 1A). We also validated significant upregulation of Myc gene expression following preconditioning peripheral nerve injury using real-time PCR (F(2,9) = 4.658, p = 0.0409, ANOVA) (Fig. 1B). At 1 d, Myc mRNA level was increased by threefold and remained elevated at 7 d after injury. Immunofluorescence staining confirmed c-MYC protein expression in predominantly nuclear compartment of DRG neurons at 1 and 7 d after injury (Fig. 1C).
Figure 1.
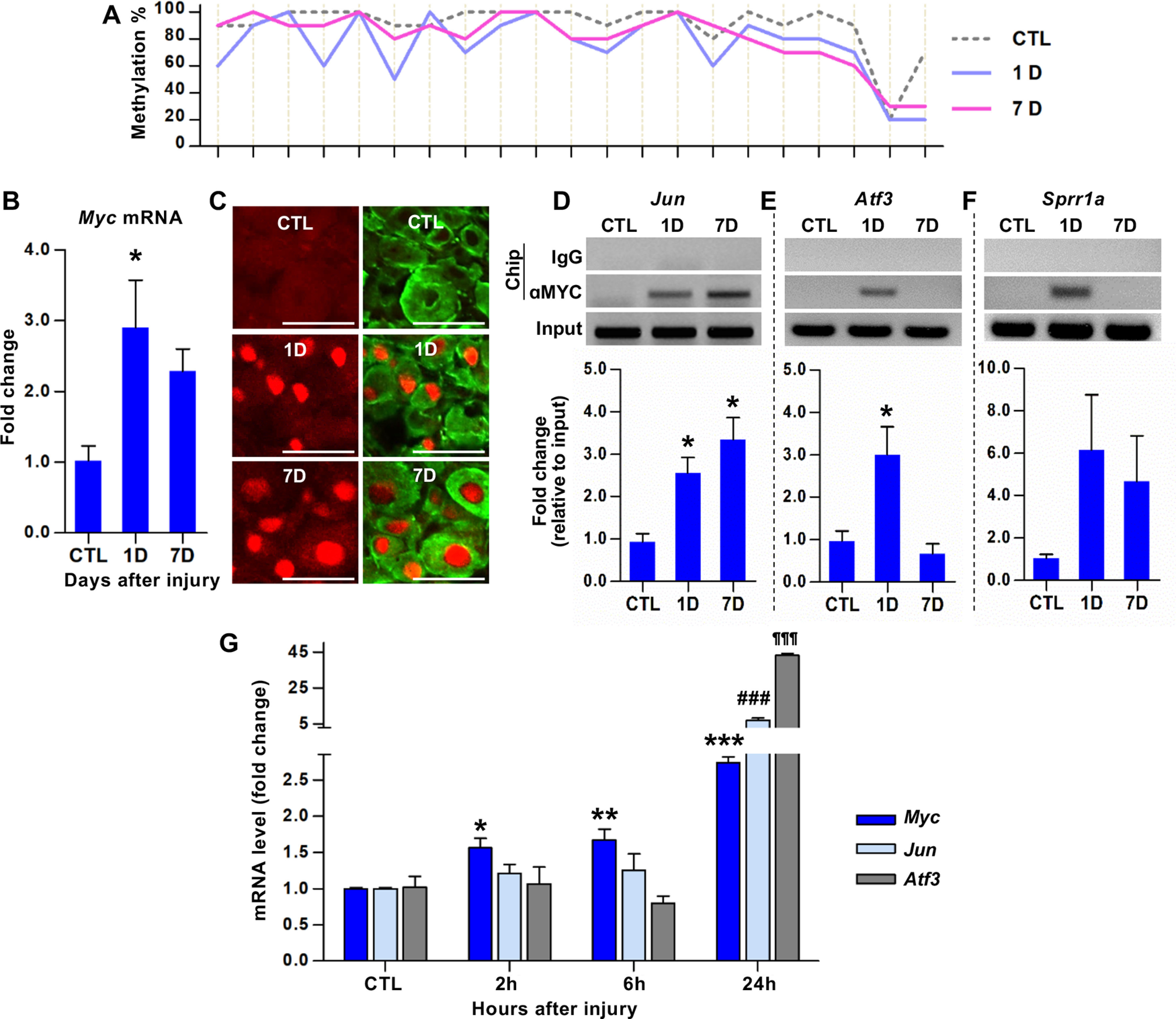
Identification of Myc as a hub transcription factor gene for a subset of the RAGs following preconditioning peripheral nerve injury. A, Bisulfite sequencing analysis of CpGs within the Myc exon 3 sequences using DNA obtained from the DRGs of animals killed at different time points following SNI. The percentage of the clones revealing methylated cytosine was shown on the y axis. Each line crossing the x axis indicates individual CpG sites. B, qRT-PCR results of the Myc gene expression in DRGs following SNI. *p < 0.05 (one-way ANOVA followed by Tukey's post hoc analysis). N = 4 animals per group. C, Representative images of DRG tissue sections immunostained with anti-c-Myc antibodies. Predominantly nuclear c-Myc immunofluorescence signals (red) were observed in DRG neurons visualized by NeuN immunoreactivity (green). Scale bars, 100 μm. D–F, ChIP analysis of c-MYC binding to the promoters of Jun (D), Atf3 (E), and Sprr1a (F). DRG samples were subjected to immunoprecipitation using antibodies against c-MYC, and the immunoprecipitated DNA fragments were amplified by qRT-PCR. *p < 0.05 (one-way ANOVA followed by Tukey's post hoc analysis). N = 3 animals per group. G, qRT-PCR results comparing the expression levels of Myc, Jun, and Atf3 in DRGs at early time points following SNI. For Myc: *p < 0.05; **p < 0.01; ***p < 0.001; compared with control (CTL). For Jun: ###p < 0.001; compared with CTL. For Atf3: ¶¶¶p < 0.001; compared with CTL. Statistical significance was determined by one-way ANOVA followed by Tukey's post hoc analysis. N = 3 animals per time point.
To determine whether Myc gene product, c-MYC, indeed functions as a transcription factor to regulate RAGs following SNI, we performed ChIP-PCR using antibodies against c-MYC. Of the 6 RAGs expected to interact with c-MYC based on the Open Regulatory Annotation database, Jun and Galanin were selected because their roles in axon regeneration had been established (Holmes et al., 2000; Mahoney et al., 2003; Raivich et al., 2004). ChIP-PCR showed robust c-MYC binding to Jun promoter at 1 and 7 d after injury (Fig. 1D). Quantification data showed that there was a statistically significant increase at 1 and 7 d following SNI (F(2,6) = 10.27, p = 0.0116, ANOVA). However, there was no significant c-MYC binding to the promoter regions of the Galanin gene. We also examined c-MYC binding to the Atf3 promoter since Myc-dependent transcriptional regulation of ATF3 had been reported (Tamura et al., 2005). The occupancy of Atf3 gene promoter by c-MYC was markedly increased at 1 d but returned to the control level by 7 d (F(2,6) = 9.042, p = 0.0155, ANOVA) (Fig. 1E). Further testing of c-MYC binding to the promoter of well-known RAGs revealed a tendency of Sprr1a promoter occupied by c-MYC at both 1 and 7 d (Fig. 1F). In contrast, promoter regions of Gap43 and Npy, other well-known RAGs, did not interact with c-MYC, suggesting that c-MYC TF is directly involved in the transcriptional regulation of a certain group of RAGs. We speculated that if Myc regulates the transcription of the RAGs of which promoter regions interact with c-MYC, the increase of Myc gene expression would precede that of the RAG expressions. As expected, Myc expression was significantly increased as early as 2 h after SNI (Fig. 1G). The increase persisted at 6 h, and a further increase was observed at 24 h. However, expression of Jun or Atf3 did not change significantly at either 2 or 6 h time point, although their expression was greatly enhanced at the 24 h time point. These results suggest that Myc upregulation occurring hours after injury leads to occupancy of the promoters belonging to a certain group of RAGs (Myc-dependent RAGs) and enhances their expression at a later time point.
Epigenetic regulation of myc gene expression
Since we observed a very early increase of Myc gene expression, we performed bisulfite sequencing using DNA obtained 2 h after injury and found marked demethylation at the same third exon sequences (Fig. 2A). This finding suggests that the preconditioning SNI may induce rapid Myc upregulation by eliciting DNA demethylation. It was reported that nerve growth factor, epidermal growth factor, and various pharmacological agents could induce early transcriptional activation of the Myc gene in PC12 cells (Greenberg et al., 1985). Among these agents, we chose db-cAMP to model the early increase of Myc gene expression following preconditioning peripheral nerve injury since cAMP increases the neuron-intrinsic capacity of axon regeneration (Qiu et al., 2002). When PC12 cells were treated with db-cAMP, Myc mRNA levels were rapidly elevated at 15 min, peaked at 30 min, and then declined (F(4,13) = 6.292, p = 0.0048, ANOVA) (Fig. 2B). Bisulfite sequencing demonstrated a considerable decrease of DNA methylation at the same third exon sequences at 30 min following db-cAMP treatment (Fig. 2C). When SAM, a methyl donor for DNA methylation promoting DNA methylation or inhibiting DNA demethylation (Shukeir et al., 2006; Massart et al., 2015), was pretreated, db-cAMP failed to induce Myc gene transcription (F(3,8) = 48.65, p < 0.0001, ANOVA) (Fig. 2D), suggesting that DNA demethylation may be responsible for the early transcriptional activation of Myc.
Figure 2.

Epigenetic regulation of Myc gene expression. A, Bisulfite sequencing analysis of CpGs within the Myc exon 3 sequences using DNA obtained from the DRGs of animals killed at a 2 h time point following SNI. The percentage of the clones revealing methylated cytosine was shown on the y axis. Each line crossing the x axis indicates individual CpG sites. B, qRT-PCR results of the Myc gene expression in cultured PC12 cells in response to treatment with db-cAMP (0.1 mm). **p < 0.01, compared with control (one-way ANOVA followed by Tukey's post hoc analysis). N = 3 or 4 independent cultures per time point. C, Bisulfite sequencing analysis of CpGs within the Myc exon 3 sequences using DNA extracted from cultured PC12 cells harvested at 30 min after db-cAMP treatment (0.1 mm). The percentage of the clones revealing methylated cytosine was shown on the y axis. Each line crossing the x axis indicates individual CpG sites. D, qRT-PCR results of the Myc gene expression in cultured PC12 cells harvested 30 min after the designated treatments. SAM was pretreated at a concentration of 500 μm to medium overnight the day before db-cAMP treatment. ***p < 0.001 (one-way ANOVA followed by Tukey's post hoc analysis). N = 3 independent cultures per experimental condition. E–G, ChIP analysis of the histone marks, H3K9ac (E), H3K9me2 (F), H3K27me3 (G), using DRG samples obtained at control (CTL) or 1 d after SNI. Immunoprecipitated DNA fragments were amplified by qRT-PCR. *p < 0.05; **p < 0.01; Student's t test. N = 4, N = 4, and N = 3 animals per group for E, F, and G, respectively.
Recent studies reported that DNA methylation status is closely associated with histone modification (Cedar and Bergman, 2009; Rose and Klose, 2014). We performed ChIP analysis to identify histone marks at the Myc exon 3 sequences with DNA demethylation. H3K9ac, which is commonly associated with open chromatin, was significantly enriched at 1 d after injury (t(6) = 4.730, p = 0.0032, unpaired t test) (Fig. 2E). On the contrary, the repressive histone mark, H3K9me2 binding to the exon 3 region was significantly decreased after injury (t(6) = 5.177, p = 0.0021, unpaired t test) (Fig. 2F). We also looked at the enrichment of another repressive histone mark, H3K27me3, but could not find any change in binding to the third exon sequences (Fig. 2G). These results indicated that the demethylation in the third exon sequences of the Myc gene may be accompanied by open chromatin structures, leading to an early increase in transcription.
Myc is necessary for the enhancement of regenerative capacity and injury-induced upregulation of a group of RAGs
To determine whether Myc is necessary for enhanced neurite outgrowth capacity following preconditioning injury, we generated mice deficient in Myc in neural lineage cells by mating Myc floxed mice with Nestin-Cre transgenic line (Hatton et al., 2006). The Myc flox/flox; Nestin-Cre mice (Mycnes-cre hereafter) were grossly not different from WT mice and there were no obvious behavioral abnormalities. Neurite outgrowth assay was performed using freshly dissected DRG neurons at 7 d after sham or SNI from either Mycfl/fl (WT) or Mycnes-cre animals (Fig. 3A). The extent of neurite outgrowth was dramatically increased by SNI in Mycfl/fl animals. SNI in Mycnes-cre also led to the increase of neurite outgrowth, but the extent of increase was approximately half of that in Mycfl/fl mice (Fig. 3A,B), indicating that deletion of Myc gene significantly attenuated the preconditioning effects on neurite outgrowth.
Figure 3.
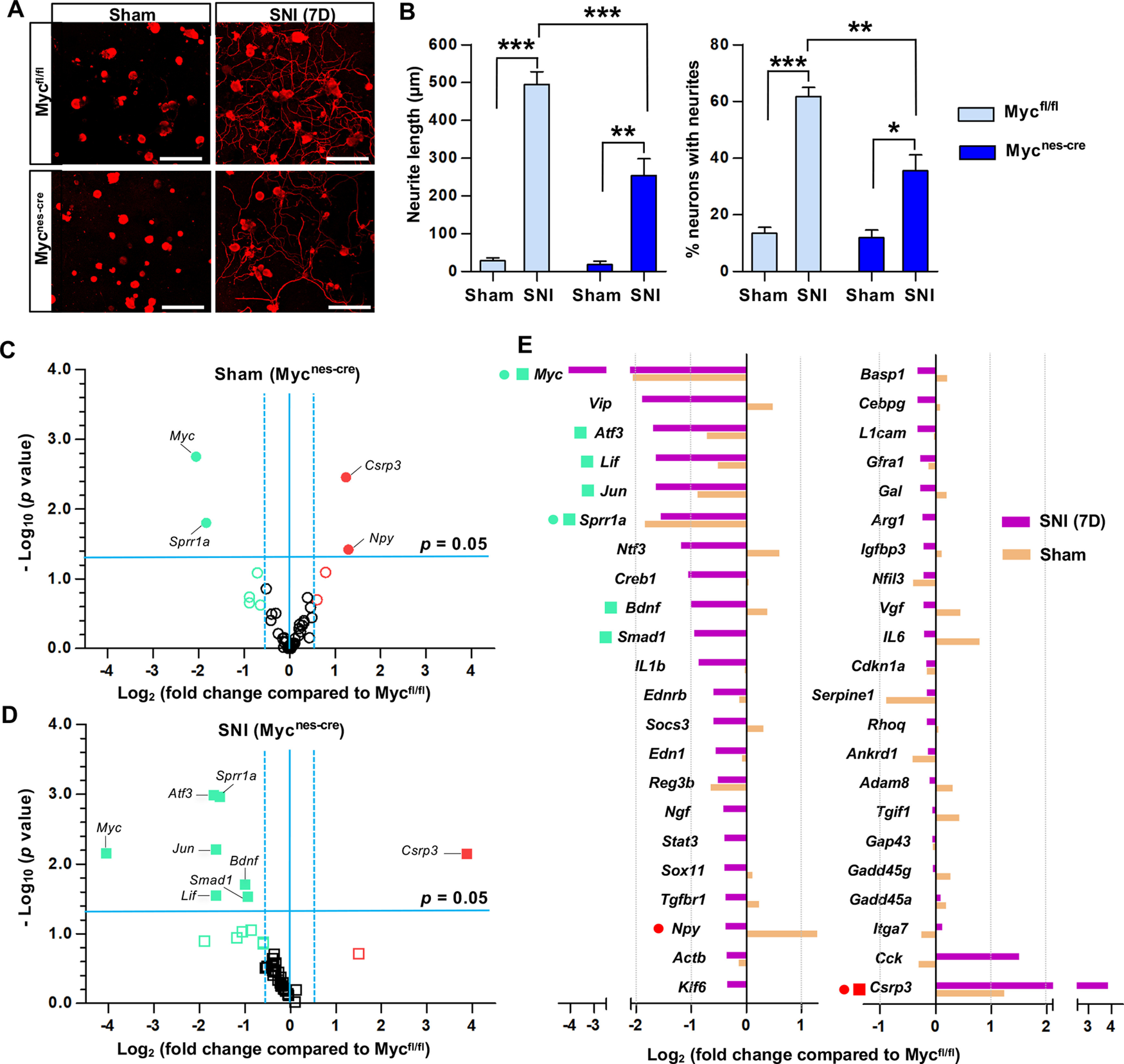
Myc is necessary for the enhancement of regenerative capacity and injury-induced upregulation of a group of RAGs. A, Representative images of cultured mouse DRG neurons. DRGs were obtained from Myc floxed mice (Mycfl/fl) or Myc flox/flox; Nestin-Cre mice (Mycnes-cre), where Myc is deleted in neural lineage cells. Mice with either genotype were sham-operated or underwent SNI, and their L4-L6 DRGs were acutely dissociated 7 d after the surgery and cultured for 15 h before fixation. Neurons and their neurites are visualized by immunoreactivity against βIII tubulin. Scale bars, 50 μm. B, Quantification graphs of the neurite length and the percentage of neurons with neurites longer than 2 times the longest length of cell bodies. *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA followed by Tukey's post hoc analysis. N = 3 animals per genotype for sham-operated groups, and N = 5 animals per genotype for SNI groups. C, D, Volcano plots of the 44 RAG expressions in DRGs measured using the PCR array comparing the WT (Mycfl/fl) and Myc-deficient (Mycnes-cre) animals either with sham operation (C) or SNI (D). The L4-L6 DRGs were dissected 7 d after sham operation or SNI. Dotted vertical lines indicate a 1.5-fold increase or decrease compared with the PBS control group. Solid horizontal line indicates p = 0.05 (unpaired t test), comparing the WT (Mycfl/fl) and Myc-deficient (Mycnes-cre) animals with either sham operation (C) or SNI (D). Red circles or squares represent the genes revealing a >1.5-fold increase in expression by Myc deletion in the sham operation (C) or SNI (D) condition, respectively. Green circles or squares represent the genes revealing a >1.5-fold decrease in expression by Myc deletion in the sham operation (C) or SNI (D) condition, respectively. Filled circles or squares represent the genes of which changes in expression by Myc deletion were statistically significant (p < 0.05). N = 3 biological replicates per experimental group. E, Quantification graph of gene expression levels of Myc-deficient animals (expressed as fold changes compared with Mycfl/fl) for the 44 RAGs measured using the PCR array. The symbols preceding the gene titles are matched with those designated in the volcano plots (C,D).
To examine the potential influence of Myc deletion on RAG induction by preconditioning SNI, we performed PCR array using a customized panel of genes consisting of 44 RAGs that were selected based on more than several previous gene expression studies in DRGs (Shin et al., 2020). As expected, the conditional deletion of Myc led to a marked decrease in Myc gene expression in DRGs from sham-operated animals (Fig. 3C,E). The majority of the selected RAGs were not significantly affected by the deletion of Myc as evidenced by the volcano plot for no injury condition (Fig. 3C). Myc deletion in sham-operated animals decreased expression of only several RAGs, including Sprr1a, while Csrp3 (Cysteine and glycine rich protein 3) and Npy increased basal expression in Myc-deficient animals without injury (Fig. 3C,E). In contrast, Myc deletion markedly shifted the volcano plot to the left side following a preconditioning injury (Fig. 3D), indicating that the upregulation of many RAGs by preconditioning SNI was blunted in Myc-deficient animals. Intriguingly, expressions of Jun, Atf3, and Sprr1a, which were bound by c-MYC transcription factor in their promoter regions following injury (Fig. 1C–E), were robustly and significantly decreased in Myc-deficient animals 7 d following SNI. Neuropoietic cytokine genes, such as Lif and Il1b, also showed a considerable decrease. In addition, expression of Bdnf and Smad1 was also significantly attenuated. However, expressions of Gap43, Npy, and Galanin genes, of which promoter regions did not interact with c-MYC, were not noticeably affected by Myc deletion following injury (Fig. 3E). Moreover, injury-induced upregulation of well-established RAGs, for example, Il-6, Gadd45a, Gadd45g, and Arg1, were not significantly changed in Mycnes-cre line. Only Csrp3 gene expression after SNI was significantly augmented in Myc-deficient animals. These results suggest that Myc is involved in the upregulation of a certain group of RAGs following a preconditioning injury, while a still significant proportion of RAGs are induced by injury independently of Myc deletion.
To confirm the influence of Myc on the injury-induced RAG transcription at the protein level, immunofluorescence staining was performed on the lumbar DRG sections 7 d following SNI. The preconditioning SNI markedly increased the expression of RAGs, such as c-JUN, ATF3, GAP-43, and GADD45α predominantly in DRG neurons. Consistent with the mRNA expression data, the percentage of DRG neurons colocalized with c-JUN or ATF3 after injury was sharply decreased in Mycnes-cre animals compared with Mycfl/fl mice (Fig. 4A–C). In contrast, injury-induced expression of GAP-43 and GADD45ɑ, of which mRNA expression was not affected by Myc deletion, persisted in the DRGs of Mycnes-cre animals (Fig. 4A,D,E).
Figure 4.
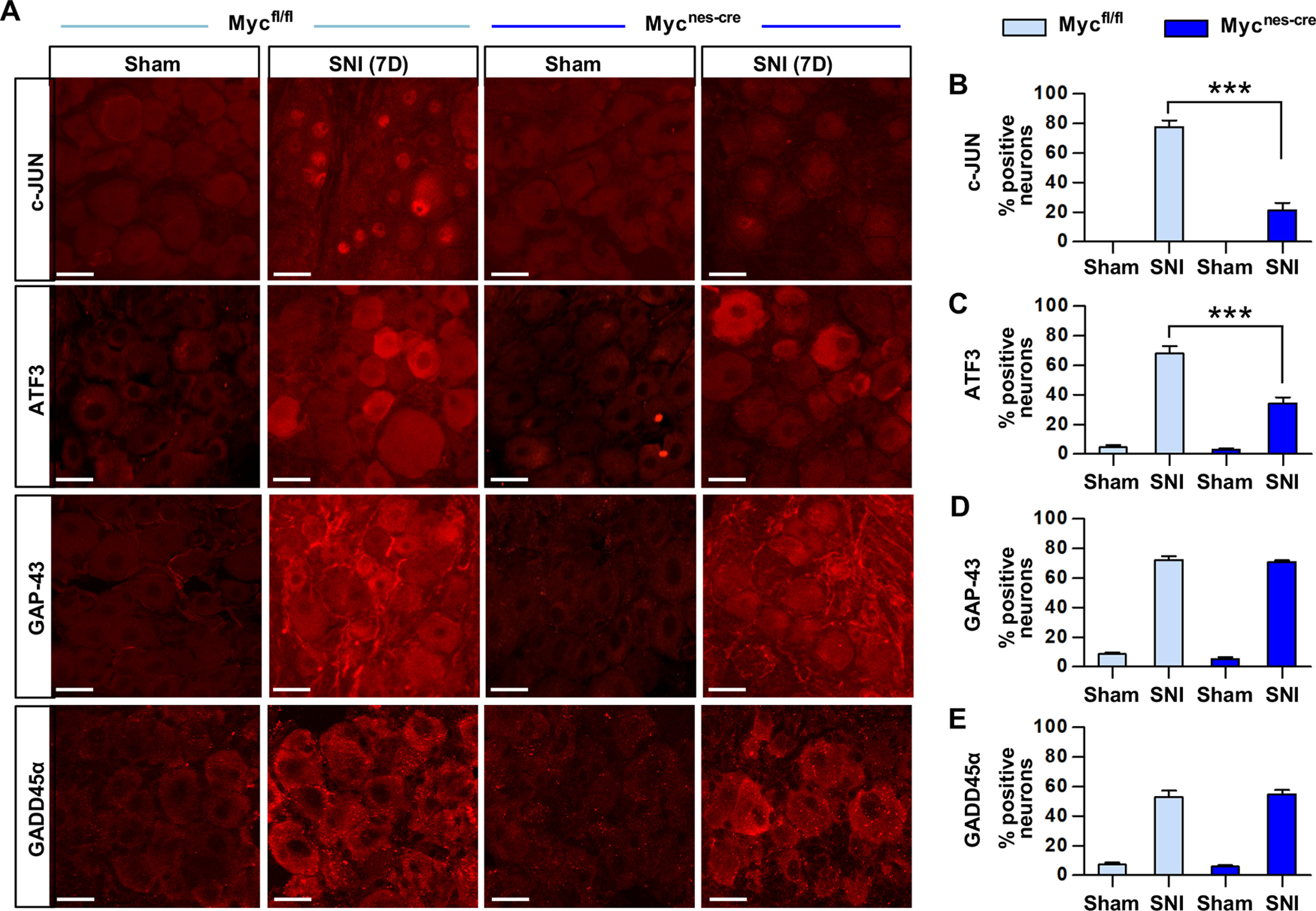
Protein expression of the RAGs in Myc-deficient mice. A, Representative images of the immunofluorescence staining on the DRG sections obtained from the WT (Mycfl/fl) and Myc-deficient (Myc Mycnes-cre) animals. Animals were killed for the immunofluorescence staining 7 d after either sham operation or SNI. Scale bars, 50 μm. B–E, Quantification graphs of the percent DRG neurons positive with c-JUN (B), ATF3 (C), GAP-43 (D), and GADD45alpha (E). ***p < 0.001 (one-way ANOVA followed by Tukey's post hoc analysis). N = 3 animals per experimental group.
Myc overexpression partially mimics the conditioning effects
Next, we tested whether Myc overexpression leads to the upregulation of RAGs and enhancement of regenerative capacity mimicking preconditioning SNI. Myc gene was delivered to rat DRGs by intraganglionic injection of AAV5. DRGs were dissected and acutely cultured to examine the extent of neurite outgrowth 28 d after injection of AAV5-Myc or AAV5-GFP as a control injection (Fig. 5A). Most DRG neurons infected with AAV5-GFP did not grow traceable neurites for 15 h after plating. In contrast, the extent of neurite outgrowth was remarkably increased from DRG neurons with AAV5-Myc (Fig. 5A,B). Compared with the neurite outgrowth of neurons collected from DRGs subjected to SNI 7 d before, the increase by AAV5-Myc was ∼60% of that achieved by preconditioning SNI, indicating that Myc overexpression partially mimics the conditioning effects.
Figure 5.
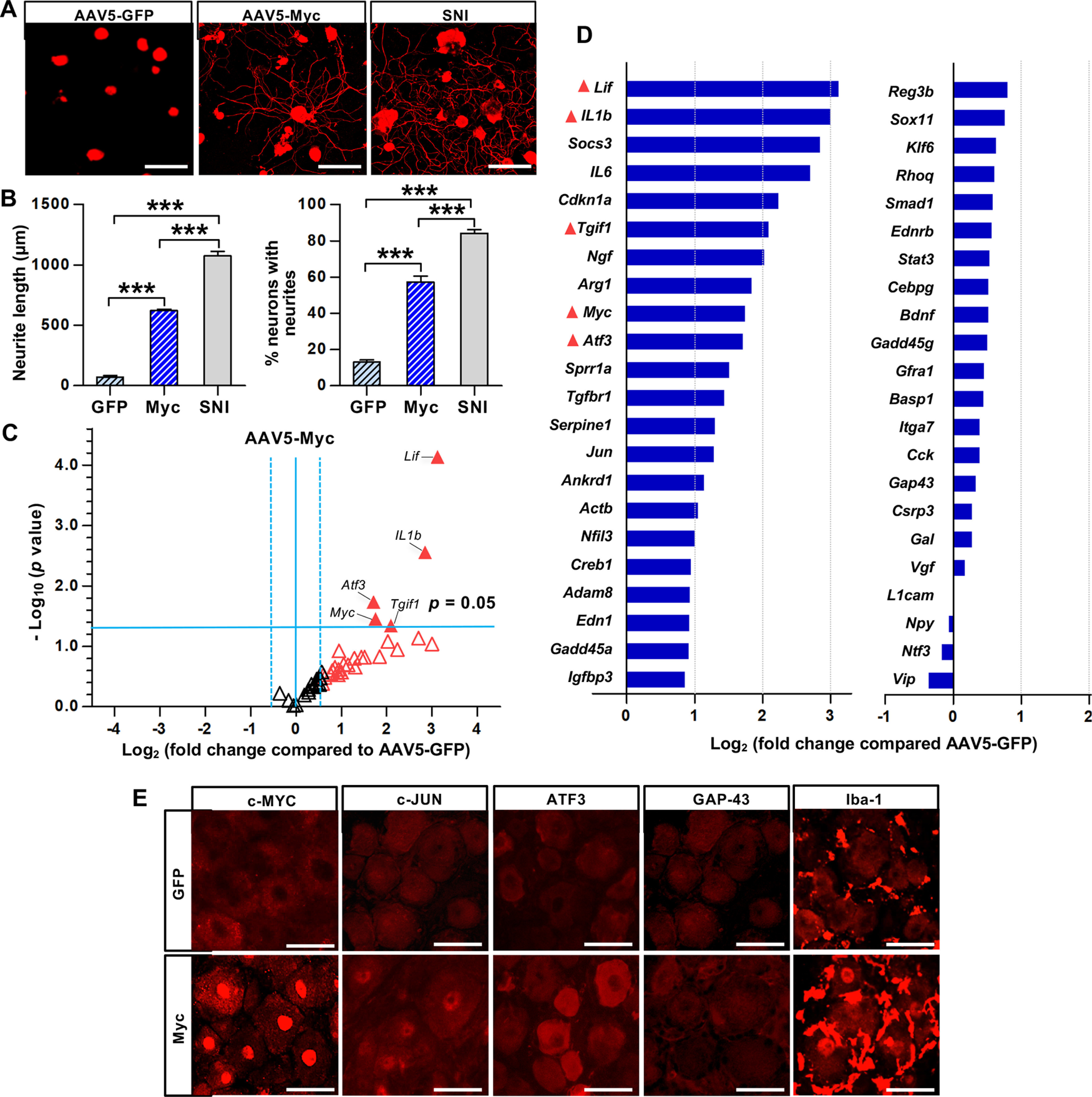
Myc overexpression enhances axon growth capacity and increases expression of the RAGs. A, Representative images of cultured rat DRG neurons visualized by βIII-tubulin staining. The L5 DRGs in rats were injected with AAV5-GFP or AAV5-Myc and dissected 28 d after the injection. The L5 DRGs were dissociated and cultured for 15 h before fixation. The L5 DRGs were acutely cultured 7 d after SNI for SNI group. Scale bars, 50 μm. B, Quantification graphs of the neurite length and the percentage of neurons with neurites longer than 2 times the longest length of cell bodies. ***p < 0.001 (one-way ANOVA followed by Tukey's post hoc analysis). N = 5 animals per group. C, Volcano plots of the 44 RAG expressions in rat L5 DRGs measured using the PCR array comparing the animals with AAV5-Myc and AAV5-GFP injections. The L5 DRGs were dissected 7 d after the intraganglionic AAV injection. Dotted vertical lines indicate a 1.5-fold increase or decrease compared with the AAV5-GFP control group. Solid horizontal line indicates p = 0.05, comparing the animals with AAV5-Myc and AAV5-GFP injections (unpaired t test). Red triangles represent the genes revealing a >1.5-fold increase in expression by AAV5-Myc injection. Filled triangles represent the genes of which changes in expression by Myc overexpression were statistically significant (p < 0.05). N = 3 biological replicates per experimental group. D, Quantification graph of the 44 RAG expression levels by AAV5-Myc injection (expressed as fold changes compared with AAV5-GFP). The symbols preceding the gene titles are matched with those designated in the volcano plots (C). E, Representative images of the immunofluorescence staining on the DRG sections obtained from animals injected with AAV5-GFP or AAV5-Myc injection. Animals were killed for the immunofluorescence staining 28 d after the AAV injection. Scale bars, 50 μm.
We also compared the pattern of RAG expression between animals with intraganglionic injection of AAV5-GFP and AAV5-Myc using the PCR array with the same panel of RAGs. The volcano plot of changes in RAG expression by Myc overexpression was almost opposite to that by Myc deletion (Fig. 5C). The majority of RAGs increased the level of expression, shifting the volcano plot to the right. Neuropoietic cytokines, Lif and Il1b, were most robustly and significantly upregulated by Myc overexpression (Fig. 5C,D). Socs3, of which expression is regulated by these cytokines, was also increased. Atf3, Jun, and Sprr1a, of which injury-induced upregulation was significantly attenuated in Myc-deficient animals (Fig. 3C–E), showed more than a twofold increase in gene expression by AAV5-Myc. In contrast, Gap43, Npy, and Galanin were unresponsive to Myc overexpression (Fig. 5D). These results indicate that Myc overexpression influences a considerable number of RAGs, many of which overlapped with the list of genes that were affected by Myc deletion.
Immunofluorescence staining verified increased immunoreactivity against c-MYC in DRG neurons, particularly within the nuclear compartment, by AAV-mediated overexpression, whereas slight immunoreactivity was noted in the cytoplasm in DRGs with control AAV5-GFP injection (Fig. 5E). As expected, increased expression of c-JUN and ATF3, but not GAP-43, was confirmed at the protein level (Fig. 5E). We have previously shown that perineuronal macrophage activation following SNI contributes to enhanced axon regeneration capacity (Kwon et al., 2013). Since we found robust upregulation of neuropoietic cytokines by Myc overexpression, we examined whether Myc overexpression could activate perineuronal macrophages. Indeed, IBA-1-positive macrophage signals surrounding DRG neurons substantially increased in DRGs with AAV5-Myc injection compared with the group with AAV5-GFP injection.
Myc overexpression prevents retraction of sensory axons following spinal cord injury
Next, we determined whether Myc overexpression promotes axon regeneration in the in vivo spinal cord injury model in female rats. Intraganglionic injection of either AAV5-GFP or AAV5-Myc was performed; and then 3 weeks later, animals were subjected to dorsal hemisection injury (Fig. 6A). In animals with AAV5-GFP injection, CTB-traced axon tips retracted a few hundred micrometers away from the caudal lesion border (Fig. 6B,H). In contrast, intraganglionic injection of AAV5-Myc markedly prevented the retraction of the axonal tips, resulting in proximity between the lesion border and the axonal front-end (Fig. 6C). There were a very small number of axons regenerating beyond the caudal lesion border, and the distance of regeneration in those axons was quite limited to <500 µm (Fig. 6H). As a positive control, we performed preconditioning SNI 1 week before spinal cord injury, and then evaluated the extent of axon regeneration at the same time point as for the animals with AAV5 injection. As previously reported, we observed a substantial number of axons growing beyond the lesion border (Fig. 6D), contrasting with the results obtained from animals injected with AA5-Myc. The mean longest distance of regeneration beyond the caudal lesion border in the SNI group was ∼1 mm, more than fivefold greater than that in the AAV5-Myc group (Fig. 6H), and the difference was statistically significant (F(2,19) = 108.9, p < 0.0001, ANOVA). The mean number of axons at the spinal cord between the lesion border and 200 μm caudal to the lesion was comparable between AAV5-Myc and SNI-preconditioned groups and much greater than that in AAV5-GFP group (Fig. 6I). Myc overexpression also significantly increased the mean number of axons compared with that of the AAV5-GFP control injection group at the spinal cord regions between 200 and 400 μm and between 400 and 600 μm away from the lesion border. Along with the changes in the length and number of regenerating axons, we also found quite striking changes in the morphology of axonal tips following AAV5-Myc injection. In animals with control AAV5-GFP injection, retracting axons frequently exhibited bulbous swelling in their axonal tips (Fig. 6E). In contrast, AAV5-Myc injection substantially prevented the formation of the retraction bulbs (Fig. 6F). In SNI-preconditioned animals, the majority of axonal tips were slim and continuous with the axonal shaft, looking like growth cones (Fig. 6G). Quantification of the percentage of the retraction bulbs revealed that AAV5-Myc injection significantly reduced the retraction bulb formation by almost 50% (F(2,19) = 40.94, p < 0.0001, ANOVA) (Fig. 6J). Myc overexpression in DRGs did not influence the formation of glial scars at the lesion site, as demonstrated by GFAP staining (Fig. 7A,B). It has been shown that macrophages at the lesion site play a significant role in retracting axons from the injury core (Horn et al., 2008). However, IBA-1 staining at the lesion site did not reveal significant difference between AAV5-GFP and AAV5-Myc injection groups (Fig. 7D,E). The preconditioning SNI also did not influence the glial microenvironment at the lesion site (Fig. 7C,F). Quantification data showed that there was no significant difference in GFAP and IBA-1 signal intensities between the three groups (GFAP, F(2,19) = 0.877, p = 0.4321, ANOVA; IBA-1, F(2,19) = 0.387, p < 0.6843, ANOVA) (Fig. 7G,H). These results suggest that the effects of Myc overexpression on preventing axonal retraction are primarily mediated by enhancing the intrinsic growth capacity of the DRG sensory neurons.
Figure 6.
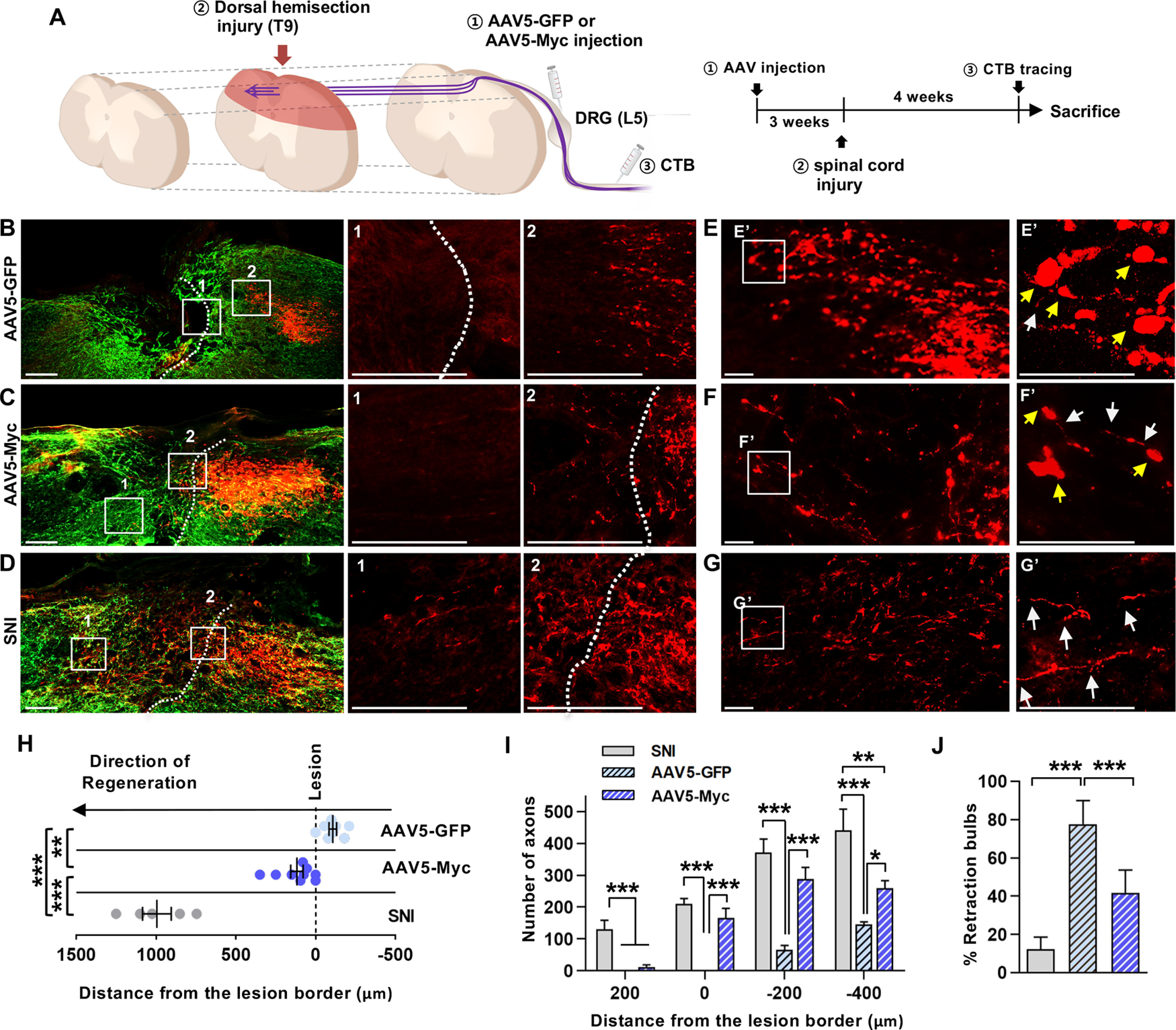
Myc overexpression prevents axon retraction following spinal cord injury. A, An experimental scheme depicting the time course of individual processes. B–D, Representative images of the parasagittal spinal cord sections from animals injected with AAV5-GFP (B) or AAV5-Myc (C), or that received preconditioning SNI (D). Red represents CTB-labeled axons. Green represents GFAP-positive glial scars. Dashed lines indicate the caudal lesion borders as determined by GFAP immunostaining. B–D, Boxed regions are magnified at the right with the corresponding numbers. Scale bars: B–D, 200 µm. E–G, Morphology of the axonal tips at the axon front. E–G, Boxed regions are magnified at the right (E′-G′). Yellow arrows indicate the axonal tips counted as the retraction bulbs. White arrows indicate the axonal tips of growth-cone like morphology. Scale bars: E–G, 50 µm. H, Quantification graph comparing the longest distance of regenerating axons from the caudal lesion border. Individual circles plot the longest distance of each animal. Negative values indicate the degree of the retraction from the lesion border. I, Quantification graph comparing the number of CTB-positive axons within counting blocks located at different distances from the lesion border. The numbers in the x axis indicate the distance of the rostral border of a counting block from the lesion border. J, Quantification graph comparing the percentage of retraction bulbs. The retraction bulbs were defined as the axons with ration between the maximum tip diameter and the axon cylindrical diameter ≥4.0. ***p < 0.001; **p < 0.01; *p < 0.05; between animals (one-way ANOVA followed by Tukey's post hoc analysis). N = 8, N = 9, and N = 5 animals for the AAV5-GFP, AAV5-Myc, and SNI groups, respectively.
Figure 7.
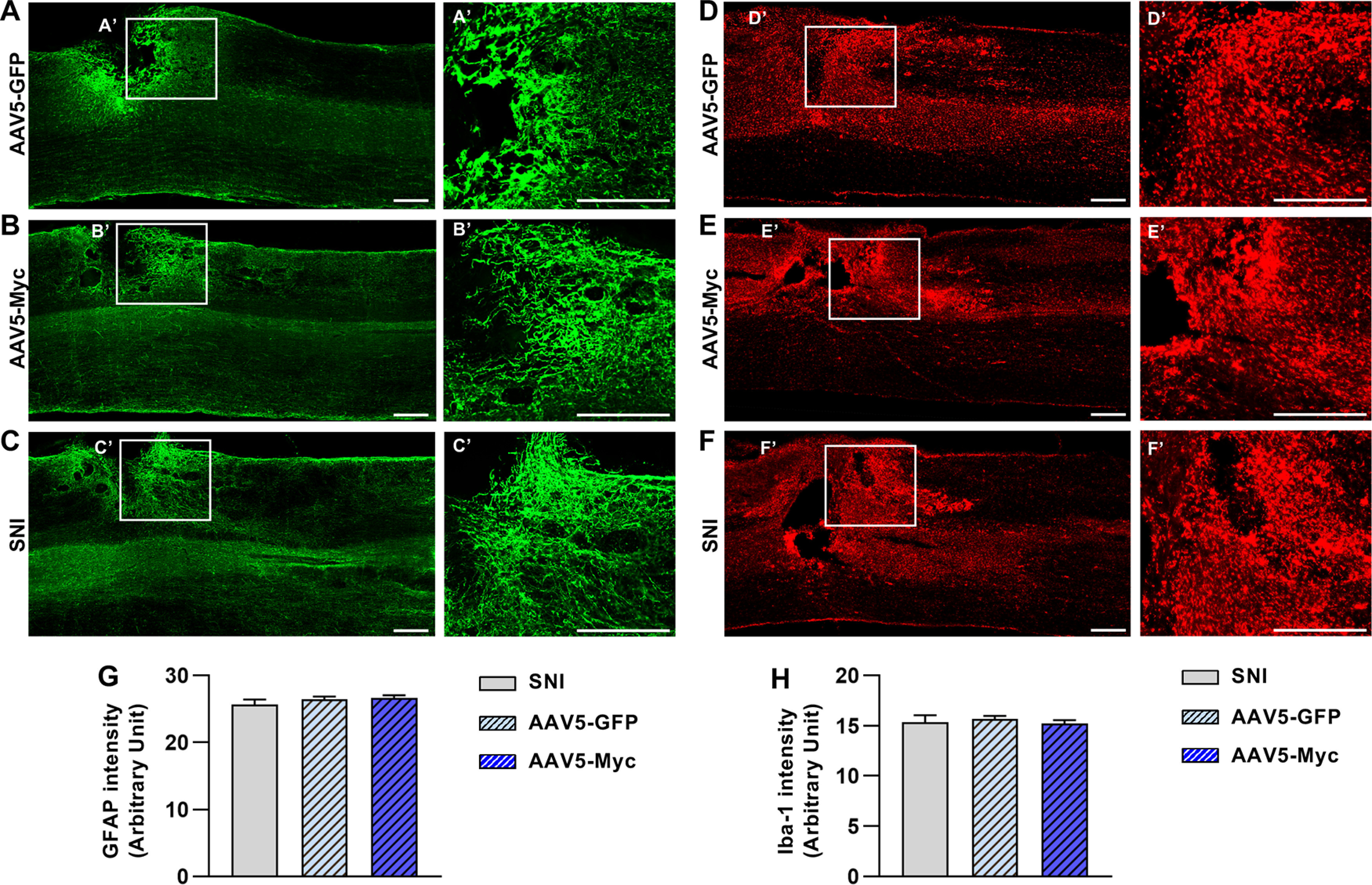
Myc overexpression in DRGs does not influence glial microenvironment at the lesioned spinal cord. A–F, Parasagittal spinal cord sections immunostained by GFAP (green; A-C) and IBA-1 (red; D-F) antibodies. Spinal cord sections were obtained from animals with AAV5-GFP (A,D), or with AAV5-Myc (B,E) injection in DRGs. C, F, Parasagittal spinal cord sections were obtained from animals that were subjected to preconditioning SNI before creating the spinal lesion. A-F, Boxed regions are magnified at the right (A'-F'). Scale bars, 500 µm. G, H, Quantification graphs of GFAP (G) and IBA-1 (H) immunoreactive intensity. N = 8, N = 9, and N = 5 animals for the AAV5-GFP, AAV5-Myc, and SNI groups, respectively.
To determine whether the effects of Myc overexpression are dependent on its transcriptional activity on the regulation of downstream RAGs, we attempted to inactivate Jun, which was persistently bound by c-MYC at both 1 and 7 d (Fig. 1C) and of which expression is robustly suppressed by Myc deletion, by delivering a JNK inhibitor, SP600125, to the DRGs where the cell bodies of the dorsal column axons are located. Three weeks after AAV5-Myc injection, an osmotic minipump containing SP600125 or vehicle control DMSO was installed just before the dorsal hemisection spinal cord injury and delivered the agents for 7 d (Fig. 8A). Inhibition of JNK resulted in the absence of regenerating axons beyond the caudal lesion border and the front-end of axons slightly retracted caudally to the lesion border (Fig. 8B,C). A more prominent observation was an increase in the formation of retraction bulbs in animals with SP600125 treatment. Axonal tips from animals in this group more frequently exhibited bulbous swelling (Fig. 8D,E). Quantification data revealed that JNK inhibition significantly decreased the mean longest distance of regenerating axons (t(4) = 5.334, p = 0.0059, unpaired t test) and that axons growing beyond the lesion border by Myc overexpression entirely disappeared by JNK inhibition (Fig. 8F,G). Furthermore, JNK inhibition markedly increased the percent retraction bulb by almost 50% (t(4) = 6.695, p = 0.0026, unpaired t test) (Fig. 8H). These data suggest that Myc-induced prevention of axonal retraction may be mediated by activation of Jun of which transcription is Myc-dependent.
Figure 8.
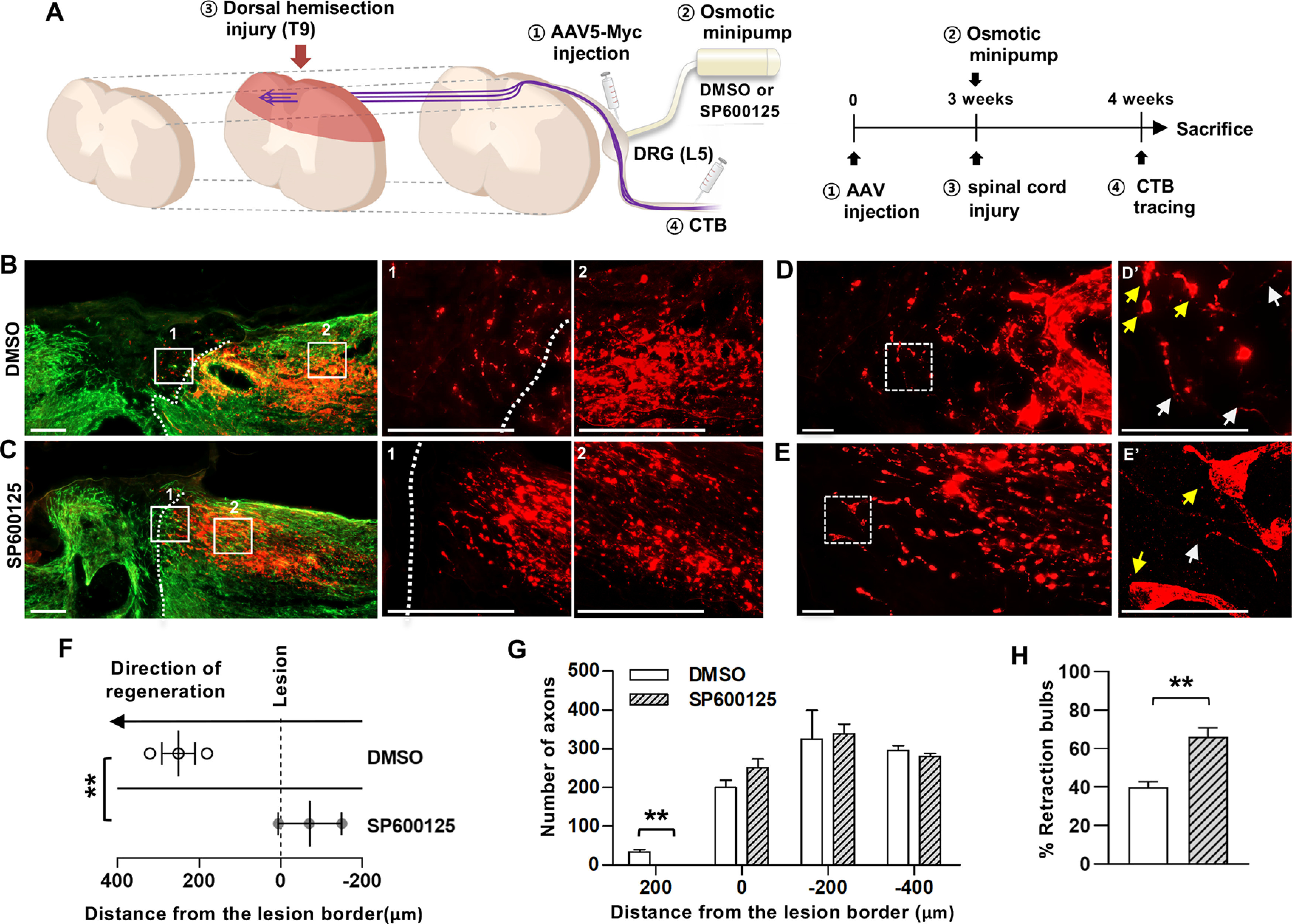
Suppression of Myc overexpression effects by JNK inhibitor. A, An experimental scheme depicting the time course of individual processes. B, C, Representative images of the parasagittal spinal cord sections from animals injected with AAV5-Myc followed by treatment of DMSO vehicle (B) or JNK inhibitor, SP600125 (C). Red represents CTB-labeled axons. Green represents GFAP-positive glial scars. Dashed lines indicate the caudal lesion borders as determined by GFAP immunostaining. B, C, Boxed regions are magnified at the right with the corresponding numbers. Scale bars: B, C, 200 µm. D, E, Morphology of the axonal tips at the axon front. D, E, Boxed regions are magnified at the right (D′-E′). Yellow arrows indicate the axonal tips counted as the retraction bulbs. White arrows indicate the axonal tips of growth-cone like morphology. Scale bars: E–G, 50 µm. F, Quantification graph comparing the longest distance of regenerating axons from the caudal lesion border. Individual circles plot the longest distance of each animal. Negative values indicate the degree of the retraction from the lesion border. G, Quantification graph comparing the number of CTB-positive axons within counting blocks located at different distances from the lesion border. The numbers in the x axis indicate the distance of the rostral border of a counting block from the lesion border. H, Quantification graph comparing the percentage of retraction bulbs. The retraction bulbs were defined as the axons with ration between the maximum tip diameter and the axon cylindrical diameter ≥4.0. **p < 0.01 between animals (one-way ANOVA followed by Tukey's post hoc analysis). N = 3 for each group.
Discussion
The main conclusion of this study is that Myc proto-oncogene plays a role as a hub TF gene to regulate a distinct subset of RAGs following preconditioning injury, contributing to the injury-induced enhancement of axon growth capacity in the DRG neurons. Several lines of experimental results support this notion. Our ChIP-PCR data demonstrated direct binding of c-MYC to the promoters of a subset of RAGs, indicating a role of direct and specific transcriptional activator for these genes. In addition, the upregulation of Myc following SNI well preceded that of the Myc-bound RAGs, such as Jun and Atf3. More importantly, deletion of Myc or Myc overexpression by AAV injection attenuated or enhanced an overlapping list of RAGs, including those directly bound by c-MYC, suggesting that the genes that are simultaneously affected by either experimental manipulation are highly likely the ones under the direct influence of Myc in the transcriptional network activated by preconditioning peripheral nerve injury (van Kesteren et al., 2011; Chandran et al., 2016). Furthermore, the in vivo effects of Myc overexpression on the axonal retraction were suppressed by JNK inhibitor, suggesting that Myc influences the cellular machinery regulating axon growth through its direct downstream RAG, June. A previous proteomics study identified c-MYC as an injury signal hub in the retinal ganglion cells and reported that overexpression of c-MYC promoted axonal regeneration after optic nerve injury (Belin et al., 2015). A more recent study reported that c-MYC regulates sensory axon regeneration via its downstream effectors, telomerase reverse transcriptase and p53 (J. J. Ma et al., 2019). However, a potential mechanism by which c-MYC regulates axon regeneration remains to be known, and the transcriptional role of the Myc gene in regulating downstream genes involved in axon regeneration was not addressed in these studies. Based on the data in the present study, we assert that Myc plays a critical role in axon regeneration via its transcriptional activity to regulate a broader spectrum of downstream effector molecules.
c-MYC is well characterized as a bHLH TF that regulates the transcription of ∼15% of all genes through the binding on the E-box sequence in the promoter regions of target genes (Dang et al., 2006; Eilers and Eisenman, 2008). It was speculated that c-MYC may function simply by amplifying gene expression as opposed to regulating the expression of specific genes (Lin et al., 2012; Nie et al., 2012). The volcano plots of the PCR array results showed that most of the RAGs tested in the array increased or decreased the level of transcription to a varying degree by overexpression (Fig. 5C) or deletion of Myc proto-oncogene in SNI condition (Fig. 3D), respectively, corroborating the role of c-MYC as a general transcriptional activator. However, our data showing prominent upregulation of several RAGs by Myc deletion or overexpression indicate that c-MYC also enhanced the transcription of specific downstream target genes. We demonstrated c-MYC binding to the promoters of several RAGs, such as Atf3, Jun, and Sprr1a. It has been reported that Atf3 is a target of the c-MYC transcription factor in the proliferation of non-neural cells in response to serum treatment (Tamura et al., 2005). It is conceivable that c-MYC also binds to the promoter of Atf3 in postmitotic neurons driving the transcription of genes required for axon growth instead of proliferation. ChIP sequencing also revealed c-MYC binding to the promoter of Jun in non-neural cells (Fernandez et al., 2003). The finding that Myc upregulation precedes that of Jun or Atf3 suggests that MYC binding to Jun or Atf3 promoter may be required for SNI-induced upregulation of these two TF RAGs. Indeed, we found that the induction of Jun and Atf3 by SNI was markedly attenuated in Mycnes-cre animals. In contrast, SNI-induced upregulation of Gap43, Npy, and Galanin, of which promoters were not occupied by MYC, was not affected by the Myc deletion. These results suggest that injury-induced transcription of a group of RAGs is Myc-dependent, while the other genes are upregulated independently of Myc proto-oncogene. Since our custom PCR array covered only 44 RAGs, it remains to be studied whether regeneration-regulating genes other than the 44 RAGs, such as Pten, Mtor, Rheb, Tsc1, Cntf, other Klf members, Hdac5, and so forth, would be Myc-dependent or Myc-independent. It is well known that many c-MYC target genes are critically implicated in metabolic regulation (Stine et al., 2015; Dejure and Eilers, 2017). It is conceivable that the regeneration-regulating genes involved in cellular metabolism, such as Pten, Mtor, Rheb, and Tsc1, may be under control of Myc transcription. Notably, Myc overexpression led to an increase in several neuropoietic cytokines accompanied by perineuronal macrophage activation. Previous studies, including our own, showed that preconditioning peripheral nerve injury induces perineuronal macrophage activation (Lu and Richardson, 1993; Kwon et al., 2013; Niemi et al., 2013). It is conceivable that Myc gene expression subsequently induces expression of chemokine or cytokine genes, which may in turn recruit macrophages around DGR neurons. Since perineuronal macrophage activation following peripheral nerve injury contributes to enhanced axon regeneration capacity (Kwon et al., 2013; Niemi et al., 2013), Myc overexpression accompanied by neuropoietic cytokines may contribute to an increase in axon growth competence in part by perineuronal macrophage activation.
The results of our neurite outgrowth assay revealed that the functional contribution of the Myc-dependent RAGs to the preconditioning effects on the competence of axonal growth was roughly half of those by the full complement of RAGs induced by SNI. Similar to the partial contribution of the Myc-dependent RAGs to the preconditioning effects, the enhancement of the axon growth competence by Myc overexpression was not as robust as that observed in the preconditioned DRG neurons. The partial functional gain was also observed in the regeneration of the spinal cord sensory axons growing from the DRG neurons with Myc overexpression. Myc overexpression resulted in a marked increase in axon numbers at the region close to the caudal lesion border and a significant decrease in the formation of retraction bulbs, indicating that the Myc overexpression counteracted the injury-induced axonal retraction processes or supported triggering initial axon growth mechanism from retracted axon tips. Axon retraction involves the disintegration of the microtubule cytoskeleton (Luo and O'Leary, 2005). Furthermore, stabilizing microtubules enables axon tips to form growth cones allowing initial growth (Erturk et al., 2007; Hellal et al., 2011; Hur et al., 2012). Therefore, it is conceivable that Myc overexpression and activation of its downstream target genes may influence the stability of the microtubules and their interaction with other cytoskeletal components. Indeed, c-MYC was found to associate with α-tubulin and microtubules (Alexandrova et al., 1995), and there is evidence that Jun and Atf3 are involved in the regulation of the microtubule stability (Yeap et al., 2010; Ahlgren et al., 2014). In contrast to the effects on the axon retraction, Myc overexpression could not support noticeable axon regeneration beyond the lesion border. This finding suggests that axonal growth across the lesion site may require activation of other RAGs that are Myc-independent. The lesion site is filled with molecules that provide inhibitory influence on growing axons. Therefore, Myc-independent RAGs may need to be mobilized to overcome the inhibitory microenvironment, allowing regenerating axons to pass the lesion core without losing growth competence.
The Myc gene was derived from the list of upstream TF genes that are expected to interact with the promoter regions of the injury-induced RAGs and whose DNA methylation is significantly changed by the preconditioning SNI. The motivation for this approach was our hypothesis that epigenetic modification may elicit an early activation of hub TF genes that in turn regulate the transcription of many downstream RAGs. It has been shown that various epigenetic mechanisms, such as histone modification and DNA methylation, can be activated within hours after injury or changes in neural activity (Guo et al., 2011; Cho et al., 2013). In this study, the preconditioning SNI elicited the DNA demethylation at the Myc exon 3 region as early as 2 h after injury. We found that the DNA demethylation was responsible for the rapid upregulation of Myc gene in response to cAMP in vitro. In addition, ChIP analysis revealed that the DNA methylation at the exon 3 region was accompanied by open chromatin histone marks. Previous studies on the DNA methylation of Myc gene in various cancers showed that the DNA sequences in the exon 3 were frequently demethylated (Ohtsuki et al., 1991; Sharrard et al., 1992; Crossen and Morrison, 1999), indicating that the demethylation in the exon 3 region is implicated in the overexpression of the Myc proto-oncogene. Therefore, our results suggest that the DNA demethylation of Myc proto-oncogene at the exon 3 region may lead to the early transcriptional activation probably via making chromatin structures open for transcriptional machinery. It is worth mentioning that our methylation assay was done using whole DRGs containing many non-neural cells. However, it is highly likely that Myc demethylation occurred in neurons because c-MYC protein expression was confined to DRG neurons and the Myc-dependent expression changes in TFs, such as c-JUN and ATF3, were observed predominantly in neurons.
Our finding that Myc regulates the transcription of only a certain group of RAGs, including TF RAGs, such as Jun and Atf3, implies a possibility that there are more upstream hub regulators that govern the expression of different groups of RAGs. Based on the current results, we speculate that, within the TF network activated by preconditioning SNI, certain hierarchical interactions may exist where a smaller number of upstream hub regulators determine the transcription of other downstream RAGs. It is conceivable that the different hub TFs may be activated by different epigenetic mechanisms that are known to be activated early after injury. Identification of more upstream hub TFs and elucidation of the epigenetic mechanism specific for individual hub TFs would advance our understanding how the preconditioning injury induces orchestrated upregulation of RAGs that governs the intrinsic capacity of axon regeneration. For therapeutic purposes, combinatorial overexpression of potential upstream hub regulators would induce sequential activation of downstream transcription factors and finally lead to the upregulation of a whole complement of RAGs. Recent studies attempted combinatorial overexpression of various transcription factors for synergistic promotion of axon growth, but the majority of the combinations failed to increase axon growth more than individual single transcription factors (Lerch et al., 2014; Fagoe et al., 2015). Dissecting out the hierarchical relationship among upstream hub TFs in the induction of RAGs should provide clues for selecting optimal TF combinations with efficient synergism to achieve more meaningful and robust axon regeneration.
Footnotes
*H.Y.S. and M.J.K. contributed equally to this work.
This work was supported by the National Research Foundation of Korea Grant NRF-2018R1A2A1A05020292 and 2019R1A5A2026045 to B.G.K., 2019R1I1A1A01059432 to M.J.K., and Ajou Research Institute for Innovation Medicine, Ajou University Medical Center Grant M-2019-C0460-00067 to D.H.H. We thank the Vector Core Facility of the University of North Carolina at Chapel Hill for rAAV viral vector production; and Professor Tae Jun Park (Department of Biochemistry and Molecular Biology, Ajou University School of Medicine, South Korea) for generous donation of full-length cDNA for human Myc gene.
H.Y. Shin's present address: Department of Pharmacology, Vivozon R&D Center, Yongin, Republic of Korea, 16914.
K. Kim's present address: Data cell, Humanscape, Yeoksam-ro 17-gil, Gangnam-gu, Seoul, Republic of Korea, 06237.
The authors declare no competing financial interests.
References
- Ahlgren H, Bas-Orth C, Freitag HE, Hellwig A, Ottersen OP, Bading H (2014) The nuclear calcium signaling target, activating transcription factor 3 (ATF3), protects against dendrotoxicity and facilitates the recovery of synaptic transmission after an excitotoxic insult. J Biol Chem 289:9970–9982. 10.1074/jbc.M113.502914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrova N, Niklinski J, Bliskovsky V, Otterson GA, Blake M, Kaye FJ, Zajac-Kaye M (1995) The N-terminal domain of c-Myc associates with alpha-tubulin and microtubules in vivo and in vitro. Mol Cell Biol 15:5188–5195. 10.1128/mcb.15.9.5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin S, Nawabi H, Wang C, Tang S, Latremoliere A, Warren P, Schorle H, Uncu C, Woolf CJ, He Z, Steen JA (2015) Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron 86:1000–1014. 10.1016/j.neuron.2015.03.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesch A, Lu P, Tsukada S, Alto LT, Roet K, Coppola G, Geschwind D, Tuszynski MH (2012) Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to camp-mediated effects. Exp Neurol 235:162–173. 10.1016/j.expneurol.2011.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla IE, Tanabe K, Strittmatter SM (2002) Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J Neurosci 22:1303–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y (2009) Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10:295–304. 10.1038/nrg2540 [DOI] [PubMed] [Google Scholar]
- Chandran V, Coppola G, Nawabi H, Omura T, Versano R, Huebner EA, Zhang A, Costigan M, Yekkirala A, Barrett L, Blesch A, Michaelevski I, Davis-Turak J, Gao F, Langfelder P, Horvath S, He Z, Benowitz L, Fainzilber M, Tuszynski M, et al. (2016) A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 89:956–970. 10.1016/j.neuron.2016.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V (2013) Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155:894–908. 10.1016/j.cell.2013.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Befort K, Karchewski L, Griffin RS, D'Urso D, Allchorne A, Sitarski J, Mannion JW, Pratt RE, Woolf CJ (2002) Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci 3:16 10.1186/1471-2202-3-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossen PE, Morrison MJ (1999) Methylation status of the 3rd exon of the c-MYC oncogene in B-cell malignancies. Leuk Res 23:251–253. 10.1016/s0145-2126(98)00167-2 [DOI] [PubMed] [Google Scholar]
- Curcio M, Bradke F (2018) Axon regeneration in the central nervous system: facing the challenges from the inside. Annu Rev Cell Dev Biol 34:495–521. 10.1146/annurev-cellbio-100617-062508 [DOI] [PubMed] [Google Scholar]
- Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F (2006) The c-Myc target gene network. Semin Cancer Biol 16:253–264. 10.1016/j.semcancer.2006.07.014 [DOI] [PubMed] [Google Scholar]
- Dejure FR, Eilers M (2017) MYC and tumor metabolism: chicken and egg. EMBO J 36:3409–3420. 10.15252/embj.201796438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami P, Saffrey P, Bruce AW, Dillon SC, Chiang K, Bonhoure N, Koch CM, Bye J, James K, Foad NS, Ellis P, Watkins NA, Ouwehand WH, Langford C, Andrews RM, Dunham I, Vetrie D (2010) Complex exon-intron marking by histone modifications is not determined solely by nucleosome distribution. PLoS One 5:e12339. 10.1371/journal.pone.0012339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M, Eisenman RN (2008) Myc's broad reach. Genes Dev 22:2755–2766. 10.1101/gad.1712408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erturk A, Hellal F, Enes J, Bradke F (2007) Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci 27:9169–9180. 10.1523/JNEUROSCI.0612-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagoe ND, Attwell CL, Kouwenhoven D, Verhaagen J, Mason MR (2015) Overexpression of ATF3 or the combination of ATF3, c-Jun, STAT3 and Smad1 promotes regeneration of the central axon branch of sensory neurons but without synergistic effects. Hum Mol Genet 24:6788–6800. 10.1093/hmg/ddv383 [DOI] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003) Genomic targets of the human c-Myc protein. Genes Dev 17:1115–1129. 10.1101/gad.1067003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Greene LA, Ziff EB (1985) Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J Biol Chem 260:14101–14110. [PubMed] [Google Scholar]
- Griffith OL, Montgomery SB, Bernier B, Chu B, Kasaian K, Aerts S, Mahony S, Sleumer MC, Bilenky M, Haeussler M, Griffith M, Gallo SM, Giardine B, Hooghe B, Van Loo P, Blanco E, Ticoll A, Lithwick S, Portales-Casamar E, Donaldson IJ, et al. (2008) ORegAnno: an open-access community-driven resource for regulatory annotation. Nucleic Acids Res 36:D107–D113. 10.1093/nar/gkm967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, Zhang K, Ming GL, Gao Y, Song H (2011) Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 14:1345–1351. 10.1038/nn.2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, de Alborán IM, Olson JM, Eisenman RN (2006) N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res 66:8655–8661. 10.1158/0008-5472.CAN-06-1621 [DOI] [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F (2011) Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331:928–931. 10.1126/science.1201148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PN. (2010) A conditioning lesion induces changes in gene expression and axonal transport that enhance regeneration by increasing the intrinsic growth state of axons. Exp Neurol 223:11–18. 10.1016/j.expneurol.2009.09.006 [DOI] [PubMed] [Google Scholar]
- Holmes FE, Mahoney S, King VR, Bacon A, Kerr NC, Pachnis V, Curtis R, Priestley JV, Wynick D (2000) Targeted disruption of the galanin gene reduces the number of sensory neurons and their regenerative capacity. Proc Natl Acad Sci USA 97:11563–11568. 10.1073/pnas.210221897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J (2008) Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci 28:9330–9341. 10.1523/JNEUROSCI.2488-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Saijilafu Zhou FQ (2012) Growing the growth cone: remodeling the cytoskeleton to promote axon regeneration. Trends Neurosci 35:164–174. 10.1016/j.tins.2011.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaki Y, Oda M, Okano M (2008) QUMA: quantification tool for methylation analysis. Nucleic Acids Res 36:W170–W175. 10.1093/nar/gkn294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MJ, Kim J, Shin H, Jeong SR, Kang YM, Choi JY, Hwang DH, Kim BG (2013) Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J Neurosci 33:15095–15108. 10.1523/JNEUROSCI.0278-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch JK, Martinez-Ondaro YR, Bixby JL, Lemmon VP (2014) cJun promotes CNS axon growth. Mol Cell Neurosci 59:97–105. 10.1016/j.mcn.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LC, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18:1427–1431. 10.1093/bioinformatics/18.11.1427 [DOI] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA (2012) Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151:56–67. 10.1016/j.cell.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Richardson PM (1993) Responses of macrophages in rat dorsal root ganglia following peripheral nerve injury. J Neurocytol 22:334–341. 10.1007/BF01195557 [DOI] [PubMed] [Google Scholar]
- Luo L, O'Leary DD (2005) Axon retraction and degeneration in development and disease. Annu Rev Neurosci 28:127–156. 10.1146/annurev.neuro.28.061604.135632 [DOI] [PubMed] [Google Scholar]
- Ma JJ, Ju X, Xu RJ, Wang WH, Luo ZP, Liu CM, Yang L, Li B, Chen JQ, Meng B, Yang HL, Zhou FQ. Saijilafu (2019) Telomerase reverse transcriptase and p53 regulate mammalian peripheral nervous system and CNS axon regeneration downstream of c-Myc. J Neurosci 39:9107–9118. 10.1523/JNEUROSCI.0419-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma TC, Willis DE (2015) What makes a RAG regeneration associated? Front Mol Neurosci 8:43. 10.3389/fnmol.2015.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar M, Cavalli V (2018) Intrinsic mechanisms of neuronal axon regeneration. Nat Rev Neurosci 19:323–337. 10.1038/s41583-018-0001-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney SA, Hosking R, Farrant S, Holmes FE, Jacoby AS, Shine J, Iismaa TP, Scott MK, Schmidt R, Wynick D (2003) The second galanin receptor GalR2 plays a key role in neurite outgrowth from adult sensory neurons. J Neurosci 23:416–421. 10.1523/JNEUROSCI.23-02-00416.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massart R, Barnea R, Dikshtein Y, Suderman M, Meir O, Hallett M, Kennedy P, Nestler EJ, Szyf M, Yadid G (2015) Role of DNA methylation in the nucleus accumbens in incubation of cocaine craving. J Neurosci 35:8042–8058. 10.1523/JNEUROSCI.3053-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, Zhao K, Levens D (2012) c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151:68–79. 10.1016/j.cell.2012.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Roldán-Hernández L, Lindborg JA, Mandell D, Zigmond RE (2013) A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J Neurosci 33:16236–16248. 10.1523/JNEUROSCI.3319-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson A, Moller K, Dahlin L, Lundborg G, Kanje M (2005) Early changes in gene expression in the dorsal root ganglia after transection of the sciatic nerve: effects of amphiregulin and PAI-1 on regeneration. Brain Res Mol Brain Res 136:65–74. 10.1016/j.molbrainres.2005.01.008 [DOI] [PubMed] [Google Scholar]
- Ohtsuki T, Nishitani K, Hatamochi A, Yawata Y, Namba M (1991) Analysis of methylation in the c-MYC gene in five human myeloma cell lines. Br J Haematol 77:172–179. 10.1111/j.1365-2141.1991.tb07973.x [DOI] [PubMed] [Google Scholar]
- Petkova TD, Seigel GM, Otteson DC (2011) A role for DNA methylation in regulation of EphA5 receptor expression in the mouse retina. Vision Res 51:260–268. 10.1016/j.visres.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttagunta R, Tedeschi A, Soria MG, Hervera A, Lindner R, Rathore KI, Gaub P, Joshi Y, Nguyen T, Schmandke A, Laskowski CJ, Boutillier AL, Bradke F, Di Giovanni S (2014) PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat Commun 5:3527. 10.1038/ncomms4527 [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Filbin MT (2002) A role for cAMP in regeneration during development and after injury. Prog Brain Res 137:381–387. 10.1016/s0079-6123(02)37029-8 [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, Lipp HP, Aguzzi A, Wagner EF, Behrens A (2004) The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43:57–67. 10.1016/j.neuron.2004.06.005 [DOI] [PubMed] [Google Scholar]
- Richardson PM, Miao T, Wu D, Zhang Y, Yeh J, Bo X (2009) Responses of the nerve cell body to axotomy. Neurosurgery 65:A74–A79. 10.1227/01.NEU.0000352378.26755.C3 [DOI] [PubMed] [Google Scholar]
- Rose NR, Klose RJ (2014) Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta 1839:1362–1372. 10.1016/j.bbagrm.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saied MH, Marzec J, Khalid S, Smith P, Down TA, Rakyan VK, Molloy G, Raghavan M, Debernardi S, Young BD (2012) Genome wide analysis of acute myeloid leukemia reveal leukemia specific methylome and subtype specific hypomethylation of repeats. PLoS One 7:e33213 10.1371/journal.pone.0033213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger JB, Verge VM, Chan KM, Webber CA (2018) The nerve conditioning lesion: a strategy to enhance nerve regeneration. Ann Neurol 83:691–702. 10.1002/ana.25209 [DOI] [PubMed] [Google Scholar]
- Sharrard RM, Royds JA, Rogers S, Shorthouse AJ (1992) Patterns of methylation of the c-myc gene in human colorectal cancer progression. Br J Cancer 65:667–672. 10.1038/bjc.1992.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HY, Kim K, Kwon MJ, Oh YJ, Kim EH, Kim HS, Hong CP, Lee JH, Lee K, Kim BG (2020) Alteration in global DNA methylation status following preconditioning injury influences axon growth competence of the sensory neurons. Exp Neurol 326:113177. 10.1016/j.expneurol.2020.113177 [DOI] [PubMed] [Google Scholar]
- Shukeir N, Pakneshan P, Chen G, Szyf M, Rabbani SA (2006) Alteration of the methylation status of tumor-promoting genes decreases prostate cancer cell invasiveness and tumorigenesis in vitro and in vivo. Cancer Res 66:9202–9210. 10.1158/0008-5472.CAN-06-1954 [DOI] [PubMed] [Google Scholar]
- Singer M, Kosti I, Pachter L, Mandel-Gutfreund Y (2015) A diverse epigenetic landscape at human exons with implication for expression. Nucleic Acids Res 43:3498–3508. 10.1093/nar/gkv153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam FJ, MacGillavry HD, Armstrong NJ, de Gunst MC, Zhang Y, van Kesteren RE, Smit AB, Verhaagen J (2007) Identification of candidate transcriptional modulators involved in successful regeneration after nerve injury. Eur J Neurosci 25:3629–3637. 10.1111/j.1460-9568.2007.05597.x [DOI] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV (2015) MYC, metabolism, and cancer. Cancer Discov 5:1024–1039. 10.1158/2159-8290.CD-15-0507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Hua B, Adachi S, Guney I, Kawauchi J, Morioka M, Tamamori-Adachi M, Tanaka Y, Nakabeppu Y, Sunamori M, Sedivy JM, Kitajima S (2005) Stress response gene ATF3 is a target of c-myc in serum-induced cell proliferation. EMBO J 24:2590–2601. 10.1038/sj.emboj.7600742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kesteren RE, Mason MR, Macgillavry HD, Smit AB, Verhaagen J (2011) A gene network perspective on axonal regeneration. Front Mol Neurosci 4:46. 10.3389/fnmol.2011.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng YL, An R, Cassin J, Joseph J, Mi R, Wang C, Zhong C, Jin SG, Pfeifer GP, Bellacosa A, Dong X, Hoke A, He Z, Song H, Ming GL (2017) An intrinsic epigenetic barrier for functional axon regeneration. Neuron 94:337–346.e336. 10.1016/j.neuron.2017.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao HS, Huang QH, Zhang FX, Bao L, Lu YJ, Guo C, Yang L, Huang WJ, Fu G, Xu SH, Cheng XP, Yan Q, Zhu ZD, Zhang X, Chen Z, Han ZG, Zhang X (2002) Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci USA 99:8360–8365. 10.1073/pnas.122231899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeap YY, Ng IH, Badrian B, Nguyen TV, Yip YY, Dhillon AS, Mutsaers SE, Silke J, Bogoyevitch MA, Ng DC (2010) c-Jun N-terminal kinase/c-Jun inhibits fibroblast proliferation by negatively regulating the levels of stathmin/oncoprotein 18. Biochem J 430:345–354. 10.1042/BJ20100425 [DOI] [PubMed] [Google Scholar]