

The clustering of TLR1/2 acts as a driver in integrating the spatial cues of ligands into cell-level activation events.
Abstract
The activation of Toll-like receptor heterodimer 1/2 (TLR1/2) by microbial components plays a critical role in host immune responses against pathogens. TLR1/2 signaling is sensitive to the chemical structure of ligands, but its dependence on the spatial distribution of ligands on microbial surfaces remains unexplored. Here, we reveal the quantitative relationship between TLR1/2-triggered immune responses and the spacing of ligand clusters by designing an artificial “phagocytic synapse” nanoarray platform to mimic the cell-microbe interface. The ligand spacing dictates the proximity of receptor clusters on the cell surface and consequently the pro-inflammatory responses of macrophages. However, cell responses reach their maximum at small ligand spacings when the receptor nanoclusters become adjacent to one another. Our study demonstrates the feasibility of using spatially patterned ligands to modulate innate immunity. It shows that the receptor clusters of TLR1/2 act as a driver in integrating the spatial cues of ligands into cell-level activation events.
INTRODUCTION
The assembly of membrane receptors and signaling molecules into nano- and micrometer-sized clusters during cell activation is a prominent phenomenon shared by a diverse set of immune cells, including T cells (1), B cells (2), and natural killer cells (3). Extensive studies revealed that the receptor clusters serve as signaling units where the molecular composition, protein interactions, and reactions are tightly regulated (4–8). In contrast, the phenomenon of receptor clustering in phagocytes, such as macrophages, is not as well understood. Some receptors, including Toll-like receptors (TLRs) (9–11), C-type lectin-like receptors (12, 13), and CD14 (14, 15), have been found to form nano- or microclusters on phagocytic cell membranes. However, the functional role of such spatial organization of receptors in innate immune regulation has only been explored in a few studies. A pioneering study on this front is the discovery of “phagocytic synapse,” which describes the large-scale clustering and segregation of Dectin-1 receptor at the interface between the macrophage cell membrane and the fungal cell wall (16). Similar phenomenon was later reported for a different receptor, Fc receptor (FcR) (17). It was proposed in both studies that the clustering of receptors and their spatial segregation from inhibitory partners provides a mechanism for sustained signaling. We reported recently that innate immune responses depend on not only the clustering of receptors but also the proximity between them. We showed that Dectin-1 and TLR2 nanoclusters must be brought to proximity of <500 nm to achieve synergistic antifungal response (18). These few studies highlight the important roles that receptor clusters play in phagocytic immune functions. However, the observations so far have been qualitative in nature and too few to establish the connection between receptor clustering and the activation of innate immune cells.
Among various groups of innate immune receptors, the heterodimer TLR1/2 plays a critical role in eliciting host defense responses against pathogens (19) and in enhancing the efficacy of antibody-based immunotherapies (20). TLR1/2 recognizes a diverse range of microbial components, particularly lipoproteins and lipopeptides, and triggers different levels of immune responses depending on the molecular structure (21–23) and the presentation form (soluble or surface bound) of ligands (24–26). It is unclear how TLR1/2 modulates the immune responses to achieve both specificity and diversity of detection. Although it has been speculated that the TLR1/2 receptor clustering might play a role (27, 28), the exact mechanisms are unknown.
On the basis of our previous study showing the importance of Dectin-1 and TLR2 proximity in immune regulation (18), we hypothesized that TLR1/2 receptor clusters act as an apparatus to differentiate not only the type of ligands but also how the ligands are spatially organized. In this study, we tested this hypothesis by developing an artificial phagocytic synapse platform. On this platform, macrophages are activated by arrays of nanodisks that are coated with TLR1/2 ligand and surrounded by FcR ligands. We tuned spacing of the ligand nanodisks while simultaneously measuring cell responses. We show that the macrophage pro-inflammatory responses, including cytokine secretion and transcription factor activation, depend monotonically on the spacing of ligand nanodisks, because the ligand spacing directly controls the proximity of activated receptor nanoclusters. The smaller the spacing, the closer the TLR1/2 nanoclusters are to one another on the cell membrane, and the more intense cell activation becomes. There is, however, an intrinsic limit. The TLR1/2 nanoclusters maintain a minimal center-to-center proximity of ≈700 nm even if ligands are densely packed. The result indicates that when the TLR1/2 clusters are adjacent, the immune responses reach the maximum. Our study establishes a quantitative dependence of cell response on the lateral arrangement of ligands. It provides direct evidence that the proximity of TLR1/2 receptor nanoclusters modulates immune responses in macrophages.
RESULTS
Fabrication of bifunctional nanoarrays
In our artificial phagocytic synapse nanoarrays, TLR1/2 binding sites were presented as arrays of nanodisks surrounded by a field of ligands for FcRs (schematic illustration in Fig. 1A). The first step in creating such bifunctional nanoarrays was to create regular arrays of gold nanodisks on glass coverslips using electron beam lithography and sequential metal vapor deposition (detailed processing flow in fig. S1). The gold nanodisks were 500 nm in diameter and 45 nm in height. Several nanoarrays were manufactured, each with a uniform center-to-center interdisk spacing. Spacings of 1.0, 1.5, 2.0, or 2.5 μm were used (Fig. 1B). A nanodisk diameter of 500 nm was chosen because our previous study showed that activated TLR2 receptors on macrophage cells form nanoclusters of 500 to 900 nm in diameter (18). Each patterned substrate used in this study contained four nanoarrays, with each of the four different nanodisk spacings. This allowed all nanoarrays to be functionalized with ligands in the same way, and therefore, cell responses on different nanoarrays could be directly compared. The second step in creating the nanoarrays was to selectively conjugate ligands for TLR1/2 and FcRs on different areas of them. Synthetic triacylated lipopeptide Pam3CSK4 (referred to as “Pam3”), which binds to TLR1/2 (29), was conjugated onto the gold nanodisks through sequential thiolation and carboxyl-amine coupling. The glass surface surrounding the Pam3-functionalized nanodisks was functionalized with immunoglobulin G (IgG), which binds to FcRs. Biotinylated IgG was tethered to a layer of polyethylene glycol (PEG) on the glass substrate through biotin-streptavidin linkage. The PEG layer acted as a cushion so that IgG was presented at a similar height as Pam3. We confirmed the selective conjugation of ligands on the nanoarrays using fluorescence microscopy. Rhodamine-labeled Pam3 was observed on the gold nanodisks and Alexa Fluor 488–labeled IgG in the surrounding areas, with negligible cross absorption (Fig. 1, C and D, and fig. S2). The conjugation density of ligands was estimated to be (5.84 ± 2.72) × 107 Pam3 molecules/μm2 on gold nanodisks and (1.00 ± 0.30) × 104 IgG/μm2 on the surrounding area (fig. S3). This observed IgG surface density is comparable to that under physiological conditions, as previous studies reported 104 to 105 IgG/μm2 on the surface of Streptococcus pyogenes after immersion in human plasma and saliva (30). We also expect that the actual conjugation density of Pam3 is lower than the estimated value because of the indirect method used for the estimation (see details in the Supplementary Materials). The topography and presence of Pam3 molecules on the gold nanodisks were further characterized using peak force infrared (PFIR) microscopy (Fig. 1, E and F). PFIR microscopy is a spectroscopic scanning probe technique that combines atomic force microscopy (AFM) with IR detection to enable simultaneous mechanical and chemical measurements of samples at high spatial resolution (31). We mapped the IR absorption on the nanoarrays at a frequency of 1660 cm−1, which corresponds to the C═O bond stretch vibrations in amide I in peptides. We confirmed that Pam3 was distributed uniformly on the gold nanodisks (Fig. 1F).
Fig. 1. Bifunctional artificial phagocytic synapse nanoarrays with tunable ligand spacing.
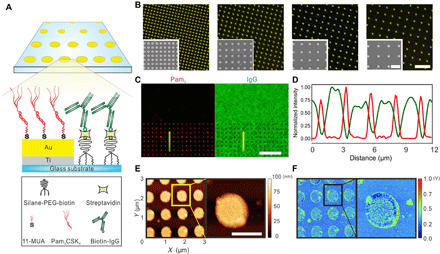
(A) Schematic illustration of the nanoarrays on which patterned Pam3 (TLR1/2 ligand) is surrounded by a field of IgG (FcRs ligand). Pam3 was conjugated onto arrays of gold nanodisks through sequential thiolation and carboxyl-amine coupling. IgG was tethered on the surrounding glass areas through streptavidin-biotin binding with PEG spacers. 11-MUA (11-mercaptoundecanoic acid). (B) Dark-field images showing gold nanodisk arrays of varied spacing (from left to right: 1.0, 1.5, 2.0, and 2.5 μm; scale bar, 5 μm). Insets: SEM images of the arrays (scale bar, 2 μm). (C) Fluorescence images showing the rhodamine-labeled Pam3 on gold nanodisks and Alexa Fluor 488–labeled IgG on the surrounding glass substrate (scale bar, 10 μm). (D) Fluorescence intensity line profiles of Pam3 and IgG along the lines marked in (C). (E) Topographic images of a representative nanoarray (scale bar, 500 nm). (F) IR absorption mapping of the bifunctional nanoarray at a frequency of 1660 cm−1.
Receptor activation and reorganization on nanoarrays
We first sought to demonstrate whether the arrays of Pam3 nanodisks would lead to the reorganization of TLR1/2 on macrophage cell membranes. To demonstrate this, we incubated RAW264.7 macrophage cells on the bifunctional nanoarrays for 30 min and examined the activation of both TLR1/2 and FcR and their spatial organization using immunofluorescence staining [Fig. 2A, (i)]. We compared these cells with several controls. Cells in one set of control experiments were activated on the nonpatterned substrates that were covalently coated with a homogeneous mixture of Pam3 and IgG [Fig. 2A, (ii)]. Cells were also incubated on bare gold nanoarrays and nonpatterned glass surfaces without ligands [Fig. 2B, (i and ii)]. Activation of TLR1/2 receptors was indicated by staining for the presence of a cytoplasmic adaptor protein called myeloid differentiation primary response 88 (MyD88). This protein is known to bind to activated TLR1/2 and to facilitate downstream signal propagation (32). Similarly, the activation of FcR was indicated by the presence and staining of phosphorylated spleen tyrosine kinase (pSyk), which facilitates FcR signaling (33). We found that on ligand-coated nonpatterned substrates, both MyD88 and pSyk appeared in nanoclusters, but these nanoclusters were distributed over the cell-substrate interface with no particular organization. In contrast, the MyD88 nanoclusters in cells on ligand-coated nanoarrays were aligned with the pattern of the Pam3 nanodisks, whereas the distribution of pSyk nanoclusters remained unchanged [2.5-μm spacing data shown in Fig. 2 (A and B) and others shown in fig. S4]. Line scan profiles of fluorescence intensity show consistent distribution of MyD88 nanoclusters on the gold nanodisks (fig. S4C), indicating uniform interaction of cells with the Pam3 nanodisks underneath.
Fig. 2. TLR1/2 reorganization and cell spreading.
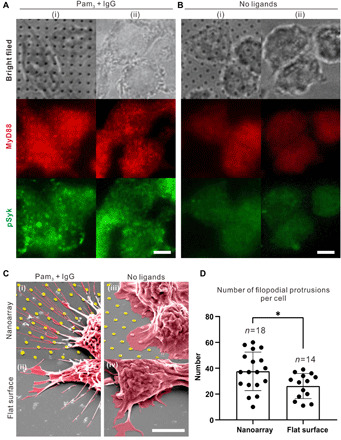
(A and B) Bright-field and fluorescence images show immunostained MyD88 and pSyk in RAW264.7 macrophage cells fixed for 30 min after spreading on nanoarrays (i) and flat glass surface (ii) with (A) and without (B) ligand functionalization (scale bar, 5 μm). (C) SEM images of cells spreading on various substrates: (i) nanoarrays with Pam3 and IgG, (ii) flat glass substrates uniformly coated with Pam3 and IgG, (iii) nanoarrays without ligands, and (iv) flat glass substrates without ligands (scale bar, 5 μm). (D) Quantification of the number of filopodial protrusions after 6 hours of cell culture on nanoarrays and flat glass substrates in the presence of Pam3 and IgG. Each data point represents result from a single cell and moved horizontally to avoid point overlay. Bar plots represent average ± SD. Statistical significance, assessed using two-tailed Mann-Whitney U test specifically for two-group comparison, is highlighted by P values as follows: *P ≤ 0.05.
In control experiments in which cells were incubated on bare nanoarrays or nonpatterned glass substrates without ligands, we observed no cluster formation for either MyD88 or pSyk (Fig. 2B). The overall immunofluorescence intensity for both proteins is also significantly lower on the substrates without ligands. The differences in immunofluorescence results mean that the recruitment of MyD88 and pSyk and their formation of nanoclusters on the bifunctional nanoarrays are not due to fluorescence enhancement effect from the gold coating on nanodisks. Instead, they indicate the successful activation of both receptors by the nanopatterned ligands. The reorganization of MyD88 indicates that the distance between activated TLR1/2 nanoclusters follows the spacing of the Pam3 nanodisks, proving the working principle of our artificial phagocytic synapse nanoarrays.
In addition to controlling receptor nanocluster organization, we found that Pam3 nanoarrays also affected cell spreading morphology, which we examined using scanning electron microscopy (SEM). Macrophage cells formed many long and thin finger-like filopodial protrusions on substrates with ligands but formed mostly ruffle-like lamellipodia on bare substrates without ligands (Fig. 2C and fig. S5). In addition, cells on ligand-coated nanoarrays formed more and longer filopodial protrusions than those on nonpatterned surfaces (Fig. 2D). Given that macrophage filopodia are known to act as tentacles for effective detection of extracellular stimuli, it is plausible that the cell filopodia must be further stretched to sense the spaced-out Pam3 nanodisks compared to a uniform substrate where Pam3 is readily available.
Transcriptional response of macrophages on nanoarrays
We next investigated how nuclear factor κB (NF-κB) signaling in macrophages depends on the spacing of the Pam3 nanodisks. NF-κB is a transcription factor that, upon activation, translocates from the cell cytoplasm to the nucleus for gene transcription regulation (34). To quantify the NF-κB translocation, we used RAW264.7 macrophage cells that stably express enhanced green fluorescent protein (EGFP)–labeled RelA, which is a subunit of the NF-κB transcription factor complex (Fig. 3A) (35). With cell nuclei also fluorescently labeled, we directly visualized the distribution of RelA in the cytoplasm and in the nucleus of the cells (Fig. 3B). We measured the fluorescence intensities of EGFP-RelA in the cytoplasm and nucleus and obtained the nuclear-to-cytoplasmic intensity ratio as a relative measure of RelA nuclear translocation (image analysis procedure shown in fig. S6). Elevated NF-κB activation is indicated by a larger RelA nuclear-to-cytoplasmic ratio. Figure 3C shows the RelA translocation results from cells on nanoarrays as a function of the spacing between Pam3 nanodisks and as a function of time after stimulation by different ligands (see statistic plots in fig. S7A).
Fig. 3. Quantification of NF-κB activation in RAW264.7 macrophages.
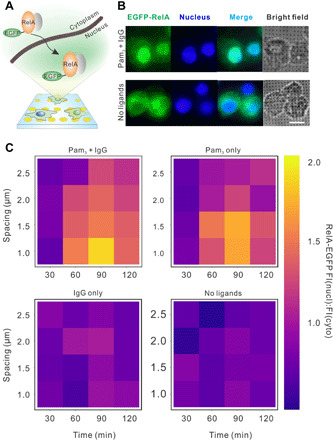
(A) Schematic illustration showing the cytoplasmic-to-nuclear translocation of NF-κB RelA upon cell activation on nanoarrays. (B) Fluorescence images of cells that stably express EGFP-RelA (green) and were labeled with nuclear stain Hoechst 33342 (blue) after 1 hour of cell culture on nanoarrays (scale bar, 10 μm). (C) Heatmaps showing NF-κB cytoplasmic-to-nuclear translocation ratio, defined as fluorescence intensity (FI) ratio of RelA in the cell nucleus to that in cytoplasm, as a function of activation time and nanoarray spacing on various substrates as indicated. Each data point represents the average value of results from a total of ≈500 cells in 10 images (fig. S6 for the statistic boxplots of the original data points).
We drew several conclusions from the results. First, NF-κB translocation is time dependent, which agrees with previous literature reports (36). Maximal NF-κB translocation on the ligand-coated nanoarrays was observed 90 min after initial stimulation. The overall kinetics exhibited no dependence on the nanodisk spacing (fig. S7B). Second, nanoarrays functionalized with both Pam3 and IgG ligands gave rise to the highest level of NF-κB signaling in cells, whereas the IgG-only nanoarrays led to minimal signaling. This indicates that TLR1/2 is mostly responsible for inducing the NF-κB signaling, but its effect is enhanced by FcR activation. This adds to a few previous reports that demonstrated the synergistic crosstalk between TLRs and FcRs (37–39). Last, NF-κB activation depends on the spacing of Pam3 nanodisks regardless of the presence of IgG. The highest level of NF-κB signaling was observed with 1-μm spacing—the smallest distance between nanodisks tested so far—and decreased with increasing nanodisk spacing. Because we have already shown that TLR1/2 nanoclusters on macrophage cell membranes are reorganized to follow the spacing of the Pam3 nanodisks, this result demonstrates the direct dependence of NF-κB signaling on the proximity between TLR1/2 nanoclusters.
Pro-inflammatory response of macrophages on nanoarrays
We detected and measured intracellular tumor necrosis factor–α (TNF-α) using immunofluorescence staining and imaging. We could not use extracellular enzyme-linked immunosorbent assays (ELISAs) that are commonly used to detect cytokines secreted into the cell culture medium, because only a tiny fraction of cells cultured on the glass substrates were on nanoarrays (<1.2 mm × 1.2 mm) compared to those on the nonpatterned area (30 mm diameter). We wished to single out this particular small fraction for analysis. To make our intracellular analysis possible, macrophage cells were incubated on nanoarrays for 6 hours in the presence of brefeldin A before fixation and staining. Brefeldin A is an inhibitor that blocks intracellular protein transport from the endoplasmic reticulum to the Golgi bodies during cell activation and thus traps secreted cytokines inside cells (40, 41). More than 1000 cells were analyzed on each nanoarray to ensure statistical significance. Representative fluorescence images are shown in Fig. 4A, and the distributions of intracellular TNF-α fluorescence intensity are indicated in Fig. 4B. It is evident that the TNF-α secretion varies depending on the presence of ligands and spacing of nanoarrays. Stimulation by both Pam3 and IgG induced the highest level of TNF-α fluorescence, whereas stimulation by Pam3 alone led to significantly higher levels of TNF-α secretion than that by IgG alone. This indicates that FcR signaling augments the TLR1/2-mediated TNF-α secretion, which is consistent with our observations of the receptor synergy in NF-κB translocation. By plotting the TNF-α fluorescence intensity (average ± SD) of individual samples as a function of array spacing (Fig. 4C), we found that TNF-α secretion decreases with increasing distance between Pam3 nanodisks upon stimulation by Pam3 or a combination of Pam3 and IgG. By comparison, cells without ligand stimulation exhibited minimal TNF-α and no dependence on the nanoarray patterns. This again agrees with the NF-κB results in suggesting that the pro-inflammatory responses of macrophages depend on the spacing between TLR1/2 nanoclusters.
Fig. 4. Quantification of TNF-α secretion in RAW264.7 macrophages.
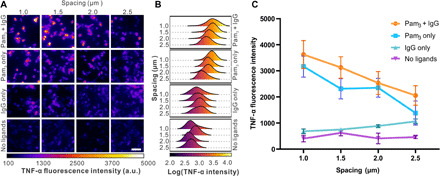
(A) Representative immunofluorescence images showing TNF-α in cells fixed after 6 hours of culture on various substrates as indicated (scale bar, 40 μm). a.u., arbitrary units. (B) Distribution plots of intracellular TNF-α intensity. For “Pam3 + IgG,” n = 334 cells (1.0 μm), 387 (1.5 μm), 352 (2.0 μm), and 468 (2.5 μm). For “Pam3 only,” n = 334 cells (1.0 μm), 463 (1.5 μm), 420 (2.0 μm), and 483 (2.5 μm). For “IgG only,” n = 351 cells (1.0 μm), 459 (1.5 μm), 427 (2.0 μm), and 468 (2.5 μm). For “No ligands,” n = 364 cells (1.0 μm), 432 (1.5 μm), 460 (2.0 μm), and 449 (2.5 μm). (C) Plots showing average intracellular TNF-α intensities as a function of nanoarray spacing with different ligand functionalization as indicated. Each data point represents average ± SD of data shown in (B) from 10 images.
However, before we can conclude that spacing between Pam3 nanodisks was the critical factor, we must consider an alternative possibility that the result might simply be the consequence of the amount of ligand in a given area. In our current design, the diameter of each nanodisk is kept constant at 500 nm. Smaller nanoarray spacing means that there are more nanodisks per unit area and therefore more Pam3 molecules per unit area for cell activation (see fig. S3 for details of nanoarray dimensions and estimated ligand amounts). We therefore needed to determine whether the increased pro-inflammatory responses observed at smaller Pam3 nanodisk spacings were really caused by the proximity of TLR1/2 receptor clusters to one another or might just be the consequence of more ligand being available in a given area for cell stimulation.
The quantitative dependence of TNF-α secretion on Pam3 spacing
To distinguish the effect of ligand spacing from that of ligand amount, we developed a second nanoarray design in which nanoarrays of different spacings presented the same amount of Pam3 and IgG per unit of surface area. This was achieved by adjusting the diameter of the gold nanodisks in proportion to their spacing such that the total area of gold surface per unit area remained constant on nanoarrays with different distance spacings between nanodisk centers (fig. S8). As with all other patterned substrates used in these experiments, each glass coverslip was fabricated to include six types of nanoarrays with spacings of 0.5, 0.75, 1.0, 1.5, 2.0, and 2.5 μm. Conjugation of Pam3 and IgG and their spatial segregation on the nanoarrays was confirmed using fluorescence microscopy as described above (Fig. 5A). The ligand conjugation density was (6.4 ± 2.2) × 107 Pam3/μm2 on the gold nanodisks and (1.2 ± 0.3) × 104 IgG/μm2 on surrounding area. We measured the TNF-α secretion of cells cultured on this new nanoarray design as a function of Pam3 spacing. As shown in Fig. 5B, within the spacing range of 0.75 to 2.5 μm, the intracellular TNF-α level increased gradually with decreasing Pam3 nanodisk spacing. Unexpectedly, the TNF-α response reached a plateau at spacings of 0.75 μm and less.
Fig. 5. Direct effects of spatial arrangement of ligands on TNF-α secretion.
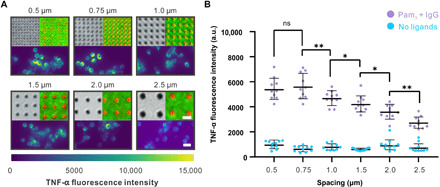
(A) Bright-field images (top left) showing the second design of nanoarrays in which the diameter of Pam3-coated nanodisks changes in proportion to spacing. Fluorescence images (top right) show the conjugation of Pam3 (red) and IgG (green) on the nanoarrays (scale bar, 1 μm). Immunofluorescence images (bottom) show the intracellular TNF-α in cells fixed after 6 hours of culture on the corresponding nanoarrays (scale bar, 20 μm). (B) Quantification of intracellular TNF-α intensity as a function of Pam3 spacing. Each point represents a mean intensity value from one image. Each data set at a given spacing represents average ± SD obtained from a total of 10 images. For “Pam3 + IgG,” n = 257 cells (0.5 μm), 184 (0.75 μm), 242 (1.0 μm), 281 (1.5 μm), 357 (2.0 μm), and 288 (2.5 μm). For “No ligands,” n = 257 cells (0.5 μm), 184 (0.75 μm), 242 (1.0 μm), 281 (1.5 μm), 357 (2.0 μm), and 288 (2.5 μm). Statistical significance, assessed using Kruskal-Wallis test with Dunn’s test as a post hoc test for multiple-group comparisons, is highlighted by P values as follows: ns P > 0.05, *P ≤ 0.05, and **P ≤ 0.01.
To understand why the macrophage cytokine response depends on the Pam3 nanodisk patterns at larger spacings but not at smaller spacings, we directly investigated the spatial distribution of TLR1/2 nanoclusters in cells cultured on the newly designed nanoarrays. Earlier, we showed qualitatively that activated TLR1/2 nanoclusters are reorganized by the Pam3 nanoarrays at spacing of 1.0 μm and larger (fig. S4A); here, we actually quantified the distance between activated receptor clusters (Fig. 6A). We used a single-particle localization algorithm (42) to track individual activated TLR1/2 nanoclusters, as indicated using MyD88 immunostaining. We measured the nearest neighbor distances between the activated nanoclusters using the R package “spatstat” (43, 44). Kernel density distributions of the MyD88 clusters within representative areas are shown in heatmaps in Fig. 6B. As expected, MyD88 on the wide-spaced arrays (1.0, 1.5, 2.0, and 2.5 μm) formed well-ordered and distinguishable foci that colocalize with the Pam3 patterns. However, on nanoarrays with spacings of 0.75 and 0.50 μm, MyD88 nanoclusters appeared randomly distributed at the cell-substrate interface, much like the MyD88 distribution in cells on nonpatterned areas. Such differences across the nanoarrays are demonstrated by the left-shifted and broadening distribution of the nearest neighbor distances of individual MyD88 cluster pairs (Fig. 6C). By plotting the mean value of nearest neighbor distances (average ± SD) as a function of nanoarray spacing, we found that MyD88 nanocluster proximity follows nanoarray spacing at >0.75 μm but reaches a plateau at array spacings of 0.75 μm and closer (Fig. 6D). The nearest neighbor distance of MyD88 nanoclusters is 673 ± 275 nm at 0.75-μm spacing, 682 ± 244 nm at 0.50-μm spacing, and, for comparison, 675 ± 249 nm on nonpatterned bifunctional areas. It is evident that a distance of ≈700 nm is the intrinsic minimal proximity of the TLR1/2 clusters. Given that the TLR2 nanoclusters have diameters within the range of 500 to 900 nm (18), a center-to-center proximity of ≈700 nm implies that the receptor clusters are likely adjacent to one another but not overlapping.
Fig. 6. Quantification of proximity of TLR1/2 clusters and its dependence on Pam3 spacing.
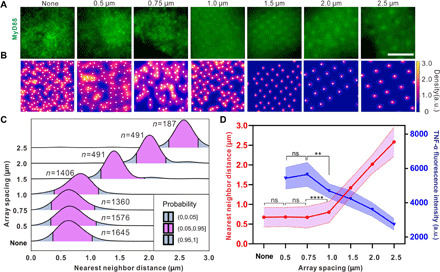
(A) Representative fluorescence images showing nanoclusters of immunostained MyD88 at the cell-substrate interface after 30 min of cell culture on the various substrates as indicated (“None” indicates nonpatterned glass surfaces with Pam3 and IgG; scale bar, 5 μm). (B) Heatmaps showing the Kernel density distribution of the same MyD88 clusters shown in (A). White dots indicate centroid position of individual nanoclusters. (C) Distribution plots of nearest neighbor distances of individual MyD88 cluster pairs obtained from 10 cells on each nanoarray. The number of cluster pairs analyzed for results of nearest neighbor distance: n = 1645 nanocluster pairs (none), 1578 (0.75 μm), 1360 (1.0 μm), 1406 (1.5 μm), 491 (1.5 μm), 491 (2.0 μm), and 187 (2.5 μm). (D) Plots showing average nearest neighbor distance of MyD88 nanoclusters (red) and average intracellular TNF-α levels (blue) in cells as a function of nanoarray spacing. Data are presented as average ± SD. Each data point of TNF-α intensity result is from 10 images containing n cells: n = 257 cells (0.5 μm), 184 (0.75 μm), 242 (1.0 μm), 281 (1.5 μm), 357 (2.0 μm), and 288 (2.5 μm). Statistical significance, assessed using Kruskal-Wallis test with Dunn’s test as a post hoc test for multiple-group comparisons, is highlighted by P values as follows: ns P > 0.05, **P ≤ 0.01, and ****P ≤ 0.0001.
We plotted the TNF-α results together with the MyD88 nanocluster proximity in Fig. 6D. One can see that the secretion of intracellular TNF-α increases as the distance between activated TLR1/2 nanoclusters decreases, and it reaches a plateau at the intrinsic proximity limit. These results demonstrate that the spatial arrangement of Pam3 on the surface is an independent factor that regulates the macrophage cell responses. When Pam3 is presented as large clusters but segregated at a distance spacing larger than the intrinsic receptor cluster proximity, TLR1/2 only becomes activated where ligand clusters are. The spatial organization of activated TLR1/2 clusters follows the pattern of the ligands, and as a result, cell responses are modulated by the lateral organization of ligand clusters. However, activated TLR1/2 nanoclusters cannot approach any closer to one another once they are immediately adjacent. This sets an intrinsic limit on the relationship between proximity and activation even in the presence of densely packed ligands.
With the modified nanoarrays, we decoupled the effects of ligand amount and ligand spacing but inevitably changed the Pam3 nanodisk size, which might also affect the cell activation. To estimate this effect, we measured the intensity of MyD88 nanoclusters on individual nanodisks as a function of nanodisk diameter (fig. S9). We found that the MyD88 intensity exhibited little changes on 375-, 500-, and 625-nm nanodisks (corresponding to 1.5-, 2.0-, and 2.5-μm spacing, respectively) and slightly lower on 250-nm nanodisks (1-μm spacing). Because the intensity of MyD88 is a direct indication of receptor activation, this means that if only ligand cluster size is considered, we should expect lower NF-κB response on 250-nm nanodisks (1-μm spacing) and similarly higher responses on other patterns. This is opposite of what we observed. Therefore, we conclude that the observed NF-κB changes on the different nanoarrays are caused by the effect of ligand spacing.
DISCUSSION
In this study, we identified the role of the proximity of TLR1/2 nanoclusters in regulating macrophage activation. By creating an artificial phagocytic synapse nanoarray platform that allows the precise control of the spacing and surface density of ligands, we established the quantitative relationship between the inflammatory responses of macrophage cells and the spacing of micropatterned ligands. We found that the TLR1/2 receptor clusters act as the driver for integrating the spatial cues of ligands into the cell-level activation. The spacing between Pam3 nanodisks dictates the spacing between TLR1/2 clusters in the cell membrane and consequently controls the cell activation. However, the intrinsic size of TLR1/2 nanoclusters also sets the upper limit of the macrophage activation. Because the TLR1/2 nanoclusters cannot approach any closer to one another once they are immediately adjacent, they maintain a minimal center-to-center proximity of ≈700 nm even if ligands are densely packed. As a result, the cell activation reaches its maximum.
The quantitative nature of our study was enabled by the phagocytic synapse nanoarray platform. The strategy of using spatially arranged ligands, in the form of micropatterned substrates or DNA nanostructures, has been used to investigate the roles of receptor clustering in cancer cell functions (45–47) and activation of immune cells, such as B cells (48) and T cells (49–52). However, our study demonstrates the use of this approach to dissect the role of TLR1/2 clusters in activation of innate immune cells. Previous studies have shown that activated TLR1/2 resides in nanodomains or lipid rafts in plasma membranes (53, 54). The speculation has been that the receptor clusters may each function as a signaling unit where molecules are brought in to interact or are excluded. In contrast, our results here demonstrate that the spatial organization of those receptor clusters in relation to one another collectively, on distance scales much larger than the range of intermolecular interactions, also plays a distinct role in regulating immune signaling.
Our findings provide new insights into understanding how innate immune receptors, such as TLR1/2, can fine-tune the level of cell activation based on the chemical and physical properties of their ligands. TLR1/2 is known to recognize a diverse range of microbial components and elicit appropriate immune responses accordingly (21–23). Studies have shown that the strength of TLR1/2 activation depends on molecular structure and the presentation form (soluble or surface-bound) of ligands (24–26). In this study, we determined that the lateral spacing of ligand clusters is an additional parameter that affects the TLR1/2 activation. Macrophage cells can alter their levels of immune activation in response to the spatial organization of TLR1/2 ligands fixed on a surface, even when other stimulation conditions, such as the total number of ligands, are the same. This finding has direct implication on understanding host cell–pathogen interactions. Many microbes have been shown to display heterogeneous distribution of cell wall components (55–57), and some microbes even alter the arrangement of wall components depending on the extracellular conditions (58, 59). In particular, we have shown recently that lipopeptides, which are ligands for TLR1/2, and polysaccharides are arranged heterogeneously on the nanoscale on the extracted yeast cell wall (60). On the basis of our findings here, it is reasonable to speculate that invading pathogens can possibly use the spatial organization of their cell wall components to modulate host cell responses.
The quantitative relationship between the spatial arrangement of Pam3 nanodisks and macrophage immune responses we found in this study indicates that it is possible to modulate immune responses by engineering the spatial organization of cell receptors. Recent studies have shown potential applications of Pam3 (61) and other TLR1/2 agonists (62–64) as adjuvants for tumor immunotherapy. It was suggested that coactivation of TLR1/2 together with FcRs may provide the strong pro-inflammatory responses needed to reverse the tumor immunosuppressive environment in antibody-based cancer immunotherapy (20, 65, 66). However, unfavorable consequences occur when coactivation is excessive (66, 67). Our results suggest that the spatial organization of stimulatory motifs is a new parameter that could be used to properly fine-tune the activation of innate immune cells.
MATERIALS AND METHODS
Fabrication of nanoarray substrates
Round glass coverslips (30 mm in diameter, no. 1.5) were cleaned in fresh piranha solution (3:1 H2SO4:H2O2) for 2 hours and dried with N2 stream. Gold nanodisk arrays were fabricated on the glass coverslips using electron beam lithography in a procedure schematically shown in fig. S1. Briefly, poly(methyl methacrylate) (PMMA) [molecular weight 495,000, 4% (w/v) in chlorobenzene; Kayaku Advanced Materials Inc.] was first spin-coated onto a glass substrate at 4000 rpm for 45 s with 1000 rpm acceleration. After baking the coated glass coverslip at 180°C on a hot plate for 2 min, it was further coated with a 6-nm-thick gold film as a conductive layer on top of the PMMA using an thermal evaporator (Boc Edwards Auto 306/FL400). After electron beam exposure for patterning, the glass coverslip was rinsed in 40 ml of KI/I2 solution (0.6 mM/0.4 mM) for 10 s before development in the mixed solvent of methyl isobutyl ketone and isopropyl alcohol (IPA) (1:3, v/v) for 90 s and then the pure IPA for 20 s. After lithography, the glass substrate was coated sequentially with a 15-nm Ti adhesion layer and a 30-nm Au layer. The metal-coated glass substrate was lifted off in acetone at 40°C for 2 hours to generate the nanodisk arrays.
Functionalization of Pam3 and IgG on nanoarrays
The fabricated nanoarray substrate was cleaned in piranha solution for 3 min, rinsed with deoinized water, and then dried under a stream of N2. Because the gold nanoarrays were dissolved in freshly made piranha solution, the piranha solution was kept at room temperature for 1.5 hours before use. For thiolation reaction, the nanoarray substrate was further cleaned in an oxygen plasma cleaner (15 W) for 5 min and then immediately immersed in a freshly prepared 5 mM 11-mercaptoundecanoic acid (Sigma-Aldrich) solution in anhydrous ethanol for approximately 12 to 18 hours. For the subsequent silanization, the nanoarray substrate was rinsed in ethanol, dried with N2 gas, and then incubated in a 95% ethanol solution containing 0.23 mM silane-PEG3400-biotin (Laysan Bio) and 0.2% (v/v) acetic acid (Sigma-Aldrich) for 4 hours at room temperature. The nanoarray substrate was rinsed in ethanol, dried under a stream of N2, and then kept under vacuum for 30 min to remove residual ethanol. To conjugate the Pam3 peptide onto the gold nanoarrays, the substrate was activated with 5 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 50 mM N-hydroxysuccinimide (NHS) (Thermo Fisher Scientific) in MES buffer (50 mM, pH 6.0) for 20 min and subsequently incubated with Pam3 (200 ng/ml; rhodamine-labeled; excitation/emission, 549/578 nm; InvivoGen) solution in phosphate-buffered saline (PBS) (1×, pH 7.4) for 2 hours. After rinsing in PBS, the nanoarray substrate was incubated in a freshly made PBS solution containing streptavidin (10 μg/ml) and bovine serum albumin (BSA; 1 mg/ml) (Sigma-Aldrich) for 30 min. After rinsing away unbound streptavidin with PBS buffer, the nanoarray substrate was incubated in a PBS solution containing biotinylated IgG (2 μg/ml; Alexa Fluor 488 labeled) and BSA (1 mg/ml) for 30 min. The functionalized substrate was extensively rinsed in PBS and used immediately for cell experiments.
Functionalization of Pam3 and IgG on nonpatterned flat glass substrate
Round glass coverslips (30 mm in diameter, no. 1.5) were cleaned in fresh piranha solution for 2 hours and dried with a stream of N2. For amination, cleaned glass coverslips were treated in an oxygen plasma cleaner (15 W) for 5 min and then immediately immersed in a freshly prepared 95% (v/v) ethanol solution containing 2% (v/v) (3-aminopropyl) triethoxysilane (Sigma-Aldrich) and 0.2% (v/v) acetic acid for approximately 2 hours at room temperature. The glass coverslips were subsequently washed with 95% (v/v) ethanol to remove excess silane compounds and kept at 110°C in an oven to remove residual water/ethanol and to enhance the formation of siloxane bonds on the glass surface. To conjugate Pam3 and IgG onto the glass surface, aminated glass coverslips were activated with 5% (v/v) glutaraldehyde (Santa Cruz Biotechnology) in 50 mM MES buffer for 2 hours and subsequently incubated with a mixture of Pam3 (200 ng/ml) and IgG solution (2 μg/ml) in PBS for 2 hours. After rinsing in PBS, the functionalized glass coverslips were used immediately for cell experiments.
PFIR microscopy
Nanoscale IR images were acquired with a homebuilt PFIR microscope as described previously (31). The setup includes an AFM component (Multimode 8 with NanoScope V controller, Bruker Nano), a quantum cascade laser (MIRcat, Daylight Solutions), and a multichannel data acquisition device (PXI-5122, National Instruments). Gold-coated AFM tips (HQ: NSC15Au, MikroMasch) were used in measurements. A parabolic mirror with a numerical aperture (NA) of 0.25 was used to focus IR beam onto the AFM tip apex. Customized LabVIEW programs were used to obtain and output IR absorption signals in real time during AFM scanning. In PFIR measurement, the peak force tapping amplitude of 150 nm and force set point of 3 to 5 nN were used. Peak force tapping frequencies of 2 and 4 kHz were used for the large-area and small-area scanning, respectively. Intensity signals from PFIR were results from local photothermal expansions, which are proportional to sample’s local IR absorptions at a certain IR frequency.
Quantification of ligand amount on nanoarray substrates
A calibration curve of rhodamine-Pam3 fluorescence intensity (excitation/emission, 549/578 nm) as a function of concentration was first obtained (fig. S3). The amount of Pam3 functionalized on the gold nanodisk arrays was obtained as the decrease in Pam3 amount in the solution before and after conjugation reaction. Briefly, the fluorescence intensity of the rhodamine-labeled Pam3 in the solution before conjugation and in the supernatant after conjugation was measured. The total amount of Pam3 in the two solutions was obtained from the calibration curve, and their difference was obtained as the amount of Pam3 conjugated on the nanoarray substrate. To estimate the amount of Pam3 that nonspecifically adsorb to the substrate, the same measurement was done but without the EDC/NHS activation step. The final amount of Pam3 covalently binding to the gold nanodisk arrays was calculated by subtracting the nonspecific binding amount from the total amount of Pam3 on the substrate. The same method was applied to estimate the amount of IgG (Alexa Fluor 488 labeled) conjugated on the areas surrounding the nanoarrays on the substrates.
Cell culture
RAW264.7 macrophage cell lines were purchased from the American Type Culture Collection. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (100 mg/ml), and 0.2 mM l-glutamine (Thermo Fisher Scientific). RAW264.7 macrophage cell line stably expressing EGFP-RelA was a gift from I. Fraser at the National Institutes of Health (NIH). The cells were cultured in the aforementioned DMEM without penicillin and streptomycin. All cell lines were kept in an incubator at 37°C with 5% CO2 and 95% relative humidity.
SEM of cells
RAW264.7 macrophages were seeded on glass substrates at a density of ~105 cells/ml. After incubation for 6 hours at 37°C, cells were washed with 0.1 M sodium phosphate buffer [0.07 M Na2HPO4 and 0.03 M NaH2PO4 (pH 7.2)] before fixing in Karnovsky’s fixative [containing 2% (w/v) paraformaldehyde and 2.5% (v/v) glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.2)] at 4°C for 12 to 18 hours. After being washed three times in 0.1 M phosphate buffer (pH 7.2) for 10 min each time, the cells were postfixed with 1% (v/v) osmium tetroxide (aqueous solution, Electron Microscopy Sciences) in the 0.1 M sodium phosphate buffer (pH 7.2) at room temperature for 1 hour. Following rinsing in Milli-Q water three times for 10 min each, the cells were dehydrated using a series of ethanol solutions with increasing volume fraction (35, 50, 75, and 95 volume %) for 15 min per rinse and washed in pure ethanol three times for 20 min each. The dehydrated sample was immersed in 100% hexamethyldisilazane (HMDS; Electron Microscopy Sciences) twice for 10 min each. Afterward, the HMDS was decanted and the sample was kept in a desiccator to air-dry at room temperature overnight. The dried sample was then mounted onto an SEM sample stub, sputter-coated with 4-nm Au/Pd alloy, and imaged with an FEI Quanta 600 SEM.
Immunofluorescence staining
RAW264.7 macrophages were seeded on the nanoarray substrates at a density of ~105 cells/ml. For immunostaining of MyD88 and pSyk, cells after 90-min incubation (37°C, 5% CO2) were washed with PBS, fixed for 10 min on ice with 4% (w/v) paraformaldehyde (PFA; Sigma-Aldrich) in PBS, and then rinsed with PBS three times for 5 min each. Cells were permeabilized with cold methanol for 10 min at −20°C, rinsed in PBS three times for 5 min each, and then passivated in blocking buffer [containing 1× PBS, 0.1% (v/v) Tween 20, 1% (w/v) BSA, and glycine (22.52 mg/ml)] for 1 hour at room temperature. Cells were then incubated with primary antibody anti-MyD88 goat IgG (2.5 μg/ml; R&D Systems Inc.) in the presence of 1% (w/v) BSA for 1 hour at room temperature, washed three times in the washing buffer (containing 1× PBS, 0.1% Tween 20, and 1% BSA), and incubated with secondary antibody rat anti-goat IgG Alexa Fluor 647 (10 μg/ml; Invitrogen) in the presence of 1% BSA for 1 hour at room temperature. In samples with also immunostained pSyk, cells after rinsing were incubated with antibody rabbit monoclonal antibody Alexa Fluor 488 (Cell Signaling Technology Inc.) for 1 hour at room temperature. The cell nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI; 0.1 μg/ml) in 1× PBS for 10 min at room temperature.
For immunostaining TNF-α, cells were seeded at a density of ~104 cells/ml and incubated for 6 hours at 37°C. To enhance the detection of TNF-α, the intracellular protein transport inhibitor brefeldin A (1000×, eBioscience) was added to the cell culture medium after 2 hours of incubation. Cells were fixed and permeabilized in the same procedure as described above, incubated with primary antibody goat anti–TNF-α (eBioscience) in the presence of 1% BSA for 1 hour at room temperature, rinsed three times in the washing buffer, and incubated with secondary antibody donkey anti-goat IgG Alexa Fluor 568 (10 μg/ml; Invitrogen) in the presence of 1% BSA for 1 hour at room temperature. Nucleus was stained with DAPI (0.1 μg/ml) in PBS for 10 min at room temperature.
Microscopy
All fluorescence imaging was performed using a Nikon Eclipse-Ti inverted microscope equipped with 1.49 NA ×100 TIRF (total internal reflection fluorescence) and 0.95 NA ×40 air objectives and an Andor iXon3 EMCCD (electron-multiplying charge-coupled device) camera. The dark-field images were acquired using a Nikon NiE upright microscope with a Hamamatsu Orca-Flash 2.8 sCMOS (scientific complementary metal oxide semiconductor) camera.
Quantification of NF-κB RelA nuclear translocation
RAW264.7 macrophages stably expressing EGFP-RelA were seeded on substrates (with or without ligands) and kept in the incubator for a varied period of time as indicated. After incubation, cells were fixed for 10 min on ice with 4% (w/v) PFA in PBS and stained with Hoechst 33342 (0.1 μg/ml; Thermo Fisher Scientific) in PBS for 10 min. Fluorescence images were captured at both DAPI and GFP channels. To quantify the NF-κB RelA nuclear translocation, the fluorescence intensities of EGFP-RelA in cell nucleus and cytoplasm were measured. The nuclear-to-cytoplasmic intensity ratio of EGFP-RelA was measured following a previously reported procedure (68). Briefly, original images (fig. S5A) were converted to binary images using automatic local thresholding (fig. S5). The nucleus mask was used as the region of interest (ROI) of the cell nucleus, while the cytoplasmic ROI was created by subtracting the nucleus mask that was obtained from the cell mask using the image calculator (fig. S5C). Each of the ROI masks was applied to the original EGFP-RelA images, resulting in separated nuclear and cytoplasmic staining images (fig. S5D). Fluorescence intensities per pixel at both channels were then measured using ImageJ. The nuclear-to-cytoplasmic fluorescence intensity ratio of EGFP-RelA was calculated to quantify NF-κB RelA nuclear translocation.
Statistical analysis
Statistic figures were plotted using both Prism 8 Software (GraphPad, USA) and R package “ggplot2” (69, 70). Data were shown as average ± SD. Mann-Whitney U test was performed for two-group comparisons. Kruskal-Wallis test with Dunn’s test as a post hoc test was performed for multiple-group comparisons. Statistical significance is indicated as follows: not significant (ns): P > 0.05; significant: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
Supplementary Material
Acknowledgments
We thank S. Zhang at the Department of Physics and Y. Yi and J. Chen at the Nanoscale Characterization Facility at Indiana University for assistance with nanofabrication, J. Powers at the IUB Light Microscopy Imaging Center for assistance with instrument use, and I. Fraser at the NIH for providing the GFP-RelA–expressing RAW264.7 macrophage cells. Funding: Research reported in this publication was supported by the National Institute of General Medical Sciences of the NIH under award number R35GM124918 to Y.Y. The PFIR work was supported by the NSF under award number CHE 1847765 and the Beckman Young Investigator Award from the Arnold and Mabel Beckman Foundation to X.G.X. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Author contributions: M.L. and Y.Y. designed research. M.L. performed research. H.W. and X.G.X. performed the PFIR measurements. W.L. provided assistance in substrate functionalization. M.L. analyzed data. M.L. and Y.Y. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabc8482/DC1
REFERENCES AND NOTES
- 1.Grakoui A., Bromley S. K., Sumen C., Davis M. M., Shaw A. S., Allen P. M., Dustin M. L., The immunological synapse: A molecular machine controlling T cell activation. Science 285, 221–227 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Batista F. D., Iber D., Neuberger M. S., B cells acquire antigen from target cells after synapse formation. Nature 411, 489–494 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Davis D. M., Chiu I., Fassett M., Cohen G. B., Mandelboim O., Strominger J. L., The human natural killer cell immune synapse. Proc. Natl. Acad. Sci. U.S.A. 96, 15062–15067 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dustin M. L., Groves J. T., Receptor signaling clusters in the immune synapse. Annu. Rev. Biophys. 41, 543–556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mossman K. D., Campi G., Groves J. T., Dustin M. L., Altered TCR signaling from geometrically repatterned immunological synapses. Science 310, 1191–1193 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Doh J., Irvine D. J., Immunological synapse arrays: Patterned protein surfaces that modulate immunological synapse structure formation in T cells. Proc. Natl. Acad. Sci. U.S.A. 103, 5700–5705 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senaratne W., Sengupta P., Jakubek V., Holowka D., Ober C. K., Baird B., Functionalized surface arrays for spatial targeting of immune cell signaling. J. Am. Chem. Soc. 128, 5594–5595 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Veatch S. L., Chiang E. N., Sengupta P., Holowka D. A., Baird B. A., Quantitative nanoscale analysis of IgE-FcεRI clustering and coupling to early signaling proteins. J. Phys. Chem. B 116, 6923–6935 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Triantafilou M., Triantafilou K., Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. 23, 301–304 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Neumann J., Ziegler K., Gelléri M., Fröhlich-Nowoisky J., Liu F., Bellinghausen I., Schuppan D., Birk U., Pöschl U., Cremer C., Lucas K., Nanoscale distribution of TLR4 on primary human macrophages stimulated with LPS and ATI. Nanoscale 11, 9769–9779 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Aaron J. S., Carson B. D., Timlin J. A., Characterization of differential Toll-like receptor responses below the optical diffraction limit. Small 8, 3041–3049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plato A., Willment J. A., Brown G. D., C-type lectin-like receptors of the dectin-1 cluster: Ligands and signaling pathways. Int. Rev. Immunol. 32, 134–156 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huysamen C., Brown G. D., The fungal pattern recognition receptor, Dectin-1, and the associated cluster of C-type lectin-like receptors. FEMS Microbiol. Lett. 290, 121–128 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitz G., Orso E., CD14 signalling in lipid rafts: New ligands and co-receptors. Curr. Opin. Lipidol. 13, 513–521 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Pfeiffer A., Böttcher A., Orsó E., Kapinsky M., Nagy P., Bodnár A., Spreitzer I., Liebisch G., Drobnik W., Gempel K., Horn M., Holmer S., Hartung T., Multhoff G., Schütz G., Schindler H., Ulmer A. J., Heine H., Stelter F., Schütt C., Rothe G., Szöllôsi J., Damjanovich S., Schmitz G., Lipopolysaccharide and ceramide docking to CD14 provokes ligand-specific receptor clustering in rafts. Eur. J. Immunol. 31, 3153–3164 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Goodridge H. S., Reyes C. N., Becker C. A., Katsumoto T. R., Ma J., Wolf A. J., Bose N., Chan A. S. H., Magee A. S., Danielson M. E., Weiss A., Vasilakos J. P., Underhill D. M., Activation of the innate immune receptor Dectin-1 upon formation of a ’phagocytic synapse’. Nature 472, 471–475 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopes F. B., Bálint Š., Valvo S., Felce J. H., Hessel E. M., Dustin M. L., Davis D. M., Membrane nanoclusters of FcγRI segregate from inhibitory SIRPα upon activation of human macrophages. J. Cell Biol. 216, 1123–1141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W., Yan J., Yu Y., Geometrical reorganization of Dectin-1 and TLR2 on single phagosomes alters their synergistic immune signaling. Proc. Natl. Acad. Sci. U.S.A. 116, 25106–25114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeuchi O., Sato S., Horiuchi T., Hoshino K., Takeda K., Dong Z., Modlin R. L., Akira S., Cutting edge: Role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169, 10–14 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Sharma N., Vacher J., Allison J. P., TLR1/2 ligand enhances antitumor efficacy of CTLA-4 blockade by increasing intratumoral Treg depletion. Proc. Natl. Acad. Sci. U.S.A. 116, 10453–10462 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang J. Y., Nan X., Jin M. S., Youn S. J., Ryu Y. H., Mah S., Han S. H., Lee H., Paik S. G., Lee J. O., Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31, 873–884 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Mogensen T. H., Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tursi S. A., Lee E. Y., Medeiros N. J., Lee M. H., Nicastro L. K., Buttaro B., Gallucci S., Wilson R. P., Wong G. C. L., Tükel Ç., Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLOS Pathog. 13, e1006315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandt K. J., Fickentscher C., Kruithof E. K., de Moerloose P., TLR2 ligands induce NF-kappaB activation from endosomal compartments of human monocytes. PLOS ONE 8, e8074301–e8074311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Bergenhenegouwen J., Plantinga T. S., Joosten L. A., Netea M. G., Folkerts G., Kraneveld A. D., Garssen J., Vos A. P., TLR2 & Co: A critical analysis of the complex interactions between TLR2 and coreceptors. J. Leukoc. Biol. 94, 885–902 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Jin M. S., Kim S. E., Heo J. Y., Lee M. E., Kim H. M., Paik S. G., Lee H., Lee J. O., Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Manukyan M., Triantafilou K., Triantafilou M., Mackie A., Nilsen N., Espevik T., Wiesmüller K. H., Ulmer A. J., Heine H., Binding of lipopeptide to CD14 induces physical proximity of CD14, TLR2 and TLR1. Eur. J. Immunol. 35, 911–921 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Triantafilou M., Gamper F. G. J., Haston R. M., Mouratis M. A., Morath S., Hartung T., Triantafilou K., Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J. Biol. Chem. 281, 31002–31011 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Kang J. Y., Lee J. O., Structural biology of the Toll-like receptor family. Annu. Rev. Biochem. 80, 917–941 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Nordenfelt P., Waldemarson S., Linder A., Mörgelin M., Karlsson C., Malmström J., Björck L., Antibody orientation at bacterial surfaces is related to invasive infection. J. Exp. Med. 209, 2367–2381 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L., Wang H., Wagner M., Yan Y., Jakob D. S., Xu X. G., Nanoscale simultaneous chemical and mechanical imaging via peak force infrared microscopy. Sci. Adv. 3, e1700255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akira S., Uematsu S., Takeuchi O., Pathogen recognition and innate immunity. Cell 124, 783–801 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Sanchez-Mejorada G., Rosales C., Signal transduction by immunoglobulin Fc receptors. J. Leukoc. Biol. 63, 521–533 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Perkins N. D., Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 8, 49–62 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Liu T., Zhang L. Y., Joo D., Sun S. C., NF-kappa B signaling in inflammation. Signal Transduct. Target. Ther. 2, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung M. H., Li N., Lao Q., Gottschalk R. A., Hager G. L., Fraser I. D. C., Switching of the relative dominance between feedback mechanisms in lipopolysaccharide-induced NF-kappa B signaling. Sci. Signal. 7, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoepel W., Newling M., Vogelpoel L. T. C., Sritharan L., Hansen I. S., Kapsenberg M. L., Baeten D. L. P., Everts B., den Dunnen J., FcγR-TLR cross-talk enhances TNF production by human monocyte-derived DCs via IRF5-dependent gene transcription and glycolytic reprogramming. Front. Immunol. 10, 0073901–0073911 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.den Dunnen J., Vogelpoel L. T. C., Wypych T., Muller F. J. M., de Boer L., Kuijpers T. W., Zaat S. A. J., Kapsenberg M. L., de Jong E. C., IgG opsonization of bacteria promotes Th17 responses via synergy between TLRs and FcγRIIa in human dendritic cells. Blood 120, 112–121 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Vogelpoel L. T., Hansen I. S., Visser M. W., Nagelkerke S. Q., Kuijpers T. W., Kapsenberg M. L., de Jong E. C., den Dunnen J., FcγRIIa cross-talk with TLRs, IL-1R, and IFNγR selectively modulates cytokine production in human myeloid cells. Immunobiology 220, 193–199 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Badovinac V. P., Harty J. T., Intracellular staining for TNF and IFN-γ detects different frequencies of antigen-specific CD8+ T cells. J. Immunol. Methods 238, 107–117 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Lovelace P., Maecker H. T., Multiparameter intracellular cytokine staining. Methods Mol. Biol. 1678, 151–166 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Parthasarathy R., Rapid, accurate particle tracking by calculation of radial symmetry centers. Nat. Methods 9, 724–726 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Gabriel E., A. Baddeley, E. Rubak, R. Turner, Spatial point patterns: Methodology and applications with R. Math. Geosci. 49, 815–817 (2017). [Google Scholar]
- 44.Baddeley A., Turner R., spatstat: An R package for analyzing spatial point patterns. J. Stat. Softw. 12, 1–42 (2005). [Google Scholar]
- 45.Chen Z., Oh D., Biswas K. H., Yu C. H., Zaidel-Bar R., Groves J. T., Spatially modulated ephrinA1:EphA2 signaling increases local contractility and global focal adhesion dynamics to promote cell motility. Proc. Natl. Acad. Sci. U.S.A. 115, E5696–E5705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verheyen T., Fang T., Lindenhofer D., Wang Y., Akopyan K., Lindqvist A., Högberg B., Teixeira A. I., Spatial organization-dependent EphA2 transcriptional responses revealed by ligand nanocalipers. Nucleic Acids Res. 48, 5777–5787 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shaw A., Lundin V., Petrova E., Fördős F., Benson E., al-Amin A., Herland A., Blokzijl A., Högberg B., Teixeira A. I., Spatial control of membrane receptor function using ligand nanocalipers. Nat. Methods 11, 841–846 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Veneziano R., Moyer T. J., Stone M. B., Wamhoff E. C., Read B. J., Mukherjee S., Shepherd T. R., Das J., Schief W. R., Irvine D. J., Bathe M., Role of nanoscale antigen organization on B-cell activation probed using DNA origami. Nat. Nanotechnol. 15, 716–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai H. G., Muller J., Depoil D., Mayya V., Sheetz M. P., Dustin M. L., Wind S. J., Full control of ligand positioning reveals spatial thresholds for T cell receptor triggering. Nat. Nanotechnol. 13, 610–617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caculitan N. G., Kai H., Liu E. Y., Fay N., Yu Y., Lohmüller T., O’Donoghue G. P., Groves J. T., Size-based chromatography of signaling clusters in a living cell membrane. Nano Lett. 14, 2293–2298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deeg J., Axmann M., Matic J., Liapis A., Depoil D., Afrose J., Curado S., Dustin M. L., Spatz J. P., T cell activation is determined by the number of presented antigens. Nano Lett. 13, 5619–5626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.J. Hellmeier, R. Platzer, A. S. Eklund, T. Schlichthärle, A. Karner, V. Motsch, E. Kurz, V. Bamieh, M. Brameshuber, J. Preiner, R. Jungmann, H. Stockinger, G. J. Schütz, J. B. Huppa, E. Sevcsik, DNA origami demonstrate the unique stimulatory power of single pMHCs as T-cell antigens. bioRxiv 2020.06.24.166850 [Preprint]. 24 June 2020. https://doi.org/10.1101/2020.06.24.166850.
- 53.Triantafilou M., Morath S., Mackie A., Hartung T., Triantafilou K., Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J. Cell Sci. 117, 4007–4014 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Triantafilou M., Lepper P. M., Olden R., Dias I. D. R., Triantafilou K., Location, location, location: Is membrane partitioning everything when it comes to innate immune activation? Mediators Inflamm. 186093 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wheeler R., Mesnage S., Boneca I. G., Hobbs J. K., Foster S. J., Super-resolution microscopy reveals cell wall dynamics and peptidoglycan architecture in ovococcal bacteria. Mol. Microbiol. 82, 1096–1109 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Dufrene Y. F., Atomic force microscopy in microbiology: New structural and functional insights into the microbial cell surface. MBio 5, e01363-01314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dague E., Alsteens D., Latge J. P., Dufrene Y. F., High-resolution cell surface dynamics of germinating Aspergillus fumigatus conidia. Biophys. J. 94, 656–660 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Formosa C., Grare M., Jauvert E., Coutable A., Regnouf-de-Vains J. B., Mourer M., Duval R. E., Dague E., Nanoscale analysis of the effects of antibiotics and CX1 on a Pseudomonas aeruginosa multidrug-resistant strain. Sci. Rep. 2, 575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Francius G., Domenech O., Mingeot-Leclercq M. P., Dufrene Y. F., Direct observation of Staphylococcus aureus cell wall digestion by lysostaphin. J. Bacteriol. 190, 7904–7909 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li W. Q., Wang H. M., Xu X. J. G., Yu Y., Simultaneous nanoscale imaging of chemical and architectural heterogeneity on yeast cell wall particles. Langmuir 36, 6169–6177 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee K., Kim S. Y., Seo Y., Kim M. H., Chang J., Lee H., Adjuvant incorporated lipid nanoparticles for enhanced mRNA-mediated cancer immunotherapy. Biomater. Sci. 8, 1101–1105 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Wang Y., Su L., Morin M. D., Jones B. T., Mifune Y., Shi H., Wang K. W., Zhan X., Liu A., Wang J., Li X., Tang M., Ludwig S., Hildebrand S., Zhou K., Siegwart D. J., Moresco E. M. Y., Zhang H., Boger D. L., Beutler B., Adjuvant effect of the novel TLR1/TLR2 agonist Diprovocim synergizes with anti-PD-L1 to eliminate melanoma in mice. Proc. Natl. Acad. Sci. U.S.A. 115, E8698–E8706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng Y., Mu R., Wang Z., Xing P., Zhang J., Dong L., Wang C., A Toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat. Commun. 10, 2272 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cen X. H., Zhu G., Yang J., Yang J., Guo J., Jin J., Nandakumar K. S., Yang W., Yin H., Liu S., Cheng K., TLR1/2 specific small-molecule agonist suppresses leukemia cancer cell growth by stimulating cytotoxic T lymphocytes. Adv. Sci. 6, 1802042 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vogelpoel L. T. C., Hansen I. S., Rispens T., Muller F. J. M., van Capel T. M. M., Turina M. C., Vos J. B., Baeten D. L. P., Kapsenberg M. L., de Jong E. C., den Dunnen J., Fc gamma receptor-TLR cross-talk elicits pro-inflammatory cytokine production by human M2 macrophages. Nat. Commun. 5, 5444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Egmond M., Vidarsson G., Bakema J. E., Cross-talk between pathogen recognizing Toll-like receptors and immunoglobulin Fc receptors in immunity. Immunol. Rev. 268, 311–327 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Sokolove J., Zhao X. Y., Chandra P. E., Robinson W. H., Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 63, 53–62 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Noursadeghi M., Tsang J., Haustein T., Miller R. F., Chain B. M., Katz D. R., Quantitative imaging assay for NF-κB nuclear translocation in primary human macrophages. J. Immunol. Methods 329, 194–200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.H. Wickham, ggplot2: Elegant Graphics for Data Analysis (Springer International Publishing, ed. 3, 2016). [Google Scholar]
- 70.R Core Team, R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabc8482/DC1