

Transcriptomic profiles of Alzheimer’s disease patients and healthy individuals were investigated using cell-free mRNA sequencing.
Abstract
The lack of accessible noninvasive tools to examine the molecular alterations occurring in the brain limits our understanding of the causes and progression of Alzheimer’s disease (AD), as well as the identification of effective therapeutic strategies. Here, we conducted a comprehensive profiling of circulating, cell-free messenger RNA (cf-mRNA) in plasma of 126 patients with AD and 116 healthy controls of similar age. We identified 2591 dysregulated genes in the cf-mRNA of patients with AD, which are enriched in biological processes well known to be associated with AD. Dysregulated genes included brain-specific genes and resembled those identified to be dysregulated in postmortem AD brain tissue. Furthermore, we identified disease-relevant circulating gene transcripts that correlated with the severity of cognitive impairment. These data highlight the potential of high-throughput cf-mRNA sequencing to evaluate AD-related pathophysiological alterations in the brain, leading to precision healthcare solutions that could improve AD patient management.
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia, affecting more than 40 million people, and is projected to triple by 2050 (1). AD is a neurodegenerative condition characterized by the accumulation of extracellular amyloid-β (Aβ) peptide, deposition of tau proteins, and intracellular formation of neurofibrillary tangles in the brain, which manifests years before clinical symptomatology (2). Multiple interrelated causes of AD including synaptic loss, oxidation, inflammation, misfolded proteins, and mitochondrial dysfunction subsequently lead to the loss of neurons and brain dysfunction (3–5). Although postmortem examination remains the gold standard for establishing AD pathology, current diagnostic guidelines use psychometric tests and clinical criteria, to establish the severity of cognitive impairment and evaluate Aβ and tau levels in cerebrospinal fluid (CSF), or use positron emission tomography (PET) brain imaging (6, 7). While medications available for patients with AD can temporarily manage the symptoms, there are no effective therapies that can cure or prevent AD. Furthermore, the development of effective therapies is hampered by the inherent difficulties associated with studying molecular changes in the brain without invasive tests. Therefore, accessible noninvasive tools that may be used to evaluate the molecular alterations in the brain of patients with AD should accelerate the development and monitoring of therapeutic strategies and the identification of biomarkers for early diagnosis and prognosis of patients with AD for precision health (8).
Gene expression profiling studies using postmortem human brain tissue samples have demonstrated that the transcriptional profile of patients with AD differs substantially from healthy individuals (9, 10). Moreover, these studies confirmed that AD pathogenesis reflects previously postulated mechanisms, as well as previously unidentified pathways that may contribute to the development of dementia (11). Considering that AD is a heterogeneous disease, characterization of cognitively impaired patients at the transcriptomic level would help elucidate the etiologies of AD and, thereby, improve the precision of AD patient management and drug development. However, difficulties associated with accessing brain tissues from patients with AD limit the applicability of tissue-based transcriptomic characterization of AD.
Recently, blood-based liquid biopsies assessing circulating nucleic acids have emerged as an alternative for noninvasive examination of molecular alterations (12–16). In particular, circulating cf-mRNA has been shown to contain transcripts that are derived from multiple organs, including the brain, and can be used to evaluate organ-specific transcriptional changes (17). The short half-life of cell-free mRNA (cf-mRNA) suggests the potential to track dynamic pathological changes. With accumulating evidence that RNA molecules cross the blood-brain barrier (18) and the identification of AD- and glioblastoma-specific transcripts in plasma of those patients (19, 20), circulating transcripts derived from the brain are promising bioanalytes for noninvasive molecular characterization of neurological disorders such as AD.
Here, we profiled plasma cf-mRNA of 126 patients with AD and 116 healthy controls with a similar age distribution. We identified gene transcripts differentially present in plasma of patients with AD, as well as genes correlated with the severity of dementia. These identified genes are enriched in biological processes associated with AD, such as synaptic dysfunction, mitochondrial dysfunction, and inflammation. Furthermore, we used genes differentially present in circulation to categorize pathological subtypes among patients with AD and built cf-mRNA–based classifiers that robustly discriminate controls from patients with AD. Collectively, our work highlights cf-mRNA profiling as a potential tool to noninvasively characterize diseases such as AD. Moreover, integrated analysis of cf-mRNA profiling with clinical information could be used for improved AD patient management and identification of new therapeutic targets for precision health.
RESULTS
cf-mRNA sequencing technical performance assessment
AD and control samples were sourced from patients across four academic centers and one commercial source. We sequenced cf-mRNA extracted from up to 1 ml of plasma obtained from 126 patients with AD and 116 non-cognitive controls (NCIs) with a similar age distribution (table S1). The average plasma cf-RNA yield did not differ between AD and control groups (P = 0.27, Student’s t test; fig. S2A), and approximately 4 ng of cf-RNA was used to generate each sequencing library. Following library preparation and sequencing runs, the median protein-coding genes identified was 11,714 [genes detected at >5 transcripts per million (TPM)] (Fig. 1A). Using external RNA spike-in mix controls ERCC (External RNA Controls Consortium) (21), the accuracy of the sequencing assay was confirmed, with the observed levels of ERCC transcripts correlating tightly with the expected spiked-in copy numbers (median ρ = 0.93, Spearman’s rank correlation; Fig. 1B). Of the 242 samples, we generated technical replicates in the first 96 samples processed. We generated two replicates by splitting RNA samples into two aliquots and subsequently generating cDNA and libraries independently. Comparison of the transcriptomic profiles between 96 pairs of technical replicates showed satisfactory correlation (median ρ = 0.84, Spearman’s rank correlation), highlighting the technical reproducibility of our approach, and was used as the rationale for including only one replicate for the remainder of the samples (fig. S2B). Last, the read distribution across exon-intron splice junctions showed that DNA contamination was negligible (Fig. 1C). Together, these results demonstrated reliable technical performance of the cf-mRNA sequencing assay for generating diverse, quantitative, and reproducible sequencing data.
Fig. 1. cf-mRNA sequencing is an accurate approach for characterizing the cf-mRNA transcriptome.
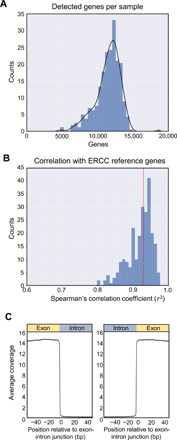
(A) Histogram of genes detected per sample (TPM > 5). (B) Histogram of Spearman’s correlation coefficient of observed versus expected copy number based on spiked-in ERCC control. (C) Aggregated coverage across all the exon-intron junctions of consistently detected genes (TPM > 5 in all NCI controls). bp, base pairs.
cf-mRNA profiling reveals AD-associated molecular changes
While AD-associated transcriptomic alterations in the postmortem brain tissues are well characterized (22–24), the cf-mRNA transcriptome of patients with AD has yet to be characterized. Profiling of AD molecular changes compared to NCI used the complete dataset, and the analysis steps are described in Fig. 2A and tables S1 and S2 for participant characteristics. Comparison of age between patients with AD and controls showed no difference between these groups (P = 0.3, Student’s t test; table S1). To account for preanalytic site-specific effects, we adjusted for source site in our analysis. Sample distribution based on the first two principal components (PCs) from a PC analysis (PCA) before and after adjustment is shown in fig. S2 (C and D) (see also Materials and Methods). Applying a negative binomial model adjusting for site using DESeq2, we identified 2591 genes differentially expressed between AD and NCI groups using raw read counts from all samples [false discovery rate (FDR) < 0.05; Fig. 2B]. The majority (2057; 79%) of genes were down-regulated, while 534 (21%) of genes were up-regulated in the circulation of patients with AD (the terms “up-regulated” and “down-regulated” are used to describe changes in the number of RNA molecules in the circulation of patients with AD compared to controls) (data file S1). Next, we used Ingenuity Pathway Analysis (IPA; ver. 47547484; QIAGEN) to evaluate the functional roles and biological processes reflected by these differentially expressed genes. The top canonical pathways identified using the genes down-regulated in patients with AD were associated with nervous system functions, including γ-aminobutyric acid (GABA) receptor signaling, cyclic adenosine monophosphate (cAMP) response element–binding protein (CREB) signaling in neurons, netrin signaling, and synaptogenesis signaling pathway (Fig. 2C and data file S1). Up-regulated genes in patients with AD were significantly enriched in pathways associated with immune response activation [e.g., interleukin-8 (IL-8) signaling and inflammasome pathway], mitochondrial activity (e.g., mitochondrial dysfunction, oxidative phosphorylation, and sirtuin signaling pathway), and proteostasis (e.g., SUMOylation and protein ubiquitination) (Fig. 2C). Moreover, genes down-regulated in patients with AD were enriched in the “nervous system development and function” category. In particular, “development of neurons,” “neurotransmission,” and “synaptic transmission” were the most enriched terms for these genes, consistent with the overall decline of neurons and synaptic connections associated with AD (fig. S3, A and B) (25). Consistently, we observed that a significant portion of genes down-regulated in cf-mRNA of patients with AD were brain-specific genes (P = 6.17 × 10−10, hypergeometric test; fig. S3C). Last, Gene Ontology biological process enrichment analysis confirmed that the genes that are down-regulated in patients with AD are associated with neuronal function, while up-regulated genes are enriched in immune response– and RNA splicing–related processes, all consistent with well-recognized AD pathophysiology (fig. S3D).
Fig. 2. Genes dysregulated in cf-mRNA of patients with AD are associated with AD pathophysiology.
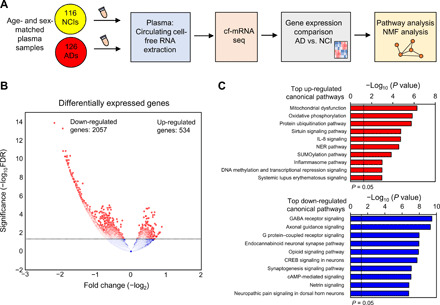
(A) Schematics of the study design. (B) Volcano plot of differentially expressed genes in cf-mRNA between AD (n = 126) and NCI controls of similar age (n = 116). FDR < 0.05 was used as the cutoff criteria. (C) Top 10 significant pathways identified using IPA canonical pathway analysis (top, up-regulated genes in AD; bottom, down-regulated genes in AD). The black vertical dotted line represents P < 0.05. NMF, non-negative matrix factorization; G protein, guanine nucleotide–binding protein; NER, nucleotide excision repair.
To further ascertain whether alterations observed in the cf-mRNA of patients with AD reflected the AD-associated transcriptional changes in the brain, we compared the differentially expressed genes identified in our study with those found in a previous RNA sequencing (RNA-seq) dataset examining transcriptional changes in the hippocampal region of patients with AD postmortem (23). We observed significant concordance between both up- and down-regulated sets of genes found in plasma and those reported in brain tissue (up-regulated genes, P = 0.017 and down-regulated genes, P < 10−5, hypergeometric test). Consistently, there was a substantial overlap of the molecular pathways identified between cf-mRNA profiling and RNA-seq of brain tissue (fig. S3E) (17). These data collectively support that circulating transcriptome captures transcriptional changes of the brain of patients with AD.
cf-mRNA profiling reveals gene signatures that correlate with AD severity and patient heterogeneity
We next explored whether genes dysregulated in the circulation of patients with AD were organized into functionally related clusters. Unsupervised decomposition analysis on DESeq counts using non-negative matrix factorization (NMF) identified six clusters of genes (Fig. 3A; see data file S2 for the complete list of genes). IPA pathway analyses revealed that these clusters are associated with the processes involved in AD onset and progression (Fig. 3B; see data file S2 for the complete list of pathways). For instance, cluster 3 is enriched in genes associated with synaptic transmission pathways, while cluster 5 is enriched in genes that are associated with immune response and neuroinflammation (Fig. 3B). We found that a heterogeneous AD patient population can be stratified into subgroups on the basis of the molecular profiles of these six gene clusters. In particular, unsupervised hierarchical clustering of all 126 patients with AD based on the magnitudes of the six gene clusters revealed five distinct groups (Fig. 3C). For example, “Group D” patients are characterized by elevated levels of cluster 5 genes (immune response and neuroinflammation; fig. S4A). The observed patient grouping was not due to sample source, age differences, or the severity of cognitive impairment (fig. S4B). These data suggest that once combined with detailed disease characteristics and patient outcome information, cf-mRNA profiling could potentially be used for noninvasive pathological subtyping of patients with AD.
Fig. 3. cf-mRNA reveals gene signatures that correlate with AD severity and patient heterogeneity of cognitive impairment in patients with AD.
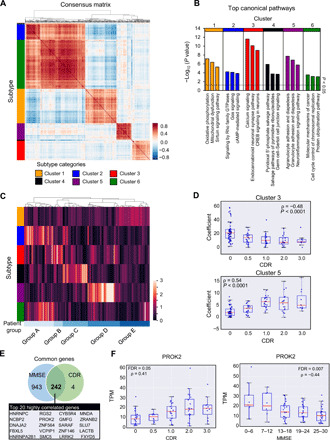
(A) Consensus NMF matrix. Unsupervised NMF clustering from 2591 differentially expressed genes. Six gene cluster subtypes are highlighted in color. (B) Top three most significant pathways (IPA) identified using gene set enrichment analysis for each cluster. The black horizontal dotted line represents significance threshold (P < 0.05). GTPases, guanosine triphosphatases. (C) Unsupervised clustering of patients with AD using their cf-mRNA profile based on NMF clusters identified in (A) revealed five groups of patients. (D) The expression of synaptic transmission and immune and inflammatory response clusters categorized by CDR rating. (E) Overlapping features between genes that correlate with Mini Mental State Exam (MMSE) and CDR scores. Top 20 genes that most highly correlated with MMSE and CDR scores are shown in the rectangular box. (F) The expression of PROK2 as a function of CDR (left) and MMSE (right) scores.
Next, to better understand the relationship between changes in these pathways/processes and the severity of dementia in patients with AD, we investigated whether any of these clusters correlate with the patient Clinical Dementia Rating (CDR) scores. The analysis revealed that the normalized expression values of two clusters of genes, clusters 3 (synaptic transmission) and 5 (“immune response and neuroinflammation”) significantly correlated with the CDR score (Fig. 3D). In particular, the synaptic transmission gene cluster showed decreased expression with increasing CDR scores (ρ = −0.47, P value of correlation P < 0.0001, Spearman’s rank correlation), and significant differences were observed even between individuals without dementia (CDR = 0) and patients with very mild dementia (CDR = 0.5) (P < 0.001, Mann-Whitney rank sum test). In contrast, the expression levels of immune response and neuroinflammation cluster increased with CDR score (ρ = 0.54, P value of correlation P < 0.0001, Spearman’s rank correlation), with most acute changes occurring between CDR stages 0 and 1.
On the basis of these observations, we assessed individual genes whose expression levels significantly correlated with disease severity. We identified 246 genes that correlated with CDR score (FDR < 0.1; fig. S5A and data file S3). Gene Ontology enrichment analyses revealed that these genes are primarily involved in biological processes such as RNA splicing, oxidative phosphorylation, and mitochondrial dysfunction (fig. S5B and data file S3), processes that are all well known to be related to AD. To ensure that the genes correlated with cognitive impairment were consistent, we repeated the same analysis using Mini Mental State Exam (MMSE) score, another widely used clinical metric for cognitive impairment assessment. A total of 1186 genes correlated with MMSE score (FDR < 0.1; fig. S5C and data file S3). As expected, genes correlated with CDR and MMSE scores overlapped significantly (top 20 genes are determined by the average rank of genes between CDR and MMSE scores for FDR values). Similarly, the molecular pathways identified using these genes also overlapped significantly (Fig. 3, E and F, and fig. S5E). For example, we identified prokineticin 2 (PROK2), a chemokine commonly dysregulated in AD (26), and leucine rich repeat kinase 2 (LRRK2), a well-known Parkinson’s disease gene that has been shown to be associated with AD tau pathology (27). In addition, we identified SLU7, a gene involved in pre-mRNA splicing, which has been shown to be dysregulated in the brain tissues of aging individuals and patients with neurodegenerative disorders (28).
Recent tissue RNA-seq data have shown that marked disease-associated transcriptomic changes are detectable in neurons from individuals with mild cognitive impairment (29). We therefore tested whether disease-associated gene signatures can be detected in the cf-mRNA of patients with AD having mild or very mild dementia. We used CDR scores to identify patients with AD having mild or very mild dementia (CDR ≤ 1, n = 36 and control n = 77). We found 1496 dysregulated genes in patients with AD having CDR ≤ 1 when compared to healthy controls (FDR < 0.05) (fig. S6A). Genes down-regulated in “early stage” patients with AD are primarily enriched in nervous system function and developmental processes (e.g., netrin signaling, CREB signaling in neurons, calcium transport, and regulation of neurogenesis) and up-regulated genes in immune response and proteostasis (e.g., protein ubiquitination, inflammasome pathway, and activation of immune response), consistent with our previous analyses (fig. S6B).
cf-mRNA–based classification of patients with AD
Next, we sought to build classifiers that discriminate patients with AD from NCI patients using machine learning algorithms. To minimize bias and overtraining, we first built a classifier exclusively using samples from the University of Kentucky by logistic regression with L2 regularization (control n = 24 and AD n = 66) (Fig. 4A). Differentially expressed genes identified in the University of Kentucky only cohort (1658 genes with FDR < 0.05) were selected as input features for the classifier. This set of genes significantly overlapped with the 2591 dysregulated genes identified using the entire cohort (i.e., 942 of the 1094 down-regulated genes identified using the University of Kentucky cohort overlap with those identified using the entire cohort, P < 10−8; 451 of the 564 up-regulated genes identified using the University of Kentucky cohort overlap with those identified using the entire cohort, P < 10−8; hypergeometric test). The classifier model was then tested on the testing set composed of the remainder of the AD (n = 60) and control samples (n = 92) derived from four independent sources. Biological pathways represented in the classifier include immune response and cellular metabolic processes (Gene Ontology analysis; data file S4), the pathways known to be associated with AD pathogenesis. The receiver operating characteristic (ROC) for the classifier in the test set is shown in Fig. 4B [area under the curve (AUC) = 0.83] and suggests that cf-mRNA signals can be used to noninvasively discern patients with AD.
Fig. 4. cf-mRNA–based classifiers distinguish patients with AD from control individuals.
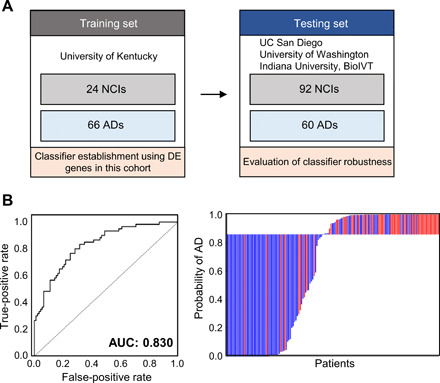
(A) Schematics of classifier establishment. DE, differentially expressed. (B) ROC curve of cf-mRNA classifier for discriminating AD against NCI; left: Waterfall plot of AD; right: control discrimination.
DISCUSSION
Here, we performed a transcriptome-wide comparison of plasma cf-mRNA profiles between patients with AD and control individuals. We first demonstrated the robust technical performance of the cf-mRNA next-generation sequencing (NGS) assay, which resulted in detection and quantification of a significant number of genes in circulation. Our proof-of-concept study showed that the circulating transcriptome has the potential to provide, in a noninvasive manner, molecular and functional information of neurodegenerative diseases such as AD. Furthermore, we showed that genes dysregulated in the plasma of patients with AD reflected biological processes and pathways known to be associated with cognitive impairment and neurodegenerative disorders. In particular, our data showed an overall decline in multiple pathways implicated in nervous system function and development (e.g., synapse loss, GABA signaling, and neurotransmission) in patients with AD, accompanied by elevated levels of genes involved in inflammation, mitochondrial dysfunction, oxidation, and proteostasis (30). Differential expression analysis showed that 79% of differentially expressed genes were down-regulated, which may be contributed from neurodegeneration and subsequent down-regulation of neuronal gene expression in patients with AD. Consistently, tissue sequencing data also showed substantially higher number of down-regulated genes in the brains of patients with AD compared to their controls (23). In addition, we showed that the genes and biological processes found to be dysregulated in the plasma of patients with AD substantially overlapped with those identified in the RNA-seq datasets from postmortem brain biopsy specimens (23). While these data suggest that gene dysregulation in the brain is reflected in the circulation, further studies are needed to confirm that these AD-associated transcriptional changes originated in the brain and not from other tissue or blood cells. Nevertheless, these results indicate that plasma cf-mRNA profiling could be used as a proxy for noninvasive molecular evaluation of brain homeostasis in patients with AD.
One potential application that may benefit from a better molecular characterization of AD is AD therapeutic drug discovery and development. cf-mRNA sequencing provides a granular characterization of the circulating transcriptomes of patients with AD, including the identification of a number of genes that are either dysregulated in patients with AD or correlated with the severity of cognitive impairment. In addition to identifying biological processes that are known to be linked with AD (e.g., 26 dysregulated genes involved in GABA signaling), we also observed reduced levels of genes associated with neurogenesis in patients with AD. The attenuation of neurogenesis-associated genes supports the recent hypothesis of adult neurogenesis being disrupted in AD (31). Furthermore, we have identified several genes that are associated with the severity of cognitive impairment including PROK2 and SLU7 (26, 28). PROK2 is a chemokine that plays a major role in neurodegeneration and has been shown to be involved in Aβ toxicity (26).
The heterogeneous nature of AD (32), as a complex neurodegenerative disease affecting multiple biological pathways and processes during onset and progression (33), represents one major hurdle for AD drug discovery and development. Another hurdle, but simultaneously an opportunity, includes the length of time over which AD develops. To date, therapeutic drugs targeting Aβ and tau proteins have shown modest results (34, 35). As a result, several drugs targeting alternative pathways that are commonly dysregulated in AD, such as inflammation and mitochondrial dysfunction, are being actively tested as potential AD treatments (34). Furthermore, the development of biomarkers to stratify patients with AD and identify likely responders would help accelerate the discovery and development of therapies for patients with AD. Since molecular characterization of patients with AD using brain biopsy is not feasible, the deployment of noninvasive tools that can evaluate the molecular dysregulations in patients with AD will substantially improve the outcomes of future clinical trials. While the precise criteria for patient eligibility before therapeutic treatment remain to be determined, our results indicate that cf-mRNA profiling could be used to measure the molecular characteristics of individual patients and may help inform precision treatment strategies and more effective patient management plans.
Despite postmortem histology remaining the gold standard for AD diagnosis, currently, tests including CSF, PET, and magnetic resonance imaging are widely used to diagnose patients with AD (36, 37). However, imaging modalities are costly, and CSF collection is invasive. Therefore, scalable, accessible, and cost-effective blood-based tests are highly desired for the diagnosis of patients with AD. To date, several protein-based blood biomarkers, including those that measure circulating levels of Aβ peptides, appear to be promising diagnostic biomarker candidates of AD. However, Aβ levels can be elevated in individuals without dementia (38–41) and have been shown to be an inconsistent predictor for the rate of cognitive decline (2). We showed that the cf-mRNA transcriptome profiling represents a new noninvasive approach for the development of classifiers to diagnose patients with AD, and our data confirmed that the cf-mRNA–based classifiers can robustly discriminate patients with AD from control individuals.
The present proof-of-concept study has several limitations that need to be addressed in future studies. First, samples used in this study were obtained from five different sources, and PCA showed relative site differences, likely because of differences in the preanalytical sample processing of these retrospective cohorts. While source-related differences were adjusted, validation of the classifiers in a well-characterized multicenter study under standardized operating procedures are needed to avoid potential site-related preanalytical effects. In addition, it is important to evaluate the performance of cf-mRNA classifiers in the intended use population of those with cognitive impairment with complete PET data to develop clinically relevant diagnostic AD classifiers. Furthermore, disease specificity of the classifiers should be evaluated using specimens from patients with other neurodegenerative diseases. Despite these qualifications, our data support the circulating transcriptome as a tool that may be used to identify transcriptional alterations of organs that are difficult to access, such as the brain. Furthermore, our results highlight the potential utility of high-throughput cf-mRNA sequencing to noninvasively characterize dynamic transcriptomic alterations associated with neurodegenerative diseases and, subsequently, potentially aid the development of other blood-based biomarkers for AD diagnosis, monitoring, and patient stratification for drug discovery and development in precision health.
MATERIALS AND METHODS
Clinical specimens
We examined a total of 242 retrospectively collected plasma specimens from five independent patient cohorts of AD and NCIs. These cohorts included: University of California, San Diego, University of Kentucky, University of Washington in St. Louis, University of Indiana, and BioIVT (fig. S1). The detailed patient demographics and clinicopathological characteristics are shown in tables S1 and S2. Written informed consent was obtained from all patients, and the study was approved by the institutional review boards of all the participating institutions.
RNA extraction, library preparation, and cf-mRNA sequencing
Plasma samples isolated from control and patients with AD were centrifuged at 12,000g, and RNA was extracted from up to 1 ml of plasma using QIAamp Circulating Nucleic Acid Kit (QIAGEN) and eluted in a volume of 15 μl. AD and NCI samples across centers were processed together across batches. ERCC RNA Spike-In Mix (Thermo Fisher Scientific, catalog no. 4456740) was added to plasma samples as an exogenous spike-in control according to the manufacturer’s instructions. Agilent RNA 6000 Pico chip (Agilent Technologies, catalog no. 5067-1513) was used to assess the integrity of extracted RNA. RNA samples were converted into cDNA, and libraries were prepared using a Swift 2S kit (Swift, catalog no. 28096), followed by whole-exome capture (Agilent). Qualitative and quantitative analysis of the NGS library preparation process were conducted by chip-based electrophoresis (Agilent Technologies, catalog no. 5067-4626) and by quantitative polymerase chain reaction (Roche, catalog no. KK4824), respectively. Sequencing was performed using Illumina NextSeq 500 platform (Illumina Inc.), using paired-end sequencing and 76-cycle sequencing. Base calling was performed on an Illumina BaseSpace platform (Illumina Inc.), using the FASTQ Generation Application. For sequencing data analysis, adaptor sequences were removed and low-quality bases were trimmed using cutadapt (v1.11). Reads shorter than 15 base pairs were excluded from subsequent analyses. Read sequences greater than 15 base pairs were aligned to the human reference genome GRCh38 using STAR (v2.5.2b) with GENCODE v24 gene models. Duplicated reads were removed using the SAMtools (v1.3.1) rmdup command. RNA-Seq by expectation-maximization (RSEM) (v1.3.0) was used to estimate the number of fragments (read counts) assigned to each gene using the deduplicated BAM files. Gene expression levels (in the unit of TPM) were calculated by normalizing RSEM. When counting the number of genes detected, we implemented a cutoff of TPM > 5. On the basis of the analysis of replicate correlation, TPM < 5 is below the limit of quantification, and therefore, we chose to consider only the observations with TPM > 5.
Brain-specific gene establishment
Tissue (cell type)–specific genes, including brain-specific genes, are defined as genes that show substantially higher expression in a particular tissue (cell type) compared to other tissue types (cell types). We first identified potential tissue (cell)–type genes using a combination of two publicly available databases of tissue (cell type) transcriptome expression levels from
Genotype-Tissue Expression (www.gtexportal.org/home/) for gene expression across 51 human tissues and Blueprint Epigenome (www.blueprint-epigenome.eu/) for gene expression across 56 human hematopoietic cell types. For each individual gene, the tissues (cell types) were ranked by their expression of that particular gene, and if the expression in the top tissue (cell type) is >5-fold higher than all the other tissues (cell types), then the gene was considered specific to the top tissue (cell type). In addition, we also required that the cf-mRNA level of the gene in consideration should be >3-fold higher than its level in the matching whole-blood sample (12 matching plasma/whole-blood samples were obtained from the San Diego Blood Bank) to rule out the possibility of circulating blood cell contamination.
Differential expression analysis, site adjustment, and pathway analysis
Differential expression analysis was implemented with DESeq2 (v1.12.4) (42) using read counts for each gene (derived from RSEM) as input. Genes with fewer than 250 total reads across the entire cohort were excluded from subsequent analyses. Normalization accounting for library sequencing depth was performed by DESeq2 according to its standard algorithm (41). Samples were obtained from five different sources described in table S2. To adjust for preanalytic variability associated with sample source, we implemented a multifactor negative binomial model “~ source + disease status,” including sample source to account for this substantial source of variation using DESeq2 (41). The site adjustment was effective as indicated by the PCA plot of the first two PCs after correction. Benjamini-Hochberg correction was used to correct for multiple testing and obtain adjusted P values (FDR cutoff of 0.05 was used to identify differentially expressed genes).
Pathway analysis was conducted using IPA software version 47547484. A complete list of differentially expressed genes and genes correlated with MMSE and CDR was uploaded to IPA, and expression analysis was used to determine pathways that are highly enriched. IPA categories including Canonical pathways and “Top diseases and bio functions” were examined.
Classifier construction and performance evaluation
To minimize bias and overtraining when evaluating classifier performance, we used the AD and NCI samples sourced from the University of Kentucky as the “training cohort” and samples from all other sources as the “test cohort.” None of the samples in the test cohort were used in any way during model training. At the feature selection step, we ran DESeq2 on the training cohort and selected the top 1658 genes differentially expressed between AD and NCI samples. The expression levels (TPM) of those 1658 genes were then used in the subsequent training of the classifiers. The training of the classifiers was implemented using the Python library scikit-learn (https://scikit-learn.org/stable/; v0.20.1). Logistic regression with L2 regularization was used. Metaparameters were determined by 15-fold cross-validation on the training cohort. Next, we applied the trained classifiers to the test cohort and obtained the predicted risk score for each sample in the test cohort. By comparing the risk score with the true disease status of the samples, we were able to plot the ROC curves and calculated the AUC. Confidence intervals for the ROC curves were calculated according to DeLong (18).
Computational transcriptome deconvolution analysis using NMF
A normalization was first implemented whereby the expression levels (DESeq2-normalized counts) of each gene were divided by its maximum value across the samples. This step is designed to rescale the expression levels among different genes so as to avoid a few highly expressed genes dominating the decomposition process. The normalized expression matrix was then subject to NMF decomposition using sklearn.decomposition.NMF within the Python library scikit-learn (https://scikit-learn.org/stable/). NMF decomposition achieves a more parsimonious representation of the data by decomposing expression matrix into the product of two matrices X = WH. X is the expression matrix with n rows (n samples) and m columns (m genes). W is the coefficient matrix with n rows (n samples) and p columns (p components). H is the loading matrix with p rows (p components) and m columns (m genes). W is, in a sense, a summarization of the original expression matrix X with fewer dimensions. H contains information about how much each gene contributes to the components. Biological interpretation of the derived components was achieved by performing pathway analysis on the top genes that contribute the most to each component. We performed patient grouping by performing hierarchical clustering on the coefficient matrix W. Hierarchical clustering was implemented using the Python library SciPy (v1.3.0) class scipy.cluster.hierarchy.linkage with parameters method = “average” and metric = “correlation.”
Statistical analysis
Risk scores derived from the multivariate logistic regression classification model were used to plot ROC curves and calculate AUCs. The AUC is calculated for each of the 15 iterations of cross-validation. Confidence intervals for the ROC curves were calculated using the method of DeLong (18). Spearman’s rank correlation was used to examine the correlation between gene expression and cognitive impairment scores. More specifically, the “stats.spearmanr” class in the SciPy library was used for the implementation of the correlation analysis. Student’s t test was used to evaluate the difference between the two variables. The hypergeometric test was used to test the significance of overlapping gene sets and implemented using the “stats.hypergeom” class in the SciPy library. The Benjamin-Hochberg method was used to correct for multiple testing and implemented with the “stats.multitest” class in the statsmodels Python library (v0.8.0). All other statistical analyses were performed using R (3.3.3, R Development Core Team; https://cran.r-project.org/) and MedCalc statistical software version 19 (MedCalc Software bvba, Ostend, Belgium).
Supplementary Material
Acknowledgments
We thank R. Rava, G. Elias, T. Wright, J. Sninsky, and T. Maddala for conceptual discussions and critical reading of the manuscript. Funding: This publication was made possible thanks to funding from the bgC3 foundation. We also thank our collaborators at University of Washington in St. Louis and their grants [Healthy Aging and Senile Dementia (P01 AG03991), Alzheimer’s Disease Research Center (P50 AG05681), and Adult Children Study (P01 AG026276)]. We thank our collaborators from Indiana University for providing samples from the National Centralized Repository for Alzheimer’s Disease and Related Dementias (NCRAD), which receives government support under a cooperative agreement grant (U24 AG021886) awarded by the National Institute on Aging [NIA and grant number U01 AT000162 from the National Center for Complementary and Alternative Medicine (NIH)]. We thank our collaborators at University of California, San Diego and collaborators from University of Kentucky (NIH grant number P30 AG028383). Author contributions: S.T., A.D.A., A.P.K., N.S.S., and A.I. performed the experiments. J.Z. performed bioinformatics analyses. S.T., A.I., and J.Z. performed data analyses and data visualization and wrote the manuscript. J.A., M.N., D.M.W., and J.B.B. facilitated access to samples. S.R.Q., M.N., and J.A. were in charge of financial support. A.I. supervised the study. Competing interests: S.T., J.Z., N.S.S., A.D.A., A.P.K., J.A., M.N., and A.I. are past or current employees at Molecular Stethoscope Inc. S.T., J.Z., N.S.S., M.N., and A.I. are named as inventors in pending patent applications related to the technologies used here filed by Molecular Stethoscope Inc. [WO2020092646A1 filed on 30 October 2019 (J.Z., N.S.S., M.N., and A.I.), WO2020087037A2 filed on 25 October 2019 (S.T., J.Z., M.N., and A.I.), and WO2019060369A1 filed on 18 September 2018 (N.S.S., M.N., and A.I.)]. S.R.Q. is a founder of Molecular Stethoscope Inc. and a member of its scientific advisory board. J.B.B. has served on advisory boards for Elan, Bristol Myers Squibb, Avanir Pharmaceuticals, Novartis, Genentech, and Eli Lilly and Company and holds stock options in CorTechs Labs Inc. and Human Longevity Inc. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Sequencing data generated in this study was deposited in Sequence Read Archive (SRA) under the accession number PRJNA574438 PRJNA574438 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA574438/). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabb1654/DC1
REFERENCES AND NOTES
- 1.Alzheimer’s Association , 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 7, 208–244 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Perez-Nievas B. G., Stein T. D., Tai H.-C., Dols-Icardo O., Scotton T. C., Barroeta-Espar I., Fernandez-Carballo L., de Munain E. L., Perez J., Marquie M., Serrano-Pozo A., Frosch M. P., Lowe V., Parisi J. E., Petersen R. C., Ikonomovic M. D., López O. L., Klunk W., Hyman B. T., Gómez-Isla T., Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardy J., Selkoe D. J., The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Heppner F. L., Ransohoff R. M., Becher B., Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Castellani R., Hirai K., Aliev G., Drew K. L., Nunomura A., Takeda A., Cash A. D., Obrenovich M. E., Perry G., Smith M. A., Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 70, 357–360 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Albert M. S., De Kosky S. T., Dickson D., Dubois B., Feldman H. H., Fox N. C., Gamst A., Holtzman D. M., Jagust W. J., Petersen R. C., Snyder P. J., Carrillo M. C., Thies B., Phelps C. H., The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 270–279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T., Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1, a006189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cummings J., Aisen P. S., Bois B. D., Frölich L., Jack C. R. Jr., Jones R. W., Morris J. C., Raskin J., Dowsett S. A., Scheltens P., Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res. Ther. 8, 39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crowley E., Di Nicolantonio F., Loupakis F., Bardelli A., Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 10, 472–484 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Lo Y. M., Chiu R. W. K., Next-generation sequencing of plasma/serum DNA: An emerging research and molecular diagnostic tool. Clin. Chem. 55, 607–608 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Sancesario G. M., Bernardini S., Alzheimer’s disease in the omics era. Clin. Biochem. 59, 9–16 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Bianchi D. W., Chiu R. W. K., Sequencing of circulating cell-free DNA during pregnancy. N. Engl. J. Med. 379, 464–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan H. C., Blumenfeld Y. J., Chitkara U., Hudgins L., Quake S. R., Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. U.S.A. 105, 16266–16271 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Vlaminck I., Valantine H. A., Snyder T. M., Strehl C., Cohen G., Luikart H., Neff N. F., Okamoto J., Bernstein D., Weisshaar D., Quake S. R., Khush K. K., Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci. Transl. Med. 6, 241ra277 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan H. C., Gu W., Wang J., Blumenfeld Y. J., El-Sayed Y. Y., Quake S. R., Non-invasive prenatal measurement of the fetal genome. Nature 487, 320–324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snyder T. M., Khush K. K., Valantine H. A., Quake S. R., Universal noninvasive detection of solid organ transplant rejection. Proc. Natl. Acad. Sci. U.S.A. 108, 6229–6234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koh W., Pan W., Gawad C., Fan H. C., Kerchner G. A., Wyss-Coray T., Blumenfeld Y. J., El-Sayed Y. Y., Quake S. R., Noninvasive in vivo monitoring of tissue-specific global gene expression in humans. Proc. Natl. Acad. Sci. U.S.A. 111, 7361–7366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.András I. E., Toborek M., Extracellular vesicles of the blood-brain barrier. Tissue Barriers 4, e1131804 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray M., Ruan J., Zhang W., Variations in the transcriptome of Alzheimer’s disease reveal molecular networks involved in cardiovascular diseases. Genome Biol. 9, R148 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Twine N. A., Janitz K., Wilkins M. R., Janitz M., Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLOS ONE 6, e16266 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The External RNA Controls Consortium , The External RNA Controls Consortium: A progress report. Nat. Methods 2, 731–734 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Alkallas R., Fish L., Goodarzi H., Najafabadi H. S., Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer’s disease. Nat. Commun. 8, 909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Annese A., Manzari C., Lionetti C., Picardi E., Horner D. S., Chiara M., Caratozzolo M. F., Tullo A., Fosso B., Pesole G., D’Erchia A. M., Whole transcriptome profiling of late-onset Alzheimer’s disease patients provides insights into the molecular changes involved in the disease. Sci. Rep. 8, 4282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nativio R., Donahue G., Berson A., Lan Y., Amlie-Wolf A., Tuzer F., Toledo J. B., Gosai S. J., Gregory B. D., Torres C., Trojanowski J. Q., Wang L.-S., Johnson F. B., Bonini N. M., Berger S. L., Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 21, 497–505 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheff S. W., Price D. A., Schmitt F. A., Mufson E. J., Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Zuena A. R., Casolini P., Lattanzi R., Maftei D., Chemokines in Alzheimer’s disease: New insights into prokineticins, chemokine-like proteins. Front. Pharmacol. 10, 622 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henderson M. X., Sengupta M., Trojanowski J. Q., Lee V. M. Y., Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 7, 183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tollervey J. R., Wang Z., Hortobágyi T., Witten J. T., Zarnack K., Kayikci M., Clark T. A., Schweitzer A. C., Rot G., Curk T., Zupan B., Rogelj B., Shaw C. E., Ule J., Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. 21, 1572–1582 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathys H., Davila-Velderrain J., Peng Z., Gao F., Mohammadi S., Young J. Z., Menon M., He L., Abdurrob F., Jiang X., Martorell A. J., Ransohoff R. M., Hafler B. P., Bennett D. A., Kellis M., Tsai L.-H., Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hara Y., McKeehan N., Fillit H. M., Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 92, 84–93 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno-Jiménez E. P., Flor-García M., Terreros-Roncal J., Rábano A., Cafini F., Pallas-Bazarra N., Ávila J., Llorens-Martín M., Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 25, 554–560 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Devi G., Scheltens P., Heterogeneity of Alzheimer’s disease: Consequence for drug trials? Alzheimers Res. Ther. 10, 122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reitz C., Toward precision medicine in Alzheimer’s disease. Ann. Transl. Med. 4, 107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kametani F., Hasegawa M., Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 12, 25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mondragón-Rodríguez S., Perry G., Zhu X., Boehm J., Amyloid Beta and tau proteins as therapeutic targets for Alzheimer’s disease treatment: Rethinking the current strategy. Int. J. Alzheimers Dis. 2012, 630182 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaw L. M., Vanderstichele H., Knapik-Czajka M., Clark C. M., Aisen P. S., Petersen R. C., Blennow K., Soares H., Simon A., Lewczuk P., Dean R., Siemers E., Potter W., Lee V. M.-Y., Trojanowski J. Q.; Alzheimer’s Disease Neuroimaging Initiative , Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark C. M., Schneider J. A., Bedell B. J., Beach T. G., Bilker W. B., Mintun M. A., Pontecorvo M. J., Hefti F., Carpenter A. P., Flitter M. L., Krautkramer M. J., Kung H. F., Coleman R. E., Doraiswamy P. M., Fleisher A. S., Sabbagh M. N., Sadowsky C. H., Reiman E. P., Zehntner S. P., Skovronsky D. M.; AV45-A07 Study Group , Use of florbetapir-PET for imaging β-amyloid pathology. JAMA 305, 275–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nabers A., Perna L., Lange J., Mons U., Schartner J., Güldenhaupt J., Saum K.-U., Janelidze S., Holleczek B., Rujescu D., Hansson O., Gerwert K., Brenner H., Amyloid blood biomarker detects Alzheimer’s disease. EMBO Mol. Med. 10, e8763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura A., Kaneko N., Villemagne V. L., Kato T., Doecke J., Doré V., Fowler C., Li Q.-X., Martins R., Rowe C., Tomita T., Matsuzaki K., Ishii K., Ishii K., Arahata Y., Iwamoto S., Ito K., Tanaka K., Masters C. L., Yanagisawa K., High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 554, 249–254 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Ovod V., Ramsey K. N., Mawuenyega K. G., Bollinger J. G., Hicks T., Schneider T., Sullivan M., Paumier K., Holtzman D. M., Morris J. C., Benzinger T., Fagan A. M., Patterson B. W., Bateman R. J., Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 13, 841–849 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mapstone M., Cheema A. K., Fiandaca M. S., Zhong X., Mhyre T. R., MacArthur L. H., Hall W. J., Fisher S. G., Peterson D. R., Haley J. M., Nazar M. D., Rich S. A., Berlau D. J., Peltz C. B., Tan M. T., Kawas C. H., Federoff H. J., Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 20, 415–418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabb1654/DC1