

Abstract
Whiteflies (Hemiptera: Sternorrhyncha: Aleyrodidae) are a superfamily of small phloem-feeding insects. They rely on their primary endosymbionts "Candidatus Portiera aleyrodidarum" to produce essential amino acids not present in their diet. Portiera has been codiverging with whiteflies since their origin and therefore reflects its host’s evolutionary history. Like in most primary endosymbionts, the genome of Portiera stays stable across the Aleyrodidae superfamily after millions of years of codivergence. However, Portiera of the whitefly Bemisia tabaci has lost the ancestral genome order, reflecting a rare event in the endosymbiont evolution: the appearance of genome instability. To gain a better understanding of Portiera genome evolution, identify the time point in which genome instability appeared and contribute to the reconstruction of whitefly phylogeny, we developed a new phylogenetic framework. It targeted five Portiera genes and determined the presence of the DNA polymerase proofreading subunit (dnaQ) gene, previously associated with genome instability, and two alternative gene rearrangements. Our results indicated that Portiera gene sequences provide a robust tool for studying intergenera phylogenetic relationships in whiteflies. Using these new framework, we found that whitefly species from the Singhiella, Aleurolobus, and Bemisia genera form a monophyletic tribe, the Aleurolobini, and that their Portiera exhibit genome instability. This instability likely arose once in the common ancestor of the Aleurolobini tribe (at least 70 Ma), drawing a link between the appearance of genome instability in Portiera and the switch from multibacteriocyte to a single-bacteriocyte mode of inheritance in this tribe.
Keywords: divergence dating; genome stasis; long-enduring taxon; molecular evolution, symbiosis; whitefly development; whitefly systematics
Significance
Whiteflies have established a mutualistic relationship with Portiera aleyrodidarum, a symbiotic bacterium. A long history of strict mother-to-offspring transmission of Portiera allows this symbiont to reflect well its host evolutionary history. Moreover, Portiera genomes usually show high synteny, but in rare cases, genomic instability is present. As the current molecular and morphological classification tools for whiteflies are limited and prone to significant errors, we used the unique characteristics of Portiera genomes to study both Portiera and whitefly evolution. This framework allowed us to propose a new working hypothesis for the evolution of the rare genomic instability in Portiera, involving a switch from multi- to a single-bacteriocyte mode of inheritance in whiteflies.
Introduction
Whiteflies are small phloem-feeding insects, which, together with aphids, scale insects, and psyllids, form the Sternorrhyncha suborder (Grimaldi and Engel 2005). Whiteflies are classified into one superfamily the Aleyrodoidea that includes one family, the Aleyrodidae. The Aleyrodidae consist of three extant subfamilies, the Udamoselinae, the Aleurodicinae, and the Aleyrodinae, and an extinct one, the Bernaeinae. The Udamoselinae subfamily contains only one genus and two species. The Aleurodicinae subfamily contains 21 extant genera, mainly distributed in Neotropical/Australasian regions (Charles 2010; Ouvrard and Martin 2020). The Aleyrodinae, with at least 142 described genera, is the most diverse and globally distributed subfamily and includes the major pest species Bemisia tabaci and Trialeurodes vaporariorum (Manzari and Quicke 2006; Ouvrard and Martin 2020). Although the extant whitefly subfamilies were reported to originate in the Middle Cretaceous (Campbell et al. 1994), the first fossils of the Aleurodicinae and Aleyrodinae subfamilies were dated to the Lower Cretaceous (Drohojowska and Szwedo 2015). During that period, whiteflies were associated with gymnosperm forests and/or proangiosperms, in contrast to extant whitefly species, which feed mainly on angiosperms. It is assumed that the emergence of angiosperms in the Lower Cretaceous opened new environmental niches and has promoted diversification and speciation of whiteflies along with their angiosperm hosts (Middle–Upper Cretaceous), leading to the emergence of the modern whitefly species (Drohojowska and Szwedo 2015).
Whiteflies, as most sternorrhynchan insects, harbor obligatory intracellular bacterial symbionts (P-endosymbionts) within specialized cells, termed bacteriocytes. Generally, these P-endosymbionts complement the restricted diets of their hosts (plant sap) and possess genomes reduced to a basic set of genes devoted to maintaining the symbiotic relationship (e.g., essential amino acids biosynthesis) and minimal cell functions (Hansen and Moran 2014; Latorre and Manzano-Marín 2017). The P-endosymbiont of whiteflies is “Candidatus Portiera aleyrodidarum” (hereafter Portiera) (Thao and Baumann 2004), which forms a monophyletic clade with “Ca. Carsonella ruddii”, the P-endosymbiont of psyllids. Based on molecular data, it has been proposed that the ancestral symbiosis was established in the Psyllinea lineage (Shcherbakov 2000), before its divergence into the Aleyrodoidea and Psylloidea lineages (Santos-Garcia et al. 2014). Because Portiera, as other P-endosymbionts, exhibits strict mother-to-offspring transmission, it has been codiverging with whiteflies since their origin. Moreover, no host-switching of Portiera has been documented (Thao and Baumann 2004; Santos-Garcia et al. 2015), even among recently diverged species belonging to the same species complex (Hsieh et al. 2014; Wang et al. 2019). Therefore, Portiera lineages reflect well both their own and their hosts phylogenetic relationships (De Vienne et al. 2013) and divergence times (Santos-Garcia et al. 2015).
Until present, only three Portiera genomes from species others than B. tabaci have been sequenced: Aleurodicus dispersus and Aleurodicus floccissimus from the Aleurodicinae subfamily, and T. vaporariorum from the Aleyrodinae. Like other P-endosymbionts, these three Portiera have maintained a genome stasis since the emergence of both the Aleurodicinae and the Aleyrodinae whitefly subfamilies, more than 135 Ma (Sloan and Moran 2013; Santos-Garcia et al. 2015). In contrast, Portiera genomes from the B. tabaci species complex, although syntenic among themselves, are extensively rearranged when compared with the other three published Portiera genomes. The genome rearrangements of Portiera from B. tabaci seem to be correlated with a massive loss of genes required for correct DNA replication and the repair machinery. These losses include the DNA polymerase III subunit epsilon dnaQ, which is required for repairing spontaneous mutations (proofreading activity) (Sloan and Moran 2013; Santos-Garcia et al. 2015). Extensive rearrangements are very uncommon events in P-endosymbionts evolution (Moran and Bennett 2014), and therefore, it is not clear if the genome instability of Portiera from B. tabaci is a unique event or a more general phenomenon present in other related and unrelated Portiera lineages.
In this work, we aimed to deepen our understanding of Portiera genome evolution and the origin of genome instability. Because P-endosymbionts gene sequences have been recognized as a valuable resource for reconstructing aphids (Martinez-Torres et al. 2001; Jousselin et al. 2009; Nováková et al. 2013; Meseguer et al. 2015, 2017) and psyllids (Hall et al. 2016) phylogenetic relationships, we used up to five Portiera genes to reconstruct the phylogeny and divergence of 42 whitefly species belonging to 25 different genera. Using this approach, we found that Portiera of Aleurolobus and Singhiella whitefly species form a monophyletic clade together with Portiera of Bemisia, the Aleurolobini tribe. Next, we conducted a polymerase chain reaction (PCR) screening to identify two alternative genome rearrangements and the presence/absence of a functional dnaQ gene along the obtained Portiera phylogeny. Although most screened Portiera presented the ancestral gene order and a functional dnaQ, all Portiera of the Aleurolobini tribe did not seem to encode a copy of dnaQ and presented different rearrangements compared with the ancestral order. At the final stage, we sequenced the genome of Portiera from Singhiella simplex, which is the most basal Aleurolobini species, to corroborate our screening. We found that the genome of Portiera from S. simplex contains a pseudogenized dnaQ and presents a new genome architecture. Also, it presents large intergenic regions and high number of repeat sequences. Finally, we discuss the possible link between the bacteriocyte transmission mode and the appearance of genome instability in Portiera.
Materials and Methods
Whitefly Collection and Genomic DNA Extraction
A total of 29 samples, accounting for 25 different whitefly species, were obtained from different sources: freshly collected adults (stored in ethanol until use), Prof. Dan Gerling’s ethanol-preserved collection, and exsiccate collection samples from the Natural History Museum (NHM) in London (nymphs were removed from dry leaves and sent in ethanol) (supplementary table 1, Supplementary Material online).
Before genomic DNA (gDNA) extractions were performed, five adult insects (or nymphs from the NHM collection) were rehydrated by consecutive passes in 70%, 50%, 30%, and 0% v/v ethanol solutions in sterile water. Whiteflies were transferred to a new 1.5-ml tube containing 80 μl lysis buffer T1 and were homogenized with 1.4-mm zirconia beads (CK14, Bertin Instruments) using a bead-beater (Minilys, Bertin Instruments). gDNA was extracted with NucleoSpin Tissue XS (Macherey-Nagel) following the manufacturer instructions. For the NHM samples, a nondestructive method was used whenever possible. Nymphs were incubated overnight (56 °C) in 80 μl lysis buffer T1 and 8 μl Proteinase K (20 μg/μl). gDNA was extracted from the lysis buffer using the NucleoSpin Tissue XS standard protocol. The nymphs were recovered, cleaned with sterile water, and stored in fresh ethanol. gDNAs from seven samples that had less than five individuals were subjected to whole-genome amplification (GenomiPhi V2, GE Healthcare), following manufacturer instructions, to ensure sufficient material.
For Illumina sequencing, S. simplex adults were accidentally collected together with Pealius mori adult whiteflies in July 2018 from Ficus benjamina (GPS coordinates 31.904511; 34.804562) and stored in ethanol. Later, whiteflies were rehydrated and sexed. Bacteriocytes in adult insects are located in the abdomen, close to the gonads (Buchner 1965). Therefore, female abdomens (50) were dissected under a stereomicroscope using autoclaved 1× phosphate-buffered saline. Abdomens were homogenized with a bead-beater, and gDNA was extracted with NucleoSpin Tissue XS, as described above. Whole-genome shotgun sequencing was performed by NovSeq 6000 using a TruSeq DNA PCR Free Library (2× 150 bp) at Macrogen Europe.
PCR Screening and Sequencing
To reconstruct Portiera phylogeny, five genes present in all insect endosymbionts showing extremely reduced genomes (Moran and Bennett 2014) were selected. These genes are widely used as bacterial phylogenetic markers and include the 16S and 23S ribosomal RNAs (rRNAs), the chaperonins groEL and dnaK, and the RNA polymerase sigma factor rpoD. We manually designed Portiera-specific universal primers using available Portiera genomes from both the Aleyrodinae (B. tabaci and T. vaporariorum) and Aleurodicinae (A. dispersus and A. floccissimus) subfamilies in UGENE v1.28.1 (Okonechnikov et al. 2012) (supplementary table 2, Supplementary Material online). Primers melting temperature (Tm), off-targets, and possible primer-dimer interactions were computed with Primer3 software implemented on https://eu.idtdna.com/calc/analyzer (last accessed October 24, 2020).
Primers (0.5 mM each) were mixed with the KAPA2G Robust HotStart ReadyMix (Kapa Biosystems) inside a DNA/RNA UV-Cleaner cabinet (UVC/T-AR). PCR was performed using the following general profile: 95 °C for 5 min, (95 °C for 30 s, Tm °C for 15 s, 72 °C for 1 min) × 35, 72 °C for 5 min. Annealing temperature (Tm) was set up for each primer set according to Primer3 predictions (supplementary table 2, Supplementary Material online). When required, the temperature was adjusted trying 5 °C above or below of the predicted Tm. PCR product size was confirmed by electrophoresis using 1% agarose gel, purified with DNA Clean & Concentrator 5 (Zymo Research), and sequenced by Sanger technology in both directions at Macrogen Europe. For each amplicon, sequences quality screening/clipping and consensus alignment was performed using the Staden Package (Bonfield and Whitwham 2010).
In parallel, we designed primers that target the DNA polymerase III subunit epsilon dnaQ. Also, we targeted two regions with different gene order in Portiera of B. tabaci, lepA-groEL (ABt) and secA-leuC (BBt), compared with the ancestral gene order found in other sequenced Portiera, groEL-rpsA (A) and leuC-leuD (B). Primer design and PCRs were conducted as described above using the predicted Tm (supplementary table 2, Supplementary Material online). PCR products were visualized by electrophoresis using 1% agarose gels. Some obtained amplicons were Sanger sequenced to validate that the correct region was amplified.
To verify species morphological identification, the 5′ region of the mitochondrial cytochrome oxidase 1 (mtCOI) gene was amplified and Sanger sequenced, when possible, for each whitefly species collected (analyses are described in supplementary Material and Methods, Supplementary Material online).
Phylogenetics, Dating, and Ancestral State Reconstruction of Portiera Lineages
To infer the phylogenetic relationship and divergence time of Portiera from the studied whitefly species, two data sets were used. The first data set incorporated sequences of Portiera 16S and 23S rRNA genes amplified in this study, 16S and 23S rRNA gene sequences generated by Thao and Baumann (2004), as well as 16S and 23S rRNA gene sequences extracted from downloaded published transcriptomes/genomes (details in the following sections). The final data set contained 59 sequences from 45 different species (including six belonging to the B. tabaci species complex). The second data set integrated the sequences of the 16S and 23S rRNA genes with those of the three protein coding genes: dnaK, rpoD, and groEL. It contained 32 sequences from 29 whitefly species, mostly obtained in this study plus few that were acquired from public transcriptomes/genomes. Orthologous genes extracted from Chromohalobacter salexigens DSM3043 (NC_007963.1) were used as outgroups in the phylogenetic analysis of both data sets (described below).
The 16S and 23S rRNA genes were aligned with R-Coffee v11.00.8cbe486 (-mode = rmcoffee -iterate = 100) (Notredame et al. 2000) and pruned with Gblocks v0.91b allowing half of gap positions (-t = d -b5 = h) (Castresana 2000). The three coding genes (dnaK, rpoD, and groEL) were codon aligned with MACSE v2.03 (-prog alignSequences -gc_def 11) (Ranwez et al. 2018) and pruned with Gblocks v0.91b (-t = c -b5 = h). The 19 obtained mtCOI gene sequences (5′ region) were aligned in the same way but using the invertebrate mitochondrial code in MACSE v2.03 and no gaps allowed in Gblocks v0.91b. Substitution saturation was assessed using the pruned alignments as an input for Xia’s test implemented in DAMBE v7.2.3 (Xia 2018) (executed under wine v1.6.2-0ubuntu14.2). BEAST v2.5.2 (Bouckaert et al. 2014) was used to infer a Bayesian posterior consensus tree and the divergence time of the different nodes for each of the two data sets outlined above. Detailed procedures of BEAST divergence dating can be found at supplementary Material and Methods, Supplementary Material online.
Results from the dnaQ screening were codified as a binary matrix. Then, the binary matrix and the topology of the Bayesian phylogenetic trees were used as input for the Ancestral Character Estimation (ace) function implemented in ape (R package) (R Core Team 2018; Paradis and Schliep 2019). The analyses were conducted twice, using each time the tree that was based on five Portiera genes or the three that was based on two Portiera genes. The presence of dnaQ on the internal nodes of both data sets was estimated using a maximum likelihood approach as a discrete character and a model assuming only gene losses. Phylogenetic trees with dnaQ presence probabilities were plotted with ape.
Whole-Genome Shotgun Sequencing, Genome Assembly, and Annotation of the S. simplex and P. mori Joint Sample
In order to obtain Portiera of S. simplex genome, a whole-genome shotgun sequencing strategy was applied. NovaSeq sequencing produced 75,274,888 raw reads that were quality screened with Trimmomatic v0.33 (TruSeq2-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:25 MINLEN:98). Possible polyGs produced by the NovaSeq platform were trimmed with fastp v0.19.7 (-g) (Chen et al. 2018). Cleaned reads were classified with Kraken v2.0.6-beta using a custom database which included several RefSeq genome databases (archaea, bacteria, viral, fungi, and protozoa), all sequenced endosymbionts from whiteflies, the genomes of B. tabaci MEAM1 and Acyrthosiphon pisum, and all complete mitogenomes of whiteflies. All reads assigned to Portiera, Halomonadaceae, or Oceanospirillales were extracted and assembled with SPAdes v3.13.0 (–sc –careful) (Bankevich et al. 2012). Three contigs larger than 60 kb (385 kb in total) and ∼100× coverage plus several contigs between 80 and 5 kb (420 kb in total) and ∼600× coverage were recovered. Kraken2 classification and coverage suggested two putative Portiera populations. To screen for possible Portiera other than that of S. simplex, all sequences obtained during the PCR screening were used as a query in a BlastN search against the obtained contigs. BlastN results confirmed that two different Portiera genomes were present. Large contigs with ∼100× coverage had perfect match to the Portiera amplified genes from P. mori. Smaller contigs with coverage of ∼600× had perfect match to the amplified Portiera genes from S. simplex. This confirmed that some P. mori individuals were collected together with S. simplex, probably due to the ability of both whitefly species to exploit Ficus benjamina as a host-tree.
As a result, the Kraken2 database was rebuilt to include the obtained contigs, and cleaned reads were reclassified. Portiera reads were reassembled separately according to their whitefly host with SPAdes v3.13.0 (–sc –careful). SSPACE v3 (-k 20 -n 35 -g 3) (Boetzer et al. 2011) and GapFiller v1.10 (-m 50 -o 10 -r 0.6 -n 50 -t 50) (Boetzer and Pirovano 2012) were used for scaffolding and gap-filling the obtained reassembly, respectively. Gap5 from the Staden package was used not only to evaluate the quality of the assemblies but also to detect the presence of chimeras and misassemblies, to join contings manually (when possible), and to check for circular contigs. The first genome to be assembled was that of Portiera from P. mori. It produced a closed circular contig without requiring any iterative mapping step. In contrast, the Portiera genome from S. simplex remained as nine contigs after several rounds of iterative mapping, discarding at each round every sequence (if present) with a significant (90% identity threshold) match to the Portiera genome of P. mori. In brief, iterative mapping was run as follows: Cleaned reads were mapped against the assembled contigs of S. simplex with Bowtie v2.3.5.1 (–very-sensitive) (Langmead and Salzberg 2012). Usearch v10 (-usearch_global -query_cov 0.5 -accel 0.5 -strand both -id 0.9) was used do discard reads without a minimum overlap of 50% and 90% identity to the contigs (Edgar 2010). Surviving reads were added to the pool of putative Portiera reads from S. simplex. The reads were mapped to the contigs with MIRA v4.9.6 (Chevreux et al. 1999), and then imported to Gap5 for manual joining/gap closure. Both final assemblies were corrected with Pilon v1.23 (–fix all, amb) (Walker et al. 2014) and the clean classified reads. Finally, the annotation of the genomes was performed with prokka v1.14.5 (Seemann 2014), using all available Portiera genomes for building the protein database of primary annotation (–proteins). Obtained annotations were manually inspected and curated in Artemis v1.5 (Rutherford et al. 2000).
Singhiella simplex and P. mori mitogenomes assembly and annotation procedures can be found at supplementary Material and Methods, Supplementary Material online.
Portiera Lineages Comparative Genomics
Proteomes of Portiera from S. simplex (ERZ1272841), P. mori (ERZ1272840), B. tabaci species—MEAM1 (NC_018677.1), MED (NC_018676.1), and Asia II 3 (NZ_CP016327.1), T. vaporariorum (LN649236.1), A. dispersus (LN649255.1), and A. floccissimus (LN734649.1) were extracted with a custom python script. Orthologous clusters of proteins (OCPs) were calculated with OrthoFinder v2.3.3 (-M msa -S mmseqs -T iqtree) (Emms and Kelly 2019). Obtained OCPs were manually curated based on protein annotations. Shared and specific OCPs were plotted with UpsetR (Conway et al. 2017). Synteny between Portiera genomes, based on 230 single-copy core OCPs (from 235), was plotted with genoPlotR (Guy et al. 2010). Finally, metabolic potential comparisons were performed with Pathway Tools v23.5 (Karp et al. 2002).
Curated OCPs were converted into a binary matrix (presence/absence) and species-specific OCPs annotated as hypothetical proteins were discarded (21 OCPs). The binary matrix and the species tree obtained with OrthoFinder v2.3.3 were used as inputs for COUNT v10.04 (Csuos 2010) to reconstruct the gene losses history during Portiera evolution. The reconstruction was performed under a posterior algorithm and allowed only gene losses. SEED profiles were computed for each Portiera using diamond (BlastP -e 1e-9 -f 100), the nonredundant NCBI database (August 08, 2020 release), and MEGAN6 (Buchfink et al. 2015; Huson et al. 2016).
Repeats and Intergenic Regions Comparisons, Molecular Evolution Analysis, Transcriptomes Assembly, and Genomic Data Retrieval
Repeats and intergenic regions were compared between Portiera lineages and different obligatory endosymbionts to correlate dnaQ presence/absence and genome instability. Synonymous (dS) and nonsynonymous (dN) substitution ratios, nucleotide substitutions per site per year (dS/t and dN/t), and omega (ω) values were used to compare evolutionary trends in Portiera lineages and whitefly mitogenomes. These values were calculated as previously described (Santos-Garcia et al. 2015) using Codeml from PAML v4.7 package (Yang 2007). Dialeurodes citri (SRR2856996) and B. tabaci SSA1 (SRR5109958) transcriptomes were assembled de novo and several Portiera and whiteflies mitochondrial genomes were downloaded to increase our data set. The full procedures of the described analysis can be found in supplementary Material and Methods, Supplementary Material online.
Results
Using Portiera Gene Sequences to Establish Phylogenetic Relationships and Estimate Divergence Time in Whiteflies
gDNA was extracted from 26 of the 29 collected samples, standing for 22 whitefly species from 17 genera (supplementary table 1, Supplementary Material online). The five sets of primers that target the Portiera genes 16S and 23S rRNAs, dnaK, rpoD, and groEL, successfully amplified in 25 samples. One sample, Bemisia reyesi JHM 7496, was excluded from further analysis because we could not amplify the target regions of the 23S rRNA and rpoD genes. We failed to obtain gDNA from three NHM collection exsiccate samples (supplementary table 1, Supplementary Material online), even when applying a whole-genome amplification approach.
Two phylogenetic trees (chronograms) were obtained using two different data sets. One tree was based on the five Portiera genes listed above (hereafter 5G-based tree). The second tree was based only on the 16S and 23S rRNA genes (hereafter 2G-based tree), which allowed us to include more species in our analyses due to the availability of published sequences (Thao and Baumann 2004). Three main characteristics were common to both trees (figs. 1 and 2): the Aleyrodinae subfamily outcompeted the Aleurodicinae subfamily in the number of analyzed species, the Aleurodicinae was represented by species from the genera Paraleyrodes and Aleurodicus, and the Aleyrodinae formed four major clusters with similar clustering patterns at the genera level (represented by “green”, “blue”, “purple”, and “orange” colors in figs. 1 and 2).
Fig. 1.

BEAST2-inferred Portiera phylogenetic tree (chronogram) based on two rRNA (16S and 23S) and three coding genes (groEL, rpoD, and dnaK) (5G-based tree). Colored branches highlight the four major clades in the Aleyrodinae subfamily. Branch lengths are displayed in million years. Period, Epoch, and Age are according to the geological time scale standards. Chromohalobacter salexigens DSM3043 was used as outgroup but is not displayed for plotting reasons.
Some variation between the trees was observed in the “orange” cluster. In the 5G-based tree, the “orange” cluster was found to be the most basal branch and only contained one species, T. vaporariorum (fig. 1). In the 2G-based tree, the “orange” cluster (this time containing five species) was integrated within the “purple” cluster and was close to the Aleyrodes clade (fig. 2). These topological inconsistencies likely result from the different taxon sampling in the two analyses. The 5G-based tree was well supported and most of the nodes presented posterior values >0.9 (fig. 1). In contrast, the 2G-based tree had a large number of nodes with posterior values below 0.8, especially at some inner branches (fig. 2). Some of the low posterior support values in the 2G-based tree were associated with potential species complexes: B. tabaci, Aleyrodes singularis/proletella and Neomaskiella andropogonis. Some inconsistencies in taxonomy were also present in both trees. For example, Aleuroviggianus adanaensis was almost identical to Tetraleurodes bicolor at the sequence level and some species from the genera Tetraleurodes, Trialeurodes, and Dialeurodes were distributed among different clades.
Fig. 2.

BEAST2-inferred Portiera phylogenetic tree (chronogram) based on two rRNA genes (16S and 23S) (2G-based tree). The sequences were generated in this work and in Thao and Baumann (2004). Colored branches highlight the four major clades in the Aleyrodinae subfamily. Branch lengths are displayed in million years. Period, Epoch, and Age are according to the geological time scale standards. Chromohalobacter salexigens DSM3043 was used as outgroup but is not displayed for plotting reasons.
Among the Aleurodicinae subfamily, Paraleyrodes minei was the first species to diverge, around 119.68 Ma (102.74–133.42 95% Highest Posterior Density or HPD) or 112.6 Ma (85.49–133.31 95% HPD) according to the 5G-based or 2G-based trees, respectively (figs. 1 and 2). The divergence of A. dispersus from A. floccissimus was estimated to be around 20.35 Ma (9.35–33.28 95% HPD) and 30.21 Ma (15.02–47.18 95% HPD) for the 5G-based and 2G-based trees, respectively (figs. 1 and 2). These dates are in agreement with previous estimates Santos-Garcia et al. (2015). In the Aleyrodinae subfamily, despite the topological differences between the two trees, the estimated time of the first cladogenetic event (the first splitting after divergence from the main branch) was similar for the “blue”, “green”, and “purple” clusters. The estimated divergence dates for the “orange” cluster were not comparable between the two data sets. However, if we consider the split between the “green” and “purple”/“orange” clusters in the 2G-based tree as the origin of the lineage leading to T. vaporariorum, then, the estimation for T. vaporariorum divergence is quite similar: 97.36 Ma (76.14–116.97 95% HPD) in the 2G-based tree and 110.26 Ma (91.43–126.3 95% HPD) in the 5G-based tree. These estimations are in agreement with previous studies (Misof et al. 2014; Santos-Garcia et al. 2015).
The most studied whitefly species, the B. tabaci species complex, was part of the “green” cluster in both trees. Our estimations of the emergence time of the Bemisia genus and the B. tabaci species complex were similar to previous estimations (Santos-Garcia et al. 2015): 44.08 Ma (31.36–57.11 95% HPD) and 7.27 Ma (3.43–11.48 95% HPD) or 47.84 Ma (31.64–64.53 95% HPD) and 11.87 Ma (5.42–19.52 95% HPD), in the 5G-based and 2G-based trees, respectively. Finally, the divergence time between B. tabaci species MEAM1 and MED was also in agreement with previous estimates (Santos-Garcia et al. 2015). Taken together, although topological differences existed between the two trees, the convergence of their divergence time estimates supports their robustness.
Tracking the Origin of Genomic Instability in Portiera
Portiera of B. tabaci lacks the DNA polymerase III proofreading subunit (dnaQ). This absence seems to be correlated with the massive rearrangements, large intergenic regions, and repetitive sequences (especially microsatellites) present in the genomes of this Portiera lineage (Sloan and Moran 2013; Santos-Garcia et al. 2015). In order to identify the evolutionary time point in which dnaQ loss and genome stability appeared, we screened our samples for the presence of dnaQ and four possible gene order configurations. We considered the two configurations groEL-rpsA (A) and leuC-leuD (B) as ancient because they are shared between Portiera from Aleurodicus and T. vaporariorum. Following this line, we considered the two other configurations, lepA-groEL (ABt) and secA-leuC (BBt), as derived ones because these rearrangements were found so far only in Portiera from the B. tabaci species complex (fig. 3).
Fig. 3.
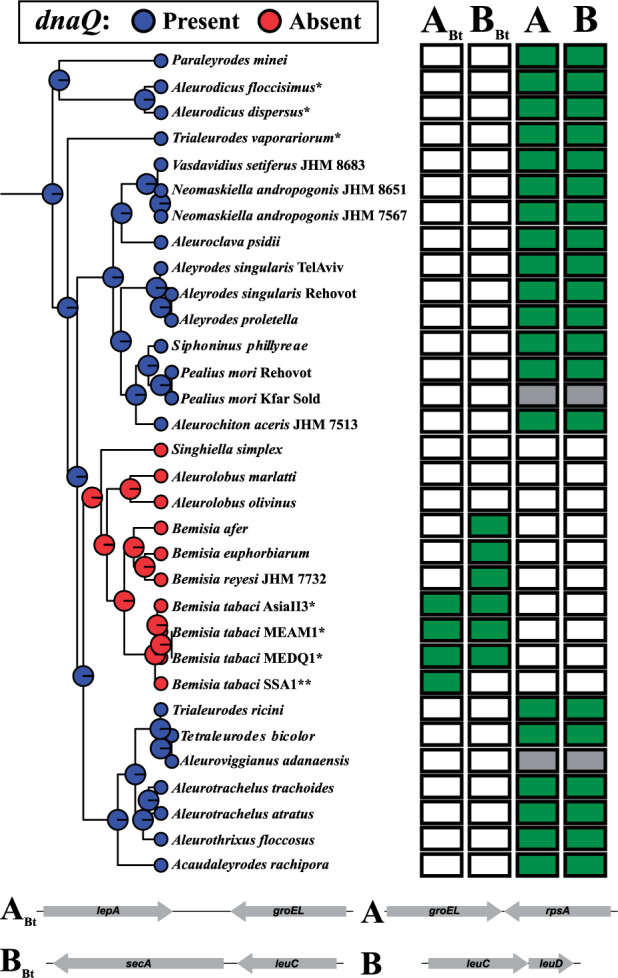
Summary of the screening for the dnaQ gene presence or absence and the gene rearrangements. Ancestral state inference was estimated using the Portiera 5G-based tree (left). Pie charts at the nodes represent the posterior probability for the presence (blue) or absence (red) of dnaQ. Note that all nodes have the probability of 1. The matrix represents the gene rearrangement amplification results (right). The letters above the matrix indicate the four possible rearrangements that were tested. Letters without index refer to the ancestral gene order found in Portiera of Aleurodicus and Trialeurodes vaporariorum. Letters with the subindex Bt refer to the gene order found in Portiera of Bemisia tabaci (bottom). White squares denotes unsuccessful amplifications, green filled squares represent successful amplifications, and gray filled squares indicate that the gene order rearrangements were not tested but the ancestral one (A and B) is assumed. *Full genome available and **no transcripts containing the BBt region were obtained.
We were able to amplify dnaQ of Portiera from all species tested except for Aleurolobus olivinus, Ale. marlatti, S. simplex, B. afer, B. euphorbiarum, and B. reyesi. In both the 5G-based and 2G-based trees, these species form a monophyletic clade together with B. tabaci, harboring three genera: Singhiella, Aleurolobus, and Bemisia. Based on the ancestral state reconstruction using the 5G-based tree, it is highly likely (posterior probability of 1) that the most recent common ancestor (MRCA) of this clade also lacked a functional dnaQ gene (fig. 3). Analysis of the 2G-based tree reached the same prediction with 0.62 posterior probability (supplementary fig. 1, Supplementary Material online). Uncertainty was too large to resolve the presence/absences of dnaQ in deeper nodes of the 2G-based tree. Still, following a maximum parsimony scenario, we hypothesize that dnaQ is likely to be present in the genome of Portiera of all whiteflies, with the exception of the Singhiella–Aleurolobus–Bemisia monophyletic clade.
Portiera of all species outside the Singhiella–Aleurolobus–Bemisia clade also presented the ancestral gene order (rearrangements A and B) (fig. 3). Portiera of Bemisia species outside the tabaci species complex only presented the BBt rearrangement, suggesting them to harbor a different rearrangement (than A or ABt) in region A. Although no transcript containing the BBt region was identified in B. tabaci SSA1, this species seems to be syntenic to other B. tabaci species (supplementary fig. 2, Supplementary Material online). In addition, the fact that the ancestral or modified A and B regions could not be amplified in both of the Aleurolobus species and S. simplex raises the possibility that several gene rearrangements took place in the A and B regions during the evolution of Portiera in the Singhiella–Aleurolobus–Bemisia clade (fig. 3).
The Genomic and Metabolomic Characterization of Portiera from S. simplex
To further elucidate the origin of Portiera genome instability and its putative effects on functionality, we sequenced the genome of Portiera from the most basal species in the Singhiella–Aleurolobus–Bemisia clade, the fig whitefly S. simplex. As explained in length in the Materials and Methods section, the sample unintentionally contained individuals of the mulberry whitefly P. mori, which shares some host-plants with S. simplex. As we were able to classify and recover complete Portiera and mitochondrial genomes from both S. simplex and P. mori, this accidental mixing had not effect on the consequent analyses.
The genome of Portiera from S. simplex was recovered as nine contigs (table 1), all ending in repetitive sequences. It is the largest Portiera genome described so far, being 134 kb larger than that of Portiera from B. tabaci (table 1). The number of coding genes was similar in Portiera of S. simplex and B. tabaci, indicating that genome expansion in Portiera of S. simplex is due to an increase in the size of the intergenic regions, which account for 40% of the genome. The genome of Portiera from S. simplex presents the lowest coding density (59.6%) and the highest number of direct (23) and inverted (17) repeats among all currently analyzed endosymbionts genomes (table 1). As was already predicted from the PCR amplification and ancestral state reconstruction results, the dnaQ gene was found to be nonfunctional (pseudogenized) in Portiera of S. simplex. The dnaQ pseudogene is located in a region that has suffered different rearrangements and an expansion of the intergenic regions (fig. 4B). Comparisons to other Portiera genomes and different obligatory endosymbionts present in mealybugs, scale insects, and cicadas indicated a clear association between the absence of a functional dnaQ and the presence of extended intergenic regions in the endosymbionts’ genomes (supplementary fig. 3, Supplementary Material online; Kruskal–Wallis test, df = 8, P value <2.2e-16 and pairwise Wilcoxon test with Benjamini–Hochberg FDR).
Table 1.
General Genomic Features of Portiera and Other Endosymbionts Lacking dnaQ, Presenting Large Intergenic Regions or Genome Instability
| Portiera TeVa | Portiera PeMo | Portiera SiSi | Portiera BeTa | Uzinura ASNER | Tremblaya PCVAL | Hodgkinia TETUND1 and TETUND2 | ||
|---|---|---|---|---|---|---|---|---|
| Accession number | LN649236 | LR744089 | CACTJB010000000 | CP003835 | NC_020135 | NC_017293 | CP007232 | CP007233 |
| Host | T. vaporariorum | P. mori | S. simplex | B. tabaci MED-Q1 | Aspidiotus nerii | Planococcus citri | Tettigades undata | |
| Genome size (bp) | 280,822 | 277,700 | 411,975 | 357,472 | 263,431 | 138,931 | 133,698 | 140,570 |
| Contigs | 1 | 1 | 9 | 1 | 1 | 1 | 1 | 1 |
| N50 (bp) | NA | NA | 17,098 | NA | NA | NA | NA | NA |
| L50 | NA | NA | 2 | NA | NA | NA | NA | NA |
| GC% | 24.69 | 24.1 | 26.18 | 26.12 | 30.2 | 58.83 | 46.77 | 46.16 |
| Genesa | 307 | 308 | 300 | 284 | 275 | 130 | 177 | 184 |
| CDS | 268 | 266 | 252 | 247 | 226 | 116 | 121 | 140 |
| Pseudogenes (CDS) | 1 | 3 | 11 | 7 | 13 | 19 | 39 | 19 |
| CDS avg. length | 989.21 ± 675.33 | 979.95 ± 672.88 | 977.88 ± 697.31 | 980.65 ± 697.56 | 965.30 ± 715.67 | 622.07 ± 591.77 | 789.69 ± 700.90 | 792.58 ± 670.70 |
| CDS avg. GC% | 23.88 ± 4.65 | 23.24 ± 5.01 | 25.33 ± 4.43 | 26.35 ± 4.01 | 29.59 ± 3.34 | 59.30 ± 3.09 | 46.15 ± 3.77 | 45.50 ± 3.81 |
| Intergenic avg. length | 62.79 ± 81.43 | 51.81 ± 63.09 | 715.44 ± 1,432.48 | 524.99 ± 871.23 | 141.63 ± 235.63 | 173.55 ± 189.19 | 105.08 ± 140 | 118.36 ± 164.76 |
| Intergenic avg. GC% | 19.12 ± 9.21 | 17.82 ± 9.37 | 22.07 ± 8.23 | 22.40 ± 7.15 | 23.60 ± 8.41 | 59.89 ± 6.15 | 47.53 ± 9.8 | 46.62 ± 7.46 |
| Coding density (%) | 91.46 | 96.60 | 59.56 | 69.60 | 89.73 | 81.26 | 90.49 | 90.06 |
| Intergenic regions (%) | 8.54 | 3.40 | 40.44 | 30.40 | 10.27 | 18.74 | 9.51 | 9.94 |
| rRNA | 3 | 3 | 3 | 3 | 3 | 6 | 3 | 3 |
| tRNA | 34 | 34 | 34 | 33 | 31 | 7 | 13 | 18 |
| tmRNA | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 |
| RnaseP RNA | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 |
| dnaQ | Yes | Yes | Pseudo | No | No | Yes | Pseudo | Yes |
| Direct repeats | 1 | 2 | 23 | 4 | 1 | 3 | 2 | 0 |
| Inverted repeats | 0 | 1 | 17 | 2 | 1 | 4b | 2 | 1 |
| Tandem repeats | 10 | 31 | 3 | 111 | 11 | 0 | 0 | 0 |
Gene features including pseudogenes.
Three of them correspond to the duplicated rRNA operons.
Fig. 4.

Portiera genomes syntenic comparisons based on 230 single-copy core genes. (A) Cladogram summarizing Portiera phylogenetic relationships based on the species tree obtained as part of the OrthoFinder pipeline. Filled circles at the nodes represent the number of coding genes estimated to be present in the MRCA using COUNT. Filled circles at the leaf tips represent the number of coding genes in each Portiera genome. Letters at the nodes list the MRCAs to allow comparison with figure 5. Portiera genomes are represented linearly. Blue boxes representing syntenic genes in the direct strand (upwards) or in the complementary strand (downwards), gray lines connect genes in the same strand, yellow lines connect genes in different strands, and twisted lines indicate inversions. The green line highlights the position of functional and nonfunctional (ψ) dnaQ genes. For Portiera of Singhiella simplex, only contigs containing core genes are represented (seven from nine contigs). (B) Magnification of synteny comparisons between Portiera of Pealius mori, S. simplex, and Bemisia tabaci AsiaII3.
Synteny evaluation analysis, based on 230 OCPs (supplementary fig. 4A, Supplementary Material online), indicated that Portiera of S. simplex presents a different genomic architecture when compared both with the ancestral Portiera and with the Portiera of B. tabaci gene order (fig. 4A). Nevertheless, a high degree of microsynteny was also observed in some genomic regions.
Portiera from S. simplex, as other sequenced Portiera, can synthesize by itself the essential amino acid threonine and the nonessential homoserine. Also, it is able to produce carotenoids, several pyruvate and folate interconversions, and proteins with Fe-S clusters. It requires the aid of the hosting cell to synthesize valine, leucine, isoleucine, phenylalanine, and tyrosine (the enzymes performing the last step of those pathways are encoded by the host), methionine (the precursor homocysteine is provided by the hosting cell), and probably histidine, as previously reported for other Portiera lineages (Luan et al. 2015; Santos-Garcia et al. 2015). We found the genome of Portiera from S. simplex to be metabolically close to that of B. tabaci (supplementary fig. 4B, Supplementary Material online). Both Portiera have lost part of the lysine biosynthetic pathway (fig. 5), which is probably complemented by the hosting cell (Luan et al. 2015). Besides, Portiera of S. simplex has lost the ability to produce tryptophan (the trpF gene is absent), but, in contrast to Portiera of B. tabaci, can still produce arginine. Portiera of S. simplex also lacks the aminoacyl-tRNA synthetases argS, asnS, and thrS (lost in all available Portiera genomes), metG and alaS (also lost in Portiera of T. vaporariorum, P. mori, and B. tabaci), and trpS (lost in Portiera of B. tabaci) (fig. 5). The tRNAIle-lysidine synthetase tilS, responsible for avoiding mischarging of methionine instead of isoleucine, was found to be uniquely pseudogenized in Portiera of S. simplex. In addition, the genome of Portiera from S. simplex has lost six genes related to the DNA replication and repair machinery (fig. 5). These genes were likely lost, together with other 12 genes, in the MRCA of the Singhiella–Aleurolobus–Bemisia clade (figs. 4A and 5).
Fig. 5.

Gene losses in Portiera lineages and their MRCAs. Posterior probabilities for the presence of the different genes in the MRCAs were obtained with COUNT. The colored sidebar in the left represents the Clusters of Orthologous Groups (COG) category assigned to each protein encoded by a lost gene. Replication and repair (L category) gene losses are accumulated in the lineage leading to Singhiella simplex and Bemisia tabaci (MRCA D). MRCAs nodes are the same as in figure 4. The genes (rows) dendrogram was computed using a binary distance and the ward. D2 clustering method. *WP_180824853: NCBI accession number of a hypothetical protein shared between all Portiera with the exception of S. simplex and B. tabaci.
Comparative Molecular Evolution among Portiera Lineages
We estimated the ratio of synonymous (S) and nonsynonymous (N) substitutions per site (dS and dN) and their omega ratio (ω = dN/dS) in 232 single-copy genes shared among Portiera lineages of six whitefly species: A. dispersus, A. floccissimus, T. vaporariorum, P. mori, S. simplex, and B. tabaci (MEAM1). After filtering, 158 orthologous shared genes were kept. To obtain the S and N per site per year (dS/t and dN/t), the values were divided by each lineage divergence time (according to the 5G-based chronogram predictions that presented high internal nodes support): 19.64 Myr for Aleurodicus, 111.29 Myr for Trialeurodes, 99.98 Myr for Pealius, and 71.34 for Singhiella–Bemisia (fig. 1). dS/t and dN/t are normalized values and allow comparisons between lineages (the branch leading to a specific Portiera genome).
Our analyses indicated that the Portiera lineages evolve at different dS/t (Kruskal–Wallis test, P value < 2.2e-16) (fig. 6A). Portiera of B. tabaci was the fastest-evolving lineage, followed by Portiera of S. simplex, whereas the slowest-evolving lineage was Portiera of P. mori (table 2). Also, dN/t values showed statistical differences among Portiera lineages (Kruskal–Wallis test, P value < 2.2e-16) (fig. 6B). Again, Portiera of B. tabaci and S. simplex were the fastest-evolving lineages, whereas Portiera of P. mori was the slowest-evolving lineage (table 2).
Fig. 6.

Synonymous (A) and nonsynonymous (B) substitutions per site per year and their ω ratios (C) estimated for 158 core shared genes between Portiera lineages of six whitefly species. Different letters indicate significant statistical differences between lineages (nonparametric Kruskal–Wallis and Wilcoxon post hoc pairwise tests). Synonymous (D) and nonsynonymous (E) substitutions per site per year and ω ratios (F) estimated for ten full mitochondrial genes from six whiteflies species. N.S., no significant difference. Different letters indicate significant statistical differences between lineages (one-way ANOVA and Tukey’s post hoc test). Organism abbreviations are as table 2.
Table 2.
Average Nucleotide Substitutions per Site per Year and ω Ratios for Portiera and Mitochondrial Lineages
| Lineage | dS/t | dN/t | Omega | |
|---|---|---|---|---|
| Portiera | A. dispersus (AlDi) | 4.79 × 10−09 | 2.35 × 10−10 | 0.1200 |
| A. floccissimus (AlFl) | 2.48 × 10−09 | 1.46 × 10−10 | 0.1490 | |
| T. vaporariorum (TeVa) | 1.02 × 10−09 | 1.43 × 10−10 | 0.2360 | |
| P. mori (PeMo) | 8.25 × 10−10 | 1.02 × 10−10 | 0.2310 | |
| S. simplex (SiSi) | 5.94 × 10−09 | 4.85 × 10−10 | 0.0914 | |
| B. tabaci (BeTa) | 1.17 × 10−08 | 9.32 × 10−10 | 0.0901 | |
| Mitochondrion | Aleurodicus (mt AlDi) | 1.54 × 10−07 | 1.98 × 10−09 | 0.0084 |
| T. vaporariorum (mt TeVa) | 1.05 × 10−07 | 2.28 × 10−09 | 0.0276 | |
| P. mori (mt PeMo) | 6.74 × 10−08 | 2.11 × 10−09 | 0.0484 | |
| S. simplex (mt SiSi) | 1.79 × 10−07 | 3.84 × 10−09 | 0.0623 | |
| B. tabaci (mt BeTa) | 1.26 × 10−07 | 3.15 × 10−09 | 0.0333 | |
| Portiera/mitochondrion | T. vaporariorum | 103.63 | 15.97 | 0.12 |
| P. mori | 81.72 | 20.72 | 0.21 | |
| S. simplex | 30.05 | 7.92 | 0.68 | |
| B. tabaci | 10.74 | 3.38 | 0.37 |
The comparison of ω ratios, used for testing if the six Portiera lineages differ in the selection forces that act on their genomes, resulted in three statistically significant groups (fig. 6C, Kruskal–Wallis test, P value < 2.2e-16): Portiera of B. tabaci and S. simplex had the lowest ω values, Portiera of A. dispersus and A. floccissimus presented intermediate ω values, and Portiera of T. vaporariorum and P. mori had the highest ω values. Most ω values were close to 0 indicating a strong purifying selection force in almost all tested genes. Still, we detected 18 genes presenting signatures of relaxed/adaptive selection (dS > 0, ω ≥ 1 and ω ≤ 10) in Portiera of T. vaporariorum (9 genes), A. floccissimus (4), P. mori (3), and A. dispersus (1) (supplementary table 4, Supplementary Material online). Some of these genes were found to be related to amino acid biosynthesis (hisH, leuC, trpC, and gatC), aminoacyl-tRNA synthetases (sufS and cysS), or to energy metabolism (atpB and cyoD).
Lastly, we compared between the nucleotide substitution rates in Portiera lineages and their insect hosts mitochondria (fig. 6D–F). Because the mitogenome of A. floccissimus is still not available, we calculated the dS/t and dN/t values of the Aleurodicus lineage using only the mitogenome of A. dispersus (dividing the values by 129.35 Ma, the estimated time when the split between the Aleurodicinae and the Aleyrodinae families occurred). Only 12 genes were included in the analysis because mitogenomes annotation was not consistent. The mitochondrial lineages presented nonsignificant dS/t and dN/t values and large within lineages variation (one-way analysis of variance [ANOVA], P value > 0.2) (fig. 6D and E and table 2). The ω values differed only between S. simplex and A. dispersus that presented the highest and lowest values, respectively (one-way ANOVA, P value < 0.02 and Tukey’s post hoc test) (fig. 6F). In all lineages, nearly all ω values were below 0.1, indicating a strong effect of purifying selection. Calculation of the dS/t ratio between the mitochondria and Portiera indicated that mitochondrial genomes are evolving prominently faster, with the ratios varying between 10-fold in B. tabaci and 100-fold in T. vaporariorum. These results agree with previous works showing that mitochondrial genomes from insects present high mutation rates (Song et al. 2012; Allio et al. 2017).
Discussion
Portiera as a Valuable Resource for Establishing Robust Phylogenetic Relationships in Whiteflies
In contrast to insect groups that rely on adult morphology, the current taxonomy of whiteflies is mostly based on the morphology of one nymphal stage (the puparium). However, this stage presents plasticity in many morphological traits that respond to various abiotic and biotic environmental factors including the identity of the plant host (Manzari and Quicke 2006; Charles 2010), eliminating in many cases the possibility of identifying a definite criterion for classification. This has led to a relatively high number of inaccuracies and misassignments in the group taxonomy (Manzari and Quicke 2006). For example, an extensive cladistic analysis suggested that around half of the 117 Aleyrodinae genera analyzed are not monophyletic (excluding monobasic genera) (Manzari and Quicke 2006). Another study used puparial morphological characters of all 20 Aleurodicinae genera and DNA sequences of nine Aleurodicinae genera, but managed to recover only 60% and 14% of the genera as monophyletic, respectively (Charles 2010). Taking all above in consideration, it is safe to state that whitefly taxonomy can significantly benefit from the development of complementary classification frameworks, especially those using molecular data.
We identified both technical and evolutionary advantages for using Portiera gene sequences for inferring the phylogenetic relationships among whiteflies, when compared with other commonly used molecular methods (mainly mtCOI gene sequences). First, in contrast to mtCOI amplicons, all designed Portiera primers had an almost perfect amplification success except for the rpoD set that failed to amplify one sample. Second, the specific targeting of Portiera genes is by itself a diagnostic tool that allows both differentiating whiteflies from similar insects (e.g., nymphal stages of psyllids) and discriminating between the two main whitefly subfamilies. Discrimination is possible because Portiera of the Aleurodicinae subfamily contain two specific insertions in the 23S rRNA gene (Thao and Baumann 2004). Third, targeting Portiera genes is especially useful when studying parasitized samples (supplementary table 1, Supplementary Material online), as the use of universal mtCOI primers is, in this case, problematic. Fourth, because Portiera is evolving slower than the mitogenome of whiteflies (table 2), its genes usually do not show phylogenetic signal saturation, making them more adequate for solving intergenerative relationships and deeper nodes than the mtCOI gene sequences (Coeur d’Acier et al. 2014). On the other hand, it is important to note that Portiera gene sequences may be limited in their ability to resolve the relationships in cases of recent speciation events or within species relationships between populations.
Portiera Phylogeny Provides New Insights on the Evolutionary History of Whiteflies
Based on nymphal morphology, the Singhiella, Aleurolobus, and Bemisia genera were reported to be paraphyletic and not closely related (Manzari and Quicke 2006). Moreover, previous studies suggested that the Singhiella genus is closer to Dialeurodes and unrelated to Bemisia (Jensen 2001; Manzari and Quicke 2006). However, the phylogeny of Portiera shows that these three genera form a monophyletic clade. Also, mtCOI phylogenetic analysis supports the monophyly of this clade (Ovalle et al. 2014; Dickey et al. 2015). We propose that the genera Singhiella, Aleurolobus, and Bemisia belong to the Aleurolobini tribe (see Manzari and Quicke 2006 for a detailed review on whitefly tribes).
An unexpected finding in our analysis was the early origin of the Paraleyrodes genus. Originally described as Aleyrodes, the nymphal stages of Paraleyrodes present typical Aleurodicinae morphological characters, such as subdorsal compound pores or legs with apical claws (Quaintance 1909). However, adults present morphological characters typical of Aleyrodinae, such as small body size and single-vein wings (Quaintance 1909; Martin 1996, 2007). Interestingly, the Paraleyrodes genus presents median ocellus, an ancestral character described in Cretaceous taxa (Drohojowska and Szwedo 2015). Our analysis supports the inclusion of the Paraleyrodes genus inside the Aleurodicinae subfamily based on its ancient origin and the presence of the 23S rRNA insertions common to the Aleurodicinae subfamily (Thao and Baumann 2004). Our estimates overlap with the calibration point used, suggesting that the Paraleyrodes genus originated in the Lower Cretaceous (100.5–145 Ma). Therefore, Paraleyrodes can be considered a long-enduring extant taxon, which may explain the retention of the middle ocellum and the mixture of morphological characteristics of both Aleyrodidae subfamilies. Although speculative, it also could be possible that other hard-to-assign Aleurodicinae genera, such as Aleuroctarthrus (presents medium ocellus) and Palaealeurodicus (does not present clawed legs), are indeed long-enduring taxa (Martin 2008). These two genera are closely related to Paraleyrodes according to cladistic analysis (Charles 2010). Also, Palaealeurodicus was placed as basal to all Aleurodicinae based on four mitochondrial genes (Charles 2010). Therefore, Paraleyrodes can be considered as sister taxon of Palaealeurodicus, which diverged before the radiation of the Aleurodicus genus (Charles 2010). Identifying such kind of long-enduring taxa could be an invaluable resource for understanding the evolution of the whitefly superfamily.
Genome Instability in Portiera of the Aleurolobini Tribe
Adaptation to an intracellular lifestyle has a significant impact on bacterial symbionts. Metabolic redundancy between the host and the endosymbiont promotes the dependency of the later on the intracellular environment of the former (Morris et al. 2012). Moreover, vertical transmission drastically reduces the endosymbiont effective population size (Ne) and the chances to acquire new genetic material, eventually leading to the generation of asexual populations. The combined effects of vertical transmission and intracellular lifestyle promote the accumulation of deleterious mutations that are otherwise pruned by selection in larger Ne, which can lead to a massive loss of genes (Moran 1996; Toft and Andersson 2010; Wernegreen 2015). The outcome of the process, known as the Muller’s Ratchet (Moran 1996), is an endosymbiont that harbors a highly reduced genome, with small intergenic regions and very few repetitive sequences (Toft and Andersson 2010; Wernegreen 2015). Common conserved elements include genes that are essential for complementing the host dietary requirements and a minimal machinery for informational flux and translation required for cell maintenance (Moran and Bennett 2014). As a consequence of a reduced or absent replication and recombination machinery, and the minimal presence of repetitive sequences, the genomes of long-standing endosymbionts are almost static (Moran and Bennett 2014). For example, only few inversions were detected in endosymbionts that have been codiverging with their host for more than 100 Myr (Patiño-Navarrete et al. 2013; Chong et al. 2019).
Most Portiera lineages do not differ from other long-standing endosymbionts and usually exhibit the “classical” reduced and static genomes (Sloan and Moran 2013; Santos-Garcia et al. 2015). One exception to this “rule” is the genome of Portiera from B. tabaci, which presents large intergenic regions, extensive rearrangements, and abundance of repetitive sequences (Sloan and Moran 2013; Moran and Bennett 2014; Santos-Garcia et al. 2015). In addition, the genome of Portiera from B. tabaci presents one of the most reduced sets of DNA replication and repair genes among known long-standing P-endosymbionts, including the loss of the DNA polymerase proofreading subunit (dnaQ) (Moran and Bennett 2014). As stated earlier, this loss has been linked to the uncommon extensive genome rearrangements, inversions, abundance of repeated sequences, large intergenic regions, and accelerated evolution found in Portiera of B. tabaci (Sloan and Moran 2013; Santos-Garcia et al. 2015). Our findings suggest that the massive loss of DNA replication and repair genes is not restricted to B. tabaci but is shared by all other members of the Singhiella–Aleurolobus–Bemisia clade (hereafter the Aleurolobini tribe for simplicity). Therefore, it is quite probable that dnaQ was already pseudogenized in the last common ancestor of this tribe, more than 70 Ma.
So far, only three genomes displaying long intergenic regions, genome instability, or the lack of functional dnaQ have been sequenced from other long-standing endosymbionts: “Ca. Uzinura diaspidicola”, “Ca.Tremblaya princeps”, and “Ca. Hodgkinia cicadicola” (Moran and Bennett 2014; Van Leuven et al. 2014; López-Madrigal et al. 2015; Łukasik et al. 2018). Only a single genome of U. diaspidicola is currently available, and therefore, it is not clear if the lack of dnaQ in this endosymbiont is associated with a significant genome instability. Relative to the genomes of Portiera from B. tabaci and S. simplex, the genome of U. diaspidicola presents lower number of repeated sequences and smaller intergenic regions. One explanation to this could be the conservation of the mutL gene in U. diaspidicola (Moran and Bennett 2014). The enzyme MutL, together with MutS, is part of the mismatch repair system that corrects mismatch events that are produced by base misincorporation and polymerase slippage (Rocha 2003). The genome of Tre. princeps presents genome instability signatures such as long intergenic regions, gene conversions, and the presence of several direct/indirect sequence repeats (López-Madrigal et al. 2015). Still, the number of repeats and the length of the intergenic regions are smaller than in Portiera of B. tabaci or S. simplex. The inactivation of the recombination machinery in Tre. princeps has been proposed as a strategy to reduce the number of homologous recombination events and their deleterious consequences in highly reduced genomes (López-Madrigal et al. 2015). However, Tre. princeps has access to a complementing recombination machinery as it harbors the endosymbiont “Ca. Moranella endobia” which has an active recombination machinery (López-Madrigal et al. 2015). For example, Tre. phenacola from the mealybug Phenacoccus peruvianus presents a chimeric genome that emerged from the fusion with its nested Sodalis endosymbiont, a process requiring a recombination machinery (Gil et al. 2018). The presence of a functional dnaQ subunit in Tre. princeps and the possible access to a complementing recombination machinery suggest that the possible causes of genome instability in Tre. princeps are different from those in Portiera.
One of the most extreme cases of genome instability was reported in H. cicadicola. In some cicada genera, which usually have a long lifespan, H. cicadicola has been split into several lineages with different genomic content within the same insect. This enforces functional complementation between the lineages for normal growth (Van Leuven et al. 2014; Łukasik et al. 2018). Although the genomic architecture of H. cicadicola seems unstable like that of Portiera, there are major differences in the relationship of these two endosymbionts with their hosts. Although Portiera is essential for whiteflies, H. cicadicola is a coprimary endosymbiont in cicadas and has been replaced several times (Łukasik et al. 2018; Matsuura et al. 2018). Therefore, the selection forces acting on both endosymbionts could be very different: strong purifying selection in the case of Portiera, whereas more relaxed, or even nonadaptive selection, in the case of H. cicadicola (Łukasik et al. 2018).
Large intergenic regions can allow Portiera with unstable genomes to better tolerate rearrangements while the expansion of repeated sequences can increase the chance of deleterious homologous recombination events (Sloan and Moran 2013). Because these Portiera show signs of gene conversion and recombination, it can be speculated that long intergenic regions and intergenic repeats are selected in their genomes to increase resilience against deleterious mutations. For example, repeated sequences mostly accumulate at the intergenic regions and pseudogenes of Portiera from B. tabaci and S. simplex suggesting strong purifying selection at the gene level. However, it could be possible that recombination is also counter selected in Portiera with unstable genomes. This could explain why Portiera lineages within the B. tabaci species complex are syntenic after, at least, 7 Myr of divergence and contain a low number of direct/indirect repeats compared with S. simplex. In the later, recombination seems still to be active. Therefore, it could be possible that after a period of genome instability and intergenic regions expansion, direct and indirect repeats are counter selected to favor more stable genomes.
In contrast, the location of tandem repeats in Portiera of T. vaporariorum (8 over 10) and P. mori (24 from 31) partially or completely overlap with coding genes. As sequence repeats in coding genes can cause gene inactivation and/or rearrangements, their existence within genes of stable Portiera genomes may reflect the presence of a minimal, but functional, DNA repair machinery that allows a more relaxed purifying selection process. In fact, Portiera of B. tabaci and S. simplex showed the lowest ω values, indicating stronger purifying selection forces acting on their genomes. Taking together, it is possible that increased resilience combined with a strong purifying selection force at the gene level has helped to maintain Portiera in the Aleurolobini tribe (Bennett and Moran 2015).
The Symbiont or the Egg: Genome Instability and Bacteriocyte Inheritance
Since the beginning of the research on insect symbiosis, it was clear that the whitefly superfamily displays a special mode of transmission of endosymbionts: whole maternal bacteriocytes migrate to the oocyte and enter through the future pedicel (Buchner 1965). In T. vaporariorum, Aleyrodes proletella, Aleurodes aceris, and Aleurochiton aceris several bacteriocytes penetrate the oocyte (from five to ten, depending on the species) (Tremblay 1959; Buchner 1965; Szklarzewicz and Moskal 2001). In contrast, in B. tabaci, Bemisia aff. gigantea, and Ale. olivinus, a single bacteriocyte is transmitted (Tremblay 1959; Buchner 1965; Coombs et al. 2007).
Although the phylogenetic relationships of B. aff. gigantea are not completely resolved, it is currently considered to be a sister clade of Aleurolobus and B. afer, and distantly related to B. tabaci (Manzari and Quicke 2006). It thus seems that all of the whitefly species with a single-bacteriocyte mode of inheritance belong to one phylogenetic group, the Aleurolobini tribe. A parsimonious explanation might be that the single-bacteriocyte mode of inheritance has evolved in the common ancestor of Aleurolobus-Bemisia, otherwise we would have to assume that it evolved multiple times in different species: Ale. olivinus, B. aff. gigantea, and B. tabaci (Tremblay 1959; Coombs et al. 2007; Luan et al. 2016, 2018; Xu et al. 2020). It would be interesting to see if the single-bacteriocyte maternal transmission pattern occurs also in S. simplex. If it does, it would suggest that the whole Aleurolobini tribe is likely to possess this derived type of bacteriocyte inheritance.
There is an apparent relationship between the emergence of a single-bacteriocyte inheritance mode and the presence of Portiera lineages with genomic instability. The single-bacteriocyte inheritance mode could potentially have a considerable impact on Portiera evolution because it drastically decreases the effective population size (Ne) compared with the inheritance of multiple bacteriocytes. The extremely low Ne probably intensified the effect of random genetic drift and accelerated the accumulation of deleterious mutations in Portiera. In addition, all the Portiera cells that are harbored in the same bacteriocyte are expected to present a homogenized allelic composition because recombination events, if happen, are limited to the bacterial cells inhabiting the same bacteriocyte. This implies a low probability for recovery from a state in which deleterious alleles are formed. At the same time, the single-bacteriocyte inheritance mode also exerts strong purifying selection at both the bacteriocyte and Portiera levels each generation as offspring harboring a bacteriocyte or Portiera with deleterious mutations will probably suffer from severe fitness costs (Luan et al. 2018). This is somewhat supported by the evidence that extant Portiera of the Aleurolobini tribe present moreover a stable gene content, with the massive gene loss events occurring only in their common ancestor. For instance, after ∼70 Myr of divergence, only five and ten genes were lost from the Portiera genomes of S. simplex and B. tabaci, respectively.
Further research on the Aleurolobini tribe is required in order to determine what occurred first: the transition from the multi- to the single-bacteriocyte inheritance mode or the switch from stable to unstable genomic architecture of Portiera. In the first case, the evolution of a different mode of transmission could have triggered the DNA replication and repair machinery loss as purifying selection was not able to maintain them under very low Ne (Lynch 2010). In addition, these losses may have been complemented by an overtake of some of their activities by the genome of the host cell (Santos-Garcia et al. 2014, 2015; Silva and Santos-Garcia 2015; Mao et al. 2018). In the alternative case, we should assume that Portiera of the Aleurolobini tribe lost its recombination and repair machinery as a consequence of a continuous genome degradation process (Bennett and Moran 2015). This increased the chances for transmitting Portiera with deleterious mutations. A multiple-bacteriocytes inheritance mode results in the transmission of mixtures that can mask the presence of bacteriocytes harboring Portiera with deleterious mutations/variations. Instead, if single bacteriocytes are inherited, the Portiera presenting deleterious mutations will reduce the fitness of the new-born carrying them and they will be counter selected. Therefore, the evolution of the single-bacteriocyte inheritance mode could have been a compensatory adaptation mechanism of the insect host to exercise an iron grip over Portiera transmission for ensuring the viability of its offspring (Campbell et al. 2018).
Conclusions
Our work brings evidence that gene sequences of the primary endosymbiont “Candidatus Portiera aleyrodidarum” provide a promising tool for establishing a robust phylogenetic framework of the whitefly superfamily. Portiera sequences can be used to establish intergenera relationships, serve as diagnostic tools by themselves, and help in the classification of problematic samples (even parasitized ones). Using the phylogenetic framework, we discovered that whitefly species from the Singhiella, Aleurolobus, and Bemisia genera form a monophyletic tribe, the Aleurolobini. We also found that Portiera in all these three genera comprise different genome rearrangements that are uncommon in primary endosymbionts. We suggest that the Portiera ancestor of the Aleurolobini tribe suffered a massive DNA replication and repair genes loss, which may have triggered the genomic instability phenomenon. We hypothesize that the appearance of genomic instability is also related to the evolutionary switch made between multi- and single-bacteriocyte mode of inheritance.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors of this work would like to honor the memory of Prof. Dan Gerling (1936–2016), a pioneering and passionate researcher of the whitefly superfamily and its natural enemies. This work was supported by the Israel Science Foundation (Grant Nos. 1039/12 to S.M. and 484/17 to E.Z.-F.). D.S.-G. was a recipient of the Golda Meir Postdoctoral Fellowship from the Hebrew University of Jerusalem. We wish to acknowledge Dr Estrella Hernández Suárez and Dr Hélène Delate for supplying whitefly samples.
Author Contributions
D.S.-G. and S.M. conceived the study. D.S.-G. performed bioinformatics analysis and collected whitefly samples. N.M.-R. performed molecular work and collected whitefly samples. D.O. supplied NHM whitefly samples and helped in whiteflies taxonomy. D.S.-G. drafted the manuscript with inputs from S.M. E.Z.-F. and D.O. reviewed and corrected advanced versions of the manuscript.
Data Availability
The relevant scripts and files generated are available in FigShare at https://doi.org/10.6084/m9.figshare.12361496 (last accessed October 24, 2020).
Contributor Information
Diego Santos-Garcia, Department of Entomology, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot, Israel.
Natividad Mestre-Rincon, Department of Entomology, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot, Israel.
David Ouvrard, Department of Life Sciences, Natural History Museum, London, United Kingdom; Entomology and Invasive Plants Unit, Plant Health Laboratory, ANSES, Montferrier-sur-Lez, France.
Einat Zchori-Fein, Department of Entomology, Newe-Ya’ar Research Center, ARO, Ramat-Yishai, Israel.
Shai Morin, Department of Entomology, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot, Israel.
Data deposition: Sanger-sequenced Portiera and mtCOI gene fragments, Novaseq raw reads, and Portiera and mitochondrial assembled genomes have been deposited at the European Nucleotide Archive (ENA) under the project number PRJEB31657.
Literature Cited
- Allio R, Donega S, Galtier N, Nabholz B. 2017. Large variation in the ratio of mitochondrial to nuclear mutation rate across animals: implications for genetic diversity and the use of mitochondrial DNA as a molecular marker. Mol Biol Evol. 34(11):2762–2772. [DOI] [PubMed] [Google Scholar]
- Bankevich A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GM, Moran NA. 2015. Heritable symbiosis: the advantages and perils of an evolutionary rabbit hole. Proc Natl Acad Sci U S A. 112(33):10169–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27(4):578–579. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. 2012. Toward almost closed genomes with GapFiller. Genome Biol. 13(6):R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfield JK, Whitwham A. 2010. Gap5-editing the billion fragment sequence assembly. Bioinformatics 26(14):1699–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R, et al. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 10(4):e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12(1):59–60. [DOI] [PubMed] [Google Scholar]
- Buchner P. 1965. Endosymbiosis of animals with plant microorganisms. Vol. 7 New York: John Wiley & Sons, Inc. [Google Scholar]
- Campbell B C, Steffen-Campbell J D, Gill R J. 1994. Evolutionary origin of whiteflies (Hemiptera: Sternorrhyncha: Aleyrodidae) inferred from 18S rDNA sequences. Insect Mol Biol. 3(2):73–88. [DOI] [PubMed] [Google Scholar]
- Campbell MA, et al. 2018. Changes in endosymbiont complexity drive host-level compensatory adaptations in cicadas. MBio 9(6):8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. [DOI] [PubMed] [Google Scholar]
- Charles E. 2010. Systematics of whiteflies (Aleyrodidae: Aleurodicinae): their distribution, phylogeny and relationship with parasitoids [PhD thesis]. London: Imperial College. [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17):i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, Suhai S. 1999. Genome sequence assembly using trace signals and additional sequence information. Comput Sci Biol Proc Ger Conf Bioinf. 99:45–56. [Google Scholar]
- Chong RA, Park H, Moran NA. 2019. Genome evolution of the obligate endosymbiont Buchnera aphidicola. Mol Biol Evol. 36(7):1481–1489. [DOI] [PubMed] [Google Scholar]
- Coeur d’Acier A, et al. 2014. DNA barcoding and the associated PhylAphidB@se website for the identification of European aphids (Insecta: Hemiptera: Aphididae). PLoS One 9(6):e97620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Lex A, Gehlenborg N. 2017. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33(18):2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs MT, Costa HS, De Barro P, Rosell RC. 2007. Pre-imaginal egg maturation and bacteriocyte inclusion in Bemisia aff. gigantea (Hemiptera: Aleyrodidae). Ann Entomol Soc Am. 100(5):736–744. [Google Scholar]
- Csuos M. 2010. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26(15):1910–1912. [DOI] [PubMed] [Google Scholar]
- De Vienne DM, et al. 2013. Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytol. 198(2):347–385. [DOI] [PubMed] [Google Scholar]
- Dickey AM, Stocks IC, Smith T, Osborne L, McKenzie CL. 2015. DNA barcode development for three recent exotic whitefly (Hemiptera: Aleyrodidae) invaders in Florida. Florida Entomol. 98(2):473–478. [Google Scholar]
- Drohojowska J, Szwedo J. 2015. Early Cretaceous Aleyrodidae (Hemiptera: Sternorrhyncha) from the Lebanese amber. Cretac Res. 52:368–389. [Google Scholar]
- Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19):2460–2461. [DOI] [PubMed] [Google Scholar]
- Emms DM, Kelly S. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20(1):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil R, et al. 2018. Tremblaya phenacola PPER: an evolutionary beta-gammaproteobacterium collage. ISME J. 12(1):124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi D, Engel M. 2005. Evolution of the insects. New York: Cambridge University Press. [Google Scholar]
- Guy L, Kultima JR, Andersson SGE. 2010. genoPlotR: comparative gene and genome visualization in R. Bioinformatics 26(18):2334–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AAG, et al. 2016. Codivergence of the primary bacterial endosymbiont of psyllids versus host switches and replacement of their secondary bacterial endosymbionts. Environ Microbiol. 18(8):2591–2603. [DOI] [PubMed] [Google Scholar]
- Hansen AK, Moran N. 2014. The impact of microbial symbionts on host plant utilization by herbivorous insects. Mol Ecol. 23(6):1473–1496. [DOI] [PubMed] [Google Scholar]
- Hsieh CH, Ko CC, Chung CH, Wang HY. 2014. Multilocus approach to clarify species status and the divergence history of the Bemisia tabaci (Hemiptera: Aleyrodidae) species complex. Mol Phylogenet Evol. 76(1):172–180. [DOI] [PubMed] [Google Scholar]
- Huson DH, et al. 2016. MEGAN community edition—interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol. 12(6):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AS. 2001. A cladistic analysis of Dialeurodes, Massilieurodes and Singhiella, with notes and keys to the Nearctic species and descriptions of four new Massilieurodes species (Hemiptera: Aleyrodidae). Syst Entomol. 26(3):279–310. [Google Scholar]
- Jousselin E, Desdevises Y, Coeur d’Acier A. 2009. Fine-scale cospeciation between Brachycaudus and Buchnera aphidicola: bacterial genome helps define species and evolutionary relationships in aphids. Proc Biol Sci. 276(1654):187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD, Paley S, Romero P. 2002. The Pathway Tools software. Bioinformatics 18(Suppl 1):S225–S232. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre A, Manzano-Marín A. 2017. Dissecting genome reduction and trait loss in insect endosymbionts. Ann N Y Acad Sci. 1389(1):52–75. [DOI] [PubMed] [Google Scholar]
- López-Madrigal S, Latorre A, Moya A, Gil R. 2015. The link between independent acquisition of intracellular gamma-endosymbionts and concerted evolution in Tremblaya princeps. Front Microbiol. 6:642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan J, Sun X, Fei Z, Douglas AE. 2018. Maternal inheritance of a single somatic animal cell displayed by the bacteriocyte in the whitefly Bemisia tabaci. Curr Biol. 28(3):459–465.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan JB, et al. 2015. Metabolic coevolution in the bacterial symbiosis of whiteflies and related plant sap-feeding insects. Genome Biol Evol. 7(9):2635–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan JB, et al. 2016. Cellular and molecular remodelling of a host cell for vertical transmission of bacterial symbionts. Proc Biol Sci. 283(1833):218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łukasik P, et al. 2018. Multiple origins of interdependent endosymbiotic complexes in a genus of cicadas. Proc Natl Acad Sci U S A. 115(2):E226–E235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010. Evolution of the mutation rate. Trends Genet. 26(8):345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzari S, Quicke DLJ. 2006. A cladistic analysis of whiteflies, subfamily Aleyrodinae (Hemiptera: Sternorrhyncha: Aleyrodidae). J Nat Hist. 40(44–46):2423–2554. [Google Scholar]
- Mao M, Yang X, Bennett GM. 2018. Evolution of host support for two ancient bacterial symbionts with differentially degraded genomes in a leafhopper host. Proc Natl Acad Sci U S A. 115(50):E11691–E11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JH. 1996. Neotropical whiteflies of the subfamily aleurodicinae established in the western palaearctic (Homoptera: Aleyrodidae). J Nat Hist. 30(12):1849–1859. [Google Scholar]
- Martin JH. 2007. Giant whiteflies (Sternorrhyncha, Aleyrodidae): a discussion of their taxonomic and evolutionary significance, with the description of a new species of Udamoselis Enderlein from Ecuador. Tijdschr Entomol. 150(1):13–29. [Google Scholar]
- Martin JH. 2008. A revision of Aleurodicus Douglas (Sternorrhyncha, Aleyrodidae), with two new genera proposed for palaeotropical natives and an identification guide to world genera of Aleurodicinae. Zootaxa 1835(1):1–100. [Google Scholar]
- Martinez-Torres D, Buades C, Latorre A, Moya A. 2001. Molecular systematics of aphids and their primary endosymbionts. Mol Phylogenet Evol. 20(3):437–449. [DOI] [PubMed] [Google Scholar]
- Matsuura Y, et al. 2018. Recurrent symbiont recruitment from fungal parasites in cicadas. Proc Natl Acad Sci U S A. 115(26):E5970–E5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meseguer AS, Coeur d’Acier A, Genson G, Jousselin E. 2015. Unravelling the historical biogeography and diversification dynamics of a highly diverse conifer-feeding aphid genus. J Biogeogr. 42(8):1482–1492. [Google Scholar]
- Meseguer AS, et al. 2017. Buchnera has changed flatmate but the repeated replacement of co-obligate symbionts is not associated with the ecological expansions of their aphid hosts. Mol Ecol. 26(8):2363–2378. [DOI] [PubMed] [Google Scholar]
- Misof B, et al. 2014. Phylogenomics resolves the timing and pattern of insect evolution. Science 346(6210):763–768. [DOI] [PubMed] [Google Scholar]
- Moran NA. 1996. Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc Natl Acad Sci U S A. 93(7):2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Bennett GM. 2014. The tiniest tiny genomes. Annu Rev Microbiol. 68(1):195–215. [DOI] [PubMed] [Google Scholar]
- Morris JJ, Lenski RE, Zinser ER. 2012. The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. MBio 3(2):e00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 302(1):205–217. [DOI] [PubMed] [Google Scholar]
- Nováková E, et al. 2013. Reconstructing the phylogeny of aphids (Hemiptera: Aphididae) using DNA of the obligate symbiont Buchnera aphidicola. Mol Phylogenet Evol. 68(1):42–54. [DOI] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28(8):1166–1167. [DOI] [PubMed] [Google Scholar]
- Ouvrard D, Martin JH. 2020. The white-files—taxonomic checklist of the world’s whiteflies (Insecta: Hemiptera: Aleyrodidae). Available from: http://www.hemiptera-databases.org/whiteflies/. Accessed October 24, 2020. doi:10.5519/0095728.
- Ovalle TM, Parsa S, Hernández MP, Becerra Lopez-Lavalle LA. 2014. Reliable molecular identification of nine tropical whitefly species. Ecol Evol. 4(19):3778–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Schliep K. 2019. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35(3):526–528. [DOI] [PubMed] [Google Scholar]
- Patiño-Navarrete R, Moya A, Latorre A, Peretó J. 2013. Comparative genomics of Blattabacterium cuenoti: the frozen legacy of an ancient endosymbiont genome. Genome Biol Evol. 5(2):351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaintance A. 1909. A new genus of Aleyrodidae, with remarks on Aleyrodes nubifera Berger and Aleyrodes citri Riley & Howard. Tech Ser US Dep Agric Bur Entomol. 12:169–174. [Google Scholar]
- R Core Team. 2018. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Ranwez V, Douzery EJP, Cambon C, Chantret N, Delsuc F. 2018. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol Biol Evol. 35(10):2582–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EPC. 2003. An appraisal of the potential for illegitimate recombination in bacterial genomes and its consequences: from duplications to genome reduction. Genome Res. 13(6):1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, et al. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16(10):944–945. [DOI] [PubMed] [Google Scholar]
- Santos-Garcia D, Vargas-Chavez C, Moya A, Latorre A, Silva FJ. 2015. Genome evolution in the primary endosymbiont of whiteflies sheds light on their divergence. Genome Biol Evol. 7(3):873–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Garcia D, et al. 2014. Small but powerful, the primary endosymbiont of moss bugs, Candidatus Evansia muelleri, holds a reduced genome with large biosynthetic capabilities. Genome Biol Evol. 6(7):1875–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069. [DOI] [PubMed] [Google Scholar]
- Shcherbakov DE. 2000. The most primitive whiteflies (Hemiptera; Aleyrodidae; Bernaeinae subfam. nov.) from the Mesozoic of Asia and Burmese amber, with an overview of Burmese amber hemipterans. Bull Nat Hist Mus Lond (Geol). 56(June):29–37. [Google Scholar]
- Silva FJ, Santos-Garcia D. 2015. Slow and fast evolving endosymbiont lineages: positive correlation between the rates of synonymous and non-synonymous substitution. Front Microbiol. 6(November):1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Moran NA. 2013. The evolution of genomic instability in the obligate endosymbionts of whiteflies. Genome Biol Evol. 5(5):783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N, Liang AP, Bu CP. 2012. A molecular phylogeny of Hemiptera inferred from mitochondrial genome sequences. PLoS One 7(11):e48778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarzewicz T, Moskal A. 2001. Ultrastructure, distribution, and transmission of endosymbionts in the whitefly Aleurochiton aceris Modeer (Insecta, Hemiptera, Aleyrodinea). Protoplasma 218(1–2):45–53. [DOI] [PubMed] [Google Scholar]
- Thao ML, Baumann P. 2004. Evolutionary relationships of primary prokaryotic endosymbionts of whiteflies and their hosts. Appl Environ Microbiol. 70(6):3401–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft C, Andersson SGE. 2010. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet. 11(7):465–475. [DOI] [PubMed] [Google Scholar]
- Tremblay E. 1959. Osservazioni sulla simbiosi endocellulare dialcuni Aleyrodidae (Bemisia tabaci Gennad., Aleurolobus olivinus Silv., Trialeurodes vaporariorum West.). Bollet Lab Entomol Agrar Filippo Silvestri Portici. 17:210–246. [Google Scholar]
- Van Leuven JT, Meister RC, Simon C, McCutcheon JP. 2014. Sympatric speciation in a bacterial endosymbiont results in two genomes with the functionality of one. Cell 158(6):1270–1280. [DOI] [PubMed] [Google Scholar]
- Walker BJ, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, et al. 2019. Insight into the microbial world of Bemisia tabaci cryptic species complex and its relationships with its host. Sci Rep. 9(1):6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernegreen JJ. 2015. Endosymbiont evolution: predictions from theory and surprises from genomes. Ann NY Acad Sci. 1360(1):16–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. 2018. DAMBE7: new and improved tools for data analysis in Molecular Biology and Evolution. Mol Biol Evol. 35(6):1550–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XR, Li NN, Bao XY, Douglas AE, Luan JB. 2020. Patterns of host cell inheritance in the bacterial symbiosis of whiteflies. Insect Sci. 27(5):938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by Maximum Likelihood. Mol Biol Evol. 24(8):1586–1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The relevant scripts and files generated are available in FigShare at https://doi.org/10.6084/m9.figshare.12361496 (last accessed October 24, 2020).