

Group transmission of viruses accelerates early replication and increases short-term viral fitness.
Abstract
The ability of viruses to infect their hosts depends on rapid dissemination following transmission. The notion that viral particles function as independent propagules has been challenged by recent observations suggesting that viral aggregates show enhanced infectivity and faster spread. However, these observations remain poorly understood. Here, we show that viral replication is a cooperative process, such that entry of multiple viral genome copies into the same cell disproportionately increases short-term viral progeny production. This cooperativity arises from the positive feedback established between replication templates and virus-encoded products involved in replication and should be a general feature of viruses. We develop a simple model that captures this effect, verify that cooperativity also emerges in more complex models for specific human viruses, validate our predictions experimentally using different mammalian viruses, and discuss the implications of cooperative replication for viral fitness.
INTRODUCTION
Whether a virus successfully invades a susceptible host should be influenced by early infection events that take place when a small number of cells are challenged with few founder viral particles (1–3). Hence, modeling how these transmitted viruses replicate might help us to better understand viral dynamics within and among hosts (4–6). According to the independent action hypothesis, the ability of a given viral particle to initiate infection is independent of the presence of other viral particles (7–9). However, groups of viral particles are often transmitted jointly to the same cell inside vesicles or as virion aggregates, potentially allowing for synergistic or antagonistic interactions among these cotransmitted viruses. These collective infectious units have been described in widely different viruses, including enteroviruses, noroviruses, rotaviruses, marseilleviruses, rhabdoviruses, baculoviruses, and lentiviruses (10–15).
Experimental evidence supports the notion that viruses benefit from coinfecting cells with multiple founder particles. For instance, in vesicular stomatitis virus (VSV), virion aggregates showed faster progeny production and higher short-term fitness than equal numbers of free virions (16). In HIV-1, factors promoting coinfection such as high virion-to-cell ratios and direct cell-to-cell spread were also found to accelerate the infection cycle (17). Similarly, a correlation between the number of genome copies initiating infection and gene expression levels has been reported for herpesvirus (18). Furthermore, en bloc delivery of pools of viral particles enclosed in vesicles was found to be an optimal interhost transmission strategy in noroviruses and rotaviruses (19). However, how early viral multiplication and fitness is determined by the number of viral genomes entering a cell remains poorly understood.
A basic question to be addressed is whether coinfecting a cell with N0 initial viral genome copies produces viral progeny more efficiently than infecting N0 cells with a single copy each. In principle, the coinfection scenario should reduce the total amount of cellular resources available to the virus, increasing virus-virus competition and reducing progeny production on a per capita basis. However, this cost might be offset if founder viral genomes interact cooperatively to accelerate infection. To explore this, we developed simple deterministic and stochastic models describing viral replication. These models revealed that viral replication is a cooperative process because it depends not only on cellular resources but also on virus-encoded products. These dependencies establish a positive nonlinear feedback between replication templates and products and, consequently, a disproportionate increase in progeny genome production as the number of founder templates increases. By coupling this intracellular process with an intercellular viral population dynamics model, we further show that cooperative replication should influence early viral dissemination and fitness. Last, we tested our predictions in more complex models tailored to specific human viruses and by performing experiments with different mammalian viruses.
RESULTS
Cooperative viral replication in a simple ordinary differential equation model
We developed an ordinary differential equation (ODE) model of viral replication using four state variables: free viral genomes (G), replicative complexes (C), viral resources (R), and finite cellular resources (E; Fig. 1A). The total number of viral genome copies was thus N = G + C. In the model, replicative complexes were formed from free viral genomes, viral resources (e.g., viral polymerase or cofactors), and cellular resources (e.g., nucleotides or energy). After replication, replicative complexes dissociated, releasing the template and viral resources, which were both reusable. Viral gene expression led to the production of new viral resources from viral genomes and cellular resources (amino acids, energy, and ribosomes). We iterated this process until cellular resources were exhausted (Fig. 1B).
Fig. 1. Cooperative viral replication revealed by a simple ODE model.
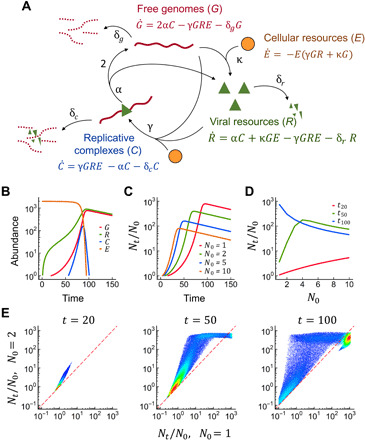
(A) Model definition (see Materials and Methods for details). Replication complexes were formed from free viral genomes and viral resources and consumed cellular resources. Complex dissociation produced a new genome copy and released the template genome, as well as reusable viral resources. New viral resources were produced from genomes and cellular resources. Parameter names are indicated, and their effects on cooperativity are shown in fig. S3. (B) Dynamics of each variable (arbitrary units) for α = 5 × 10−1 t−1, κ = 5 × 10−4 U−1 t−1, γ = 10−6 U−2 t−1, and δg = δc = δr = 10−2 t−1. Simulations were started from N0 = G0 free viral genomes, using C0 = 0, R0 = 0, and E0 = 2000 U. (C) Dynamics of per capita progeny production, Nt/N0, for different N0 values. (D) N0 versus Nt/N0 at different time points. (E) Nt/N0 for N0 = 1 versus N0 = 2 at three time points for 100,000 random sets of parameters (each set represented with a dot) within the following ranges: α (0 to 1), κ (0 to 5 × 10−4), γ (0 to 5 × 10−6), δg (0 to 3 × 10−2), δc (0 to 3 × 10−2), and δr (0 to 3 × 10−2). The red dashed line indicates equal per capita progeny production for N0 = 1 and N0 = 2. Dots above this line indicate cooperativity. Larger values of δ relative to the other parameters often resulted in abortive infections, while a general increase or decrease in all parameters affected infection time but not cooperativity (fig. S2). Graphs for N0 = 1 versus N0 = 5 and N0 = 1 versus N0 = 10 are shown in fig. S1.
This model revealed that viral replication should be a cooperative process, as indicated by the fact that early viral genome production per capita (Nt/N0) increased with the number of founder genomes per cell (N0). This initial synergy manifested as a disproportionate acceleration of progeny production (Fig. 1C). In contrast, at later stages of the infection cycle, increasing the number of founder genomes became costly because, as expected, the limited amount of cellular resources meant that per capita progeny production necessarily dropped as the number of founder genomes increased (same Nt at end point and, hence, lower Nt/N0; Fig. 1C). At intermediate time points, there was an optimal N0 that maximized per capita viral progeny production (Fig. 1D). Examination of 100,000 random combinations of our model parameters showed that this result was robust to parameter choice (Fig. 1E and figs. S1 and S2). Systematic parameter scanning showed that replication became increasingly cooperative as the production of replication complexes or viral resources became slower and when viral genomes, resources and replication complexes degraded or disassembled faster (fig. S3).
Cooperativity stemmed from the positive nonlinear feedback established between viral genome copy number and virus-encoded product abundance. This concerns a defining property of viruses since viral proteins are generally required to produce more viral genomes. To illustrate this, we modified the ODE model such that replication was still dependent on cellular resources but was no longer dependent on viral products. Free replication templates (G), replicative complexes (C), and resources (R and E) were considered as above, but R was defined as cell-encoded resources instead of viral products. As expected, when the dependence of viral replication on virus-encoded products was removed, replication was no longer cooperative (fig. S4).
Cooperative viral replication in a stochastic model
Since at the very initial stages of infection the number of viral genomes and viral resources was small, the homogeneous concentration of reactants assumed in mass action kinetics may not be realized. To better model initial randomness, we transformed our ODE model into a Markov jump process using the Gillespie algorithm (20). This led to strong variation in replication time profiles (fig. S5A), consistent with previous single-cell experiments (21–23). Despite this variation, increasing the number of founder viruses again accelerated mean progeny production as a result of cooperative replication (fig. S5, B and C). Qualitatively similar results were obtained with 100,000 random parameter combinations, again demonstrating generality (fig. S5D). In addition, the stochastic model allowed us to investigate how N0 determined the probability that the infection did not die out prematurely (infectivity). We found that infectivity deviated from the independent action hypothesis and became increasingly cooperative as degradation rates increased (fig. S5E). This effect emerged because, when the initial number of viral components was small, there was a high chance that some essential component was degraded before replication could proceed. By accelerating early replication, infections initiated from a larger number of founder viral genomes were less likely to experience this abortive scenario.
Fitness effects of cooperative replication in a multiscale population dynamics model
Since transmitting multiple viruses to a given cell initially enhanced viral replication but was costly afterward, a time-integrated approach was needed to assess how N0 influenced viral fitness. In turn, this required considering a viral population dynamics model that incorporated intercellular transmission. To achieve this, we first implemented in our basic ODE model the release of extracellular infectious particles (V) from viral genomes and viral resources. Then, we built an age-structured discretized susceptible-infected population model (Fig. 2A). Using this multiscale approach, we competed a founder population of N0 viral genomes coinfecting a single cell against a founder population of N0 genomes infecting one cell each. We observed that early cooperative replication tended to confer a population-level fitness benefit to the N0 founders infecting the same cell (Fig. 2B). The result became much more variable but similar on average when the deterministic intracellular ODE model was replaced with a stochastic intracellular replication model (Fig. 2C and fig. S6). Scanning a wide range of parameter values showed that the benefit of initial coinfection increased when rapid cell death limited viral progeny production (Fig. 2D). This suggests that early cooperative replication may help overcome antiviral responses such as gene expression arrest and apoptosis, consistent with recent work with VSV (16). On the other hand, initial coinfection was no longer beneficial when postreplicative stages such as release of mature viral particles and intercellular transmission were slow compared to replication and limited population spread (Fig. 2D).
Fig. 2. Fitness effect of cooperative replication in a multiscale model.
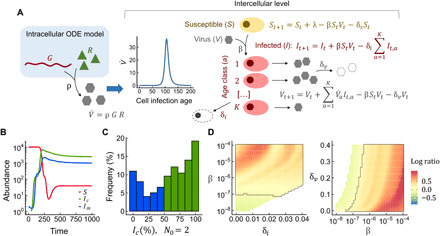
(A) Model definition (see Materials and Methods for details). The ODE model described in Fig. 1A was used to simulate the intracellular dynamics. Infectious particles (V) were produced from free viral genomes and cellular resources and were released from infected cells. This yielded a distribution of the rate at which infectious particles were released as a function of cell infection time (age), which was plugged into an intercellular difference equations model with K discrete infected cell ages. Infected cells (I) were produced from susceptible cells (S) and infectious particles at rate β. Infected cells, susceptible cells, and infectious particles died at rates δi, δs, and δv, respectively. (B) We competed N0 viral genomes of a virus subpopulation c that initially coinfected a single cell against N0 viral genomes of a subpopulation m distributed in N0 cells. The two competitors differed only in these initial conditions. The number of cells infected with c and m viruses (Ic and Im, respectively) for N0 = 2 is shown. (C) Distribution of the fraction of Ic cells at equilibrium for N0 = 2 in 1000 simulations in using a stochastic intracellular model. Colored areas indicate simulations in which initial coinfection was beneficial (green) or detrimental (blue), excluding abortive infections. This distribution deviated significantly from neutrality (binomial test, P < 0.001). Data for N0 = 5 and N0 = 10 are shown in fig. S6. (D) Heatmaps showing how the ratio of the two competitors at equilibrium, log10(Ic/Im), depended on infectivity (β), infected cell death rate (δi), and viral particle degradation rate (δv). The black contours indicate the parameter region for which c was fitter than m. White areas correspond to parameter combinations producing abortive infections at the population level.
Cooperative effects in previous models for well-studied human viruses
Our approach captured a general feature of virus replication but ignored virus-specific details. Previous work has modeled thoroughly the infection cycle of certain viruses (24, 25). To test for cooperativity in a more realistic scenario, we used two previous deterministic ODE models describing the entire infection cycles of HIV-1 (26) and hepatitis C virus (HCV) (27), as well as a stochastic model for influenza virus A (IVA) (23). We examined the dynamics of the number of progeny infectious particles (Vt) as a function of the number of founder particles per cell (V0) using the parameter values reported in the original publications. For the HIV-1 and IVA models, at early time points, Vt/V0 increased with V0, qualitatively reproducing the results obtained with our simple model (Fig. 3). In the IVA stochastic model, increasing V0 had a disproportionate effect on infectivity. For instance, the percentage of IVA abortive infections dropped from 90.8% for V0 = 1 to 33.6% for V0 = 5, whereas, for V0 = 5, the expected percentage under the independent action hypothesis was 0.9085 = 61.7%. The HCV model exhibited no cooperative effects for progeny particle production, but a more in-depth analysis revealed that replication was indeed cooperative since increasing the number of founder genomes per cell (N0) resulted in higher per capita production of progeny genomes (Nt/N0). The effects of cooperative replication were lost at the level of HCV progeny particle production because virion assembly was slow compared to replication (fig. S7), an observation that is compatible with our model.
Fig. 3. Cooperative effects in previous models describing the infection cycles of HIV-1, HCV, and IVA.
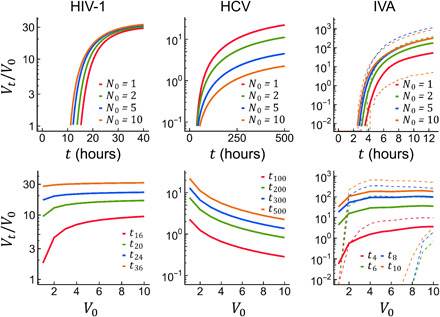
The per capita production of viral genomes, Vt/V0, is shown for different time points and V0 values. The HIV-1 and HCV models were based on a deterministic ODE system. For the stochastic IVA model, the average Vt/V0 obtained from 3000 replicate simulations is shown, including productive and abortive infections. Dashed lines indicate the 10th and 90th percentiles.
Experimental evidence for cooperative effects in different viruses
To test model predictions experimentally, we used five mammalian viruses: VSV, respiratory syncytial virus (RSV), coxsackievirus B3 (CVB3), human adenovirus 5 (hAdV5), and vaccinia virus (VacV). The five viruses were titrated by the plaque or foci assay under the same conditions and used to inoculate cells at varying multiplicities of infection (m0), defined as the initial ratio of infectious units to cells. The average number of founder infectious units per infected cell was calculated as V0 = m0/(1 − e−m0), that is, the mean m0 assuming a Poisson distribution of the number of infectious units per cell and excluding noninfected cells (i.e., ). At different time points within the first infection cycle, viruses were collected and titrated to obtain the per capita progeny production (Vt/V0), where Vt is the number of progeny infectious units produced at time t per initially infected cell, i.e., Vt = mt/(1 − e−m0). To extend our analysis, we also considered previously published data obtained for IVA (28). We then plotted Vt/V0 as a function of V0. As predicted by our cooperative replication model, the observed Vt/V0 was maximized at some intermediate V0 value for all viruses (Fig. 4). A possible exception was VacV, for which the optimal V0 was significantly higher than but very close to 1.0. Overall, these data support the conclusion that, in most mammalian viruses, progeny production is enhanced by challenging cells with multiple infectious particles, up to a certain point where the benefits of cooperativity are outweighed by the costs of sharing limited cellular resources.
Fig. 4. Experimental test for cooperative effects in different viruses.
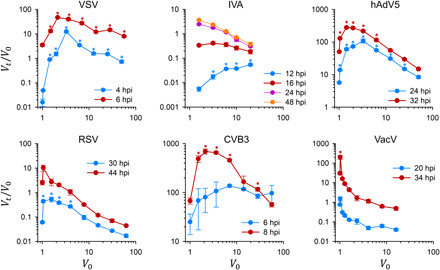
For each virus, the progeny per capita (Vt/V0) at different time points within the first infection cycle is shown as a function of the average number of initial infectious units per cell (V0). Earlier time points were not productive for each of the assayed V0 values, while later times resulted in a decreasing function of Vt/V0 due to competition for limiting cellular resources. V0 was calculated as m0/(1 − e−m0), where the m0 is the multiplicity of infection. Asterisks indicate Vt/V0 values significantly greater than the Vt/V0 value obtained for the leftmost V0 (Welch’s test of log-transformed data with Dunn-Sidak correction for multiple tests, P < 0.05).
DISCUSSION
A general conclusion from our basic models, more complex virus-tailored models, and experimental data is that initiating the cellular infection with multiple viral genome copies disproportionately increases viral replication rate. In addition, this reduces the probability of abortive infection. We have also identified the conditions under which cooperative replication should confer the virus a net fitness benefit. First, there should be an optimal number of founder particles per cell dictated by the opposite effects of cooperative replication and competition for limited cellular resources. Second, the fitness benefits afforded by cooperative replication should be more marked when viral replication is slow compared to degradation rates and antiviral responses since this reduces the time window available for viral progeny production. Third, early cooperative replication is expected to confer a durable fitness benefit if subsequent stages of the viral infection cycle, such as virion assembly, release, and intercellular transmission, do not take place at much lower rates than replication.
Another factor limiting the beneficial fitness effects of cooperative replication is that high levels of coinfection through multiple infection cycles tend to promote the emergence of defective viruses, which function as social cheaters that spread at the expense of fully functional viruses (29). To avoid these negative fitness effects, collective spread could be restricted to specific episodes, such as, for instance, interhost transmission. In addition, according to our model, the effects of cooperative replication should depend on certain viral features. For instance, whereas enveloped viruses start releasing progeny before cell death, in nonenveloped viruses, progeny particles are released by lysis in a single burst that delineates the end of the infection cycle, potentially reducing the impact of cooperative replication on the overall dynamics of viral spread. However, some nonenveloped viruses also exhibit early nonlytic release of progeny particles (10, 12). These predictions could be addressed in future experimental work. Our model could also be extended in several directions. For instance, considering intracellular spatial effects may be particularly relevant. In many viruses, replication is compartmentalized into discrete viral factories (30). On one hand, this intracellular spatial structure might limit interactions (and, hence, cooperativity) among founder viral genomes entering a cell, but, on the other hand, cooperativity might be promoted within each factory by locally increasing the concentration of viral components. It is also possible that viruses in different factories might cooperate indirectly by rearranging cellular resource distribution and metabolism or by jointly blocking innate immunity. A quantitative characterization of the intracellular spatial structure of viral replication will be needed to test these effects.
MATERIALS AND METHODS
Viral replication ODE model
The model is schematically represented and described in Fig. 1A and captures the essential dynamics of virus replication within cells through the following system of four coupled differential equations
We first assumed that free viral genomes inside the cell (G) produce virus-encoded resources (R) using resources from cell (E) in a lumped reaction occurring at rate κ. Then, these newly synthesized viral products interacted with viral genomes at a rate γ to form replicative complexes (C) that used cellular resources to create new viral genome copies. During replication, viral components remained sequestered in C (with G and R acting as templates and polymerases or other virus-encoded replication factors, respectively) and dissociated at rate α while doubling the amount of G molecules involved in the replicative process. Moreover, we compromised viral replication success by subjecting viral components to an exponential decay using linear rates with δg, δr, and δc rate constants for G, R, and C, respectively. For simplicity, we assumed no E production during the infection and that every reaction consumed the same amount of E (i.e., an E unit disappeared per each G or R units produced). Model implementation and data analysis were performed in R. We used the deSolve R package for numerical integration using the LSODA algorithm. In all runs, we set initial conditions to N0 = G0 + C0 = G0, R0 = 0, C0 = 0, and E0 = 2000.
Nonviral replication ODE model
By removing the κGE term from the R production differential equation in our model, we transformed our virus replication model into the following model (fig. S4)
In this model, R was no longer virus-encoded products but reusable cellular resources, while E remained as consumable cellular resources. Replication was modeled as in the virus model, with the exception that replication complexes were formed by the interaction of genomes with a constant, cell-dependent amount of R. To compare both models, we set G0 = 1 and C0 = 0 and searched for R0 and E0 values in the nonviral model for which the maximum growth time point of genomes was shared in both models when keeping the rest of parameters and initial conditions unchanged. It was necessary to ignore degradation rates to find a unique solution to this fitting.
Stochastic model
We converted our deterministic ODE model for virus replication into a continuous-time Markov process that we simulated using the exact Gillespie stochastic algorithm implemented in the adaptivetau R package. The following set of chemical reactions represents this conversion
The same set of initial conditions as above was used, and reaction propensities were calculated using the same values for the stochastic rate constants as for the deterministic model.
Multiscale model
To construct the multiscale model shown in Fig. 2, we first added virion production to the intracellular ODE model. For this, we assumed that progeny viral particles (V) were created and released from the cell when viral genomes (G) and virus-encoded resources (R) interacted at rate ρ. This assumption kept the model simple, while also reflecting the general rule that viruses incorporate many of their products into virions. Both G and R were removed from the cell as they were incorporated into new viral progeny. The intracellular ODE model was thus modified accordingly
To model the intercellular viral population dynamics, we considered free extracellular viral particles (V), susceptible cells (S), and infected cells (I). Since cells infected at different times produced a different amount of virus progeny, the model was structured according to time since infection, using K nonzero infected cell age classes. Hence, we considered simultaneously two time scales: cell infection age (a) and absolute time (t). Free viruses and susceptible cells interacted at rate β (infectivity) to produce newly infected cells It,0, which then aged with time. Specifically
The rate at which free viral particles were produced in each cell age class, , was given by the intracellular ODE model. These rates where then plugged into a simple, target cell–limited, population dynamics model
Susceptible cells were introduced into the population at rate λ and died at rate δs. Free viral particles decayed exponentially at rate δv. For simplicity, we chose a constant death rate δi for infected cells.
Fitness effects of initial coinfection in the multiscale model
Using the above multiscale scheme, we competed two subpopulation of viruses. The first subpopulation (denoted with subindex c) initially coinfected a single cell with N0 viral genomes. This cell produced progeny particles denoted Vc at a rate νca, where a is cell infection age. The second subpopulation (subindex m) was constituted by N0 initial viral genomes infecting one cell each, which produced progeny particles Vm at a rate νma. To obtain νca and νma, we solved the intracellular model using the deSolve R package, as above, with N0 = G0 + C0 = G0 for c viruses and N0 = 1 for m viruses. The other initial conditions and parameter values were R0 = 0, C0 = 0, E0 = 2000, K = 2000, V0 = 0, α = 5 × 10−1 t−1, κ = 5 × 10−4 U−1 t−1, γ = 10−6 U−2 t−1, ρ = 3 × 10−4 virions U−2 t−1, and δg = δc = δr = 10−2 t−1. The distinction between the intracellular dynamics of c and m viruses (i.e., different numbers of founder genomes per cell) was applied to the first infection cycle only. In subsequent cycles, the two viral populations were treated equally, assuming that all cells were infected with a single copy of one or the other virus and produced viral progeny at the same rate, νma. The number of cells infected by the progeny of each viral subpopulation at time t was Ict − Ic0 and Imt − Im0, respectively, where subindex 0 denotes initial conditions. The system of equations describing these competitions was as follows
The initial conditions were S0 = 104, Vc0 = 0, Vm0 = 0, Im0 = Im0,0 = N0, and Ic0 = Ic0,0 = 0. We used Ic0 = 0 because the virus production dynamics of the single cell initially infected with N0 copies of the c virus was described using a separate term νca = t (1 − δi)t (notice that, in this first cycle, a = t). Parameter values were λ = 25 cells t−1, β = 3 × 10−6 virions−1 t−1, δs = 2 × 10−3 t−1, δi = 7 × 10−3 t−1, and δv = 4 × 10−2 t−1. The system was solved using a custom R script considering K = 2000 nonzero classes in the infection time interval [0 + Δt, 1000]. We used the Ic/Im ratio at equilibrium (10,000 time units) to assess the fitness effects of initial aggregation. This ratio was equivalent to the Vc/Vm ratio but tended to converge faster.
Implementation of first-cycle stochastic effects in the multiscale model
We simulated viral particle production for each initial infected cell using the stochastic model described above but incorporating a virion production reaction
Virion production rates vca=t and vma=t,n with n ∈ {1, …, N0} were obtained by applying centered finite differences to simulated cumulative virus production over K + 2 age classes, and the boundary values vca=0 = vma=0,n = 0 were incorporated. To account for stochastic initial infected cell death, we considered that a particular infected cell can only be productive before reaching certain age class aδi ~ Exp(λ = δi), so that va ≥ aδi = 0. After applying this consideration, we included stochastically determined vca=t and into the model as separate terms (i.e., independent on Ict, a and Imt, a) to describe the first infection cycle of a single cell coinfected with N0 viral genomes, versus N0 singly infected cells. Subsequent cycles were modeled using a unique rate , which was calculated from 10,000 stochastic simulations initiated with N0 = 1 viral genomes without considering stochastic infected cell death. Therefore, the last two equations in the above multiscale competition model were modified as follows
All parameters and initial conditions used for both the intracellular stochastic model and the multiscale population model remained the same except for Im0 = 0 (note that, in this case, initial singly infected cells were described by ).
Implementation of the HIV-1 model
We tested for cooperativity in a previously published quantitative deterministic ODE model describing the entire cellular infection cycle of HIV-1, from virion-receptor binding to viral progeny maturation (26). A comprehensive description of the model equations and parameters is available from the original publication. This model includes a nonlinear regulation of viral transcription mediated by Tat and Rev viral proteins. Since Tat is a transactivator that enhances transcription of HIV-1 proviral DNA, this protein should create a positive feedback loop similar to that found in our simple ODE model between G and R production. Another possible source of cooperativity is Rev, which is required to transport viral RNA genomes and incompletely spliced RNAs from the nucleus to the cytoplasm and should increase translation of virus structural proteins, as well as the availability of viral RNA genomes in the cytoplasm to produce new viral particles. Authors calibrated the model using HIV-1 kinetic data from the literature. To test viral dynamics for different numbers of initial particles per cell, we modified the initial conditions for the Vfree variable representing initial extracellular virions accordingly. To obtain the total number of progeny genomes (Nt), we summed mRNAg, mRNAcg, RNAmem, Vprevirion, Vbud, and Vmat model variables representing full-length RNAs in the nucleus-, cytoplasm-, and membrane-assembled previrion complexes, budding virions, and mature virions, respectively. To obtain the number of progeny viral particles (Vt), we considered Vmat only. The model was implemented in R using the deSolve package.
Implementation of the HCV model
This model describes the HCV infection cycle, starting from positive RNA viral genomes in the cytoplasm and ending with the release of viral progeny (27). We, hence, assumed V0 = N0. In contrast to earlier models for HCV replicons, this model considers a more realistic dynamics of the complete virus replicating in Huh-7 cells. To calibrate this model, authors not only used kinetic data from literature but also obtained new data by model fitting to experiments based on the cell culture system for HCV genotype 2a (JFH1 strain). To test for cooperative replication, we changed initial conditions provided by the authors for positive RNA in the cytoplasm ( = 1 molecule) and HCV core protein (S = 180 molecules) by multiplying them by the desired N0. The total production of progeny viral genomes (Nt) was calculated by adding , TC, Rp5B, Rip, dsRNA, Rids, , and Vcreated model variables representing positive RNA genomes free in the cytoplasm, in translational complexes, in RNA/NS5B complexes, in replicative complexes, as free double-stranded RNA, and in double-stranded RNA complexes, as free positive RNA genomes inside vesicular membrane structures or as part of released virion progeny, respectively. Vcreated was used to count progeny viral particles (Vt). The model was implemented in R using the deSolve package.
Implementation of the IVA model
This stochastic model comprises the complete virus infection cycle from virus binding to virion release (23). In IVA, the production of infectious progeny is conditional to the availability of a full set of genome segments and proteins necessary for viral particle formation and, consequently, loss of any single segment leads to nonproductive infection. To vary the number of initial particles per cell, we modified the initial number of extracellular virions accordingly (Vex). To calculate the number of progeny genomes, we first added, for each segment i, the model variables , , , , , and Vrel, which correspond to negative RNA genomes free in the nucleus, forming a complex with viral RNA-dependent RNA polymerases, as nuclear viral ribonucleoproteins (vRNPs), as nuclear M1-vRNP complexes, as cytoplasmic nuclear export protein (NEP)–M1-vRNP complexes, or inside released virions, respectively. The effective number of progeny genomes was equal to the number of copies of the least abundant segment. The number of progeny infectious particles was obtained directly from Vrel. The model was implemented using the MATLAB scripts created and kindly provided by F. S. Heldt (Sensyne Health). Following the recommendations made in the original publication, we performed 3000 replicate simulations for each condition tested.
Cell culturing
HeLa-H1 cells were purchased from the American Type Culture Collection (CRL-1958) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), nonessential amino acids, penicillin (10 U/ml), streptomycin (10 μg/ml), and amphotericin B (250 ng/ml) at 37°C in a 5% CO2 humidified incubator. Cells were tested mycoplasma free by polymerase chain reaction.
Viruses
VSV was recovered from a complementary DNA clone. CVB3 Nancy was recovered from an infectious clone obtained from M. Vignuzzi (Pasteur Institute, France). A green fluorescent protein–encoding hAdV5 was provided by R. Alemany (Bellvitge Biomedical Research Institute, Spain). A VacV recombinant virus encoding T7 phage RNA polymerase was provided by G.W. Wertz (University of Alabama, USA). RSV strain A2-Line19F encoding mKate was provided by R. Geller (Universitat de València, Spain).
Virus titration by the plaque assay
For VSV, VacV, and CVB3, HeLa-H1 cells cultured in six-well plates were inoculated with 200 μl of virus suspensions. Incubation times of 45 min were used for VSV and CVB3. VacV was incubated for 2 hours, gently rocking culture plates every 30 min. Cell monolayers were then overlaid with DMEM supplemented with 2% FBS and 0.5% agar for VSV, 0.8% noble agar for CVB3, and no agar for VacV. Infected cell monolayers were fixed at 24 hours post-inoculation (hpi) for VSV, 48 hpi for CVB3, and 72 hpi for VacV using 10% formaldehyde and stained with 2% crystal violet in 10% formaldehyde to count plaques manually.
Virus titration by fluorescence microscopy
As RSV and hAdV5 formed plaques inefficiently, titrations were carried out by determining the number of infection foci using fluorescence microscopy. For RSV, HeLa-H1 cell monolayers were cultured in 12-well plates, inoculated with 100 μl of viral suspensions, and incubated for 2 hours, shaking every 30 min. For hAdV5, cells in 12-well plates were inoculated with 500 μl of virus suspensions and incubated for 4 hours without shaking. The inoculum was then removed, and cells were overlaid with DMEM culture media supplemented with 2% FBS. Fluorescence imaging of cells was performed in an IncuCyte S3 live-cell analysis system (Essen BioScience) housed inside a humidified tissue culture incubator at 37°C and 5% CO2. Images were acquired with the 4× objective using phase contrast and green channel for hAdV5 or red channel for RSV.
Viral growth assays
Virus infections were carried out in HeLa-H1 monolayers cultured in 12-well plates and inoculated in triplicate with 100 μl (except for hAdV5, for which we used 500 μl) at different multiplicities of infection. Cells were incubated with these inocula as in virus titrations, inocula were removed by aspiration, monolayers doubly washed with phosphate-buffered saline, and infected cultures incubated in DMEM supplemented with 2% FBS for the indicated times. For VSV, RSV, and VacV, raw culture supernatants were collected directly. For hAdV5 and CVB3, we subjected cells to three freeze-thaw cycles to also collect intracellular infectious particles.
Virus concentration
In contrast to the other viruses analyzed, RSV did not reach sufficiently high titer to inoculate cells at high multiplicity of infection. To overcome this, we concentrated the virus by centrifugation. For this, HeLa-H1 cells cultured in eight T175 flasks were infected at approximately 1 plaque-forming units per cell. After 60 hpi, culture supernatants were collected and centrifuged at 3000g for 5 min at 4°C to remove cellular debris. Then, the virus suspension was centrifuged at 50,000g for 90 min at 4°C, and the pelleted viral particles were resuspended in 1 ml of DMEM supplemented with 10% dimethyl sulfoxide to improve virus conservation at −70°C.
Analysis of previous IVA data
Experimental data similar to those obtained for VSV, CVB3, RSV, hAdV5, and VacV were obtained from a previous publication (28) in which viral titers of IVA Pan/99-WT (H3N2) were determined in MDCK (Madin-Darby canine kidney) cells at different times after inoculating cells at different multiplicities of infection. We downloaded these data from the GitHub source provided by the authors (https://github.com/njacobs627/Pan99_IVGs_Spatial_Structure). Since only means and SEM of log-transformed data were available, we back-transformed these two parameters to linear scale using the bt.log function included in the fishmethods R package. Per capita progeny production was calculated, assuming that each confluent cell monolayer in a six-well plate contained 106 MDCK cells.
Supplementary Material
Acknowledgments
We thank E. Segredo Otero for helpful discussions, F. S. Heldt for the IVA MATLAB script, and M. Vignuzzi, R. Alemany, and R. Geller for providing the viruses. Funding: This work was funded by ERC Consolidator Grant 724519 (Vis-a-Vis) and the Spanish Ministerio de Ciencia, Innovación y Universidades (grant BFU2017-84762-R). I.A.-M. was funded by a Ph.D. fellowship from the Spanish Ministerio de Ciencia, Innovación y Universidades. J.-V.B. was funded by Prometeo Grant 2016/122 from the Generalitat Valenciana. Author contributions: I.A.-M. designed the research, performed experiments, and analyzed data. J.-V.B. performed experiments. R.S. obtained funding, designed the research, and wrote the article. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in paper are presented in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabd4942/DC1
REFERENCES AND NOTES
- 1.Zwart M. P., Elena S. F., Matters of size: Genetic bottlenecks in virus infection and their potential impact on evolution. Annu. Rev. Virol. 2, 161–179 (2015). [DOI] [PubMed] [Google Scholar]
- 2.McCrone J. T., Lauring A. S., Genetic bottlenecks in intraspecies virus transmission. Curr. Opin. Virol. 28, 20–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutiérrez S., Michalakis Y., Blanc S., Virus population bottlenecks during within-host progression and host-to-host transmission. Curr. Opin. Virol. 2, 546–555 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Graw F., Perelson A. S., Modeling viral spread. Annu. Rev. Virol. 3, 555–572 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumberger P., Frey F., Schwarz U. S., Graw F., Multiscale modeling of virus replication and spread. FEBS Lett. 590, 1972–1986 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Gog J. R., Pellis L., Wood J. L. N., McLean A. R., Arinaminpathy N., Lloyd-Smith J. O., Seven challenges in modeling pathogen dynamics within-host and across scales. Epidemics 10, 45–48 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Druett H. A., Bacterial invasion. Nature 170, 288 (1952). [DOI] [PubMed] [Google Scholar]
- 8.Zwart M. P., Elena S. F., Testing the independent action hypothesis of plant pathogen mode of action: A simple and powerful new approach. Phytopathology 105, 18–25 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Zwart M. P., Hemerik L., Cory J. S., de Visser J. A. G. M., Bianchi F. J. J. A., Van Oers M. M., Vlak J. M., Hoekstra R. F., Van der Werf W., An experimental test of the independent action hypothesis in virus-insect pathosystems. Proc. Biol. Sci. 276, 2233–2242 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altan-Bonnet N., Extracellular vesicles are the Trojan horses of viral infection. Curr. Opin. Microbiol. 32, 77–81 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanjuán R., Collective infectious units in viruses. Trends Microbiol. 22, 402–412 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mutsafi Y., Altan-Bonnet N., Enterovirus transmission by secretory autophagy. Viruses 10, 139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanjuán R., Thoulouze M. I., Why viruses sometimes disperse in groups. Virus Evol. 5, vez014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguilera E. R., Pfeiffer J. K., Strength in numbers: Mechanisms of viral co-infection. Virus Res. 265, 43–46 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolgin E., The secret social lives of viruses. Nature 570, 290–292 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Andreu-Moreno I., Sanjuán R., Collective infection of cells by viral aggregates promotes early viral proliferation and reveals a cellular-level Allee effect. Curr. Biol. 28, 3212–3219.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boullé M., Müller T. G., Dähling S., Ganga Y., Jackson L., Mahamed D., Oom L., Lustig G., Neher R. A., Sigal A., HIV cell-to-cell spread results in earlier onset of viral gene expression by multiple infections per cell. PLOS Pathog. 12, e1005964 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen E. M., Kobiler O., Gene expression correlates with the number of herpes viral genomes initiating infection in single cells. PLOS Pathog. 12, e1006082 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santiana M., Ghosh S., Ho B. A., Rajasekaran V., Du W.-L., Mutsafi Y., De Jésus-Diaz D. A., Sosnovtsev S. V., Levenson E. A., Parra G. I., Takvorian P. M., Cali A., Bleck C., Vlasova A. N., Saif L. J., Patton J. T., Lopalco P., Corcelli A., Green K. Y., Altan-Bonnet N., Vesicle-cloaked virus clusters are optimal units for inter-organismal viral transmission. Cell Host Microbe 24, 208–220.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillespie D. T., A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 22, 403–434 (1976). [Google Scholar]
- 21.Timm A., Yin J., Kinetics of virus production from single cells. Virology 424, 11–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulte M. B., Andino R., Single-cell analysis uncovers extensive biological noise in poliovirus replication. J. Virol. 88, 6205–6212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heldt F. S., Kupke S. Y., Dorl S., Reichl U., Frensing T., Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza A virus infection. Nat. Commun. 6, 8938 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin J., Redovich J., Kinetic modeling of virus growth in cells. Microbiol. Mol. Biol. Rev. 82, e00066-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Canini L., Perelson A. S., Viral kinetic modeling: State of the art. J. Pharmacokinet. Pharmacodyn. 41, 431–443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shcherbatova O., Grebennikov D., Sazonov I., Meyerhans A., Bocharov G., Modeling of the HIV-1 life cycle in productively infected cells to predict novel therapeutic targets. Pathogens 9, 225 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aunins T. R., Marsh K. A., Subramanya G., Uprichard S. L., Perelson A. S., Chatterjee A., Intracellular hepatitis C virus modeling predicts infection dynamics and viral protein mechanisms. J. Virol. 92, e02098-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobs N. T., Onuoha N. O., Antia A., Steel J., Antia R., Lowen A. C., Incomplete influenza a virus genomes occur frequently but are readily complemented during localized viral spread. Nat .Commun. 10, 3526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genoyer E., López C. B., The impact of defective viruses on infection and immunity. Annu. Rev. Virol. 6, 547–566 (2019). [DOI] [PubMed] [Google Scholar]
- 30.den Boon J. A., Ahlquist P., Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 64, 241–256 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabd4942/DC1