

Abstract
Motivation
As the cost of sequencing decreases, the amount of data being deposited into public repositories is increasing rapidly. Public databases rely on the user to provide metadata for each submission that is prone to user error. Unfortunately, most public databases, such as non-redundant (NR), rely on user input and do not have methods for identifying errors in the provided metadata, leading to the potential for error propagation. Previous research on a small subset of the NR database analyzed misclassification based on sequence similarity. To the best of our knowledge, the amount of misclassification in the entire database has not been quantified. We propose a heuristic method to detect potentially misclassified taxonomic assignments in the NR database. We applied a curation technique and quality control to find the most probable taxonomic assignment. Our method incorporates provenance and frequency of each annotation from manually and computationally created databases and clustering information at 95% similarity.
Results
We found more than two million potentially taxonomically misclassified proteins in the NR database. Using simulated data, we show a high precision of 97% and a recall of 87% for detecting taxonomically misclassified proteins. The proposed approach and findings could also be applied to other databases.
Availability and implementation
Source code, dataset, documentation, Jupyter notebooks and Docker container are available at https://github.com/boalang/nr.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Researchers use BLAST on the non-redundant (NR) database on a daily basis to identify the source and function of a protein/DNA sequence. The NR database encompasses protein sequences from non-curated (low quality) and curated (high quality) databases. It contains NR sequences from GenBank translations (i.e. GenPept) together with sequences from other databases [Refseq (Pruitt et al., 2007), PDB (Berman et al., 2003), SwissProt (Boeckmann et al., 2003), PIR (Wu et al., 2003) and PRF]. NR removes 100% identical sequences and merges the annotations and sequence IDs.
We have identified three root causes for annotation errors in the public databases: user metadata submission, contamination error in the biological samples and computational methods. NCBI relies on the accuracy of the metadata provided by researchers that are depositing sequencing data into the database. Data are deposited into NCBI into Biosamples and Bioprojects as raw data, genome assemblies and transcriptomes. Biosamples contain metadata describing the data type, scope, organism, publication, authors and attributes, which include cultivar, biomaterial provider, collection date, tissue, developmental stage, geographical location, coordinates and additional notes. This metadata is then propagated to the sequences that are deposited. For example, if data for DNA sequences were deposited by a plant researcher studying soybeans obtained from a soybean roots, then all sequences tied to that metadata will be labeled with the organism name Glycine max. If the researcher had in fact been working on Glycine soja then this would result in a misassignment of all Glycine max sequences.
The second key challenge that all large databases have is the issue of contamination (Schnoes et al., 2009). For example, if the aforementioned hypothetical soybean research did not remove the soybean root nodules during sample processing, then the tissue sample could also contain DNA from Nitrogen fixating soil bacteria that infect nodules leading to contamination of the sequences and ultimately the sequence database. NCBI is aware of the potential for contamination in sequence databases and describes potential sources of contamination that include: DNA recombination techniques (vectors, adaptors, linkers and PCR primers, transposon and insertion sequences) and sample impurities (organelle, DNA/RNA and multiple organisms). NCBI encourages the use of programs to try to reduce issues with contamination. Specifically, they recommend screening for contamination using VecScreen (VecScreen; https://www.ncbi.nlm.nih.gov/tools/vecscreen/) and BLAST for the sequences used in sequencing library preparation. More broadly, they recommend BLAST to screen out bacterial, yeast and Escherichia coli sequences and BLASTing against the NR database to identify potential contaminating sequences. Unfortunately, despite efforts to reduce contamination, sequences still end up in the NR database that is incorrectly taxonomically classified. This can limit our ability to identify contamination of future sequence submissions, as BLASTing against the database could propagate these types of errors as the database grows in size (Schnoes et al., 2009). The contamination problem is not unique to NCBI but can be found in all large databases. A large-scale study of complete and draft bacterial and archea genomes in the NCBI RefSeq database revealed that 2250 genomes are contaminated by human sequences (Breitwieser et al., 2019). Breitwieser et al. reported 3437 spurious protein entries that are currently present in NR and TrEMBL protein databases.
The third key challenge is that there are errors in the annotations due to the computational error in tools that are based on homology to existing sequences to predict the annotations (Schnoes et al., 2009). These errors have caused annotation accuracy and database quality issues over the years. Annotation errors are not limited to contamination or computationally predicted one. For instance, there exists evidence that suggests some of the experimentally derived annotations may be incorrect (Schnoes et al., 2009).
Therefore, it will be beneficial for researchers to utilize a quality control method to detect misclassified sequences and propose the most probable taxonomic assignment.
To address these well-known problems, there are two approaches in the literature: phylogenetic-based approach and functional approach. For the first approach, Kozlov et al. (2016) have proposed a phylogeny-aware method to detect and correct misclassified sequences in public databases. They utilized the Evolutionary Placement Algorithm (EPA) to identify mislabeled taxonomic annotation. Edgar (2018) has studied taxonomy annotation error in rRNA databases. They showed that the annotation error rate in SILVA and Greengenes databases is about 17%. They also used the phylogenetic-based approach.
In the second approach, it is a common technique for quality control and data cleaning to utilize domain knowledge in the form of ontologies (Chu et al., 2015). Gene Ontology (Ashburner et al., 2000) has been suggested to infer aspects of protein function based on sequence similarity (Holliday et al., 2017). The MisPred Nagy and Patthy (2013) and FixPred (Nagy and Patthy, 2014) programs are used to address the identification and correction of misclassified sequences in the public databases. The FixPred and MisPred methods are based on the principle that an annotation is likely to be erroneous if its feature violates our knowledge about proteins (Nagy et al., 2008). MisPred (Nagy and Patthy, 2013) is a tool developed to detect incomplete, abnormal, or mispredicted protein annotations. There is a web interface to check the protein sequence online. MisPred uses protein-coding genes and protein knowledge to detect erroneous annotations at the protein function level. For example, they have found for a subset of protein databases that violation of domain integrity accounts for the majority of mispredictions. Modha et al. (2018) have proposed a pipeline to pinpoint taxonomic error as well as identifying novel viral species. There is another web-server for exploratory analysis and quality control of proteome-wide sequence search (Medlar et al., 2018) that requires a protein sequence in a FASTA format. European Bioinformatics Institute (EMBL-EBI) developed InterPro (InterPro; http://www.ebi.ac.uk/interpro/) to classify protein sequences at the superfamily, family and subfamily levels. UniProt has also developed two prediction systems, UniRule and the Statistical Automatic Annotation System (SAAS) (SAS; https://www.uniprot.org/help/saas), to annotate UniProtKB/TrEMBL protein database automatically. CDD is a Conserved Domain Database for the functional annotation of proteins (Marchler-Bauer et al., 2011).
Exploring public sequence databases and curating annotations at large-scale are challenging. Previous research on the NR database focused on a small subset of the NR database and analyzed annotation error due to the computational requirements. There has been a study (Schnoes et al., 2009) on misclassification levels for molecular function for a model set of 37 enzyme families. To the best of our knowledge, the amount of misclassification in the entire database has not been well quantified.
Here, we attempt to address these limitations in detecting and correcting annotations at large-scale and make the following contributions:
We utilize a genomics-specific language, BoaG, that uses the Hadoop cluster (Bagheri et al., 2019), to explore annotations in the NR database that is not available in other works.
We also present a heuristic-based method to detect misclassified taxonomic assignments in the NR database that is low-cost and easy to use. We automatically generate a phylogenetic tree from a list of taxonomic assignments and use the tree, along with frequency, the provenance (database of origin) of each taxonomic annotation and clustering information from NR at 95% similarity to identify potential misclassification and propose the most probable taxonomic assignment.
The technique proposed in this work could be generalized to apply to other public databases and different kinds of annotations like protein functions. In this work, we address the taxonomic annotation error in protein databases. We also tested our approach on the RNA dataset introduced in the literature.
We have identified ‘29 175 336’ proteins in the NR database that have more than one distinct taxonomic assignments, among which ‘2 238 230’ (7.6%) are potentially taxonomically misclassified. We also found that the total number of potential misclassifications in clusters at 95% similarity, above the genus level, is ‘3 689 089’ out of 88 M clusters, which are 4% of the total clusters. This percentage of misclassifications in NR has a significant impact due to the potential for error propagation in the downstream analysis (Mukherjee et al., 2015).
The rest of the paper is organized as follows. In Section 2, we present methods and materials for dataset generation and our approach. In Section 3, we discuss the results of taxonomically misclassified proteins within sequences and in NR 95%. We also present the correcting approach for detected sequences. In Section 4, we conclude with suggestions for the future.
2 Materials and methods
In this section, we will describe the overview architecture of our detection and correction approach. Then, we describe the dataset generation and how we generate a phylogenetic tree from taxonomic assignments. Next, we discuss our detection algorithm to find misclassified sequences. Then, we describe our approach to propose taxonomic assignments for the sequences identified as misclassified. Finally, we will describe the sensitivity analysis on changing the different parameters to propose the taxonomic assignments.
2.1 An overview of the method
Figure 1 shows an overview of our approach. The NCBI’NR database files were downloaded from (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/) on October 20, 2018. Taxonomic information was obtained from XML files on NCBI (https://ftp.ncbi.nlm.nih.gov/blast/temp/DB\_XML/). CD-HIT (Fu et al., 2012) (version v4.6.8-2017-1208) was used to cluster NR protein sequences into clusters at 95% similarity using the following parameters (-n 5 -g 1 -G 0 -aS 0.8 -d 0 -p 1 -T 28 -M 0). These parameters use a word length of 5 and require that the alignment of the short sequences is at least 80% of its length. The data acquisition, preprocessing and clustering took about 3 days. The detection and correction part took about 8 h.
Fig. 1.
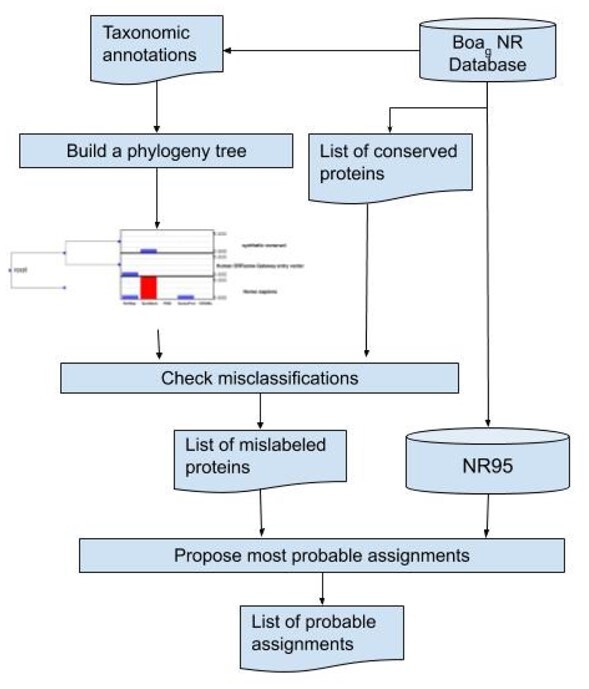
Overview architecture of the proposed method to detecting taxonomically misclassified sequences in the NR database. Diagram shows the raw dataset and steps for the proposed work
We took the NR protein FASTA files that have the definition lines containing annotations from different databases and generate the BoaG format that took about 2 h. Each definition line in the raw data includes protein ID, protein name followed by an organism name in square brackets, e.g. ‘>AAB18559 unnamed protein product [E. coli str. K-12 substr. MG1655]’. BoaG is a domain-specific language that uses a Hadoop-based infrastructure for biological data (Bagheri et al., 2019). A BoaG program is submitted to the BoaG infrastructure. It is compiled and executed on a distributed Hadoop cluster to execute a query on the BoaG-formatted database of the raw data. BoaG has aggregators that can be run on the entire database or a subset of the database taking advantage of protobuf-based schema design optimized for a Hadoop cluster for both the data and the computation. These aggregators are similar to but not limited to aggregators traditionally found in SQL databases and NoSQL databases like MongoDB. A BoaG script requires fewer lines of code, provides storage efficiency and automatically parallelized large-scale analysis.
2.1.1 Dataset generation
To describe our dataset, let denotes the protein and clustering dataset in our study: . Here, is a set of all the proteins in the NR database. represents a set of all clusters at 95% similarity. and in our dataset are about 174 M and 88 M, respectively. τ is a set of taxonomic assignment for proteins, and is a set of functions in the NR database. In this work, we focus on exploring taxonomic assignments.
Definition 1.
Cluster. We define cluster as a set of protein sequences such that their sequence are 95% similar and their sequence length is 80% similar. Every particular cluster, Cj, has k members:
(1)
In Definition 1, each protein sequence belongs to exactly one cluster at 95% similarity, and each cluster has one representative sequence. If a protein is not identical in sequence and length, it will fall into a cluster with no other member.
2.1.2 Generating phylogenetic tree from taxonomic assignments
We get the list of taxonomic assignments that originate from different databases (manually reviewed and computationally created) and build a phylogenetic tree by utilizing the ETE3 library (Huerta-Cepas et al., 2016). This library utilizes the NCBI taxonomy database that is updated frequently.
Definition 2.
Annotation List. Each phylogeny tree is associated with one particular protein, Pi and has the set of taxonomic assignments that originate from different databases. Here, denotes annotation number j for protein Pi:
(2)
For example, the protein sequence AAB18559 has taxonomic assignments of ‘511 145’ and ‘723 603’ that each appeared once.
Definition 3.
Provenance. For the particular protein Pi, we define the provenance of annotation as a set of databases that the annotation originates from:
(3)
In Definition 3, annotations from GenBank, trEMBL and PDB are calculated computationally, while annotations from RefSeq and SwissProt are manually reviewed. For example, meaning that the tax id ‘511 145’ for the sequence AAB18559 originates from the GenBank database.
Definition 4.
Annotation Probability
We define probability for each taxonomic assignment based on the frequency of each annotation divided by total taxonomic assignments from different databases as follows:
(4)
In Definition 4, represents the annotation that calculated computationally (Comp) from databases i.e. GenBank, trEMBL, PDB and denotes the reviewed (Rev) one from RefSeq, SwissProt. One annotation might originate from both reviewed and computational created databases. We use a conservative weighting factor, w, to denote the importance of the experimental annotation (manually reviewed) in which w is an integer number and .
The upper bound for total proteins, i.e. , is 174 M at the time we downloaded NR. Each leaf node, Va, in the phylogenetic tree is annotated with the information described in the Definitions 2, 3 and 4. There are list of frequencies and provenances, shows as top bar, since one particular taxonomic annotation might originate from different databases:
| (5) |
For particular protein Pi, we define most probable annotation (MPA) as as an annotation with the highest probability among the set of annotations. In addition, we define least probable annotation (LPA), with the lowest probability, that potentially might be misclassified as , in which .
Definition 5.
Conserved Proteins. We define a conserved protein as a protein that has more than 10 distinct taxonomic assignment. List of these conserved proteins are shown in our repository (https://github.com/boalang/nr).
(6)
2.2 Approach to detect taxonomic misclassification
Algorithm 1 The NR misassignment detection algorithm. Input comes from the BoaG query (Supplementary Material)
1: procedure DetectMisassignments
2: ▹ m = 174 M proteins
3: while do
4: .
5: If misassigned(phylo) && not conserved(Pi) then
6: print (misassignment found in Pi)
7: procedure PhyloTreePi
8: ▹ used to generate phylogeny tree
9: ▹ From taxa list
10: for in do
11:
12: return phyloTree.
Our approach is as follows: first, we run a BoaG query (Supplementary Fig. S1) on the NR database. This query runs on the full NR database in the Hadoop cluster. The Algorithm 1 describes the detection approach for misclassified sequences. It iterates over the entire NR database. In line 4, it takes a protein Pi and generates a phylogeny tree from the set of taxonomic assignments for Pi. Then, in line 5, it checks if it has a misclassification. If the lowest common ancestor (LCA) is the root level, it means there is a considerable distance between taxonomic assignments for that particular protein sequence. Therefore, there is a potential misassignment among the list of the taxonomic assignments due to the contamination in the sample, error in the computational method, or data entry by the researchers who deposited the sequence. We call this a root violation or conflict. We also consider superkingdom, phylum, class, order and family violations. In addition, we looked at the highly conserved proteins to remove false positives because conserved proteins might appear in species that are far from each other, i.e. belong to different domains in the phylogeny tree. We did not remove the list of conserved proteins in the dataset, since they contain taxonomic information that were utilized for proposing taxonomic assignment for the misclassified sequences. Assume Pi belongs to Cj. Once we detected the violation in Pi, we look at the cluster Cj and consider the most frequent taxonomic assignment as the correct taxa. Details are shown in Section 2.3.
The Algorithm 1 requires time. Here, is the size of proteins in the NR database and is the upper bound of number of taxonomic assignments per proteins. In line 5, misassigned(phylo) verifies if the LCA of the generated tree shows a root violation or any other violations. The expression checks if the protein sequence is a conserved one (Equation 6). This requires time because this is a straight-forward fetch, and we have the pointer to the root of the tree to check the LCA. In line 5, to check that a protein is not in a conserved list, Definition 5, it requires a membership test and takes time. This conserved list is a precomputed list from our dataset that is shown in our repository. We wrote a multi-threaded Python code, and the total run time for the algorithm was 7 h for the entire NR database on an iMac (Retina 5 K, 27-inch, Late 2015) with core i7 and 32 GB RAM. For the second procedure, in line 11, the algorithm requires to calculate the probability of each leaf in the generated phylogenetic tree.
Algorithm 2 Annotation correction: The MPA for the misclassified sequences. Input from the BoaG query (Supplementary Material)
1: procedure mostProbable
2: ▹ Most probable taxa
3: if then
4: return ().
5: else
6: in which
7: .
8: return .
9: procedure ClusterMostProbableclustr, p, c
10: if then
11: for in do
12:
13: ▹ Most probable taxa
14: if then
15: return
16: else
17: return null ▹ Cannot fix misclassification
2.3 The most probable taxonomic assignment for detected misclassifications
For the detected misclassified sequences, we defined criteria to propose the most probable taxonomic assignment (MPA). First, we ran a BoaG query (Supplementary Fig. S2) to retrieve the annotations and clustering information at 95% similarity. As shown in Definition 4, we considered provenance or database of origin, frequency of annotations to calculate the probable taxonomic assignment (MPA), which is the highest probability. Let’s assume that Pi belongs to cluster Cj. If the algorithm does not find the MPA within a certain threshold, probability p, then we look at the cluster of 95% similarity that the sequence belongs to. Second, we found the most probable taxonomic assignment in Cj. If a particular taxonomic assignment was the most frequent one in Cj then we return that annotation as the MPA for the protein sequence Pi. For example, in cluster Cj, 7 sequences out of 10 sequences have a specific annotation. Then, we consider this annotation to be the MPA protein sequence Pi with 70% confidence.
Details are shown in the Algorithm 2. In line 2, for a particular protein Pi, it returns the most frequent taxonomic assignment within a certain threshold p. Let’s assume we want a taxonomic assignment that appears more than 70% of the time. If the algorithm does not find the MPA, it checks the cluster Cj with 95% similarity that this sequence belongs to and finds the one with a certain probability, p and a cluster size, c (line 7). In line 9, ClusterMostProbable takes the cluster id and finds the most probable taxonomic assignment in the cluster (line 13).
The Algorithm 2 requires time, Definition 2, to find the top(1) or maximum probability of an annotation in the list of annotations.
2.4 Simulated and literature dataset
To evaluate the performance of our taxonomic misclassification approach, we generated a simulated dataset. We took a subset of one million proteins of the reviewed dataset, i.e. RefSeq database and randomly misclassified 50% of the proteins in the sample by adding a taxonomic assignment from another phylum or kingdoms. Then, we tested if the approach can detect these sequences. We also tested our approach for detecting misclassified sequences and correcting them on the real-world data, presented in the literature (Edgar, 2018; Kozlov et al., 2016). These works have focused on the RNA dataset, and they quantified misclassified RNA sequences. We also used CD-HIT to cluster RNA databases based on 95% sequence similarity. Further details on the simulated dataset, scripts and data files can be accessed from https://github.com/boalang/nr.
2.5 Sensitivity analysis
We define sensitivity analysis as a way that an input parameter affects the output of the proposed approach. Here, probability based on annotation frequencies and the cluster size are the two input parameters that affect what percentages of detected misclassified sequences that we can fix, i.e. MPA, as shown in Algorithm 2 on the NR dataset. The algorithm will not give the same suggestion for changes in parameters. For example, if we change the cluster size, number of proteins in the cluster, it may or may not find correct taxa. We conducted a sensitivity analysis based on the probability of each annotation that we defined in Definition 4 and the size of the cluster of 95% that the sequence belongs to. We run the algorithm to find the most probable taxonomic assignments (MPA) with different clusters size, c and with different probabilities, p. As it is shown in (Supplementary Fig. S3), with a probability of 0.4 and without giving more weight to the annotations that verified experimentally, we could provide a most probable taxonomic assignment to about 60% of the proteins that we detected as misclassified. We also extended sensitivity analysis by giving more weight to the experimental taxonomic assignment with the probability of 0.4 we could provide the most probable taxonomic assignment for more than 80% of the sequences that were identified as a misclassification.
3 Results
In this section, we present the number of proteins that are misclassified taxonomically. We also present the performance of our work on the simulated dataset and the datasets presented in the literature. Then, we describe our findings on misassignments in the clusters. Next, we present correcting taxonomic misclassification. Finally, we discuss a case study that we explored deeply to identify a subset of clusters that contain sequences with a taxonomic misclassification.
3.1 Detected taxonomically misclassified proteins
We found ‘29 175 336’ proteins in the NR database that have more than one distinct taxonomic assignments. The rest of the proteins have identical taxonomic assignments, even though they originate from different databases. The total number of potential taxonomically misclassified sequences is ‘2 238 230’ out of ‘29 175 336’ (7.6%) at the time of download. This percentage of NR is significant because of the error propagation in the downstream analysis (Mukherjee et al., 2015). Table 1 shows the number of violations in the protein sequences in NR at the superkingdom to the family level that have been detected by applying distance in the phylogenetic tree. The second column shows the number of total proteins that have a certain number of taxonomic assignments. For example, there are ‘17 496 167’ protein sequences in NR that have 2 taxonomic assignments in which ‘30 237’ of them have potential root violations and ‘47 271’, ‘202 205’, ‘59 606’, ‘177 132’, ‘290 065’ have kingdom, phylum, class, order and family violations, respectively. For the NR datasets, we did a sample study of 1000 samples and manually found 5.5% misassignment. The potentially misclassified sequences detected by the approach was around 7.6% that is consistent with the total number that was manually found, i.e. 5.5%.
Table 1.
Detected misclassified taxonomic proteins in the NR database
| taxa | Total | root | Kingdom | Phylum | Class | Order | Family |
|---|---|---|---|---|---|---|---|
| 2 | 17 496 167 | 30 237 | 47 271 | 202 205 | 59 606 | 177 132 | 290 065 |
| 3 | 5 921 066 | 14 376 | 19 666 | 107 705 | 38 575 | 104 709 | 236 515 |
| 4 | 2 132 971 | 4673 | 21 587 | 64 801 | 17 662 | 47 914 | 94 054 |
| 5 | 1 022 482 | 3143 | 9469 | 34 322 | 10 050 | 27 295 | 53 276 |
| 6 | 642 760 | 2509 | 5662 | 24 136 | 7333 | 23 324 | 37 998 |
| 7 | 388 794 | 1572 | 3959 | 12 972 | 5905 | 13 488 | 27 221 |
| 8 | 262 682 | 1121 | 2803 | 5988 | 5375 | 10 075 | 16 340 |
| 9 | 190 756 | 783 | 2647 | 3825 | 3173 | 7557 | 12 681 |
| 10 | 156 767 | 667 | 1843 | 3805 | 2451 | 6413 | 11 327 |
| >10 | 960 891 | 10 940 | 23 232 | 30 048 | 38 679 | 46 391 | 107 679 |
The first two bold rows show the highest potential misassignments because if a protein has two or three taxonomic assignments and shows a root or kingdom violation, it is more likely to be misclassified.
Table 1 shows proteins that have less than 10 taxonomic assignments. The last row shows all other proteins with more than 10 assignments. The first two bold rows show the highest potential misassignments because if a protein has two or three taxonomic assignments and shows a root or kingdom violation, it is more likely to be misclassified. The full list of detected misclassified proteins, and detailed analysis are shown in our GitHub repository. We did not report the genus conflict since the probability of a false-positive misclassification is much higher compared to other taxonomic levels of conflict, such as root and superkingdom.
Figure 2 shows one example of a detected misclassified protein, with an id of NP_001026909. Since the LCA in this tree is the root, it means those taxonomic assignments belong to a different kingdom. Leaves are annotated with a frequency of each taxonomic assignment as a bar chart from all reviewed and unreviewed databases i.e. RefSeq (Pruitt et al., 2007), GenBank (Benson et al., 2009), PDB (Berman et al., 2003), UniProtSwissProt (Boeckmann et al., 2003) and UniProtTrEMBL (Consortium, 2014), respectively. As it is shown in the annotations, there are potential misassignments even though the key IDs originate from the reviewed databases, i.e. RefSeq and SwissProt. In this example, synthetic construct is the misassignment and the MPA for this protein is Homo sapiens.
Fig. 2.

Phylogenetic tree generated for sequence ID NP_001026909. Taxonomic assignments originate from GenBank, trEMBL, PDB, RefSeq and SwissProt database
We also explored some clusters in depth as a case study and identified proteins that are taxonomically misclassified as Glycine, which are in fact contamination in the sample (Supplementary Section S1.6).
3.2 Performance on simulated and real-world dataset
Our approach to detecting taxonomically misclassified proteins on the simulated dataset showed 87% recall and 97% precision. We define true positive (TP) as sequences that misclassified in the sample, and our approach retrieves those sequences. False positives (FP) are sequences that do not have misassignments, but our approach classified them as misclassified sequences. False negative (FN) is a reviewed sequence which the algorithm incorrectly classifies as correct (not misclassified), while it is misclassified. Some of these false negatives are due to changes in the taxonomies over time. Some taxonomic IDs might be obsolete, deleted, or get merged into other tax ids. We also found that some of the trees generated by NCBI API have the root named ‘Cellular Organisms’ with rank equal to ‘no rank’, that did not fall in any of the taxonomic ranking. We use the following formula to calculate precision and recall (precision = ).
We extended our experiment and added more than two random assignments to the proteins and the precision increased. The reason is that adding more random assignments increases the distance among tax IDs in the phylogeny tree and hence increases the chance of detection by the approach. We also tested our approach on the dataset presented by (Edgar, 2018) in which they explored the Greengenes and the SILVA database for taxonomic error. Our methods reproduced their finding on annotation conflicts among SILVA and Greengenes (McDonald et al., 2012) database. We did not run their approach on the simulated dataset since it was designed to detect misassignments in rRNA sequences, not proteins. For evaluating our work, we looked for similar works that focused on detecting taxonomic misassignments. However, their approach was hard-coded for RNA sequences. Therefore, we modified our approach to test on their dataset. The proposed work focuses on inconsistencies among the list of taxonomies, and it can be applied to the RNA sequences as well. We clustered their dataset at 95% similarity and used the same consensus-based technique to detect conflicts between sequences and clusters. The phylogeny-aware technique proposed by Kozlov et al. (2016), called SATIVA, identifies and corrects misclassified sequences for RNA databases . They utilized the EPA to detect misclassified sequences. In their approach, a reference tree is created. Then, to estimate the most likely placements of the query sequence in the reference tree, they use EPA. We took their RNA dataset and cluster the sequences at 95% similarity, then utilized our technique to check if the annotation of each sequence has a conflict with a cluster that the sequence belongs to. There is a difference between the NR dataset and the RNA dataset presented by Kozlov et al. in terms of the number of taxonomic annotation. In their experiment, they have one taxonomic label for each sequence; however, in the NR database, there are several annotations for each protein sequence. Therefore, their technique is not designed to detect misclassification in a set of given annotations. In terms of running time, the clustering at 95% is less expensive than running sequence alignment and generating phylogeny-tree and verifying each query sequence. Therefore, our approach is scalable for large-scale sequence databases. In general, examining the distance on the phylogenetic tree of multiple annotations for the shorter sequences performs better compared to the alignment-based approaches with the reference databases. Table 2 shows the standard values for precision and recall, as well as the running time comparison. Our approach to detect misassignments on the sample RNA dataset has a lower recall. This is due to the relatively smaller datasets that caused some clusters to have few members and made it challenging to detect misclassified sequences.
Table 2.
Accuracy of detecting misassignments and the comparison with work presented in SATIVA (Kozlov et al., 2016)
| Precision |
Recall |
Runtime |
|||
|---|---|---|---|---|---|
| SATIVA | Proposed | SATIVA | Proposed | SATIVA | Proposed |
| 0.93 | 0.98 | 0.98 | 0.90 | 116 min | 12 min |
Note: The best values are highlighted.
3.3 Detected misassignments in clusters
There are ‘12 960 476’ clusters at 95% similarity that have two taxonomic assignments in which ‘17 099’ of them have potential root violations and ‘92 526’, ‘263 844’, ‘100 560’, ‘267 251’ and ‘461 795’ have kingdom, phylum, class, order and family violations, respectively. The number of root violations for 2 tax assignments in clusters is less than sequences because there are protein sequences that do not belong to any clusters at 95% similarity. In total 64 M out of 174 M proteins (36%) in the NR database are unclustered (Supplementary Table S1). The total number of potential misclassifications for clusters at 95% similarity, without genus level, is ‘3 689 089’ out of ‘25 159 866’ clusters that have more than 1 taxa, which are 15% of total clusters. Detail numbers of misclassified sequences in the clusters along with an example of detected taxonomically assigned annotations in the cluster are shown in the Supplementary Materials.
3.4 Correcting taxonomic misclassification
Each protein sequence belongs to one and only one cluster. We analyzed the set of top three taxonomic annotations of each sequence and compared them with the top three taxonomic annotations of the cluster the sequence belongs to. For example, top three taxonomic assignment for sequence with id AAA32344 is ’10 743’, ‘1 182 665’ and ‘656 390’. This sequence falls in the cluster-id 8 461 728, and the top tax ids in this cluster are ‘562’, ‘83 334’ and ‘621’. We consider this as a conflict between sequence AAA32344 and cluster 8 461 728. All three annotations are different; therefore, we consider this case as three conflicts. If two annotations out of three are different, we classify this as two conflicts. If one taxonomic annotation is different from the two sets, we classify it as one conflict. Finally, if the three annotations are identical, there is no conflict. Different percentages of conflicts from the subset of one million sequences are shown in Supplementary Fig. S5.
Table 3 shows several examples of the protein sequences that we have found to be misclassified in the NR database. The first column represents the sequence id, and the second column is the cluster id corresponds to the sequence. The third column shows the original taxonomic assignment, and the forth column is the proposed taxonomy based on the consensus information from the clusters of the NR database at 95% similarity. The last column is Confidence Score (CS), a number between 0 and 1, shows how confident we are in proposing new taxonomic assignment based on the consensus information from the clusters at 95% similarity. This score calculated from the clusters’ information as top taxonomic assignment, i.e. most frequent one, in the cluster divided by total taxa in the cluster. The assumption here is that the consensus of multiple independent sequence annotations can catch simple misannotation errors. For example, protein sequence with id YP_950729 has Staphylococcus virus PH15 as its taxonomic assignment. It falls in cluster id 83178931 and the recommended annotation is firmicutes. We also conducted similar analysis on the dataset by SATIVA, and could reproduce the proposed taxa based on the consensus information from the clusters. For the dataset by Edgar (2018) since the number of sequences was small, we could not get clusters with enough members to suggest annotations.
Table 3.
Proposed taxa for the detected misclassified sequences in NR
| Protein ID | Cluster ID | Original taxa | Proposed taxa | CS |
|---|---|---|---|---|
| AAB18559 | 18 982 245 | Uncultured actinobacterium | Escherichia coli | 1 |
| AAT83007 | 21 005 513 | Mycobacteroides abscessus | Cutibacterium acnes | 0.8 |
| CCW09133 | 9 901 357 | Streptococcus pneumoniae | Bacillus cereus | 0.5 |
| KFV03115 | 13 041 247 | Tauraco erythrolophus | Pelodiscus sinensis | 0.4 |
| YP_950729 | 83 178 931 | Staphylococcus virus PH15 | firmicutes | 0.8 |
Note: Last column shows the confidence score (CS).
3.5 Running time
We conducted an analysis of the RNA dataset presented by SATIVA with different samples of sequences. Firstly, we took 100 sequences and ran SATIVA in the sample. Next, we took 500 sequences. In two other experiments, we took 1000 and 2000 additional sequences and recorded the running time. Figure 3 shows the comparison in terms of running time between proposed work and the SATIVA method. The most time-consuming part of our approach is the clustering time (run by CD-HIT). By adding more sequences, the runtime slightly increased. In contrast, for the SATIVA method, as we increase the number of sequences, the running time increases significantly. The computational expensive part of the SATIVA approach is the phylogenetic methods (EPA) it uses. The comparison between the proposed approach and SATIVA method has been made on the local system iMac (Retina 5 K, 27-inch and Late 2015) with core i7 and 32 GB RAM.
Fig. 3.
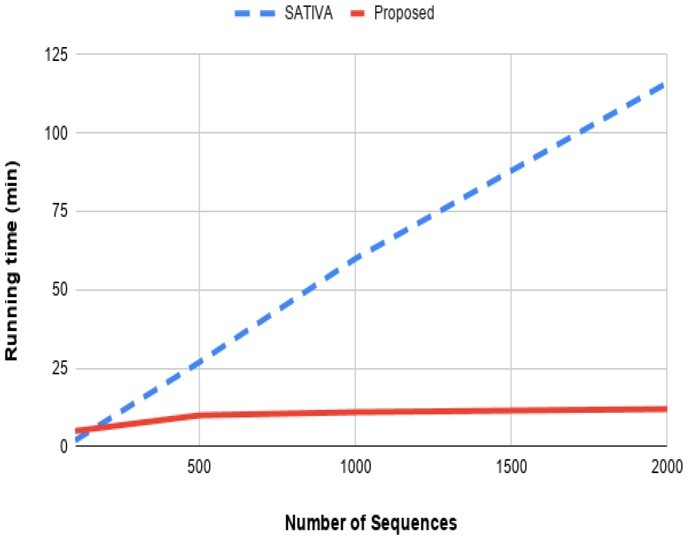
Compare running time of the proposed work with the SATIVA method. We used dataset from the SATIVA paper
4 Discussion and conclusion
In this work, we addressed taxonomically misclassified sequences in the large publicly available databases by utilizing our domain-specific language and Hadoop-based infrastructure. We focused on the misassignments at the taxonomic level, and similar to MisPred (Nagy and Patthy, 2013), we utilize the current knowledge of organismal classification, to detect annotation errors. Similar to (Holliday et al., 2017), we utilized this form of knowledge-based reasoning for quality control and detect annotation errors. Compared to other works, our work differs in that we do not need to run sequence similarity to explore annotations and find taxonomic inconsistency for each query sequence in the NR database. Instead, first, we clustered the NR proteins at the data generation phase and this is a one-time cost and used the clustering information later to detect annotation error and propose the MPAs. In this work, we proposed a heuristic method to find inconsistencies in the metadata, i.e. taxonomic assignments. In our method, we proposed the most probable taxonomic assignment for each protein sequence. We applied this method to the entire database. We also provided a Python implementation that could be used for analyzing a list of annotations for any protein of interest and find the misclassification. The violations reported in this paper in Table 1 are the upper bound of the misassignments. The more stringent filter includes hypothetical protein and membrane protein functions in the list of conserved protein, which will lower the number of identified misclassification. We use open-source CD-HIT clustering software only at the data generation phase, and we could utilize any other clustering software. Steinegger and Söding, (2018) have built a novel clustering tool that clusters a huge protein database in linear time . Since this one-time cost happens only in the data generation phase, our approach to detect misassignments and propose the most probable taxonomic assignment is scalable.
4.1 Applications and limitations
At 95% similarity, 64 M sequences in the NR remain unclustered. Therefore, if a particular protein remains unclustered, there is not enough consensus information to correct annotation for that protein. A solution for this might be to take the EPA approach (Kozlov et al., 2016) for these sequences that remains as future work. The proposed technique to detect misassignments may fail with recent horizontal gene transfer (HGT) events since HGT is not transferred from parent to offspring. However, the consensus information from the clusters might reveal annotation errors. The proposed heuristic technique and findings could also be applied to other databases. Current work focuses on detecting and correcting misassignments at the level of taxonomic assignments, and we do not address protein function annotations.
5 Conclusion
Misclassification can lead to significant error propagation in the downstream analysis. In this work, we proposed a heuristic approach to detect misclassified taxonomic assignments and find the MPAs for misclassified sequences. This method will be a valuable tool in cleaning up on large-scale public databases. The technique we proposed could be extended in the form of ontologies to address other annotation errors like protein functions.
Supplementary Material
Acknowledgements
This study was supported by the National Science Foundation under Grant CCF-15-18897, CNS-15-13263 and CCF-19-34884 and the VPR office at Iowa State University. The listed funders played no role in the design of the study, data generation, implementation, or in writing the manuscript. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF grant number ACI-1548562.
Funding
This study was supported by the National Science Foundation under Grant CCF-15-18897, CNS-15-13263 and CCF-19-34884 and the VPR office at Iowa State University
Financial Support: none declared.
Conflict of Interest: none declared.
Contributor Information
Hamid Bagheri, Department of Computer Science, Ames, IA 50011, USA.
Andrew J Severin, Genome Informatics Facility, Iowa State University, Ames, IA 50011, USA.
Hridesh Rajan, Department of Computer Science, Ames, IA 50011, USA.
References
- Ashburner M. et al. (2000) Gene ontology: tool for the unification of biology. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri H. et al. (2019) Shared data science infrastructure for genomics data. BMC Bioinformatics, 20, doi: https://doi.org/10.1186/s12859-019-2967-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D.A. et al. (2009) Genbank. Nucleic Acids Res., 37, D26–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M. et al. (2003) The protein data bank. In: Protein Structure. pp. 394–410, CRC Press. [Google Scholar]
- Boeckmann B. et al. (2003) The Swiss-Prot protein knowledgebase and its supplement trembl in 2003. Nucleic Acids Res., 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitwieser F.P. et al. (2019) Human contamination in bacterial genomes has created thousands of spurious proteins. Genome Res., 29, 954–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X. et al. (2015) Katara: a data cleaning system powered by knowledge bases and crowdsourcing. In: Proceedings of the 2015 ACM SIGMOD International Conference on Management of Data, Association for Computing Machinery, Melbourne, Victoria, Australia, pp. 1247–1261.
- Consortium,U. (2014) Uniprot: a hub for protein information. Nucleic Acids Res., 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. (2018) Taxonomy annotation and guide tree errors in 16s RRNA databases. Peer J., 6, e5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L. et al. (2012) Cd-hit: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday G.L. et al. (2017) Evaluating functional annotations of enzymes using the gene ontology. In: The Gene Ontology Handbook. Humana Press, New York, NY, pp. 111–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J. et al. (2016) Ete 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol., 33, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov A.M. et al. (2016) Phylogeny-aware identification and correction of taxonomically mislabeled sequences. Nucleic Acids Res., 44, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A. et al. (2011) Cdd: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res., 39, D225–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D. et al. (2012) An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J., 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlar A.J. et al. (2018) Aai-profiler: fast proteome-wide exploratory analysis reveals taxonomic identity, misclassification and contamination. Nucleic Acids Res., 46, W479–W485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modha S. et al. (2018) Victree: an automated framework for taxonomic classification from protein sequences. Bioinformatics, 34, 2195–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S. et al. (2015) Large-scale contamination of microbial isolate genomes by illumina phix control. Stand. Genomic Sci., 10, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A., Patthy L. (2013) Mispred: a resource for identification of erroneous protein sequences in public databases. Database, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A., Patthy L. (2014) FixPred: a resource for correction of erroneous protein sequences. Database, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A. et al. (2008) Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinformatics, 9, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K.D. et al. (2007) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res., 35, D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoes A.M. et al. (2009) Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol., 5, e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M., Söding J. (2018) Clustering huge protein sequence sets in linear time. Nat. Commun., 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.H. et al. (2003) The protein information resource. Nucleic Acids Res., 31, 345–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.