

sHDL is a promising antithrombotic agent delivery system due to its antithrombotic effect itself and improved drug delivery.
Abstract
Antiplatelet agents offer a desirable approach to thrombosis prevention through the reduction of platelet reactivity. However, major bleeding events greatly attenuate the clinical outcomes of most antithrombotic agents. Therefore, the development of safer and more effective strategies to prevent vascular occlusion and avoid bleeding is urgently needed. A reconstituted nanoparticle, synthetic high-density lipoprotein (sHDL), which mimics the native HDL, has been established as clinically safe and is easily manufactured on a large scale. In this study, we propose that the delivery of the antiplatelet drug ML355, a selective inhibitor of 12(S)-lipoxygenase (12-LOX), by sHDL will efficiently inhibit thrombosis by targeting ML355 to the intended site of action, improving the pharmaceutical profile and harnessing the innate antithrombotic efficacy of the sHDL carrier. Our data show that ML355-sHDL exhibits more potent inhibition of thrombus formation in both small arterioles and larger arteries in mice without impairing the normal hemostasis in vivo.
INTRODUCTION
Thrombosis is a serious health problem underlying several cardiovascular pathologies, including stroke and myocardial infarction (1). Current antithrombotic therapies include (i) anticoagulants, which limit blood clotting; (ii) antiplatelets, which inhibit platelet activation and aggregation (2, 3); and (iii) thrombolytics, which work to dissolve the thrombus once formed (4–6). However, adverse effects associated with antithrombotic agents including high risk of hemorrhagic bleeding, slow absorption, poor bioavailability, short half-life, and narrow therapeutic windows have severely limited the agents’ broad application in the clinic (7–10). With an estimated 48% of U.S. citizens living with cardiovascular disease (11), safer and more effective antithrombotic therapies are needed.
Nanoparticle-based therapies have received notable attention as they offer rapid and targeted delivery of antithrombotic agents to the site of injury and promise to overcome many of the clinical obstacles faced by conventional therapeutic approaches (12, 13). Among them, several thrombus-targeted nanoparticles have been developed, including iron oxide (14, 15), polymer-based (16–18), and cell membrane–coated (19) nanoparticles, to encapsulate a variety of antithrombotic drugs, including antiplatelet agents (20–22), anticoagulants (23, 24), and thrombolytics (14, 16, 25, 26). Despite these advances, most of the existing platforms suffer from particle heterogeneity and complicated fabrication processes that are difficult to scale (27, 28). Here, we develop and use synthetic high-density lipoprotein (sHDL) nanoparticles as a drug delivery vehicle (29, 30). Compared to other types of nanoparticles, sHDL offers the advantage of proven clinical safety and established large-scale manufacturing processes (28, 31). Furthermore, in addition to their pleiotropic atheroprotective effects (32–35), much preclinical and clinical evidence has shown that both native HDL and sHDL have direct influence on attenuating platelet reactivity and inhibiting thrombus formation (35–37). Among them, HDL3, the major HDL subfraction in the blood, was proven to inhibit thrombin-induced platelet aggregation (38). The infusion of the plasma-purified HDL CSL-111 (80 mg/kg) to individuals with type 2 diabetes mellitus resulted in the widespread attenuation of platelet activation and a 50% reduction in thrombus formation under flow (39). Similarly, the administration of a recombinant ApoA1 Milano–based sHDL, ETC-216, reduced platelet aggregation and thrombus formation on an occlusive platelet-fibrin–rich thrombus rat model (35). These findings suggest that sHDL holds promise in acting as an effective antithrombotic vehicle for delivery and exerting a synergistic inhibitory effect on platelet hyperactivity when delivering antiplatelet agents.
As a proof of concept, we plan to investigate the effect of sHDL on human platelet function in vitro and in well-established mouse models of thrombosis and hemostasis in vivo. Specifically, we will test the ability of encapsulating ML355 in nano-based delivery systems to achieve targeted drug delivery at the site of vascular injury to enhance the drug’s therapeutic index and to further expand its broader therapeutic applications against thrombosis and other diseases (40, 41).
To fulfill this aim, we developed sHDL nanoparticles that consist of apoA1 mimetic peptide 22A (22–amino acid peptide) and phospholipids. Our sHDL platform exhibited the sustained release of ML355 in vitro and a superior pharmacokinetic profile in vivo. Incubation of sHDL with isolated human platelets resulted in the robust uptake of sHDL by platelets and the attenuation of platelet function in vitro. Intravenous administration of sHDL blank or loaded with ML355 in mice resulted in the rapid uptake of sHDL by platelets and the retention within platelets in vivo for up to 72 hours following intravenous administration. Given the intrinsic antithrombotic properties of endogenous HDL, we hypothesized that sHDL would also have antithrombotic properties. We found that sHDL alone was capable of attenuating platelet thrombosis in vivo and that our previously unidentified generated ML355-sHDL exhibited synergetic antithrombotic effects of both sHDL and ML355 as examined in a laser-induced thrombosis mouse model, without impairing the normal hemostatic property of platelets. Together, these results demonstrate that the entrapment of an antithrombotic agent by sHDL provides an effective, feasible, and rapidly translatable strategy for the prevention of thrombotic events (presented in fig. S1).
RESULTS
Preparation and characterization of ML355-sHDL
ML355, a selective 12(S)-lipoxygenase (12-LOX) inhibitor, has an estimated solubility of <5 μg/ml in pH 7.4 phosphate-buffered saline (PBS) (42). Its low water solubility requires the addition of solubilizing agents when administered orally and intravenously. Despite ML355’s effective antithrombotic effects observed in previous animal studies (41, 43), sustained strong inhibition of thrombus formation in the laser-induced cremaster arteriole model required frequent dosing. This dosing regimen could be ascribed to nonspecific delivery. To increase the site selectivity of the drug, we encapsulated ML355 into sHDL (illustrated in Fig. 1A), termed ML355-sHDL. sHDL is composed of ApoA1 mimetic peptide (22A) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), both of which are clinically validated biomaterials with a long history of safe use in humans (28). The discoidal structure of sHDL was retained upon ML355 loading as was observed by transmission electron microscopy (Fig. 1B). Dynamic light scattering (Fig. 1C) and gel permeation chromatography (Fig. 1D) revealed a homogeneous size distribution of 10.3 ± 1.6 nm and 10.6 ± 0.7 nm for both sHDL and ML355-sHDL, respectively, suggesting a minimal impact of drug loading on the size and homogeneity of ML355-sHDL. The optimal formulation of ML355-sHDL was determined to be DMPC (20 mg/ml), 22A (10 mg/ml), and ML355 (0.5 mg/ml). In addition, the solubility of ML355 was enhanced roughly 100-fold after sHDL encapsulation (42). Compared to the ML355 in solution, ML355 release from sHDL was investigated in vitro under physiological conditions (pH 7.4 PBS at 37°C) and displayed a sustained release profile. In addition, the presence of serum in the release medium had no notable effect on the ML355 release from ML355-sHDL, indicating the stability of ML355-sHDL in serum (Fig. 1E).
Fig. 1. Preparation and characterization of ML355-sHDL.
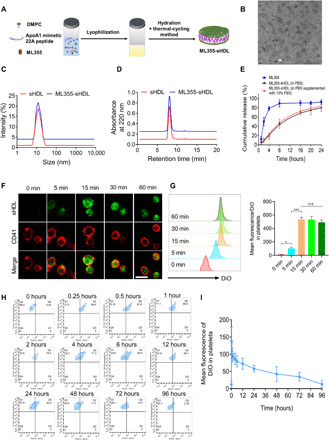
(A) Illustration of the synthesis of ML355-sHDL composed of an ApoA1 mimetic 22-mer peptide (22A), 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), and ML355 using the thermal-cycling method. (B) Negative-stained transmission electron microscopy image of ML355-sHDL; scale bar, 20 nm. (C) Dynamic light scattering of blank sHDL and ML355-sHDL. (D) Gel permeation chromatography of blank sHDL and ML355-sHDL. (E) Release of ML355 from sHDL at PBS and PBS cumulative supplemented with 10% fetal bovine serum (FBS). Data are means ± SD (n = 3). (F) Platelet uptake and intracellular distribution of DiO-sHDL in platelets imaged by confocal microscopy (scale bar, 2 μm). Washed mouse platelets were suspended in 1 ml of Tyrode’s buffer at 3 × 106 platelets/ml and incubated with DiO-sHDL (sHDL at 50 μg/ml and DiO at 2.5 μg/ml) for indicated lengths of time (5, 15, 30, and 60 min) at 37°C. Platelets were then washed with Tyrode’s buffer twice to remove free DiO-sHDL and then fixed with 4% paraformaldehyde, followed by staining with Alexa Fluor 647–conjugated anti-mouse CD41 antibody. Platelet membrane is shown in red, and sHDL particle accumulation in platelets is shown in green. (G) Representative flow cytometry histograms and data analysis of mean DiO fluorescence intensity in platelets by flow cytometry. Data are means ± SD (n = 3). *P < 0.05 and N.S. (no significant difference). (H) CD41-PE–positive platelets in the blood versus DiO-sHDL. (I) Quantification of DiO-sHDL uptake by CD41-PE–positive platelets in blood. Data are means ± SD (n = 4).
To assess the intercellular uptake of sHDL by platelets, washed mouse platelets were collected and prepared as described previously (43, 44). Washed mouse platelets were incubated with DiO-sHDL (sHDL at 50 μg/ml and DiO at 2.5 μg/ml) for 5, 15, 30, and 60 min, respectively, and the uptake of DiO-sHDL by platelets was monitored by fluorescent microscopy. The Alexa Fluor 647 anti-CD41 antibody was used to label the membrane of mouse platelets. The laser scanning confocal images showed that sHDL was internalized by platelets after 5 min of incubation. The fluorescence signal (Fig. 1F) was detected after 5 min of incubation, and the signal reached a maximum after 15 min of incubation. This suggested that sHDL was specifically internalized by mouse platelets and that uptake was saturated after 15-min incubation (30-min incubation did not further increase the fluorescence signal in mouse platelets). The sHDL uptake profile in mouse platelets was also quantitatively verified by flow cytometry (shown in Fig. 1G). In addition, our data (fig. S2) showed that preactivated platelets were able to uptake sHDL to a similar extent when compared with resting platelets. We next sought to determine the specific uptake of sHDL by platelets in vivo using flow cytometry to analyze whole blood. Both male and female C57BL/6J mice (n = 4) were dosed with DiO-sHDL (sHDL at 50 mg/kg and DiO at 2.5 mg/kg) via tail vein injection, and blood was collected at predetermined time points. Platelets were stained with CD41-PE (phycoerythrin) antibody. Platelet uptake of sHDL over time is shown in Fig. 1H. The fluorescence signal of sHDL in platelets rapidly increased in vivo and then gradually decreased after 15 min through 96 hours after administration (Fig. 1I). These results indicate that sHDL is internalized by platelets in vivo and is retained for up to 72 hours. Internalization of sHDL by platelets was further confirmed in vivo by injecting dual-labeled DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate)–488–sHDL (labeling the 22A peptide in sHDL with Alexa Fluor 488 dye and the lipid bilayer in sHDL with cell-labeling fluorophore DiI) in mice. Our results showed that both lipid and peptide were internalized in consistent with our in vitro platelet uptake of sHDL. We observed that red blood cells and neutrophils were able to uptake sHDL in vivo in mice following sHDL treatment. However, the sHDL uptake by platelets is notably higher than in red blood cells and neutrophils in circulation (see fig. S3).
ML355-sHDL treatment inhibits human platelet aggregation in vitro
To examine whether sHDL or ML355-sHDL modulates the platelet functions and inhibitory effects on platelet aggregation to a greater extent than that of ML355 alone, we performed in vitro studies on isolated, washed human platelets. Among these results shown in Fig. 2A, ML355-sHDL showed the strongest inhibition of platelet aggregation relative to the control (0.4 ± 0.2% aggregation at 0.1 nM thrombin, P < 0.001, and 20.3 ± 2.4% aggregation at 0.25 nM thrombin, P < 0.01), followed by ML355 (5.6 ± 2.5% aggregation at 0.1 nM thrombin, P < 0.01, and 40.4 ± 6.8% aggregation at 0.25 nM thrombin, P < 0.01) and sHDL (20.8 ± 5.2% aggregation at 0.1 nM thrombin, P < 0.01, and 70.7 ± 9.1% aggregation at 0.25 nM thrombin, P < 0.05). At 0.5 nM thrombin, only ML355 (76.4 ± 4.7%, P < 0.05) and ML355-sHDL (56.4 ± 3.6%, P < 0.001) showed inhibitory effects on platelet aggregation relative to the control, while sHDL did not significantly inhibit thrombin-induced platelet aggregation (81.2 ± 3.0%). In contrast, the inhibition of platelet aggregation at the highest concentration of thrombin (1 nM) was not observed for any of the treatments. Together, these data suggest that sHDL exerts an inhibitory effect on thrombin-induced platelet activation, and ML355 entrapment by sHDL (ML355-sHDL) exhibits a preferred inhibitory profile on platelet activation relative to either sHDL or ML355.
Fig. 2. ML355-sHDL treatment inhibits both human and mouse platelet aggregation.
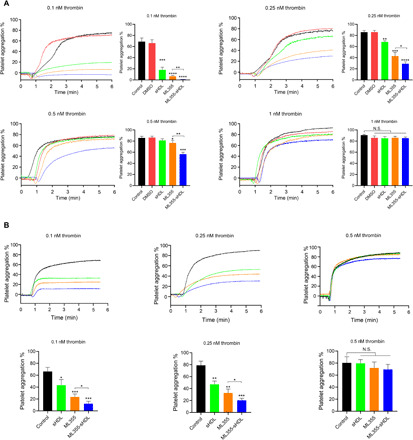
(A) Washed human platelets were incubated with different ML355 formulations for 15 min, including dimethyl sulfoxide (DMSO) (equivalent amount used to dissolve ML355), ML355 (10 μM), sHDL (100 μg/ml), or ML355-sHDL (sHDL at 100 μg/ml and ML355 at 10 μM). Untreated platelets were included as control. After different treatments, platelet aggregation was measured by the addition of thrombin at various concentrations. Compared to both control and DMSO group, both ML355 and sHDL treatment inhibited platelet aggregation. Furthermore, encapsulation of ML355 into sHDL exhibited an improved inhibitory effect on human platelet aggregation at multiple thrombin concentrations compared to treatment with either ML355 or sHDL. Inhibition of aggregation was overwhelmed for all formulations by a high concentration (1 nM) of thrombin. Data are means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, and N.S. (no significant difference) relative to control or as otherwise indicated. (B) Mice were intravenously administered with saline control, ML355 (1.5 mg/kg), sHDL (50 mg/kg), or ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg) for 24 hours followed by platelet isolation. Washed mouse platelets were subjected to aggregation analysis by incubation with various concentrations of thrombin. Platelets isolated from mice treated with ML355-sHDL exhibited less aggregation following incubation with thrombin (0.1 and 0.25 nM thrombin) than platelets from the mice treated with other formulations. Inhibition of aggregation was overwhelmed for all formulations by a high concentration (0.5 nM) of thrombin. Data are means ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001, and N.S. (no significant difference) relative to control or as otherwise indicated.
Platelet aggregation was inhibited in mice treated with ML355-sHDL
To determine the regulatory effect of sHDL on mouse platelet aggregation ex vivo, mice were intravenously treated with ML355, sHDL, or ML355-sHDL. After 24 hours, platelets were isolated from the blood and subjected to the aggregation assay. Similar to in vitro results, all groups effectively inhibited platelet aggregation compared to platelets from vehicle control–treated mice at both 0.1 and 0.25 nM thrombin (Fig. 2B). Among them, ML355-sHDL exhibited better inhibition of thrombin-induced platelet aggregation. Specifically, at 0.1 nM thrombin stimulation, platelets from mice treated with ML355-sHDL exhibited the least amount of aggregation (12.2 ± 4.1%, P < 0.001), platelets isolated from mice treated with ML355 exhibited 20.3 ± 3.5% aggregation (P < 0.001), and platelets isolated from mice treated with sHDL exhibited 43.3 ± 9.4% aggregation (P < 0.05); at 0.25 nM thrombin stimulation, platelets from mice treated with ML355-sHDL only exhibited 20.3 ± 3.5% aggregation (P < 0.001), platelets isolated from mice treated with ML355 showed 32.6 ± 6.6% aggregation (P < 0.01), and platelets isolated from mice treated with sHDL had 47.1 ± 5.5% aggregation (P < 0.05). Again, the inhibitory effects of ML355-sHDL, ML355, and sHDL on platelet aggregation were overcome by high concentrations of thrombin (0.5 nM), which is consistent with our in vitro platelet aggregation results and suggests that the normal hemostatic responses to acute bleeding injuries remain intact and unaffected with both ML355-sHDL and sHDL treatment.
Pharmacokinetics of ML355-sHDL
Given the long circulation time of sHDL nanoparticles in vivo, we sought to examine whether ML355 pharmacokinetics were improved by the incorporation of ML355 into sHDL. The plasma concentration of ML355 was determined by liquid chromatography–mass spectrometry (LC-MS) following intravenous administration of ML355-sHDL or ML355. The concentration curves in fig. S4 show that ML355-sHDL (3 mg/kg, intravenously) has a longer circulation time compared to ML355 alone, suggesting that the encapsulation of ML355 into sHDL extends its blood circulation time.
In vivo examination of sHDL’s thrombus targeting ability
sHDL’s significant inhibition of platelet aggregation and long circulation time in vivo led us to investigate whether sHDL could specifically target thrombi. Here, we tested whether DiO-sHDL could accumulate at the site of a thrombus in mice using an endothelial damage–induced thrombus model (43). DiO-sHDL was administered through an intravenous injection, and after 24 hours, before thrombus formation, platelet marker DyLight 647–conjugated rat anti-mouse platelet GP1bβ antibody was administered. Following laser ablation and thrombus formation, we found that DiO-sHDL and DyLight 647–conjugated rat anti-mouse platelet GP1bβ antibody were colocalized within the newly formed thrombi, suggesting that sHDL may facilitate the targeted delivery of antithrombotic agents (Fig. 3A and movie S1). In addition, the colocalization of sHDL within the thrombus was examined under confocal intravital microscopy and further evaluated using a three-dimensional constructed model of the thrombus. As shown in Fig. 3B, sHDL (green) mostly accumulated within the core regions of the thrombus, which consists of densely packed platelets (red). Furthermore, our results also indicate that sHDL adheres to the injured endothelium surrounding the thrombus.
Fig. 3. sHDL targets thrombus in vivo.
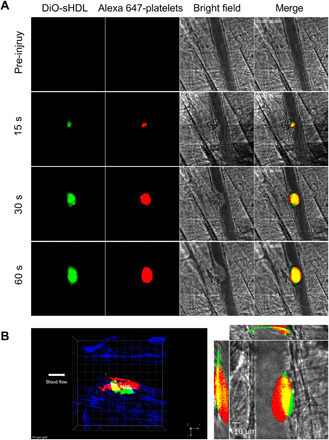
Male mice were pretreated with DiO-sHDL via intravenous injection with sHDL (50 mg/kg) and DiO (2.5 mg/kg). After 24 hours, mice were anesthetized and surgically prepared as described in detail in Materials and Methods. DyLight 647–conjugated rat anti-mouse platelet GP1bβ antibody (0.1 μg/g, X649, EMFRET Analytics) was administered by jugular vein cannula before vascular injury. Multiple independent thrombi (8 to 10) were induced in the arterioles (30 to 50 μm diameter) per mouse (three mice in each group) by a laser ablation system as described in Materials and Methods. Images of thrombus formation at the site of injured arterioles were acquired in real time under 63× water-immersion objective with a Zeiss Axio Examiner Z1 fluorescence microscope. (A) Sequence of intravital fluorescence microscopic images recorded over 1 min showing accumulation of fluorescently labeled DiO-sHDL in a forming thrombus indicated by fluorescently labeled platelets. (B) Left: Representative three-dimensional confocal images of DiO-sHDL (green) and platelet accumulation (red) in stable thrombi recorded under confocal intravital microscopy. Right: Representative middle section of thrombus shown under a three-direction view.
Laser-induced cremaster arteriole thrombosis model
Now that we have shown sHDL’s ability to reduce platelet aggregation, improve ML355’s circulation time, and target thrombi in vivo, we sought to determine ML355-sHDL’s ability to inhibit thrombus formation in vivo. Using the laser injury cremaster arterial thrombosis model, we studied the dynamics of fluorescence-labeled platelet accumulation in growing thrombi. Thrombus formation was quantitatively assessed by real-time measurements of platelet recruitment at the site of injury in mice pretreated with different formulations 24 hours before vessel injury. As shown in representative images of thrombus formation in Fig. 4A and quantitative analysis of dynamic platelet recruitment within thrombi in Fig. 4B, all of the treatment conditions (ML355, sHDL, and ML355-sHDL) exhibited varying levels of inhibition of thrombus formation. Notably, encapsulation of ML355 within sHDL (ML355-sHDL) exhibited synergetic inhibition on thrombus formation.
Fig. 4. ML355-sHDL efficiently inhibits thrombus formation in laser-induced cremaster arteriole thrombosis models.
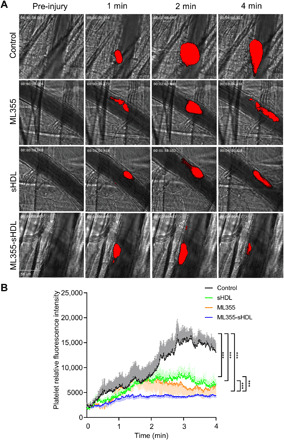
Male mice (n = 4) were pretreated intravenously with saline control, sHDL (50 mg/kg), ML355 (1.5 mg/kg), or ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg). After 24 hours, mice were anesthetized and surgically prepared as described in detail in Materials and Methods. Multiple independent thrombi were induced and recorded as described above. (A) Representative captured thrombi from each group at different time points. (B) Dynamics of platelet accumulation in thrombi assessed by relative mean fluorescence intensity of platelet averaged by 8 to 10 thrombi per mouse, three mice in each group. All thrombi were statistically analyzed for the change of fluorescence intensity over the course of thrombus formation after subtracting fluorescent background defined on an uninjured section of the vessel using the SlideBook program. Data are means ± SD and compared by two-way analysis of variance (ANOVA) analysis for statistical difference. ***P < 0.001.
FeCl3-induced carotid artery thrombosis model
To confirm the inhibitory effect of ML355, sHDL, or ML355-sHDL treatment on thrombus formation and vessel occlusion in a large artery with severe injury, mice treated with ML355, sHDL, or ML355-sHDL, respectively, were studied using a FeCl3-induced carotid artery thrombosis model. Following the vascular injury induced by FeCl3 application, platelets started to adhere to the injury site in the carotid artery and quickly formed visible platelet aggregates, resulting in the formation of stable thrombi (Fig. 5A). Thrombi were stable, grew to larger sizes, and reached vessel occlusion. There was no reopening of the occluded vessel in the control group. Thrombus growth was delayed and unstable in mice treated with either ML355 or sHDL, which resulted in a delay in vessel occlusion time. Thrombus growth and vessel occlusion were severely impaired in mice treated with ML355-sHDL as compared to other groups, which proved consistent with our microvascular thrombosis model [mean vessel occlusion time in ML355, sHDL, and ML355-sHDL was 11.0 ± 2.3 min (P < 0.01), 11.3 ± 1.3 min (P < 0.01), and 21.4 ± 6.4 min (P < 0.001), respectively, while mean vessel occlusion time in control mice was only 7.1 ± 1.2 min].
Fig. 5. ML355-sHDL efficiently delays vessel occlusion in FeCl3-induced carotid artery thrombosis model.
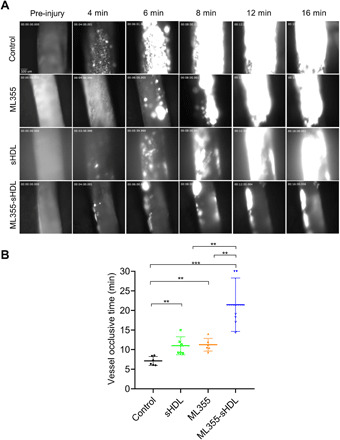
Both female and male mice (n = 6) were pretreated intravenously with saline control, sHDL (50 mg/kg), ML355 (1.5 mg/kg), or ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg). After 24 hours, mice were anesthetized and surgically prepared as described in detail in Materials and Methods. (A) Representative images of thrombosis in carotid artery in response to FeCl3 injury. FeCl3 (10%) was topically applied on the right carotid artery for 3 min, images of thrombi were recorded in real time, and vessel occlusion was determined by the secession of blood flow. (B) Carotid artery vessel occlusion time in each group. The mean vessel occlusion time in the control mice, ML355, sHDL, and ML355-sHDL was 7.1 ± 1.2 min, 11.0 ± 2.3 min, 11.3 ± 1.3 min, and 21.4 ± 6.4 min, respectively. Data are means ± SD (n = 6). **P < 0.01, ***P < 0.001.
The evaluation of hemostasis in vivo
Determination of phosphatidylserine exposure on platelets after administration of different ML355 formulation shown in Fig. 6A suggested that neither ML355, sHDL, nor ML355-sHDL treatment increases platelet phosphatidylserine exposure, as its levels are comparable to the levels on resting platelets, indicating that sHDL treatment is not likely to cause platelet activation or platelet clearance. This result was further supported by our data showing that sHDL had no significant effect on platelet counts in mice (fig. S5). We also examined the effect of sHDL on coagulation and hemostatic clot formation using thromboelastography (TEG) and tail-bleeding assays. Our results from the TEG analysis of whole blood collected from mice treated by different formulations (Fig. 6, B to F) showed that sHDL treatment in mice did not significantly alter the hemostatic parameters of clotting, indicating that ML355, sHDL, and ML355-sHDL treatment did not compromise the coagulation. Furthermore, our study showed that sHDL, ML355, and ML355-sHDL did not prolong tail-bleeding time and had no effect on blood loss evaluated by measuring hemoglobin (Fig. 6, G and H).
Fig. 6. ML355-sHDL does not affect the phosphatidylserine exposure on platelet surface, coagulation system, and hemostatic action.
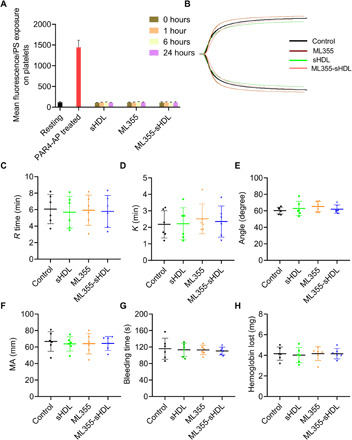
Both female and male mice (n = 6) were pretreated intravenously with saline control, sHDL (50 mg/kg), ML355 (1.5 mg/kg), or ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg). All treated groups were subjected to the following different examinations. (A) At different time points after administration (1, 6, and 24 hours), the phosphatidylserine (PS) exposure over the time course in platelets from different groups was quantified by flow cytometry. Blood collected from untreated mice was taken as resting platelets (negative control), and blood stimulated with protease-activated receptor 4–activating peptide (PAR4-AP) (200 μM) was used as activated platelets (positive control). (B) Representative tracings indicating different treatments were presented. Some major coagulation parameters were analyzed. (C) R time, time to formation of the initial fibrin threads (min). (D) K time, the time until the clot reaches a certain strength (min). (E) Alpha (α) angle, the rapidity with which the clot forms (degree). (F) Maximum amplitude (MA), clot’s maximum strength (mm). (G) Bleeding times and (H) blood loss (quantified as milligrams of hemoglobin) were quantified.
DISCUSSION
sHDL infusion has previously been demonstrated as an effective approach to inhibit platelet activation and arterial thrombosis in both mice (33, 35) and diabetic patients (39). The novelty of our approach is to use sHDL as a delivery vehicle for antithrombotic agents to enable the delivery of higher levels of the drug to sites of injury or inflammation while avoiding the off-target accumulation, thus achieving improved antithrombotic efficacy through synergistic effects without compromising hemostasis. Through this proof-of-concept study, we hope to further strengthen the antithrombotic application of sHDL. In addition to its proven antithrombotic effect, sHDL is a biomimetic nanoparticle used to mimic the in vivo biological activity of endogenous HDL, which is well recognized as protective in cardiovascular and chronic inflammatory diseases. We have previously loaded various therapeutics into sHDL for atherosclerosis and cancer applications (29, 45, 46). Thus, the development of sHDL as an antithrombotic drug delivery vehicle is a sensible approach to improve the therapeutic potential of these drug molecules. ML355, a selective 12-LOX inhibitor, was previously assessed as a promising antiplatelet therapeutic in vivo (43). Despite efficient thrombus inhibition of orally administrated ML355 in multiple thrombosis models in our previous studies, the targeted delivery of ML355 may further enhance its efficacy and decrease any unforeseen off-target effects.
In this study, we developed and investigated the effect of sHDL on platelet function in vitro and thrombus formation in vivo using mouse models of thrombosis and hemostasis. Furthermore, we encapsulated ML355 in our nano-based delivery system for targeted drug delivery at the site of vascular injury to enhance the drug’s therapeutic efficacy to explore the potential application of the advantages of sHDL in thrombotic disease. Preparation and characterization of ML355-sHDL showed that ML355 was encapsulated in sHDL, which displayed a typical discoidal structure and uniform distribution with nanoscale size. ML355-sHDL exhibited sustained release of ML355. Both in vitro human platelet uptake and ex vivo mouse platelet uptake studies demonstrated that platelets specifically endocytosed sHDL. Moreover, sHDL and platelets were colocalized in our endothelial injury–induced thrombi in mice. Specific mechanisms of sHDL uptake and accumulation in thrombi have yet to be identified. Possible mechanisms for platelet uptake of sHDL could be mediated by the following, but are not limited to (i) passive uptake of sHDL by platelets in blood, which is the fundamental principle for nonspecific nanoparticle-directed drug delivery; (ii) active internalization mediated by some unidentified proteins expressed on the surface of platelets, like scavenger receptors or glycoproteins; and (iii) bonding of sHDL to the other components involved in thrombus formation, like von Willebrand factor.
In addition, we found that sHDL exhibited a significant antiplatelet effect itself, inhibiting thrombin-induced platelet aggregation, which was consistent with previous reports (35, 39). Moreover, the encapsulation of ML355 within sHDL (ML355-sHDL) showed improved antiplatelet effects by combining the antiplatelet effects of ML355 and sHDL. Last, a laser injury–induced cremaster arteriole thrombus was used to investigate the antithrombotic efficacy of ML355-sHDL in vivo. The inhibition of thrombus formation was observed in mice treated with either ML355 or sHDL. The antithrombotic effects of sHDL here are consistent with the previous finding (35). While the antithrombotic effects of sHDL in vivo appear to be due to the direct inhibitory effects on platelet activation (32, 47), other possible mechanisms, including modulation of vascular endothelial cell function (48), cholesterol efflux (34, 49), and prevention of von Willebrand factor self-association and subsequent platelet adhesion, may play additional roles in its effect in the vessel (33). ML355-sHDL exhibits stronger thrombotic inhibition compared to ML355 and sHDL alone. The stronger thrombotic effect of ML355-sHDL is partially due to the combination of ML355 and sHDL working together, creating a synergistic effect. In addition, by encapsulating ML355 in sHDL, sHDL limits wide drug exposure in the blood, delivering more of the drug into platelets that can then accumulate within the thrombus, thus enhancing the drug’s therapeutic index. Last, tail vein bleeding results suggested that ML355-sHDL effectively inhibits thrombosis without impairing hemostasis.
Together, the results presented here demonstrate the utility of targeting 12-LOX in the platelet with ML355 delivered by sHDL for the prevention of platelet activation and thrombus formation and warrant further development of ML355-sHDL for clinical translation. sHDL not only exerts its own antithrombotic effects but also additionally functions as an effective delivery vehicle for antithrombotic agents such as ML355. While the efficient uptake of sHDL by platelets was demonstrated in our study, we still observed some uptake by other blood cells including leukocytes and red blood cells. While targeting platelets with ML355-sHDL is beneficial, more selective targeting may be achieved in the future by incorporating aptamers that exhibit high binding affinity to glycoproteins specifically expressed on the platelet surface. While the current study is focused on models of arterial thrombosis, future studies will need to assess the feasibility of delivering antithrombotic agents by sHDL for other types of thrombosis including prevention of venous thromboembolisms. In addition, future studies will focus on the underlying mechanisms by which sHDL regulates vessel hemostatic conditions, including endothelial protection, prevention of von Willebrand factor self-association, and inhibition of tissue factor. Furthermore, the platelet targeting property of sHDL will be optimized through various modifications such as integration of targeting ligands and functional lipids to continue to improve specificity and efficacy for preventing occlusive thrombotic events, leading to a reduction in pathologic conditions such as myocardial infarction and stroke.
Antiplatelet drugs, either administered as a mono- or polytherapy, are the first-choice therapy for the clinical treatment of cardiovascular disease and prevention of arterial thrombotic events. Current treatment options in clinical use have been limited to primarily cyclooxygenase-1, the adenosine diphosphate (ADP) receptor (P2Y12), and integrin receptor (αIIbβ3). Despite effectively reduced morbidity and mortality in the clinic, there are limitations associated with oral and intravenous administration of the currently approved antiplatelet agents, including an increased risk of bleeding, and delayed onset of action due to the requirement for in vivo conversion (like thienopyridines), irreversible inhibition (like thienopyridines) and delayed offset, as well as suboptimal platelet inhibition due to poor target specificity (50). Therefore, developing safer antiplatelet agents to rapidly and potently curtail thrombotic-associated events without increasing the risk of bleeding events in the gut and brain remains an unmet clinical need. Recent advances have identified a number of newer platelet pharmacological targets, including targeting surface receptors (glycoproteins and G protein–coupled receptors), oxygenases, and phosphodiesterases (2). However, most of those novel antiplatelet drugs are still in preclinical or early clinical stages of development. Among them, our preclinical studies have previously demonstrated the potential therapeutic benefits of selectively targeting 12-LOX with ML355 without notably impairing hemostasis (43). While oral administration is always preferred, some agents have low or variable bioavailability and large patient to patient variability in pharmacokinetics, which could lead to side effects especially when administered in an acute disease setting. Our current study shows that the preferential uptake of sHDL by platelets has served as a means of direct platelet targeting strategy, thus enhancing the antiplatelet effects of ML355. ML355-sHDL showed favorable synergistic antithrombotic effects without increasing the bleeding risk in addition to a targeted delivery to the site of platelet-rich thrombi. In summary, delivering antiplatelet agents using sHDL as a vehicle may be a promising approach for the prevention of thrombotic events associated with cardiovascular disease such as heart attacks and strokes and may improve clinical outcomes.
MATERIALS AND METHODS
Materials
ApoA1 mimetic peptide 22A, PVLDLFRELLNELLEALKQKLK, was synthesized by GenScript Inc. (Piscataway, NJ). Peptide purity was determined to be >95% by reversed-phase high-performance liquid chromatography (HPLC). Phospholipid DMPC was purchased from NOF America Corporation. ML355 was synthesized by the National Institutes of Health (NIH) Molecular Libraries Program, Bethesda, MD. For the washed human platelet aggregation assay, ML355 was dissolved in dimethyl sulfoxide (DMSO) as a stock solution at 10 mM. For animal studies, ML355 was dissolved in a vehicle [5% DMSO, 10% Solutol, 20% polyethylene glycol 300 (PEG300), and 65% PBS] and administrated to animals via intravenous injection immediately. All other materials were obtained from commercial sources.
Preparation of washed human platelets
All studies involving human subjects have been reviewed and approved by University of Michigan Institutional Review Board. Written informed consent was obtained from all healthy donors before the blood draws. Platelets were isolated as described previously (43, 44).
Experimental animals
All experimental procedures in this study were approved by the Institutional Animal Care and Use Committee at University of Michigan. Both male and female C57BL/6 wild-type mice (10 to 12 weeks old) were purchased from The Jackson Laboratory and housed at 22° ± 1°C in a 12-hour:12-hour light-dark cycle at the University of Michigan.
Preparation and characterization of ML355-sHDL
ML355-sHDL was prepared using a lyophilization method that we previously developed (29). Briefly, DMPC and ApoA1 mimetic 22A peptide were dissolved in acetic acid, and ML355 was dissolved in DMSO. The solution was mixed at a 20:10:0.5 weight ratio and freeze-dried for 24 hours. The lyophilized powder was hydrated in PBS and cycled between 55° and 4°C (3 min for each cycle, and three thermal cycles) to obtain ML355-sHDL. The pH of the ML355-sHDL was adjusted to 7.4 by NaOH. The solution was passed through sterile filters (0.22 μm) and stored frozen at −20°C until use. The final concentrations for 22A, DMPC, and ML355 were determined by LC-MS to be 20, 10, and 0.5 mg/ml, respectively. In vitro release of ML355 from sHDL was investigated using Float-A-Lyzer G2 dialysis devices with 3000-Da molecular weight cutoff (Spectrum) (51, 52). Briefly, 1 ml of ML355-sHDL was introduced into the dialysis membrane tube and then incubated in 50 ml of PBS with constant stirring (PBS buffer at 37°C) that contained 0.5% SDS to increase the solubility of ML355 in the external PBS medium to maintain sink conditions. In addition, ML355 (in DMSO) was tested in the above release medium to obtain a free drug release profile. To test the effect of serum on ML355 release from sHDL, 10% fetal bovine serum (FBS) was added with ML355-sHDL and further incubated in PBS buffer supplemented with 10% FBS. At predetermined intervals, buffer was drawn and replaced with an equal volume of fresh medium. The concentration of ML355 was measured by HPLC (42). For observation of sHDL uptake by platelet in vitro, DiO (Thermo Fisher Scientific) was encapsulated into sHDL to obtain DiO-sHDL using the same method above, and the final DiO-sHDL formulation contained 22A peptide (10 mg/ml) and DiO (0.5 mg/ml). For investigation of biodistribution of sHDL in different major blood cell types in vivo, DiI-488-sHDL was prepared by dual-labeling the 22A peptide in sHDL with Alexa Fluor 488 dye using Invitrogen protein labeling kit (A10235) and labeling the lipid bilayer in sHDL with cell-labeling fluorophore DiI (V22889) following the manufacturer’s instructions.
sHDL uptake by washed mouse platelets
Washed mouse platelets were resuspended in Tyrode’s buffer at 3 × 106 platelets/ml. The mouse platelet suspension was incubated with DiO-sHDL (sHDL at 50 μg/ml and DiO at 2.5 μg/ml) for predetermined durations (5, 15, 30, and 60 min) at 37°C. After incubation, mouse platelets were washed with Tyrode’s buffer twice and fixed with 4% paraformaldehyde followed by staining with Alexa Fluor 647–conjugated anti-mouse CD41 antibody (BioLegend, #133934) before imaging with a confocal microscope (Nikon A1). For quantitative measurement of sHDL uptake by mouse platelets, after incubation with DiO-sHDL for different time points, mouse platelets were washed and mean fluorescence intensity of DiO in platelets was quantitatively analyzed by flow cytometry (ZE5 Cell Analyzer, Bio-Rad) (53). Washed mouse platelets were treated with protease-activated receptor 4–activating peptide (PAR4-AP) (AYPGKF; AnaSpec, Fremont, CA, USA) to induce platelet activation and then incubated with DiO-sHDL as described above, and sHDL uptake by activated platelets was determined and compared to unstimulated resting platelets.
sHDL distribution in major blood cells in vivo
Both male and female C57BL/6J mice (n = 4) were intravenously dosed with DiI-488-sHDL (DiI at 0.5 mg/kg and Alexa Fluor 488 at 0.5 mg/kg). At different time points (from 5 min up to 24 hours), whole blood was collected and mean fluorescence intensity of both lipid tracer DiI and peptide tracer Alexa Fluor 488 in each cell type was analyzed by flow cytometry. Briefly, 5 μl of heparinized whole blood was collected and labeled with platelet marker Alexa Fluor 647 anti-mouse CD41 antibody (BioLegend, #133934), red blood cell marker Alexa Fluor 647 anti-mouse TER-119 antibody (BioLegend, #116218), and neutrophil marker Alexa Fluor 647 anti-mouse Ly-6G antibody (BioLegend, #116218), followed by flow cytometry as described previously (54).
In vitro washed human platelet aggregation test
Human platelets were prepared and aggregation was assayed as described previously (43, 44, 55). Platelets were incubated with DMSO (equivalent volume to dissolve ML355) and treated with control, ML355 (10 μM), sHDL (100 μg/ml), or ML355-sHDL (sHDL at 100 μg/ml and ML355 at 10 μM) for 15 min. In addition, untreated platelets were included as control. Platelet aggregation was induced by various doses of thrombin (0.1 to 1 nM) as reported in previous studies (43). Aggregation was measured in response to thrombin with a lumi-aggregometer (Model 700D; Chrono-Log) under stirring conditions at 1100 rpm at 37°C.
Ex vivo–washed mice platelet activation inhibition test
Male and female C57BL/6J mice were divided into four groups (n = 4) and intravenously dosed with the following formulations: (i) control vehicle (equivalent volume), (ii) ML355 (1.5 mg/kg), (iii) sHDL (50 mg/kg), or (iv) ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg). Twenty-four hours after administration, mice were sacrificed under terminal anesthesia. Blood was drawn from the inferior vena cava using a syringe containing sodium citrate. Mouse platelet preparation was performed as previously described (43, 44). Murine platelets were resuspended at 3 × 108 platelets/ml in Tyrode’s buffer. Mouse platelet aggregation was induced by various doses of thrombin (0.1 to 0.5 nM) and measured with a lumi-aggregometer using the same method described above.
Pharmacokinetic study
Male and female C57BL/6J mice were divided into two groups (n = 4) and dosed with the following formulations: intravenous administration of ML355 (3 mg/kg) or intravenous administration of ML355-sHDL (sHDL at 100 mg/kg and ML355 at 3 mg/kg). Plasma drug concentration was determined at different time points (0.25, 2, 8, 24, and 48 hours) and assessed by LC-MS analysis as previously described (42).
In vivo thrombus targeting property of sHDL
Male C57BL/6J mice were chosen in this section due to the growing thrombus induced by the laser injury cremaster arterial thrombosis model. Male C57BL/6J mice (n = 3) were intravenously dosed with DiO-sHDL (sHDL at 50 mg/kg and DiO at 2.5 mg/kg). Twenty-four hours after administration, mice were anesthetized by an intraperitoneal injection of ketamine-xylazine (100 and 10 mg/kg, respectively) and the cremaster arteriole was externalized. The cremaster muscle was prepared and perfused with preheated bicarbonate-buffered saline throughout the experiment. DyLight 647–conjugated rat anti-mouse platelet GP1bβ antibody (0.1 μg/g; X649, EMFRET Analytics) was administered by jugular vein cannula before vascular injury. Multiple independent thrombi (8 to 10 thrombi per mouse) were induced in the arterioles (30 to 50 μm diameter) using a laser ablation system (Ablate! photoablation system; Intelligent Imaging Innovations). Images of thrombus formation at the site of injured arterioles were acquired in real time with a Zeiss Axio Examiner Z1 fluorescence microscope equipped with a 63× water-immersion objective and a high-speed scientific complementary metal-oxide semiconductor (sCMOS) camera. Furthermore, thrombus composition was examined under confocal intravital microscopy as described (43, 55).
Laser-induced cremaster arteriole thrombosis model
Male C57BL/6J mice (n = 3) were intravenously dosed with the following treatments: (i) saline control (equivalent volume), (ii) ML355 (1.5 mg/kg), (iii) sHDL (50 mg/kg), or (iv) ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg). Twenty-four hours after administration, mice were anesthetized and surgically prepared as above described. DyLight 647–conjugated rat anti-mouse platelet GP1bβ antibody (0.1 μg/g; X649, EMFRET Analytics) was administered by jugular vein cannula before vascular injury. Multiple independent thrombi (8 to 10) were induced in the arterioles (30 to 50 μm diameter) in each mouse (three mice per group) with a laser ablation system. All captured images were analyzed for change in fluorescence intensity over time of thrombus formation by subtracting fluorescent background defined on an uninjured section of the vessel using the SlideBook program. To monitor and compare dynamic thrombus formation among different treatment groups, the relative fluorescence intensity of Alexa Flour 647–labeled platelets (recruited within thrombus) was plotted using the mean fluorescence at each time point. Data were evaluated for significance with two-way analysis of variance (ANOVA) and Mann-Whitney test for nonparametric data using Prism 6 software (GraphPad, La Jolla, CA, USA).
FeCl3-induced carotid artery thrombosis model
Male and female C57BL/6J mice (n = 6) were intravenously injected with vehicle control (equivalent volume of saline), ML355 (1.5 mg/kg), sHDL (50 mg/kg), or ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg), respectively. Twenty-four hours after administration, mice were anesthetized as described above and tail veins were injected with DyLight 488 anti-GPIb (1 μg/g; Emfret, EIbelstadt, Germany) to label circulating resting platelets in mice before intravital microscopy imaging. The mice were placed on a heating pad, and the right common carotid artery was prepared under the dissecting microscope. Then, the mice were placed on the microscopic stage and blood flow in the carotid artery was visualized under 10× air objective using a Zeiss Axio Examiner Z1 upright fluorescence microscope. Carotid artery injury was induced by topically placing a 10% FeCl3 saturated Whatman paper for 3 min under recording. Images of platelet adhesion and the dynamics of thrombus formation were recorded for 30 min using a high-speed sCMOS camera using SlideBook 6.0. Vessel occlusion was defined by formation of an occlusive thrombus, and cease of blood flow for 1 or 30 min was taken as the vessel occlusion time if the carotid artery failed to occlude during the recording.
Phosphatidylserine exposure on platelets
Male and female C57BL/6J mice (n = 6) were intravenously dosed with the following treatments: (i) saline control (equivalent volume), (ii) ML355 (1.5 mg/kg), (iii) sHDL (50 mg/kg), or (iv) ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg). At different time points after administration (1, 6, and 24 hours), the phosphatidylserine exposure over time course in platelets from different groups was quantified by flow cytometry. Briefly, whole blood was collected from the saphenous vein and incubated with PE anti-mouse CD41 antibody (BioLegend, #133906) for platelet labeling and annexin V, Alexa Fluor 647 conjugate (phosphatidylserine exposure) (Thermo Fisher Scientific). The mean fluorescence intensity of annexin in platelets was quantified by flow cytometry. In addition, blood collected from mice before treatment was taken as resting platelets (negative control) and blood stimulated with PAR4-AP (200 μM) was used as activated platelets (positive control).
Mouse tail-bleeding assay
Male and female C57BL/6J mice (n = 6) were intravenously dosed with the following treatments: (i) vehicle control (equivalent volume), (ii) ML355 (1.5 mg/kg), (iii) sHDL (50 mg/kg), or (iv) ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg). The tail-bleeding assay was performed after 24 hours of treatment as previously described (43, 55). Briefly, mice were anesthetized as described above and placed on a heating pad. Five millimeters of tail tip was excised, and the tails were immediately immersed in 14 ml of sterile saline at 37°C. Bleeding time was recorded as the cessation of blood flow from the tail for at least a minute using a stopwatch. The amount of blood loss from tail tip was quantified by measuring hemoglobin using Drabkin’s reagent (Sigma-Aldrich). Briefly, blood samples were pelleted at 500g for 10 min at room temperature, and the pellet was resuspended in 5 ml of Drabkin’s reagent and incubated at room temperature for 15 min. The amount of hemoglobin lost was quantified by comparing the absorbance of the samples at 540 nm using a SpectraMax i3 microplate reader (Molecular Devices LLC, San Jose, CA) to a standard curve of bovine hemoglobin in Drabkin’s reagent.
Coagulation tests by TEG
Male and female C57BL/6J mice (n = 6) were intravenously injected with vehicle control (equivalent volume of saline), ML355 (1.5 mg/kg), sHDL (50 mg/kg), or ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg), respectively. Twenty-four hours after administration, mice were anesthetized by intraperitoneal injection of ketamine-xylazine mixture as described above. Whole blood was collected via vena cava using a 25-gauge syringe containing 3.8% sodium citrate (blood to citrate volume ratio is 1:9). Citrated blood (340 μl) was mixed with 20 μl of CaCl2 (0.2 M), and viscoelastic properties of whole blood clot formation were studied under low shear stress using the Haemoscope TEG 5000 Thrombelastograph Hemostasis Analyzer (Haemonetics Corp., Braintree, Massachusetts, USA) according to the manufacturer’s instructions. Major coagulation parameters including R time (time to formation of the initial fibrin threads), Alpha angle (the rapidity with which the clot forms), K time (the time until the clot reaches a certain strength), and maximum amplitude (clot’s maximum strength) were analyzed and compared among different groups.
Platelet counts in mouse whole blood
Male and female C57BL/6J mice (n = 6) were intravenously dosed with the following treatments: (i) vehicle control (equivalent volume of saline), (ii) ML355 (1.5 mg/kg), (iii) sHDL (50 mg/kg), or (iv) ML355-sHDL (sHDL at 50 mg/kg and ML355 at 1.5 mg/kg). Twenty-four hours after administration, mice were anesthetized as described above. Blood sample was collected from the saphenous vein. Complete blood counts were performed using a Hemavet 950 analyzer (Drew Scientific Inc., Oxford, CT, USA).
Statistical analysis
Unpaired and paired two-tailed Student’s t tests and two-way ANOVA were used to compare between experimental groups with Prism 6.0 software (GraphPad). Where appropriate, the statistical test used is contained in the figure legend. Data represent mean values ± SD. Differences were considered significant when *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
Funding: This study was supported by NIH GM113832 (A.S.), HL134569 (A.S.), GM131835 (M.H.), Cayman Chemicals Biomedical Research Institute—CaBRI (M.H.), T32-GM07767 (K.H.), and University of Michigan Frankel Cardiovascular Center McKay Award G024210 (R.A.). Author contributions: H.H., R.A., A.S., and M.H. conceived the idea. H.H., R.A., and L.L. performed experiments. All authors designed, analyzed, and discussed data. All authors wrote the paper with contributions and critical revisions from all coauthors. Competing interests: M.H. is a consultant and equity holder for Veralox Therapeutics, although no work presented here is associated with the relationship with Veralox Therapeutics. M.H. is an inventor for the ML355 patent issued on 30 April 2019 by the U.S. patent and trademark office, U.S. patent no. US10,266,488 (Japan patent no. 6463366). A.S., M.H., R.A., and H.H. are the inventors for “Compositions and methods for treating cardiovascular related disorders,” filed by the University of Michigan (U.S. Provisional Patent Application No. 63/093,839, filed on 20 October 2020). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabd0130/DC1
REFERENCES AND NOTES
- 1.Mackman N., Triggers, targets and treatments for thrombosis. Nature 451, 914–918 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yeung J., Li W., Holinstat M., Platelet signaling and disease: Targeted therapy for thrombosis and other related diseases. Pharmacol. Rev. 70, 526–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbate R., Cioni G., Ricci I., Miranda M., Gori A. M., Thrombosis and acute coronary syndrome. Thromb. Res. 129, 235–240 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Dunne P. D. J., Laursen S. B., Laine L., Dalton H. R., Ngu J. H., Schultz M., Rahman A., Anderloni A., Murray I. A., Stanley A. J., Previous use of antithrombotic agents reduces mortality and length of hospital stay in patients with high-risk upper gastrointestinal bleeding. Clin. Gastroenterol. Hepatol. 17, 440–447.e2 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Eikelboom J. W., Hirsh J., Spencer F. A., Baglin T. P., Weitz J. I., Antiplatelet drugs: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 141, e89S–e119S (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mega J. L., Simon T., Pharmacology of antithrombotic drugs: An assessment of oral antiplatelet and anticoagulant treatments. Lancet 386, 281–291 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Weitz J. I., Eikelboom J. W., Samama M. M., New antithrombotic drugs: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 141, e120S–e151S (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watson R. D. S., Chin B. S. P., Lip G. Y. H., Antithrombotic therapy in acute coronary syndromes. BMJ 325, 1348–1351 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine G. N., Ali M. N., Schafer A. I., Antithrombotic therapy in patients with acute coronary syndromes. Arch. Intern. Med. 161, 937–948 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Ageno W., Gallus A. S., Wittkowsky A., Crowther M., Hylek E. M., Palareti G., Oral anticoagulant therapy: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 141, e44S–e88S (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benjamin E. J., Muntner P., Alonso A., Bittencourt M. S., Callaway C. W., Carson A. P., Chamberlain A. M., Chang A. R., Cheng S., Das S. R., Delling F. N., Djousse L., Elkind M. S. V., Ferguson J. F., Fornage M., Jordan L. C., Khan S. S., Kissela B. M., Knutson K. L., Kwan T. W., Lackland D. T., Lewis T. T., Lichtman J. H., Longenecker C. T., Loop M. S., Lutsey P. L., Martin S. S., Matsushita K., Moran A. E., Mussolino M. E., O’Flaherty M., Pandey A., Perak A. M., Rosamond W. D., Roth G. A., Sampson U. K. A., Satou G. M., Schroeder E. B., Shah S. H., Spartano N. L., Stokes A., Tirschwell D. L., Tsao C. W., Turakhia M. P., Van Wagner L. B., Wilkins J. T., Wong S. S., Virani S. S.; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee , Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 139, e56–e528 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Flores A. M., Ye J., Jarr K.-U., Hosseini-Nassab N., Smith B. R., Leeper N. J., Nanoparticle therapy for vascular diseases. Arterioscler. Thromb. Vasc. Biol. 39, 635–646 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varna M., Juenet M., Bayles R., Mazighi M., Chauvierre C., Letourneur D., Nanomedicine as a strategy to fight thrombotic diseases. Future Sci. OA 1, FSO46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Y.-H., Wu S.-Y., Wu T., Chang Y.-J., Hua M.-Y., Chen J.-P., Magnetically targeted thrombolysis with recombinant tissue plasminogen activator bound to polyacrylic acid-coated nanoparticles. Biomaterials 30, 3343–3351 (2009). [DOI] [PubMed] [Google Scholar]
- 15.McCarthy J. R., Sazonova I. Y., Erdem S. S., Hara T., Thompson B. D., Patel P., Botnaru I., Lin C. P., Reed G. L., Weissleder R., Jaffer F. A., Multifunctional nanoagent for thrombus-targeted fibrinolytic therapy. Nanomedicine 7, 1017–1028 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawata H., Uesugi Y., Soeda T., Takemoto Y., Sung J.-H., Umaki K., Kato K., Ogiwara K., Nogami K., Ishigami K., Horii M., Uemura S., Shima M., Tabata Y., Saito Y., A new drug delivery system for intravenous coronary thrombolysis with thrombus targeting and stealth activity recoverable by ultrasound. J. Am. Coll. Cardiol. 60, 2550–2557 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Korin N., Kanapathipillai M., Matthews B. D., Crescente M., Brill A., Mammoto T., Ghosh K., Jurek S., Bencherif S. A., Bhatta D., Coskun A. U., Feldman C. L., Wagner D. D., Ingber D. E., Shear-activated nanotherapeutics for drug targeting to obstructed blood vessels. Science 337, 738–742 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Jin H.-j., Zhang H., Sun M.-l., Zhang B.-g., Zhang J.-w., Urokinase-coated chitosan nanoparticles for thrombolytic therapy: Preparation and pharmacodynamics in vivo. J. Thromb. Thrombolysis 36, 458–468 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Xu J., Wang X., Yin H., Cao X., Hu Q., Lv W., Xu Q., Gu Z., Xin H., Sequentially site-specific delivery of thrombolytics and neuroprotectant for enhanced treatment of ischemic stroke. ACS Nano 13, 8577–8588 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Wei Z., Xin G., Wang H., Zheng H., Ji C., Gu J., Ma L., Qin C., Xing Z., Niu H., Huang W., The diosgenin prodrug nanoparticles with pH-responsive as a drug delivery system uniquely prevents thrombosis without increased bleeding risk. Nanomedicine 14, 673–684 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Jin S., Wang Y., Zhu H., Wang Y., Zhao S., Zhao M., Liu J., Wu J., Gao W., Peng S., Nanosized aspirin-Arg-Gly-Asp-Val: Delivery of aspirin to thrombus by the target carrier Arg-Gly-Asp-Val tetrapeptide. ACS Nano 7, 7664–7673 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Chen Y., Cui G., Zhao M., Wang C., Qian K., Morris-Natschke S., Lee K.-H., Peng S., Synthesis, nano-scale assembly, and in vivo anti-thrombotic activity of novel short peptides containing l-Arg and l-Asp or l-Glu. Bioorg. Med. Chem. 16, 5914–5925 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Palekar R. U., Jallouk A. P., Myerson J. W., Pan H., Wickline S. A., Inhibition of thrombin with PPACK-nanoparticles restores disrupted endothelial barriers and attenuates thrombotic risk in experimental atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 36, 446–455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang A. C. L., Chang M.-Y., Tang Z. C. W., Li H.-J., Hwang G.-L., Hsieh P. C. H., Treatment of acute thromboembolism in mice using heparin-conjugated carbon nanocapsules. ACS Nano 6, 6099–6107 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Zhou J., Guo D., Zhang Y., Wu W., Ran H., Wang Z., Construction and evaluation of Fe3O4-based PLGA nanoparticles carrying rtPA used in the detection of thrombosis and in targeted thrombolysis. ACS Appl. Mater. Interfaces 6, 5566–5576 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Bi F., Zhang J., Su Y., Tang Y.-C., Liu J.-N., Chemical conjugation of urokinase to magnetic nanoparticles for targeted thrombolysis. Biomaterials 30, 5125–5130 (2009). [DOI] [PubMed] [Google Scholar]
- 27.He H., Liu L., Morin E. E., Liu M., Schwendeman A., Survey of clinical translation of cancer nanomedicines—Lessons learned from successes and failures. Acc. Chem. Res. 52, 2445–2461 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Kuai R., Li D., Chen Y. E., Moon J. J., Schwendeman A., High-density lipoproteins: Nature’s multifunctional nanoparticles. ACS Nano 10, 3015–3041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Y., Yuan W., Yu B., Kuai R., Hu W., Morin E. E., Garcia-Barrio M. T., Zhang J., Moon J. J., Schwendeman A., Chen Y. E., Synthetic high-density lipoprotein-mediated targeted delivery of liver X receptors agonist promotes atherosclerosis regression. EBioMedicine 28, 225–233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song Q., Huang M., Yao L., Wang X., Gu X., Chen J., Chen J., Huang J., Hu Q., Kang T., Rong Z., Qi H., Zheng G., Chen H., Gao X., Lipoprotein-based nanoparticles rescue the memory loss of mice with Alzheimer’s disease by accelerating the clearance of amyloid-beta. ACS Nano 8, 2345–2359 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Rader D. J., Apolipoprotein A-I infusion therapies for coronary disease: Two outs in the ninth inning and swinging for the fences. JAMA Cardiol. 3, 799–801 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Barter P., The role of HDL-cholesterol in preventing atherosclerotic disease. Eur. Heart J. 7, F4–F8 (2005). [Google Scholar]
- 33.Chung D. W., Chen J., Ling M., Fu X., Blevins T., Parsons S., Le J., Harris J., Martin T. R., Konkle B. A., Zheng Y., López J. A., High-density lipoprotein modulates thrombosis by preventing von Willebrand factor self-association and subsequent platelet adhesion. Blood 127, 637–645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy A. J., Bijl N., Yvan-Charvet L., Welch C. B., Bhagwat N., Reheman A., Wang Y., Shaw J. A., Levine R. L., Ni H., Tall A. R., Wang N., Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat. Med. 19, 586–594 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li D., Weng S., Yang B., Zander D. S., Saldeen T., Nichols W. W., Khan S., Mehta J. L., Inhibition of arterial thrombus formation by ApoA1 Milano. Arterioscler. Thromb. Vasc. Biol. 19, 378–383 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Lerch P. G., Spycher M. O., Doran J. E., Reconstituted high density lipoprotein (rHDL) modulates platelet activity in vitro and ex vivo. Thromb. Haemost. 80, 316–320 (1998). [PubMed] [Google Scholar]
- 37.Pajkrt D., Lerch P. G., van der Poll T., Levi M., Illi M., Doran J. E., Arnet B., van den Ende A., ten Cate J. W., van Deventer S. J., Differential effects of reconstituted high-density lipoprotein on coagulation, fibrinolysis and platelet activation during human endotoxemia. Thromb. Haemost. 77, 303–307 (1997). [PubMed] [Google Scholar]
- 38.Nofer J.-R., Walter M., Kehrel B., Wierwille S., Tepel M., Seedorf U., Assmann G., HDL3-mediated inhibition of thrombin-induced platelet aggregation and fibrinogen binding occurs via decreased production of phosphoinositide-derived second messengers 1,2-diacylglycerol and inositol 1,4,5-tris-phosphate. Arterioscler. Thromb. Vasc. Biol. 18, 861–869 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Calkin A. C., Drew B. G., Ono A., Duffy S. J., Gordon M. V., Schoenwaelder S. M., Sviridov D., Cooper M. E., Kingwell B. A., Jackson S. P., Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation 120, 2095–2104 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Ma K., Xiao A., Park S. H., Glenn L., Jackson L., Barot T., Weaver J. R., Taylor-Fishwick D. A., Luci D. K., Maloney D. J., Mirmira R. G., Imai Y., Nadler J. L., 12-lipoxygenase inhibitor improves functions of cytokine-treated human islets and type 2 diabetic islets. J. Clin. Endocrinol. Metab. 102, 2789–2797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X.-J., Cheng X., Yan Z.-Z., Fang J., Wang X., Wang W., Liu Z.-Y., Shen L.-J., Zhang P., Wang P.-X., Liao R., Ji Y.-X., Wang J.-Y., Tian S., Zhu X.-Y., Zhang Y., Tian R.-F., Wang L., Ma X.-L., Huang Z., She Z.-G., Li H., An ALOX12–12-HETE–GPR31 signaling axis is a key mediator of hepatic ischemia–reperfusion injury. Nat. Med. 24, 73–83 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Luci D. K., Jameson J. B., Yasgar A., Diaz G., Joshi N., Kantz A., Markham K., Perry S., Kuhn N., Yeung J., Kerns E. H., Schultz L., Holinstat M., Nadler J. L., Taylor-Fishwick D. A., Jadhav A., Simeonov A., Holman T. R., Maloney D. J., Synthesis and structure–activity relationship studies of 4-((2-hydroxy-3-methoxybenzyl) amino)benzenesulfonamide derivatives as potent and selective inhibitors of 12-lipoxygenase. J. Med. Chem. 57, 495–506 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adili R., Tourdot B. E., Mast K., Yeung J., Freedman J. C., Green A., Luci D. K., Jadhav A., Simeonov A., Maloney D. J., Holman T. R., Holinstat M., First selective 12-LOX inhibitor, ML355, impairs thrombus formation and vessel occlusion in vivo with minimal effects on hemostasis. Arterioscler. Thromb. Vasc. Biol. 37, 1828–1839 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeung J., Tourdot B. E., Fernandez-Perez P., Vesci J., Ren J., Smyrniotis C. J., Luci D. K., Jadhav A., Simeonov A., Maloney D. J., Holman T. R., McKenzie S. E., Holinstat M., Platelet 12-LOX is essential for FcγRIIa-mediated platelet activation. Blood 124, 2271–2279 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadiyala P., Li D., Nuñez F. M., Altshuler D., Doherty R., Kuai R., Yu M., Kamran N., Edwards M., Moon J. J., Lowenstein P. R., Castro M. G., Schwendeman A., High-density lipoprotein-mimicking nanodiscs for chemo-immunotherapy against glioblastoma multiforme. ACS Nano 13, 1365–1384 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuai R., Ochyl L. J., Bahjat K. S., Schwendeman A., Moon J. J., Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Badimon L., Vilahur G., LDL-cholesterol versus HDL-cholesterol in the atherosclerotic plaque: Inflammatory resolution versus thrombotic chaos. Ann. N. Y. Acad. Sci. 1254, 18–32 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Ruiz M., Frej C., Holmér A., Guo L. J., Tran S., Dahlbäck B., High-density lipoprotein–associated apolipoprotein M limits endothelial inflammation by delivering sphingosine-1-phosphate to the sphingosine-1-phosphate receptor 1. Arterioscler. Thromb. Vasc. Biol. 37, 118–129 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Badrnya S., Assinger A., Volf I., Native high density lipoproteins (HDL) interfere with platelet activation induced by oxidized low density lipoproteins (OxLDL). Int. J. Mol. Sci. 14, 10107–10121 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Meijden P. E. J., Heemskerk J. W. M., Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 16, 166–179 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Wang Y.-C., Liu X.-Q., Sun T.-M., Xiong M.-H., Wang J., Functionalized micelles from block copolymer of polyphosphoester and poly(ɛ-caprolactone) for receptor-mediated drug delivery. J. Control. Release 128, 32–40 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Meng Z., Meng L., Wang K., Li J., Cao X., Wu J., Hu Y., Enhanced hepatic targeting, biodistribution and antifibrotic efficacy of tanshinone IIA loaded globin nanoparticles. Eur. J. Pharm. Sci. 73, 35–43 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Novakowski S., Jiang K., Prakash G., Kastrup C., Delivery of mRNA to platelets using lipid nanoparticles. Sci. Rep. 9, 552 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z., Zhang N., Shao B., Panicker S. R., Fu J., McEver R. P., Replacing the promoter of the murine gene encoding P-selectin with the human promoter confers human-like basal and inducible expression in mice. J. Biol. Chem. 291, 1441–1447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adili R., Voigt E. M., Bormann J. L., Foss K. N., Hurley L. J., Meyer E. S., Veldman A. J., Mast K. A., West J. L., Whiteheart S. W., Holinstat M., Larson M. K., In vivo modeling of docosahexaenoic acid and eicosapentaenoic acid-mediated inhibition of both platelet function and accumulation in arterial thrombi. Platelets 30, 271–279 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabd0130/DC1