

MS-derived oligodendrocytes conserve their ability to fully interact with axons and glial cells in vivo.
Abstract
Remyelination failure in multiple sclerosis (MS) is associated with a migration/differentiation block of oligodendroglia. The reason for this block is highly debated. It could result from disease-related extrinsic or intrinsic regulators in oligodendroglial biology. To avoid confounding immune-mediated extrinsic effect, we used an immune-deficient mouse model to compare induced pluripotent stem cell–derived oligodendroglia from MS and healthy donors following engraftment in the developing CNS. We show that the MS-progeny behaves and differentiates into oligodendrocytes to the same extent as controls. They generate equal amounts of myelin, with bona fide nodes of Ranvier, and promote equal restoration of their host slow conduction. MS-progeny expressed oligodendrocyte- and astrocyte-specific connexins and established functional connections with donor and host glia. Thus, MS oligodendroglia, regardless of major immune manipulators, are intrinsically capable of myelination and making functional axo-glia/glia-glia connections, reinforcing the view that the MS oligodendrocyte differentiation block is not from major intrinsic oligodendroglial deficits.
INTRODUCTION
Remyelination occurs in multiple sclerosis (MS) lesions but its capacity decreases over time (1–3). Failed remyelination in MS leads to altered conduction followed by axon degeneration, which, in the long run, results in severe and permanent neurological deficits (4). MS lesions may or may not harbor immature oligodendroglia (oligodendrocyte progenitors and pre-oligodendrocytes), with these cells failing to differentiate into myelin-forming cells, suggesting that oligodendrocyte differentiation is blocked (5–7). So far, the mechanism underlying this block is poorly understood. It may result from adverse environmental conditions or the failed capacity of oligodendrocyte progenitors/pre-oligodendrocytes to migrate or mature efficiently into myelin-forming cells or even a combination of these conditions, all of which may worsen with aging. It has been shown that increasing remyelination either through manipulating the endogenous pool (8, 9) or by grafting competent myelin forming oligodendroglia (10, 11) or both (12) can restore the lost axonal functions, improve the clinical scores, and protect from subsequent axonal degeneration in experimental (13, 14) or clinical (3) settings.
There are multiple ways to investigate the oligodendroglial lineage in disease. Cells can be studied in postmortem tissue sections or purified from postmortem adult human brain for in vitro and transcriptomic/proteomic analysis. In this respect, in vitro experiments highlighted the heterogeneity of the adult human oligodendrocyte progenitor population in terms of antigen and microRNA expression, suggesting that remyelination in the adult human brain involves multiple progenitor populations (15). Moreover, single-cell transcriptomics characterized in detail the heterogeneity of human oligodendroglial cells, emphasizing changes in MS, with some subpopulations expressing disease-specific markers that could play a role in disease onset and/or aggravation (16, 17).
Yet, this MS signature could preexist or be acquired early at disease onset. Moreover, most of these MS postmortem analyses or experimental models cannot overlook the involvement of extrinsic factors such as immune factors that might add more complexity toward understanding the behavior of MS oligodenroglial cells.
Little is known about the biology of the MS oligodendroglial lineage, primarily due to the impossibility, for ethical reasons, to harvest oligodendroglial populations from patients and study the diseased cells and their matching controls in vitro or in vivo after cell transplantation. While cell-cell interactions and cell heterogeneity in diseased conditions generate more complexity when comparing control and pathological samples, the induced pluripotent stem cell (iPSC) technology provides a unique opportunity to study homogeneous populations of human oligodendroglial cells and gain further insights into monogenetic diseases and multifactorial diseases, such as MS. The iPSC technology has unraveled differences in oligodendroglia biology, in Huntington’s disease (18), and schizophrenia (19, 20), indicating that these cells can contribute autonomously to multifactorial diseases outcome. However, so far, little is known about the potential contribution of MS oligodendroglia to failed remyelination. While senescence affects iPSC–neural precursor cells (NPCs) derived from patients with primary progressive MS (PPMS) (21), only few preliminary reports alluded to the fate of PPMS (22, 23) or relapsing-remitting (RRMS) (24) iPSC-derived oligodendroglia after experimental transplantation and did not study per se their capacity to differentiate into functional myelin-forming cells. We exploited a robust approach (25) to generate large quantities of iPSCs-derived O4+ oligodendroglial cells from skin fibroblasts (hiOLs) of three RRMS and three healthy subjects, including two monozygous twin pairs discordant for the disease. As a critical feature of the pluripotent-derived cells should be their ability to fully integrate and function in vivo, we compared the capacity of healthy and MS-hiOL derivatives to integrate and restore axo-glial and glial-glial functional interactions after engraftment in the developing dysmyelinated murine central nervous system (CNS). Our data show that in noninflammatory conditions, the intrinsic properties of iPSC-oligodendroglial cells to differentiate, myelinate, and establish functional cell-cell interactions in vivo are not altered in MS, making them candidates of interest for personalized drug/cell therapies as pluripotency maintains MS oligodendroglial cells in a genuine “nonpathological” state.
RESULTS
Fibroblasts were isolated from three control and three patients with MS and reprogrammed into iPSC. Pluripotent cells were differentiated into NPCs and further into O4+ hiOLs for 12 days in vitro under glial differentiation medium (GDM) conditions as previously described (25). hiOL cells were selected using flow cytometry for O4 before transplantation. Because our aim was to study the intrinsic properties of MS cells, we chose to engraft O4+ hiOLs in the purely dysmyelinating Shi/Shi:Rag2−/− mouse model to avoid confounding immune-mediated extrinsic effects.
MS hiOLs do not exhibit aberrant survival or proliferation in vivo
We first questioned whether MS-hiOLs differed from control-hiOLs wild type (WT) in their capacity to survive and proliferate in vivo. To this aim, we grafted MS- and control-hiOLs in the forebrain of neonatal Shi/Shi:Rag2−/− mice. MS cells engrafted (one injection per hemisphere) in the rostral forebrain, spread primarily through white matter, including the corpus callosum and fimbria, as previously observed using control human fetal (11, 26, 27) and iPSC (25, 28) progenitors. With time, cells also spread rostrally to the olfactory bulb and caudally to the brain stem and cerebellum (fig. S1). Examining engrafted brains at 8, 12, and 16 weeks postgraft (wpg), we found that MS-hiOLs expressing the human nuclear marker STEM101 and the oligodendroglial-specific transcription factor OLIG2 maintained a slow proliferation rate at all times (5 to 19% of STEM+ cells), with no difference in Ki67+ MS-hiOLs compared to control (Fig. 1, A and C). Moreover, immunostaining for cleaved Caspase3 at 8 wpg indicated that MS cells survived as well as control-hiOLs (Fig. 1, B and D). Evaluation of the cell density of human cells based on STEM positivity at each stage revealed no significant difference between grafted MS-hiOLs and control cells (fig. S2).
Fig. 1. MS hiOLs do not exhibit aberrant survival or proliferation in the developing Shi/Shi:Rag2−/− brain.
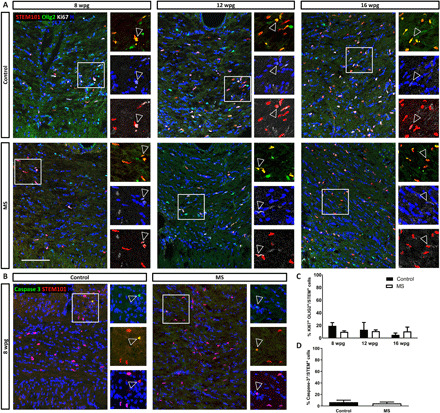
(A and C) Immunodetection of the human nuclei marker STEM101 (red) combined with OLIG2 (green) and the proliferation marker Ki67 (white) shows that a moderate proportion of MS-hiOLs sustains proliferation (empty arrowheads in the insets) following transplantation in their host developing brain, with no significant difference in the rate of proliferation between MS- and control-hiOLs over time. (B and D) Immunodetection of the apoptotic marker Caspase3 (green) indicates that MS-hiOLs survive as well as control-hiOLs 8 wpg. Two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison or Mann-Whitney t tests were used for the statistical analysis (n = 3 to 4 mice per group). Error bars represent SEMs. H, Hoechst dye. Scale bars, 100 μm.
MS-hiOLs do not show a differentiation block over time
Because MS-hiOLs and control cells proliferated and survived to the same extent, we next questioned whether their differentiation potential into mature oligodendrocytes could be affected. We used the human nuclei marker STEM101 to detect all human cells in combination with SOX10, a general marker for the oligodendroglial lineage, and CC1 as a marker of differentiated oligodendrocytes. We found that the number of MS oligodendroglial cells (SOX10+) increased slightly but significantly with time, most likely resulting from sustained proliferation (Fig. 2, A and B). Moreover, they timely differentiated into mature CC1+ oligodendrocytes with a fourfold increase at 12 wpg and a fivefold increase at 16 wpg when compared to 8 wpg and with no difference with control-hiOLs (Fig. 2, B and C).
Fig. 2. Differentiation of MS-hiOLs into mature oligodendrocytes is timely regulated in the corpus callosum of the developing Shi/Shi:Rag2−/− brain.
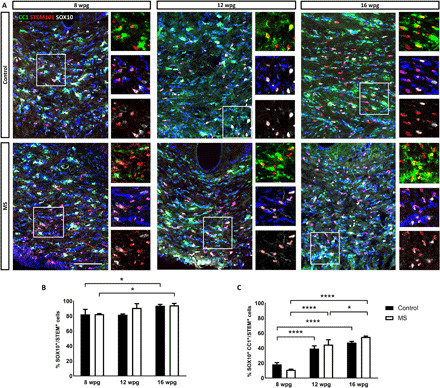
(A) Combined immunodetection of human nuclei marker STEM101 (red) with CC1 (green) and SOX10 (white) for control (top) and MS-hiOLs (bottom) at 8, 12, and 16 wpg. (B and C) Quantification of SOX10+/STEM+ cells (B) and CC1+ SOX10+ over STEM+ cells (C). While the percentage of human oligodendroglial cells increased only slightly with time, the percentage of mature oligodendrocytes was significantly time regulated for both MS- and control-hiOLs. Two-way ANOVA followed by Tukey’s multiple comparison tests were used for the statistical analysis of these experiments (n = 3 to 4 mice per group). Error bars represent SEMs. *P < 0.05 and ****P < 0.0001. Scale bar, 100 μm.
MS-hiOLs do not show an aberrant pattern of myelination
The absence of abnormal MS-hiOL differentiation did not exclude a potential defect in myelination potential. We further investigated the capacity of MS-hiOLs to differentiate into myelin-forming cells. We focused our analysis on the core of the corpus callosum and fimbria. MS-hiOLs, identified by the human nuclear and cytoplasmic markers (STEM101 and STEM121), evolved from a bipolar to multibranched phenotype (Fig. 3A and fig. S3: compare 4 wpg to 8 and 12 wpg) and differentiated progressively into myelin basic protein–positive (MBP+) cells associated, or not, with T-shaped MBP+ myelin-like profiles of increasing complexity (Fig. 3A and figs. S3 and S4B). Myelin-like profiles clearly overlapped with NF200+ axons (fig. S4A) and formed functional nodes of Ranvier expressing ankyrin G and flanked by paranodes enriched for CASPR (fig. S4B) or neurofascin (fig. S4C), as previously observed with control-hiOLs (25).
Fig. 3. MS-hiOL–derived progeny extensively myelinates the dysmyelinated Shi/Shi:Rag2−/− corpus callosum.
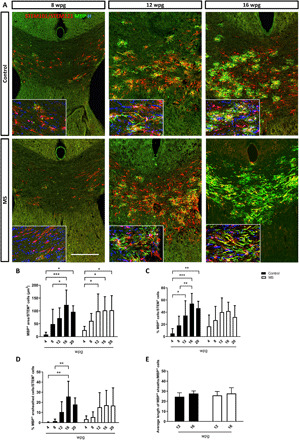
(A) Combined detection of human nuclei (STEM101) and human cytoplasm (STEM 121) (red) with MBP (green) in the Shi/Shi Rag2−/− corpus callosum at 8, 12, and 16 wpg. General views of horizontal sections at the level of the corpus callosum showing the progressive increase of donor-derived myelin for control- (top) and MS- (bottom) hiOLs. (B) Evaluation of the MBP+ area over STEM+ cells. (C and D) Quantification of the percentage of (C) MBP+ cells and (D) MBP+ ensheathed cells. (E) Evaluation of the average sheath length (μm) per MBP+ cells. No obvious difference was observed between MS and control-hiOLs. Two-way ANOVA followed by Tukey’s multiple comparison tests were used for the statistical analysis of these experiments (n = 6 to 14 mice per group). Error bars represent SEMs. *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bar, 200 μm. See also figs. S3 and S5.
We further analyzed, in depth, the myelinating potential of MS-hiOLs, applying automated imaging and analysis, which provided multiparametric quantification of MBP as established in vitro (29) for each donor hiOL (three controls and three RRMS) at 4, 8, 12, 16, and 20 wpg in vivo (Fig. 3, B to D). We first examined the MBP+ surface area generated by the STEM+ cell population (Fig. 3B). While MS-hiOLs generated very low amount of myelin at 4 wpg, they generated significantly more myelin at 12, 16, and 20 wpg, with similar findings for control-hiOLs, highlighting the rapid progress in the percentage of myelin producing STEM+ cells in MS group over time. Detailed MBP+ surface area generated by the STEM+ cell population per donor is presented in fig. S5 and shows differences among hiOLs in the control and MS groups, respectively.
We also quantified the percentage of STEM+ cells expressing MBP and the percentage of MBP+ with processes associated with linear myelin-like features, which we called MBP+ ensheathed cells. Both parameters increased significantly with time for control-hiOLs, reaching a plateau at 16 wpg. The same tendency was achieved for MS-hiOLs with no significant differences between the control- and MS-hiOL groups (Fig. 3, C and D).
Myelin sheath length is considered to be an intrinsic property of oligodendrocytes (30). We analyzed this paradigm in our MS cohort at 12 and 16 wpg, time points at which sheaths were present at a density compatible with quantification. For those time points, we found that the average MS MBP+ sheath length was equivalent to that of control with 25.86 ± 0.98 and 27.74 ± 1.52 μm for MS-hiOLs and 24.52 ± 1.48 and 27.65 ± 0.96 μm for control-hiOLs at 12 and 16 wpg, respectively (Fig. 3F). In summary, our detailed analysis of immunohistochemically labeled sections indicates that MS-hiOLs did not generate abnormal amounts of myelin in vivo when compared to control-hiOLs.
Moreover, the myelinating potential of MS-hiOLs was further validated after engraftment in the developing spinal cord (4 weeks of age). Immunohistological analysis 12 wpg revealed that STEM+ cells not only populated the whole dorsal and ventral columns of the spinal cord with preferential colonization of white matter but also generated remarkable amounts of MBP+ myelin-like internodes that were found on multiple spinal cord coronal sections (fig. S6), thus indicating that their myelination potential was not restricted to only one CNS structure.
MS-derived myelin is compacted and rescues the slow transcallosal conduction of the MBP deficient host
The presence of normal amounts of donor MBP+ myelin-like structures in the shiverer forebrain does not exclude potential structural anomalies. Therefore, we examined the quality of MS derived myelin at the ultrastructural level at 16 wpg in the Shi/Shi:Rag2−/− forebrain. In the corpus callosum of both MS and control-hiOLs grafted mice, we detected numerous axons surrounded by electron dense myelin, which at higher magnification was fully compacted compared to the uncompacted shiverer myelin (Fig. 4, A to F) (25, 31). Moreover, MS myelin reached a mean g ratio of 0.76 ± 1.15 comparable to that of control myelin (0.75 ± 1.56) (Fig. 4G) and thus a similar myelin thickness. This argues in favor of (i) MS-hiOLs having the ability to produce normal compact myelin and thus its functional normality and (ii) a similar rate of myelination between the two groups and, consequently, an absence of delay in myelination for MS-hiOLs.
Fig. 4. MS-hiOLs produce compact myelin in the dysmyelinated Shi/Shi:Rag2 −/− corpus callosum.
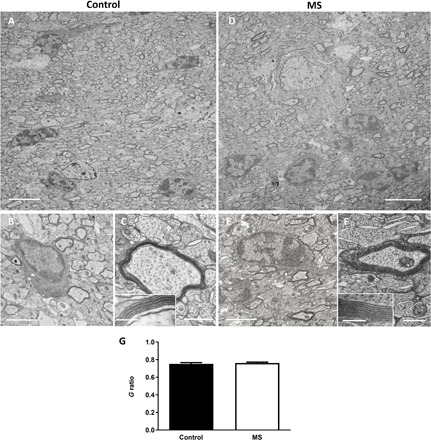
(A to F) Ultrastructure of myelin in sagittal sections of the core of the corpus callosum 16 wpg with control-hiOLs (A to C) and MS-hiOLs (D to F). (A and D) General views illustrating the presence of some electron dense myelin, which could be donor derived. (B, C, E, and F) Higher magnifications of control (B and C) and MS (E and F) grafted corpus callosum validate that host axons are surrounded by thick and compact donor derived myelin. Insets in (C) and (F) are enlargements of myelin and show the presence of the major dense line. No difference in compaction and structure is observed between the MS and control myelin. (G) Quantification of g-ratio revealed no significant difference between myelin thickness of axons myelinated by control- and MS-hiOLs. Mann-Whitney t tests were used for the statistical analysis of this experiment (n = 4 mice per group). Error bars represent SEMs. Scale bars, (A and D) 5 μm , (B and E) 2 μm, and (C and F) 500 nm [with 200 and 100 nm, respectively in (C) and (F) insets].
Myelin compaction has a direct impact on axonal conduction with slower conduction in shiverer mice compared to WT mice (10, 32). We therefore questioned whether newly formed MS-hiOL–derived myelin has the ability to rescue the slow axon conduction velocity of shiverer mice in vivo (Fig. 5). As previously performed with fetal glial-restricted progenitors (11), transcallosal conduction was recorded in vivo at 16 wpg in mice grafted with MS- and control-hiOLs and compared with nongrafted shiverer and WT mice. As expected, conduction in nongrafted shiverer mice was significantly slower compared to WT mice. However, axon conduction velocity was rescued by MS-hiOLs and, to the same extent, by control-hiOLs.
Fig. 5. MS-hiOLs improve transcallosal conduction of the dysfunctional Shi/Shi:Rag2−/− axons to the same extent than control-hiOLs.
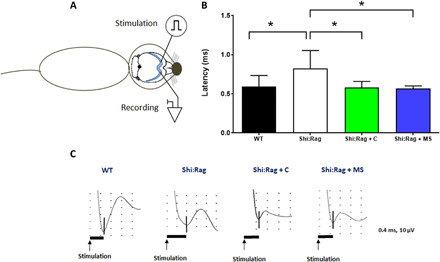
(A) Scheme illustrating that intracallosal stimulation and recording are performed in the ipsi- and contralateral hemisphere, respectively. (B) N1 latency was measured following stimulation in different groups of Shi/Shi:Rag2−/−: intact or grafted with control or MS-hiOLs and WT mice at 16 wpg. MS-hiOL–derived myelin significantly restored transcallosal conduction latency in Shi/Shi:Rag2−/− mice to the same extent than control-derived myelin (P = 0.01) and close to that of WT levels. One-way ANOVA with Dunnett’s multiple comparison test for each group against the group of intact Shi/Shi:Rag2−/− was used. Error bars represent SEMs. *P < 0.05. (C) Representative response profiles for each group. Scales in Y axis is equal to 10 μV and in the X axis is 0.4 ms.
Grafted MS-hiOLs show typical cell stage–specific electrophysiological properties
Rodent oligodendrocyte progenitors and oligodendrocytes can be distinguished by cell stage–specific electrophysiological properties (33, 34). To assess the electrophysiological properties of oligodendroglial lineage cells derived from human grafted control- and MS-hiOLs, red fluorescent protein (RFP)–hiOLs were engrafted in the Shi/Shi:Rag2−/− forebrain and recorded with a K-gluconate–based intracellular solution in acute corpus callosum slices at 12 to 15 wpg (Fig. 6A). As previously described for rodent cells, hiOLs in both groups were identified by their characteristic voltage-dependent current profile recognized by the presence of inward Na+ currents and outwardly rectifying steady-state currents (Fig. 6B). We found that ~60 and ~44% of recorded cells were oligodendrocyte progenitors derived from MS and control progenies, respectively. No significant differences were observed in the amplitude of Na+ currents measured at −20 mV (Fig. 6D) or steady-state currents measured at +20 mV between MS- and control-derived oligodendrocyte progenitors (Isteady = 236.70 ± 19.45 pA and 262.10 ± 31.14 pA, respectively; P = 0.8148, Mann Whitney U test). We further confirmed the identity of these cells by the combined expression of SOX10 or OLIG2 with STEM101/121 and the absence of CC1 in biocytin-loaded cells (Fig. 6F, top). The remaining recorded cells (MS and control) did not show detectable Na+ currents after leak subtraction and were considered to be differentiated oligodendrocytes by their combined expression of SOX10, STEM101/121, and CC1 in biocytin-loaded cells (Fig. 6F, bottom). The I-V curve of these differentiated oligodendrocytes displayed a variable profile that gradually changed from voltage dependent to linear as described for young and mature oligodendroglial cells in the mouse (33). Figure 6C illustrates a typical linear I-V curve of fully mature MS-derived oligodendrocytes. No significant differences were observed in the amplitude of steady-state currents measured at +20 mV between MS- and control-derived oligodendrocytes (Fig. 6E). Overall, the electrophysiological profile of oligodendrocyte progenitors and oligodendrocytes derived from control and MS was equivalent and showed similar characteristics to murine cells (33, 34).
Fig. 6. Grafted MS-hiOLs show typical cell stage–specific electrophysiological properties.
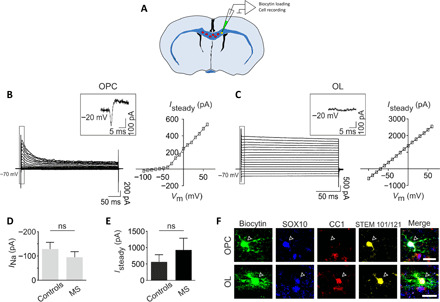
(A) Schematic representation of the concomitant Biocytin loading and recording of single RFP+ hiOL derivative in an acute coronal brain slice prepared from mice engrafted with hiOLs (control or MS) and analyzed at 12 to 14 wpg. (B and C) Currents elicited by voltage steps from −100 to +60 mV in a control-oligodendrocyte progenitor (B, left) and a MS-oligodendrocyte (C, left). Note that the presence of an inward Na+ current obtained after leak subtraction in the oligodendrocyte progenitor, but not in the oligodendrocyte (insets). The steady-state I-V curve of the oligodendrocyte progenitor displays an outward rectification (B, right) while the curve of the oligodendrocyte has a linear shape (C, right). (D) Mean amplitudes of Na+ currents measured at −20 mV in control and MS iPSCs-derived oligodendrocyte progenitors (n = 8 and n = 9, respectively, for four mice per condition; P = 0.743, Mann-Whitney U test). (E). Mean amplitudes of steady-state currents measured at +20 mV in control and patient differentiated iPSC-derived oligodendrocytes (n = 10 and n = 6 for 3 and four mice, respectively; P = 0.6058, Mann-Whitney U test). (F) A control iPSC-derived oligodendrocyte progenitor loaded with biocytin and expressing OLIG2, STEM101/121, and lacking CC1 (top) and an MS iPSC–derived oligodendrocyte loaded with biocytin and expressing SOX10, CC1, and STEM101/121 (bottom). Scale bar, 20 μm.
MS- and control-hiOL progeny functionally connect to other glial cells
Studies with rodents have reported that oligodendrocytes exhibit extensive gap-junctional intercellular coupling between other oligodendrocytes and astrocytes (35). Whether oligodendrocytes derived from grafted human cells can be interconnected with cells in the adult host mouse brain was not known, and whether MS-hiOLs maintain this intrinsic property was also not addressed. Because biocytin can pass through gap junctions, we inspected biocytin-labeled cells for dye coupling (Figs. 6A and 7, A and B).
Fig. 7. Grafted hiOL derivatives are functionally connected to murine cells.
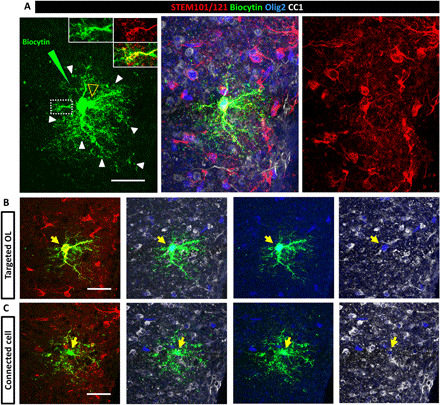
(A) Z-stack identifying a target and connected cell. One single grafted human RFP+ cell (per acute slice) was loaded with biocytin by a patch pipette and allowed to rest for 30 min. The white arrowheads and insets in (A) illustrate biocytin diffusion up to the donut-shaped tip of the human oligodendrocyte processes. Another biocytin-labeled cell (empty yellow arrowhead) was revealed at different morphological level indicating diffusion to a neighboring cell and communication between the two cells via gap junctions. (B and C) Split images of (A) showing the target (B) and connected (C) cell separately at different levels. Immunolabeling for the combined detection of the human markers STEM101/121 (red), OLIG2 (blue), and CC1 (white) indicated that the target cell is of human origin (STEM+) and strongly positive for OLIG2 and CC1, a mature oligodendrocyte, and that the connected cell is of murine origin (STEM-) and weakly positive for OLIG2 and CC1, most likely an immature oligodendrocyte. Scale bars, 30 μm. See also fig. S7.
We found that two of seven MS-derived oligodendrocytes (~29%) and 5 of 21 control-derived oligodendrocytes (~24%) were connected with a single neighboring cell, which was either human or murine (Fig. 7), except in one case where three mouse cells were connected to the biocytin-loaded human cell. These findings reveal that gap junctional coupling can occur between cells from the same or different species, and MS-hiOLs can functionally connect to other glial cells to the same extent as their control counterparts.
To validate the presence of glial-glial interactions, we investigated whether the grafted hiOL-derived progeny had the machinery to be connected to one another via gap junctions. To this end, we focused on oligodendrocyte-specific Cx47 and astrocyte-specific Cx43 as Cx43/47 channels, which are important for astrocyte/oligodendrocyte cross talk during myelination and demyelination (36, 37). Combined immunolabeling for hNOGOA, CC1, OLIG2, and Cx47 revealed that MS-derived oligodendrocyte cell bodies and processes were decorated by Cx47+ gap junction plaques, which were often shared by exogenous MS-derived oligodendrocytes or by MS and endogenous murine oligodendrocytes (fig. S7A). In addition, colabeling exogenous myelin for MBP and Cx43 identified the presence of several astrocyte-specific Cx43 gap junction plaques between human myelin internodes, highlighting contact points between astrocyte processes and axons at the human-murine chimeric nodes of Ranvier (fig. S7B).
Last, colabeling of hNOGOA, with Cx47 and the astrocyte-specific Cx43, revealed coexpression of oligodendrocyte- and astrocyte-specific connexins at the surface of MS-derived oligodendrocyte cell bodies and at the level of T-shaped myelin-like structures (fig. S7C), thus implying connections between human oligodendrocytes and murine and/or human astrocytes, as a small proportion of the grafted hiOLs differentiated into astrocytes. Immunolabeling for human glial fibrillary acidic protein (GFAP), and Cx43 showed that these human astrocytes were decorated by Cx43+ aggregates, as observed in the host subventricular zone (fig. S8A).
Furthermore, immunolabeling for human GFAP, mouse GFAP, and Cx43 indicated that Cx43+ gap junctions were shared between human and mouse astrocytes as observed at the level of blood vessels (fig. S8B). These data validate interconnections between the grafted-derived human glia (MS and controls) with murine host glial cells and confirm their interconnection with the pan-glial network.
DISCUSSION
Two main hypotheses have been considered in understanding MS pathology and etiology: the outside-in hypothesis highlighting the role of immune regulators and environmental inhibitors as extrinsic key players in MS pathology and possibly its repair failure or the inside-out hypothesis pointing to the intrinsic characteristics of neuroglia including oligodendroglial cells as the main contributors in the MS scenario. Single-cell transcriptomic analysis revealed the presence of disease-specific oligodendroglia expressing susceptibility genes in MS brains (16) and altered oligodendroglia heterogeneity in MS (17). The question remains open as to whether these altered oligodendroglial phenotypes are acquired in response to the disease environment or whether they reflect intrinsic traits of the MS oligodendroglial population. On the other hand, the whole exome sequencing analysis in 132 patients from 34 multi-incident families identified 12 candidate genes of the innate immune system and provided the molecular and biological rational for the chronic inflammation, demyelination, and neurodegeneration observed in patients with MS (38) and revealed the presence of epigenetic variants in immune cells and in a subset of oligodendrocytes contributing to risk for MS (39).
While none of these hypotheses have been fully proven or rejected, research efforts for a better understanding of this multifactorial disease have continued. Impaired remyelination or oligodendrocyte differentiation block in MS is still considered a potentially disease-relevant phenotype (40, 41). Many histological and experimental studies suggest that impaired oligodendrocyte progenitor to oligodendrocyte differentiation may contribute to limited remyelination in MS, although some reports question the contribution of newly generated oligodendrocytes to remyelination (17, 42, 43). Understanding MS oligodendrocyte biology has been challenging mainly due to the following reasons: (i) oligodendroglial cells are not easily accessible to be studied in vivo; (ii) dynamic remyelination observed in patients with MS, which points to their individual remyelination potential, is inversely correlated with their clinical disability (3), highlighting even more complexity in oligodendrocyte heterogeneity between patients with MS; and (iii) exclusion of the role of immune system players in understanding MS oligodendrocyte biology being inevitable in most of clinical or experimental studies.
In such a complex multifactorial disease, one of the most accessible and applicable approaches to overcome these problems is the generation of large quantities of disease and control oligodendroglia using the iPSC technology, and to investigate their genuine behavior in vivo after engraftment in a B and T cell–free system. Using a very efficient reprogramming method (25), and the purely dysmyelinating Shi/Shi:Rag2−/− mouse model to avoid confounding immune-mediated extrinsic effects, we show that MS-hiOLs derivatives survive, proliferate, migrate, and timely differentiate into bona fide myelinating oligodendrocytes in vivo as efficiently as their control counterparts. Nicaise and colleagues reported that iPSC-NPCs from PPMS cases did not provide neuroprotection against active CNS demyelination compared to control iPSC-NPCs (44) and failed to promote oligodendrocyte progenitor genesis due to senescence without affecting their endogenous capacity to generate myelin-forming oligodendrocytes (21, 22). However, their myelinating potential was not evaluated against control cells. Generation of iPSC-oligodendrocyte progenitors from patients with PPMS or RRMS has also been reported by other groups, yet with no evidence for their capacity to become functional oligodendrocytes in vivo (23, 24). Thus, so far, no conclusion could be made regarding the potential impact of disease severity (PPMS verses RRMS) on the functionality of the iPSC-derived progeny.
We compared side by side, and at different time points after engraftment, hiOLs from patients with RRMS and controls including two pairs of homozygous twins discordant for disease. We found no significant difference in their capacity to timely differentiate (according to the human tempo of differentiation) and efficiently myelinate axons in the shiverer mouse in terms of the percentage of MBP+ cells generated, amount of myelin produced, length of MBP+ sheaths, and the ultrastructure and thickness of myelin sheaths. MS-hiOLs also reconstructed nodes of Ranvier expressing nodal components key to their function. We not only verified that the grafted MS-hiOLs derivatives were anatomically competent but also established their functionality at the electrophysiological level using (i) in vivo recordings of transcallosal evoked potentials and (ii) ex vivo recordings of the elicited current-voltage curves of the grafted MS-hiOLs verses controls. Our data show that the grafted MS-hiOLs were able to rescue the established delayed latency of shiverer mice to the same extent as control cells, as previously reported for human fetal glial progenitors grafted in the same model (11). Moreover, at the single-cell level, MS-hiOL–derived oligodendrocyte progenitors and oligodendrocytes did not harbor aberrant characteristics in membrane currents compared to control cells ex vivo. Thus, iPSC-derived human oligodendroglial cells shift their membrane properties with maturation as previously observed in vitro (45) and these properties are not impaired in MS.
The absence of differences among control and MS-derivatives might be due to different causes. One might consider that pluripotency induction could by in vitro manipulation, erase cell epigenetic traits and/or reverse cells to an embryonic state, and as a result, modulate their intrinsic characteristics. Yet, several reports have highlighted differences in the behavior of diseased iPSC-derived oligodendrocytes in comparison to those from healthy controls using the same technology in multifactorial diseases such as schizophrenia (19, 20), Huntington’s disease (18), and others (46). In this regard, direct reprogramming of somatic cells into the desired cell type, bypassing the pluripotent stage, could be an attractive alternative. However, so far only mouse fibroblasts have been successfully directly converted into oligodendroglial cells, and with relatively low efficiency (47, 48).
iPSCs were transduced with three transcription factors to generate hiOLs in a fast and efficient way (25). While we cannot rule out that the use of these three transcription factors may have obscured differences between MS and controls, results for controls are quite comparable to our previously published data based on human fetal oligodendrocyte progenitor engraftment in the Shi/Shi:Rag2−/− developing forebrain (49) or fetal NPC engrafted in the Shi/Shi:Rag2−/− demyelinated spinal cord (50), suggesting that transduction with the three transcription factors does not overly modify the behavior of the grafted human cells. It could also be argued that the absence of differences between control and MS monozygous twins is not surprising given their equal genetic background. Yet, comparing controls with nonsibling MS hiOLS (compare C1 with RRMS2 and RRMS3; C2 with RRMS1, RRMS2, and RRMS3; and C3 with RRMS1 and RRMS2) revealed no defect in myelination for MS cells as well.
Analysis of hiOLs from each donor showed differences within each group. This could result from phenotypic instability, heterogeneity among donors, or disease subtype. Yet, the clinical history of each patient suggests a certain homogeneity among the MS disease phenotype, all being RRMS. In addition, the equal survival and proliferation rates between both groups argue in favor of cell stability. These confounding observations sustain that differences in terms of myelination are most likely due to heterogeneity among individuals rather than phenotypic instability or disease subtype.
While most preclinical transplantation studies have focused on myelination potential as the successful outcome of axo-glia interactions, less is known about the capacity of the grafted cells to fulfill glial-glial interactions in the pan-glial syncytium, which could ensure maintenance of newly generated myelin (51) and cell homeostasis (52). Oligodendrocytes are extensively coupled to other oligodendrocytes and oligodendrocyte progenitors through the homologous gap junctions Cx47 (35). These intercellular interactions between competing oligodendroglial cells influence the number and length of myelin internodes and the initiation of differentiation (53, 54). Oligodendrocytes are also coupled to astrocytes through heterologous gap junctions such as Cx32/Cx30 and Cx47/Cx43 (55). Disruption of oligodendrocytes from each other and from astrocytes, i.e., deconstruction of pan-glial network, has been observed in experimental models of demyelination (unpublished data) and frequently reported in MS and neuromyelitis optica (37, 56, 57). Mutations in Cx47 and Cx32 result in developmental CNS and PNS abnormalities in leukodystrophies (58, 59). Moreover, experimental ablation of Cx47 results in aberrant myelination (60) and significantly abolished coupling of oligodendrocytes to astrocytes (35).
In view of the major role of Cx-mediated gap junctions among oligodendrocytes and between oligodendrocytes and astrocytes during myelin formation (55), we asked whether the MS-hiOL progeny was capable of making functional gap junctions with other glial cells, and integrating into the host panglial network. We show that grafted MS-hiOLs, in common with rodent oligodendrocytes, express Cx47 that was frequently shared not only between the human and murine oligodendrocytes (through Cx47-Cx47) but also in conjunction with the astrocyte Cx43 (via Cx47/Cx43). The dye-coupling study highlighted that MS-hiOLs, similar to control cells, were capable of forming functional gap junctions with neighbor murine or human glial cells, indicating that MS-hiOLs retained the intrinsic property, not only to myelinate host axons but also to functionally integrate into the host pan-glial network. While our study focused mainly on oligodendroglial cells, a small proportion of the grafted hiOLs differentiated into astrocytes expressing Cx43. These human astrocytes were detected associated with blood vessels or the subventricular zone, where they were structurally gap-junction coupled to mouse astrocytes as observed after engraftment of human fetal glial restricted progenitors (61).
Together, our data highlight that human skin–derived glia retain characteristics of embryonic/fetal brain–derived glia as observed for rodent cells (10). In particular, we show that MS-hiOLs timely differentiate into mature oligodendrocytes, functionally myelinate host axons and contribute to the human-mouse chimeric pan-glial network as efficiently as control-hiOLs. These observations favor a role for extrinsic rather than intrinsic oligodendroglial factors in the failed remyelination of MS. The International Multiple Sclerosis Genetics Consortium after analyzing the genomic map of more than 47,000 MS cases and 63,000 control subjects, implicated microglia, and multiple different peripheral immune cell populations in disease onset (62). Moreover, neuroinflammation appears to block oligodendrocyte differentiation and to alter their properties and thereby aggravate the autoimmune process (63). Furthermore, MS lymphocytes are reported to exhibit intrinsic capacities that drive myelin repair in a mouse model of demyelination (64). On the other hand, a recent study highlighted the presence of disease-specific oligodendroglia in MS (16, 17). However, it should be considered that most of the data in the later were collected using single nuclei RNA sequencing of postmortem tissues from MS or control subjects of different ages that were suffering from other disorders ranging from cancer to sepsis and undergoing various treatment, and so died for different reasons, that may have influenced the type or level of RNA expression by the cells. In addition, the presence of genetic variants that alter oligodendrocyte function in addition to that of immune cells was also found (39). While this oligodendrocyte dysfunction contributes to MS risk factor, whether it is involved in other aspects of MS such as severity, relapse rate, and rate of progression is not yet known.
Numerous factors may cause the failure of oligodendrocyte progenitor maturation comprising factors such as axonal damage and/or altered cellular and extracellular signaling within the lesion environment (65) without neglecting aged-related environmental and cellular changes (40). Although the cells generated in this study are more of an embryonic nature, and did not experienced the kind of inhibitory environment that is present in MS, our data provide valuable findings in the scenario of MS pathology highlighting that RRMS-hiOLs, regardless of major manipulators of the immune system, do not lose their intrinsic capacity to functionally myelinate and interact with other neuroglial cells in the CNS under nonpathological conditions. Whether RRMS-hiOLs or oligodendroglial cells directly reprogrammed from MS fibroblasts would behave similarly well, if challenged with neuropathological inflammatory conditions as opposed to conditions wherein the immune system is intact (presence of T and B cells), or whether they would reflect intrinsic aging properties will require further investigation.
In summary, our findings provide valuable insights not only into the biology of MS oligodendroglia but also their application for cell-based therapy and should contribute to the establishment of improved preclinical models for in vivo drug screening of pharmacological compounds targeting the oligodendrocyte progenitors, oligodendrocytes, and their interactions with the neuronal and pan-glial networks.
MATERIALS AND METHODS
Experimental design
We examined side by side the molecular, cellular, and functional behavior of MS hiOLs with their control counterparts after their engraftment in a dysmyelinating animal model to avoid the effect of major immune modulators. We used three MS and three control hiOLs including two monozygous twin pairs discordant for the disease. We performed in vivo studies in mouse with sample size between three to six animals per donor/time point/assay required to achieve significant differences. Numbers of replicates are listed in each figure legend. Animals were monitored carefully during all the study time, and animal welfare criteria for experimentation were fully respected. All experiments were randomized with regard to animal enrollment into treatment groups. The same experimenter handled the animals and performed the engraftment experiments to avoid errors. The data were analyzed by a group of authors.
Animals
Shiverer mice were crossed to Rag2 null immunodeficient mice to generate a line of Shi/Shi:Rag2−/− dysmyelinating-immunodeficient mice to (i) prevent rejection of the grafted human cells and allow detection of donor-derived WT myelin and (ii) investigate the original behavior of MS-derived oligodendrocytes in a B cell/T cell–free environment. Mice were housed under standard conditions of 12-hour light/12-hour dark cycles with ad libitum access to dry food and water at the ICM animal facility. Experiments were performed according to European Community regulations and INSERM ethical committee (authorization 75-348; 20/04/2005) and were approved by the local Darwin ethical committee.
Human cells
Fibroblasts were obtained under informed consent from three control and three RRMS subjects including two monozygous twin pairs discordant for the disease. They were reprogrammed into iPSCs using the replication incompetent Sendaï virus kit (Invitrogen) according to manufacturer’s instructions. Table S1 summarizes information about the human cell lines used in this study. The study was approved by the local ethical committees of Münster and Milan (AZ 2018-040-f-S, and Banca INSpe).
Human iPSCs were differentiated into NPC by treatment with small molecules as described (66, 67). Differentiation of NPCs into O4+ oligodendroglial cells used a poly-cistronic lentiviral vector containing the coding regions of the human transcription factors Sox10, Olig2, and Nkx6.2 (SON) followed by an IRES-pac cassette, allowing puromycin selection for 16 hours (25). For single-cell electrophysiological recordings, the IRES-pac cassette was replaced by a sequence encoding RFP. Briefly, human NPCs were seeded at 1.5 × 105 cells per well in 12-well plates, allowed to attach overnight and transduced with SON lentiviral particles and protamine sulfate (5 μg/ml) in fresh NPC medium. After extensive washing, viral medium was replaced with glial induction medium (GIM). After 4 days, GIM was replaced by differentiation medium (DM). After 12 days of differentiation, cells were dissociated by accutase treatment for 10 min at 37°C, washed with phosphate-buffered saline (PBS) and resuspended in PBS/0.5% bovine serum albumin (BSA) buffer, and singularized cells were filtered through a 70-μm cell strainer (BD Falcon). Cells were incubated with mouse immunoglobulin M (IgM) anti–O4-APC antibody (Miltenyi Biotech) following the manufacturer’s protocol, washed, resuspended in PBS/0.5% BSA buffer (5 × 106 cells/ml), and immediately sorted using a FACS Aria cell sorter (BD Biosciences). Subsequently, human O4+ hiOLs were frozen and stored in liquid nitrogen. Media details were provided in (25). hiOLS from each donor was assayed individually (no cell mix) and studied as follows for forebrain engraftment: immunohistochemistry (all donors, three to seven mice per time point), electron microscopy (C1 and RRMS1, four mice per donor at 16 wpg), in vivo electrophysiology (C1 and RRMS1, six mice per donor and eight mice per medium at 16 wpg), dye coupling, and ex-vivo electrophysiology (C1-RFP and RRMS3-RFP, six to seven mice per donor at 16 wpg). For spinal cord engraftment: immuno-histochemistry (C1 and RRMS3, 3 and 4 mice respectively at 12 wpg).
MS patient history
RRMS1: Disease duration at biopsy was 11 years. Started on Rebif 22 and switched to Rebif 44 because of relapses. Relapse was treated with bolus of cortisone 20 to 30 days before biopsy and then switched to natalizumab.
RRMS2: Disease duration at biopsy was 16 months. Relapse at disease onset. On Rebif 22 from disease onset until biopsy with no episodes. A new lesion was identified 3 months after biopsy. At the time of biopsy, the patient reported cognitive difficulties, no motor dysfunctions.
RRMS3: Disease duration at biopsy was 15 months. Relapse 6 months before biopsy with dysesthesias and hypoesthesia right thigh and buttock. Active lesion identified by magnetic resonance imaging at day 10. On Rebif smart 44 mcg, 50 days later, and skin biopsy 4 months later. A new gadolinium negative temporal lesion identified 2 months after biopsy and the patient switched to Tecfidera.
Cell transplantation
To assay hiOL contribution to forebrain developmental myelination, newborn Shi/Shi:Rag2−/− pups (n = 148) were cryo-anesthetized, and control and RRMS hiOLs were transplanted bilaterally, rostral to the corpus callosum. Injections (1 μl in each hemisphere and 105 cells/μl) were performed 1 mm caudally, 1 mm laterally from the bregma, and to a depth of 1 mm as previously described (49, 68). Animals were sacrificed at 4, 8, 12, 16, and, when indicated, 20 wpg for immunohistological studies and at one time point for electron microscopy (16 wpg), ex vivo (12 to 15 wpg), and in vivo (16 wpg) electrophysiology.
To assay the fate of hiOLs in the developing spinal cord, 4-week-old mice (n = 4) were anesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg) (Alcyon) and xylazine (10 mg/kg) (Alcyon) and received a single injection at low speed (1 μl/2 min) of hiOLs (1 μl, 105 cells/μl) at the spinal cord thoracic level using a stereotaxic frame equipped with a micromanipulator and a Hamilton syringe. Animals were sacrificed at 12 wpg for immunohistological studies.
Postmortem analysis
Immunohistochemistry
Shi/Shi:Rag2−/− mice grafted with control and RRMS hiOLs (n = 3 to 6 per group, donor and time point) were sacrificed by transcardiac perfusion-fixation with 4% paraformaldehyde in PBS. Tissues were postfixed in the same fixative for 1 hour and incubated in 20% sucrose in 1× PBS overnight before freezing at −80°C. Serial horizontal brain and spinal cord cross sections of 12 μm thickness were performed with a cryostat (CM3050S, Leica). Transplanted hiOLs were identified using anti-human cytoplasm [1:100; STEM121; Takara, Y40410, IgG1], anti-human nuclei (1:100; STEM101; Takara, Y40400, IgG1), and anti-human NOGOA (1:50; Santa Cruz Biotechnology, sc-11030, goat) antibodies. In vivo characterization was performed using a series of primary antibodies listed in tableS2. For MBP staining, sections were pretreated with ethanol (10 min, room temperature). For glial-glial interactions, oligodendrocyte-specific connexin was detected with anti-connexin 47 (1:200; Cx47; Invitrogen, 4A11A2, IgG1) and astrocyte-specific connexin, with anti-connexin 43 (1:50; Cx43; Sigma-Aldrich, C6219, rabbit), and sections were pretreated with methanol (10 min, −20°C). Secondary antibodies conjugated with fluorescein isothiocyanate, tetramethyl rhodamine isothiocyanate (SouthernBiotech), or Alexa Fluor 647 (Life Technologies) were used, respectively, at 1:100 and 1:1000. Biotin-conjugated antibodies followed by AMCA AVIDIN D (1:20; Vector, A2006). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml; Sigma-Aldrich) (1:1000). Tissue scanning, cell visualization, and imaging were performed with a Carl Zeiss microscope equipped with ApoTome 2.
Electron microscopy
For electron microscopy, Shi/Shi:Rag2−/− mice grafted with control and RRMS hiOLs (n = 4 per group) were perfused with 1% PBS followed by a mixture of 4% paraformaldehyde/5% glutaraldehyde (Electron Microscopy Sciences) in 1% PBS. After 2-hour postfixation in the same solution, 100-μm-thick sagittal sections were cut and fixed in 2% osmium tetroxide (Sigma-Aldrich) overnight. After dehydration, samples were flat-embedded in Epon. Ultra-thin sections (80 nm) of the median corpus callosum were examined and imaged with a HITACHI 120 kV HT-7700 electron microscope.
Electrophysiology in vivo
Electrophysiological recordings were performed in mice grafted with MS- and control-hiOLs, and compared with nongrafted intact or medium injected Shi/Shi:Rag2−/− mice and WT mice 16 weeks after injection (n = 4 to 6 per group) as described (11). Briefly mice were anesthetized with 2 to 4% isoflurane performed under analgesia (0.1 mg/kg buprecare) and placed in a stereotaxic frame (D. Kopf, Tujunga, CA, USA). Body temperature was maintained at 37°C by a feedback-controlled heating blanket (CMA Microdialysis). Electrical stimulation (0.1 ms at 0 to 0.1 mA) was applied using a bipolar electrode (FHC- CBBSE75) inserted to a depth of 200 μm into the left cortex at 2 mm posterior to bregma and 3 mm from the midline. At the same coordinates in the contralateral hemisphere, homemade electrodes were positioned for recording local field potentials (LFPs) generated by transcallosal electric stimulation. Electrical stimulation and evoked LFPs were performed by the data acquisition system apparatus (Neurosoft, Russia), and signals were filtered at 0.01 to 1 000 Hz. Each response latency (in ms) was measured as the time between the onset of stimulus artifact to the first peak for each animal. A ground electrode was placed subcutaneously over the neck.
Electrophysiology in acute brain slices
Slice preparation and recordings
Acute coronal slices (300 μm) containing corpus callosum were made from Shi/Shi:Rag2−/− mice grafted with control (n = 7) and RRMS (n = 6) RFP+ hiOLs. They were prepared from grafted mice between 12 and 15 wpg as previously described (69). Briefly, slices were performed in a chilled cutting solution containing 93 mM N-methyl-d-glucamine, 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM Hepes, 25 mM glucose, 2 mM urea, 5 mM Na-ascorbate, 3 mM Na-pyruvate, 0.5 mM CaCl2, and 10 mM MgCl2 (pH 7.3 to pH 7.4; 95% O2 and 5% CO2) and kept in the same solution for 8 min at 34°C. Then, they were transferred for 20 min to solution at 34°C containing 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 20 mM glucose, 5 mM Na-pyruvate, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.3 to pH 7.4; 95% O2 and 5% CO2). Transplanted RFP+ hiOLs were visualized with a 40× fluorescent water-immersion objective on an Olympus BX51 microscope coupled to a CMOS digital camera (TH4-200 OptiMOS) and an light-emitting diode light source (CoolLed p-E2, Scientifica, UK) and recorded in voltage-clamp mode with an intracellular solution containing 130 mM K-gluconate, 0.1 mM EGTA, 2 mM MgCl2, 10 mM Hepes, 10 mM γ-aminobutyric acid, 2 mM Na2-adenosine 5′-triphosphate, 0.5 mM Na-guanosine 5′-triphosphate, 10 mM Na2-phosphocreatine, and 5.4 mM biocytin (pH 7.23). Holding potentials were corrected by a junction potential of −10 mV. Electrophysiological recordings were performed with Multiclamp 700B and Pclamp10.6 software (Molecular Devices). Signals were filtered at 3 kHz, digitized at 10 kHz, and analyzed off-line.
Immunostainings and imaging of recorded slices
For analysis of recorded cells, one single RFP+ cell per hemisphere was recorded in a slice and loaded with biocytin for 25 min in whole-cell configuration. After gently removing the patch pipette, biocytin was allowed to diffuse for at least 10 min before the slice was fixed 2 hours in 4% paraformaldehyde at 4°C. Then, the slice was rinsed three times in PBS for 10 min and incubated with 1% Triton X-100 and 10% normal goat serum (NGS) for 2 hours. After washing in PBS, slices were immunostained for SOX10, CC1, and STEM101/121. Tissues were incubated with primary antibodies for 3 days at 4°C. Secondary antibodies were diluted in 2% NGS and 0.2% Triton X-100. Tissues were incubated with secondary antibodies for 2 hours at room temperature. Biocytin was revealed with secondary antibodies using DyLight-488 streptavidin (Vector Laboratories, Burlingame, USA, 1:200). Images of biocytin-loaded cells were acquired either with a Carl Zeiss microscope equipped with ApoTome 2 or a LEICA SP8 confocal microscope (63× oil objective; numerical aperture, 1.4; 0.75-μm Z-step) and processed with National Institutes of Health ImageJ software (70).
Automated quantification of oligodendrocyte myelination in vivo
We adapted the heuristic algorithm from (29) to identify STEM+ MBP+ OLs in tissue sections. The foundations of the quantitative method remained the same. A ridge-filter extracted sheath-like objects based on intensity and segments associated to cell bodies using watershed segmentation. Two additional features adapted the workflow beyond its original in vitro application. First, we added functionality to allow colocalization of multiple fluorescent stains, as we needed to quantify triple positive STEM+/MBP+/DAPI+ cell objects. Second, because oligodendrocyte sheaths are not parallel and aligned in situ as they are in dissociated nanofiber cell cultures, we adapted the algorithm to report additional metrics about MBP production locally and globally that do not rely on the dissociation of sheaths in dense regions.
Cell nuclei were identified using watershed segmentation of DAPI+ regions and then colocalized pixel-wise with STEM+ objects. The DAPI+ nuclei were then used as local minima to seed a watershed segmentation of the STEM+ stain to separate nearby cell bodies. Last, the identified STEM+ cell bodies were colocalized with overlapping MBP+ sheath-like ridges to define ensheathed cells. We reported the area of MBP overlapping with STEM fluorescence in colocalized regions associated with individual cells, as well as the number of single, double, and triple fluorescently labeled cells. In addition, different cellular phenotypes were noted in situ that were then captured with the adapted algorithm. Qualitatively, we observed cells with expansive MBP production without extended linear sheath-like segments that were not observed in previous applications of the algorithm. These cells were denoted as “tuft” cells, and were quantitatively defined as STEM+/MBP+/DAPI+ cells without fluorescent ridges that could be identified as extended sheath-like objects.
The myelination potential of three control and 3 MS hiOLs was evaluated at 4, 8, 12, 16, and 20 wpg (n = 2 to 7 per line and per time point; n = 6 to 14 per time point). For each animal, six serial sections at 180-μm intervals were analyzed. The percentage of MBP+ cells (composed of ensheathed or tuft cells) was evaluated. Total MBP+ area per STEM+ cells and the average length of MBP+ sheaths per MBP+ cells were analyzed.
Other quantification
Cell survival, proliferation, and differentiation in vivo
The number of STEM101+ grafted cells expressing Caspase3, or Ki67, or SOX10 and CC1 was quantified in the core of the corpus callosum at 8, 12, and 16 wpg. For each animal (n = 3 per group), six serial sections at 180-μm intervals were analyzed. Cell counts were expressed as the percentage of total STEM101+ cells.
Myelination by electron microscopy
G ratio (diameter of axon/diameter of axon and myelin sheath) of donor-derived compact myelin was measured as previously described (10). Briefly, the maximum and minimum diameters of a given axon and the maximum and minimum axon plus myelin sheath diameter were measured with the ImageJ software at a magnification of 62,000 for a minimum of 70 myelinated axons per animal. Data were expressed as the mean of the maximal and minimal values for each axon for mice from each group (n = 4 mice per group). Myelin compaction was confirmed at a magnification of 220,000.
Statistical analysis
Data are presented as means + SEM. Statistical significance was determined by two-tailed Mann Whitney U test when comparing two statistical groups, and with one-way or two-way analysis of variance (ANOVA) followed by Tukey’s or Dunnett’s (in vivo electrophysiology) multiple comparison tests for multiple groups. Because electrophysiological data in brain slices do not follow a normal distribution after a D’Agostino-Pearson normality test, we also performed two-tailed Mann-Whitney U test for comparison between groups. Statistics were done in GraphPad Prism 5.00 and GraphPad Prism 8.2.1 (GraphPad Software Inc., USA). See the figure captions for the test used in each experiment.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Progressive MS Alliance [PMSA; collaborative research network PA-1604-08492 (BRAVEinMS)] to G.M., J.P.A., A.B.-V.E., and T.K., the National MS Society (NMSS RG-1801-30020 to T.K. and A.B.-V.E.), INSERM and ICM grants to A.B.-V.E., the German Research Foundation (DFG CRC-TR-128B07 to T.K.), and the Italian Multiple Sclerosis Foundation (FISM) (project no. “Neural Stem Cells in MS” to G.M.). M.C.A. was supported by grants from Fondation pour l’aide à la recherche sur la Sclérose en Plaques (ARSEP) and a sub-award agreement from the University of Connecticut with funds provided by grant no. RG-1612-26501 from National Multiple Sclerosis Society. During this work, S.M. was funded by European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). B.G.-D. and M.J.F.L. were supported by the PMSA, PA-1604-08492 and the National MS Society (RG-1801-30020), respectively. B.M.-S. was supported by a Ph.D. fellowship from the French Ministry of Research (ED BioSPC). A.B. and M.C.A. thank respective imaging facilities, ICM Quant and IPNP NeurImag and their respective funding sources ‘Institut des Neurosciences Translationnelles’ ANR-10-IAIHU-06 Fondation Leducq. Author contributions: Conceptualization: S.M. and A.B.-V.E. Methodology: S.M., L.S., B.M.-S., Y.K.T.X., B.G.-D., M.J.F.L., D.R., L.O., K.-P.K., H.R.S., J.P.A., T.K., G.M., T.E.K., M.C.A., and A.B.V.-E. Formal analysis: S.M., B.M-S., Y.K.T.X., M.C.A., and A.B.-V.E. Writing: S.M. and A.B.V.-E, with editing and discussion from all coauthors Funding acquisition: S.M. and A.B.V.-E. Supervision: A.B.V.-E. Competing interests: T.K. has a pending patent application for the generation of human oligodendrocytes. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabc6983/DC1
REFERENCES AND NOTES
- 1.Goldschmidt T., Antel J., König F. B., Brück W., Kuhlmann T., Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 72, 1914–1921 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Patrikios P., Stadelmann C., Kutzelnigg A., Rauschka H., Schmidbauer M., Laursen H., Sorensen P. S., Brück W., Lucchinetti C., Lassmann H., Remyelination is extensive in a subset of multiple sclerosis patients. Brain 129, 3165–3172 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Bodini B., Veronese M., García-Lorenzo D., Battaglini M., Poirion E., Chardain A., Freeman L., Louapre C., Tchikviladze M., Papeix C., Dollé F., Zalc B., Lubetzki C., Bottlaender M., Turkheimer F., Stankoff B., Dynamic imaging of individual remyelination profiles in multiple sclerosis. Ann. Neurol. 79, 726–738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohno N., Ikenaka K., Axonal and neuronal degeneration in myelin diseases. Neurosci. Res. 139, 48–57 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Chang A., Tourtellotte W. W., Rudick R., Trapp B. D., Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 346, 165–173 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Kuhlmann T., Miron V., Cui Q., Wegner C., Antel J., Brück W., Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131, 1749–1758 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Wolswijk G., Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J. Neurosci. 18, 601–609 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mozafari S., Sherafat M. A., Javan M., Mirnajafi-Zadeh J., Tiraihi T., Visual evoked potentials and MBP gene expression imply endogenous myelin repair in adult rat optic nerve and chiasm following local lysolecithin induced demyelination. Brain Res. 1351, 50–56 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Smith K. J., McDonald W. I., Blakemore W. F., Restoration of secure conduction by central demyelination. Trans. Am. Neurol. Assoc. 104, 25–29 (1979). [PubMed] [Google Scholar]
- 10.Mozafari S., Laterza C., Roussel D., Bachelin C., Marteyn A., Deboux C., Martino G., Baron-Van Evercooren A., Skin-derived neural precursors competitively generate functional myelin in adult demyelinated mice. J. Clin. Invest. 125, 3642–3656 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Windrem M. S., Schanz S. J., Guo M., Tian G.-F., Washco V., Stanwood N., Rasband M., Roy N. S., Nedergaard M., Havton L. A., Wang S., Goldman S. A., Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell 2, 553–565 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laterza C., Merlini A., De Feo D., Ruffini F., Menon R., Onorati M., Fredrickx E., Muzio L., Lombardo A., Comi G., Quattrini A., Taveggia C., Farina C., Cattaneo E., Martino G., iPSC-derived neural precursors exert a neuroprotective role in immune-mediated demyelination via the secretion of LIF. Nat. Commun. 4, 2597 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Mei F., Lehmann-Horn K., Shen Y.-A. A., Rankin K. A., Stebbins K. J., Lorrain D. S., Pekarek K., Sagan S. A., Xiao L., Teuscher C., von Büdingen H.-C., Wess J., Lawrence J. J., Green A. J., Fancy S. P., Zamvil S. S., Chan J. R., Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife 5, e18246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verden D., Macklin W. B., Neuroprotection by central nervous system remyelination: Molecular, cellular, and functional considerations. J. Neurosci. Res. 94, 1411–1420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leong S. Y., Rao V. T. S., Bin J. M., Gris P., Sangaralingam M., Kennedy T. E., Antel J. P., Heterogeneity of oligodendrocyte progenitor cells in adult human brain. Ann. Clin. Transl. Neurol. 1, 272–283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falcão A. M., van Bruggen D., Marques S., Meijer M., Jäkel S., Agirre E., Samudyata, Floriddia E. M., Vanichkina D. P., ffrench-Constant C., Williams A., Guerreiro-Cacais A. O., Castelo-Branco G., Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 24, 1837–1844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jäkel S., Agirre E., Falcão A. M., van Bruggen D., Lee K. W., Knuesel I., Malhotra D., ffrench-Constant C., Williams A., Castelo-Branco G., Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osipovitch M., Martinez A. A., Mariani J. N., Cornwell A., Dhaliwal S., Zou L., Chandler-Militello D., Wang S., Li X., Benraiss S.-J., Agate R., Lampp A., Benraiss A., Windrem M. S., Goldman S. A., Human ESC-derived chimeric mouse models of huntington’s disease reveal cell-intrinsic defects in glial progenitor cell differentiation. Cell Stem Cell 24, 107–122.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McPhie D. L., Nehme R., Ravichandran C., Babb S. M., Ghosh S. D., Staskus A., Kalinowski A., Kaur R., Douvaras P., Du F., Ongur D., Fossati V., Eggan K., Cohen B. M., Oligodendrocyte differentiation of induced pluripotent stem cells derived from subjects with schizophrenias implicate abnormalities in development. Transl. Psychiatry 8, 230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Windrem M. S., Osipovitch M., Liu Z., Bates J., Chandler-Militello D., Zou L., Munir J., Schanz S., McCoy K., Miller R. H., Wang S., Nedergaard M., Findling R. L., Tesar P. J., Goldman S. A., Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21, 195–208.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicaise A. M., Wagstaff L. J., Willis C. M., Paisie C., Chandok H., Robson P., Fossati V., Williams A., Crocker S. J., Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 116, 9030–9039 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douvaras P., Wang J., Zimmer M., Hanchuk S., O’Bara M. A., Sadiq S., Sim F. J., Goldman J., Fossati V., Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem Cell Reports 3, 250–259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.García-León J. A., Kumar M., Boon R., Chau D., One J., Wolfs E., Eggermont K., Berckmans P., Gunhanlar N., de Vrij F., Lendemeijer B., Pavie B., Corthout N., Kushner S. A., Dávila J. C., Lambrichts I., Hu W.-S., Verfaillie C. M., SOX10 single transcription factor-based fast and efficient generation of oligodendrocytes from human pluripotent stem cells. Stem Cell Reports 10, 655–672 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song B., Sun G., Herszfeld D., Sylvain A., Campanale N. V., Hirst C. E., Caine S., Parkington H. C., Tonta M. A., Coleman H. A., Short M., Ricardo S. D., Reubinoff B., Bernard C. C. A., Neural differentiation of patient specific iPS cells as a novel approach to study the pathophysiology of multiple sclerosis. Stem Cell Res. 8, 259–273 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Ehrlich M., Mozafari S., Glatza M., Starost L., Velychko S., Hallmann A.-L., Cui Q.-L., Schambach A., Kim K.-P., Bachelin C., Marteyn A., Hargus G., Johnson R. M., Antel J., Sterneckert J., Zaehres H., Schöler H. R., Evercooren A. B.-V., Kuhlmann T., Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. U.S.A. 114, E2243–E2252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seilhean D., Gansmüller A., Baron-Van Evercooren A., Gumpel M., Lachapelle F., Myelination by transplanted human and mouse central nervous system tissue after long-term cryopreservation. Acta Neuropathol. 91, 82–88 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Windrem M. S., Nunes M. C., Rashbaum W. K., Schwartz T. H., Goodman R. A., McKhann G. II, Roy N. S., Goldman S. A., Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat. Med. 10, 93–97 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Wang S., Bates J., Li X., Schanz S., Chandler-Militello D., Levine C., Maherali N., Studer L., Hochedlinger K., Windrem M., Goldman S. A., Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12, 252–264 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y. K. T., Chitsaz D., Brown R. A., Cui Q. L., Dabarno M. A., Antel J. P., Kennedy T. E., Deep learning for high-throughput quantification of oligodendrocyte ensheathment at single-cell resolution. Commun. Biol. 2, 116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bechler M. E., Byrne L., Ffrench-Constant C., CNS myelin sheath lengths are an intrinsic property of oligodendrocytes. Curr. Biol. 25, 2411–2416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gansmüller A., Lachapelle F., Baron-Van Evercooren A., Hauw J. J., Baumann N., Gumpel M., Transplantations of newborn CNS fragments into the brain of shiverer mutant mice: Extensive myelination by transplanted oligodendrocytes. II. Electron microscopic study. Dev. Neurosci. 8, 197–207 (1986). [DOI] [PubMed] [Google Scholar]
- 32.Ruff C. A., Ye H., Legasto J. M., Stribbell N. A., Wang J., Zhang L., Fehlings M. G., Effects of adult neural precursor-derived myelination on axonal function in the perinatal congenitally dysmyelinated brain: Optimizing time of intervention, developing accurate prediction models, and enhancing performance. J. Neurosci. 33, 11899–11915 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kukley M., Nishiyama A., Dietrich D., The fate of synaptic input to NG2 glial cells: Neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J. Neurosci. 30, 8320–8331 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahel A., Ortiz F. C., Kerninon C., Maldonado P. P., Angulo M. C., Nait-Oumesmar B., Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Front. Cell. Neurosci. 9, 77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maglione M., Tress O., Haas B., Karram K., Trotter J., Willecke K., Kettenmann H., Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32. Glia 58, 1104–1117 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Basu R., Sarma J. D., Connexin 43/47 channels are important for astrocyte/ oligodendrocyte cross-talk in myelination and demyelination. J. Biosci. 43, 1055–1068 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papaneophytou C., Georgiou E., Kleopa K. A., The role of oligodendrocyte gap junctions in neuroinflammation. Channels (Austin) 13, 247–263 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vilariño-Güell C., Zimprich A., Martinelli-Boneschi F., Herculano B., Wang Z., Matesanz F., Urcelay E., Vandenbroeck K., Leyva L., Gris D., Massaad C., Quandt J. A., Traboulsee A. L., Encarnacion M., Bernales C. Q., Follett J., Yee I. M., Criscuoli M. G., Deutschländer A., Reinthaler E. M., Zrzavy T., Mascia E., Zauli A., Esposito F., Alcina A., Izquierdo G., Espino-Paisán L., Mena J., Antigüedad A., Urbaneja-Romero P., Ortega-Pinazo J., Song W., Sadovnick A. D., Exome sequencing in multiple sclerosis families identifies 12 candidate genes and nominates biological pathways for the genesis of disease. PLOS Genet. 15, e1008180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Factor D. C., Barbeau A. M., Allan K. C., Hu L. R., Madhavan M., Hoang A. T., Hazel K. E. A., Hall P. A., Nisraiyya S., Najm F. J., Miller T. E., Nevin Z. S., Karl R. T., Lima B. R., Song Y., Sibert A. G., Dhillon G. K., Volsko C., Bartels C. F., Adams D. J., Dutta R., Gallagher M. D., Phu W., Kozlenkov A., Dracheva S., Scacheri P. C., Tesar P. J., Corradin O., Cell type-specific intralocus interactions reveal oligodendrocyte mechanisms in MS. Cell 181, 382–395.e21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franklin R. J. M., ffrench-Constant C., Regenerating CNS myelin – from mechanisms to experimental medicines. Nat. Rev. Neurosci. 18, 753–769 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Stangel M., Kuhlmann T., Matthews P. M., Kilpatrick T. J., Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat. Rev. Neurol. 13, 742–754 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Yeung M. S. Y., Djelloul M., Steiner E., Bernard S., Salehpour M., Possnert G., Brundin L., Frisén J., Dynamics of oligodendrocyte generation in multiple sclerosis. Nature 566, 538–542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duncan I. D., Radcliff A. B., Heidari M., Kidd G., August B. K., Wierenga L. A., The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. U.S.A. 115, E11807–E11816 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nicaise A. M., Banda E., Guzzo R. M., Russomanno K., Castro-Borrero W., Willis C. M., Johnson K. M., Lo A. C., Crocker S. J., iPS-derived neural progenitor cells from PPMS patients reveal defect in myelin injury response. Exp. Neurol. 288, 114–121 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Livesey M. R., Magnani D., Cleary E. M., Vasistha N. A., James O. T., Selvaraj B. T., Burr K., Story D., Shaw C. E., Kind P. C., Hardingham G. E., Wyllie D. J. A., Chandran S., Maturation and electrophysiological properties of human pluripotent stem cell-derived oligodendrocytes. Stem Cells 34, 1040–1053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chanoumidou K., Mozafari S., Baron-Van Evercooren A., Kuhlmann T., Stem cell derived oligodendrocytes to study myelin diseases. Glia 68, 705–720 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Najm F. J., Lager A. M., Zaremba A., Wyatt K., Caprariello A. V., Factor D. C., Karl R. T., Maeda T., Miller R. H., Tesar P. J., Transcription factor-mediated reprogramming of fibroblasts to expandable, myelinogenic oligodendrocyte progenitor cells. Nat. Biotechnol. 31, 426–433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang N., Zuchero J. B., Ahlenius H., Marro S., Ng Y. H., Vierbuchen T., Hawkins J. S., Geissler R., Barres B. A., Wernig M., Generation of oligodendroglial cells by direct lineage conversion. Nat. Biotechnol. 31, 434–439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marteyn A., Sarrazin N., Yan J., Bachelin C., Deboux C., Santin M. D., Gressens P., Zujovic V., Baron-Van Evercooren A., Modulation of the innate immune response by human neural precursors prevails over oligodendrocyte progenitor remyelination to rescue a severe model of pelizaeus-merzbacher disease. Stem Cells 34, 984–996 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Buchet D., Garcia C., Deboux C., Nait-Oumesmar B., Baron-Van Evercooren A., Human neural progenitors from different foetal forebrain regions remyelinate the adult mouse spinal cord. Brain 134, 1168–1183 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Tress O., Maglione M., May D., Pivneva T., Richter N., Seyfarth J., Binder S., Zlomuzica A., Seifert G., Theis M., Dere E., Kettenmann H., Willecke K., Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J. Neurosci. 32, 7499–7518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kamasawa N., Sik A., Morita M., Yasumura T., Davidson K. G., Nagy J. I., Rash J. E., Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: Implications for ionic homeostasis and potassium siphoning. Neuroscience 136, 65–86 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chong S. Y. C., Rosenberg S. S., Fancy S. P. J., Zhao C., Shen Y.-A. A., Hahn A. T., McGee A. W., Xu X., Zheng B., Zhang L. I., Rowitch D. H., Franklin R. J. M., Lu Q. R., Chan J. R., Neurite outgrowth inhibitor Nogo-A establishes spatial segregation and extent of oligodendrocyte myelination. Proc. Natl. Acad. Sci. U.S.A. 109, 1299–1304 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenberg S. S., Kelland E. E., Tokar E., De la Torre A. R., Chan J. R., The geometric and spatial constraints of the microenvironment induce oligodendrocyte differentiation. Proc. Natl. Acad. Sci. U.S.A. 105, 14662–14667 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vejar S., Oyarzún J. E., Retamal M. A., Ortiz F. C., Orellana J. A., Connexin and pannexin-based channels in oligodendrocytes: Implications in brain health and disease. Front. Cell. Neurosci. 13, 3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masaki K., Early disruption of glial communication via connexin gap junction in multiple sclerosis, Baló’s disease and neuromyelitis optica. Neuropathology 35, 469–480 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Rash J. E., Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: Pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 168, 982–1008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kleopa K. A., Scherer S. S., Molecular genetics of X-linked Charcot-Marie-Tooth disease. Neuromolecular Med. 8, 107–122 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Uhlenberg B., Schuelke M., Rüschendorf F., Ruf N., Kaindl A. M., Henneke M., Thiele H., Stoltenburg-Didinger G., Aksu F., Topaloğlu H., Nürnberg P., Hübner C., Weschke B., Gärtner J., Mutations in the gene encoding gap junction protein α12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am. J. Hum. Genet. 75, 251–260 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Odermatt B., Wellershaus K., Wallraff A., Seifert G., Degen J., Euwens C., Fuss B., Büssow H., Schilling K., Steinhäuser C., Willecke K., Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 23, 4549–4559 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han X., Chen M., Wang F., Windrem M., Wang S., Shanz S., Xu Q., Oberheim N. A., Bekar L., Betstadt S., Silva A. J., Takano T., Goldman S. A., Nedergaard M., Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12, 342–353 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.International Multiple Sclerosis Genetics Consortium , Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirby L., Jin J., Cardona J. G., Smith M. D., Martin K. A., Wang J., Strasburger H., Herbst L., Alexis M., Karnell J., Davidson T., Dutta R., Goverman J., Bergles D., Calabresi P. A., Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 10, 3887 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.El Behi M., Sanson C., Bachelin C., Guillot-Noël L., Fransson J., Stankoff B., Maillart E., Sarrazin N., Guillemot V., Abdi H., Cournu-Rebeix I., Fontaine B., Zujovic V., Adaptive human immunity drives remyelination in a mouse model of demyelination. Brain 140, 967–980 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Franklin R. J. M., ffrench-Constant C., Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 9, 839–855 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Reinhardt P., Glatza M., Hemmer K., Tsytsyura Y., Thiel C. S., Höing S., Moritz S., Parga J. A., Wagner L., Bruder J. M., Wu G., Schmid B., Röpke A., Klingauf J., Schwamborn J. C., Gasser T., Schöler H. R., Sterneckert J., Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLOS ONE 8, e59252 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ehrlich M., Hallmann A.-L., Reinhardt P., Araúzo-Bravo M. J., Korr S., Röpke A., Psathaki O. E., Ehling P., Meuth S. G., Oblak A. L., Murrell J. R., Ghetti B., Zaehres H., Schöler H. R., Sterneckert J., Kuhlmann T., Hargus G., Distinct neurodegenerative changes in an induced pluripotent stem cell model of frontotemporal dementia linked to mutant TAU protein. Stem Cell Reports 5, 83–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lachapelle F., Duhamel-Clerin E., Gansmüller A., Baron-Van Evercooren A., Villarroya H., Gumpel M., Transplanted transgenically marked oligodendrocytes survive, migrate and myelinate in the normal mouse brain as they do in the shiverer mouse brain. Eur. J. Neurosci. 6, 814–824 (1994). [DOI] [PubMed] [Google Scholar]
- 69.Ortiz F. C., Habermacher C., Graciarena M., Houry P.-Y., Nishiyama A., Nait Oumesmar B., Angulo M. C., Neuronal activity in vivo enhances functional myelin repair. JCI Insight 5, e123434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabc6983/DC1