

Abstract
The discovery of multi-targeting ligands of bacterial enzymes is an important strategy to combat rapidly spreading antimicrobial resistance. Bacterial DNA gyrase and topoisomerase IV are validated targets for the development of antibiotics. They can be inhibited at their catalytic sites or at their ATP binding sites. Here we present the design of new hybrids between the catalytic inhibitor ciprofloxacin and ATP-competitive inhibitors that show low nanomolar inhibition of DNA gyrase and antibacterial activity against Gram-negative pathogens. The most potent hybrid 3a has MICs of 0.5 µg/mL against Klebsiella pneumoniae, 4 µg/mL against Enterobacter cloacae, and 2 µg/mL against Escherichia coli. In addition, inhibition of mutant E. coli strains shows that these hybrid inhibitors interact with both subunits of DNA gyrase (GyrA, GyrB), and that binding to both of these sites contributes to their antibacterial activity.
Keywords: antibacterial, ciprofloxacin, DNA gyrase, dual inhibitor, hybrid
1. Introduction
Bacterial resistance poses a major threat to global health, so new therapies against bacterial infections are urgently needed. One of the approaches to address this problem is to target multiple bacterial macromolecules [1]. Among the enzymes that enable multitargeting are the bacterial type IIA topoisomerases: DNA gyrase and topoisomerase IV. These enzymes are well-established targets for antibacterial drug discovery and they have important roles in DNA replication, transcription, repair, and recombination, through altering DNA topology during these processes [2]. They are homologous enzymes, where DNA gyrase consists of two GyrA subunits plus two GyrB subunits, and topoisomerase IV consists of two ParC subunits plus two ParE subunits, thus forming the heterotetrameric A2B2 and C2E2 complexes, respectively. The GyrA/ParC subunits contain catalytic sites that bind to DNA, while the GyrB/ParE subunits contain ATP binding sites, and provide the energy required for the catalytic reaction through ATP hydrolysis [3,4]. The fluoroquinolones are catalytic site inhibitors that have been successfully used in clinical applications since the introduction of nalidixic acid, and they remain an important class of antibacterial agents for the treatment of Gram-positive and Gram-negative bacterial infections [5,6]. On the other hand, the only inhibitor of GyrB that interacts with the ATP binding site that reached the clinic was novobiocin, although therapeutic use was discontinued due to toxicity and the emergence of target-based resistance [7].
Mutations conferring resistance to novobiocin in Escherichia coli GyrB are most commonly found in the Arg136 residue, although other residues have also been identified where their mutation can lead to novobiocin resistance (Asp73, Gly77, Ile78, Thr165) [8]. Fluoroquinolones such as ciprofloxacin, levofloxacin, and moxifloxacin are generally effective against infections caused by Gram-positive and Gram-negative pathogens, and hence they are widely used in medicine. However, resistance to these agents has emerged and continues to spread despite recommendations to limit their use [9,10]. Indeed, resistance to fluoroquinolones has been detected for most bacterial infections treated with these antibiotics. In particular, the difficult to treat infections here include: (i) urinary tract infections caused by E. coli; (ii) respiratory infections caused by Streptococcus pneumoniae, which is a leading cause of community-acquired pneumonia; (iii) intra-abdominal infections caused by resistant E. coli, Salmonella spp., and Shigella; (iv) infections of the skin and skin structures through methicillin-resistant Staphylococcus aureus, where fluoroquinolones resistance is spread worldwide; and (v) gonococcal infections through Neisseria gonorrhoeae, where ciprofloxacin resistance already appeared in the late 1990s [11].
The mechanisms behind this fluoroquinolones resistance include: (i) chromosomal mutations that cause increased antibiotic efflux or reduced uptake, and hence reduced intracellular accumulation; (ii) plasmid-acquired genes that encode drug-modifying enzymes, efflux pumps, or target protection proteins; and (iii) most commonly and clinically significant, gene mutations at the target site [5,12]. Mutations can occur within the quinolone-resistance-determining regions of GyrA and/or ParC. The most common mutation sites in E. coli GyrA are Ser83 and Asp87, which are the key amino-acid residues for fluoroquinolone binding, and the corresponding homologous positions in ParC, as Ser80 and Glu84 [11,13].
For these reasons, there is an urgent need for development of new drugs that circumvent common fluoroquinolone resistance mechanisms. Due to the structural similarities between DNA gyrase and topoisomerase IV, dual-targeting inhibitors can be developed that simultaneously inhibit GyrA and ParC or GyrB and ParE. The development of bacterial target-based resistance to such inhibitors will be less likely because resistance-conferring mutations would need to occur simultaneously at both targets, which is unlikely to happen [1,7,14]. In the present study, we used a different approach to design dual-targeting antimicrobial compounds. Specifically, we aimed to inhibit both the catalytic and the ATP binding sites of the same target protein.
In recent years, we have investigated and reported on several structural types of ATP-competitive GyrB/ParE inhibitors [15,16,17,18,19]. Recently, we also reported the discovery of GyrB inhibitor/ ciprofloxacin hybrids [20]. These previously designed hybrids showed weak antibacterial activities, which were shown to be mainly due to interactions with the GyrA and/or ParC subunits. To overcome this difficulty, we focused on our recently developed balanced dual GyrB/ParE ATP-competitive inhibitors 1a and 1b (Figure 1). These inhibitors have potent antibacterial activity against several Gram-positive and Gram-negative bacterial strains [21]. In this paper, we present new hybrids between ciprofloxacin and 1a or 1b. By combining these molecules, we have reached superior antibacterial activities due to the interactions of these hybrids with both subunits, as GyrA and GyrB or ParC and ParE. Importantly, the compounds are effective against Gram-negative strains of bacteria that belong to the group of resistant ‘ESKAPE’ pathogens that pose a major threat to society and health (i.e., Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter spp.) [22].
Figure 1.

Structure of E. coli DNA gyrase (Protein Data Bank (PDB) entry: 6RKW [23]). The two GyrA subunits are in gray, and the binding site of GyrA/ParC inhibitor ciprofloxacin is represented by the upper black dashed rectangle. The two GyrB subunits are in yellow, and the binding site of GyrB/ParE inhibitors 1a and 1b is represented by the lower black dashed rectangle. The general structure of GyrA/ParC and GyrB/ParE inhibitor hybrids is shown in the middle.
2. Materials and Methods
2.1. General Information—Chemistry
Chemicals were obtained from Acros Organics (Geel, Belgium), Sigma-Aldrich (St. Louis, MO, USA), and Apollo Scientific (Stockport, UK), and were used without further purification. Analytical thin-layer chromatography was performed on silica gel plates (Merck 60 F254; 0.25 mm), with visualization with UV light (at 254 nm and 366 nm) and spray reagent ninhydrin. Column chromatography was carried out on silica gel 60 (particle size, 240–400 mesh). Analytical reversed-phase HPLC analysis was performed on a liquid chromatography system (1260 Infinity II LC; Agilent Technologies Inc., Santa Clara, CA, USA). A C18 column was used (3.5 μm, 4.6 mm × 150 mm; XBridge; Waters, Milford, MA, USA), with a flow rate of 1.5 mL/min and a sample injection volume of 10 µL. The mobile phase consisted of acetonitrile (solvent A) and 0.1% formic acid in 1% acetonitrile in ultrapure water (solvent B). The gradient (defined for solvent A) was: 0–1.0 min, 25%; 1.0–6.0 min, 25–98%; 6.0–6.5 min, 98%; 6.5–7.5 min, 98–25%; 7.5–10.5 min, 25%. Ultrapure water was obtained with a Milli-Q Advantage A10 water purification system (Millipore, Merck, Burlington, MA, USA). Melting points were determined on a hot stage microscope (Reichert) and are uncorrected. 1H NMR spectra were recorded at 400 MHz (Bruker AVANCE III 400 spectrometer; Bruker Corporation, Billerica, MA, USA) in DMSO-d6 solutions, with tetramethylsilane as the internal standard. Infrared (IR) spectra were recorded (Thermo Nicolet Nexus 470 ESP FT-IR spectrometer; Thermo Fisher Scientific, Waltham, MA, USA). Mass spectra were obtained using a compact mass spectrometer (Advion expression; Advion Inc., Ithaca, NY, USA). High-resolution mass spectrometry was also performed (Exactive Plus Orbitrap; Thermo Fisher Scientific, Waltham, MA, USA). Detailed synthetic procedures and analytical data for all of the compounds are in Appendix A. 1H NMR spectra and HPLC chromatograms can be found in Supplementary Materials.
2.2. Molecular Docking
Molecular docking calculations were performed using Schrödinger Release 2020-1 (Schrödinger, LLC, New York, NY, USA, 2020). The crystal structures of S. aureus DNA gyrase A in complex with moxifloxacin (PDB entry: 5CDQ [24]) and S. aureus DNA gyrase B in complex with 1b (PDB entry: 6TCK [21]) were retrieved from the Protein Data Bank. The proteins were prepared using Protein Preparation Wizard, with the default settings. The receptor grid was calculated for the ligand-binding site, and the designed hybrids were docked using the Glide XP protocol, as implemented in Schrödinger Release 2020-1 (Glide, Schrödinger, LLC, New York, NY, USA, 2020). Figures were prepared with PyMOL [25].
2.3. Determination of Inhibitory Activities on Escherichia coli DNA Gyrase and Topoisomerase IV
The assay for determination of the IC50 values was performed according to previously reported procedures [26].
Briefly, inhibitory activities were determined using supercoiling (for DNA gyrase) and relaxation (for topoisomerase IV) assay kits (Inspiralis, Norwich, UK) on streptavidin-coated 96-well microtiter plates from Thermo Scientific Pierce (Thermo Fisher Scientific, Waltham, MA, USA). First, the plates were rehydrated with Wash Buffer and the biotinylated oligonucleotide TFO1 was then immobilized. After washing off the unbound oligonucleotide with Wash Buffer, the enzyme assay was performed. Into each well was added 24 µL of mixture containing 6 µL of Assay Buffer (containing ATP), 0.75 µL of relaxed (for E. coli DNA gyrase supercoiling assay) or supercoiled (for E. coli topoisomerase IV relaxation assay) pNO1 plasmid and 17.25 µL of water. Additionally, 3 µL of a solution of the inhibitor in 10% DMSO containing 0.008% Tween 20 and 3 µL of enzyme (1.5 U) in Dilution Buffer were also added to the wells. Reaction solutions were incubated at 37 °C for 30 min. The TF buffer was added to terminate the enzymatic reaction and after additional incubation for 30 min at room temperature, which allowed for triplex formation (biotin–oligonucleotide–plasmid), the unbound plasmid was washed off using TF buffer. Promega Diamond dye in T10 buffer was then added. After another 15 min incubation at room temperature in the dark, the fluorescence (excitation: 485 nm, emission: 535 nm) was measured with an automated microplate reader (SynergyTM H4, BioTek, Winooski, VT, USA). Initial screening was done at 10 µM and 1 µM concentrations of inhibitors and for the active inhibitors at these concentrations, IC50 values were determined using seven concentrations of tested compounds. The test concentration range in E. coli DNA gyrase assay was 0.028–10 µM for compounds 3a and 3b, 0.0028–1 µM for 7a, 7b, and 11a, and 0.0011–0.4 for 11b. Compounds were diluted using 0.375-fold serial dilution steps of the given compound. The test concentration range in E. coli topoisomerase IV assay was 0.063–4 µM for compound 3a and 0.156–10 µM for compounds 7a, 7b, and 11b. Here, the compounds were diluted using 2-fold serial dilution steps of the given compound. GraphPad Prism 6.0 software was used to calculate the IC50 values, which were determined in at least two independent measurements, and their means are given as the final result. Novobiocin was used as the positive control. Dose-response curves can be found in Supplementary Materials.
2.4. Determination of Antibacterial Activities
The following clinical microbiology control strains were obtained from American Type Culture Collection (ATCC) via Microbiologics Inc. (St. Cloud, MN, USA): A. baumannii (ATCC 17978); E. coli (ATCC 25922); K. pneumoniae (ATCC 10031); P. aeruginosa (ATCC 27853); and Enterobacter cloacae spp. cloacae (ATCC 13047). E. coli MG1655 originated from the laboratory collection of Dr. Csaba Pál. The GyrA and GyrB mutant strains of E. coli MG1655 were constructed using pORTMAGE [27] recombineering (Addgene plasmid #120418; http://n2t.net/addgene:120418; RRID:Addgene_120418), according to the published protocol [28]. E. coli K-12 BW25113 single-gene knockout mutant lines ΔdapF, ΔmrcB, ΔsurA, ΔacrB and ΔtolC originated form the Keio collection copy owned by the laboratory of Dr. Csaba Pál [29].
Cation-adjusted Mueller Hinton II broth (MHBII) was used for growth of the bacteria under standard laboratory conditions, for antimicrobial susceptibility tests, and for selection of resistant variants. To prepare the MHBII broth, 22 g MHBII powder (containing 3 g beef extract, 17.5 g acid hydrolysate of casein, 1.5 g starch; Becton, Dickinson and Co., Franklin Lakes, NJ, USA) was dissolved in 1 L water. MHBII agar was prepared by addition of 14 g agar (Bacto; Molar Chemicals, Halásztelek, Hungary) to 1 L broth.
Minimum inhibitory concentrations (MICs) were determined using a standard serial broth microdilution technique, according to the Clinical and Laboratory Standards Institute guidelines [30]. Bacterial strains were inoculated onto MHBII agar plates and grown overnight at 37 °C. Next, three individual colonies from each strain were inoculated into 1 mL MHBII medium and propagated at 37 °C overnight, with agitation at 250 rpm. For Enterococcus sp., the cells were plated in BHI agar plates, and BHI broth was used to determine the MICs. To perform the MIC assays, 12-step serial dilutions using two-fold dilution steps of the given compound (each dissolved in 100% DMSO) were generated in 96-well microtiter plates (Corning Inc., Corning, NY, USA). The concentrations used were: 64, 32, 16, 8, 4, 2, 1, 0.5, 0.25, 0.125, 0.0625, and 0.03125 µg/mL. Following the dilutions, each well was seeded with 5 × 104 bacterial cells. Each measurement was performed in three parallel replicates, and to avoid possible edge effects in the microwell plates, the outside rows (A, H) were filled with sterile medium. Following the inoculations, the plates were covered with the lids and wrapped in polyethylene plastic bags, to minimize evaporation but to allow O2 transfer. The plates were incubated at 37 °C under continuous shaking at 150 rpm for 18 h. After incubation, the OD600 of each well was measured using a microplate reader (Synergy 2; Biotek, Winooski, VT, USA). The MICs were defined as the antibiotic concentrations which inhibited the growth of the bacterial cultures; i.e., the drug concentration where the average OD600 increment of the three technical replicates was below 1.5-fold the background OD increment.
3. Results and Discussion
3.1. Design
Our first series of dual GyrA and GyrB inhibitor hybrids was designed by combining the GyrA inhibitor ciprofloxacin with benzothiazole-based GyrB inhibitors [20]. Although the GyrB inhibitors used in the first series of hybrids showed potent DNA gyrase inhibition, they showed only low antibacterial activity [17]. When the hybrids were tested against bacteria, we showed that in the bacteria they only bound to GyrA, and not to GyrB, which means that the observed antibacterial activity was mainly due to the ciprofloxacin part that interacted with GyrA. In the present series, we combined our balanced dual-targeting GyrB and ParE inhibitors 1a and 1b with potent antibacterial activity against Gram-positive and Gram-negative strains with ciprofloxacin [21]. The new hybrids were prepared either by direct fusion of inhibitors 1a and 1b with ciprofloxacin, or by linking the two molecules with linkers of different lengths (i.e., glycine in 7a, β-alanine in 7b or 2-ethoxyethyl in 11a and 11b). The design of the new dual GyrA and GyrB inhibitors is shown in Figure 1 and Figure 2.
Figure 2.

Docking binding modes of the representative hybrid 3a (cyan sticks) in the catalytic site of GyrA (S. aureus GyrA in gray; DNA in orange; PDB entry: 5CDQ [24]) and in the ATP-binding site of GyrB (S. aureus GyrB in yellow; PDB entry: 6TCK [21]). For clarity, only amino-acid residues forming hydrogen bonds (dashed lines) are presented as sticks. The magnesium ion in GyrA is a green sphere, while the structural water in GyrB is a red sphere.
Molecular docking showed that all designed hybrids 3a, 3b, 7a, 7b, 11a and 11b can bind to either GyrA or GyrB. As an example, the docking binding mode of a representative hybrid 3a in GyrA and GyrB active sites is presented in Figure 2. Docking of 3a to the catalytic site of GyrA reproduced the binding conformation of the fluoroquinolone portion, as observed for moxifloxacin in the crystal structure. Important hydrogen bonds were formed with Ser84, Arg122 and a magnesium ion, while π-stacking interactions were formed with DNA bases. The GyrB part of hybrid 3a made additional hydrophobic contacts with Asn474–476 (Figure 2). At the ATP binding site of GyrB, the pyrrolamide moiety of 3a interacted with Asp81 and a structural water molecule, as observed for 1b in the crystal structure. The carbonyl group connecting the benzothiazole moiety to the piperidine nitrogen atom of ciprofloxacin formed a hydrogen bond with Arg144. Furthermore, the ciprofloxacin part of 3a pointed towards the bulk solvent and did not interfere with the binding (Figure 2). Similar binding modes were obtained also for other designed GyrA/GyrB inhibitor hybrids.
3.2. Chemistry
The fused hybrids 3a and 3b were prepared in the one-step synthesis presented in Scheme 1. Compounds 1a and 1b [21] were coupled to ciprofloxacin (2) using the reagents 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt), to obtain the final compounds 3a and 3b.
Scheme 1.

Reagents and conditions: (a) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 1-hydroxybenzotriazole (HOBt), N-methylmorpholine (NMM), N,N-dimethylformamide (DMF), rt, 15 h.
The synthesis of the linked compounds 7a and 7b is shown in Scheme 2. First, ciprofloxacin (2) was coupled to N-(tert-butoxycarbonyl)glycine (4a) or 3-(N-(tert-butoxycarbonyl)amino)propanoic acid (4b) using EDC- and HOBt-promoted coupling, to obtain compounds 5a and 5b. The Boc protecting groups of 5a and 5b were removed by acidolysis, and the obtained intermediates 6a and 6b were coupled to 1a to obtain the desired compounds 7a and 7b.
Scheme 2.

Reagents and conditions: (a) EDC, HOBt, NMM, DMF, rt, 15 h; (b) 4 M HCl in 1,4-dioxane, 1,4-dioxane, rt, 3 h; (c) 1a, EDC, HOBt, NMM, DMF, rt, 15 h.
The compounds 11a and 11b were synthesized according to Scheme 3. The reaction between 2-(2-((tert-butoxycarbonyl)amino)ethoxy)ethyl methanesulfonate (8) and ciprofloxacin (2) using triethylamine (Et3N) in DMF and water yielded intermediate 9. The Boc protecting group of 9 was removed with HCl in 1,4-dioxane to obtain compound 10. In the final step, compound 10 was coupled to 1a or 1b, to obtain the final compounds 11a and 11b.
Scheme 3.

Reagents and conditions: (a) Et3N, DMF/H2O, 80 °C, 72 h; (b) 4 M HCl in 1,4-dioxane, 1,4-dioxane, rt, 3 h; (c) 1a or 1b, EDC, HOBt, NMM, DMF, rt, 15 h.
3.3. Enzyme Inhibition and Antibacterial Activities
Six new hybrids were prepared and tested for their inhibitory activities against DNA gyrase and topoisomerase IV from E. coli in supercoiling and relaxation assays, respectively (Table 1). The antibacterial activities of the hybrids were tested against Gram-negative bacteria from the ESKAPE group of pathogens, and are shown in Table 2.
Table 1.
Inhibitory activities of the new hybrid compounds against E. coli DNA gyrase and topoisomerase IV.
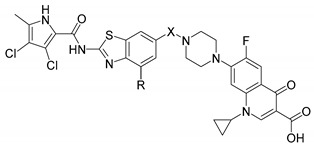
| ||||
|---|---|---|---|---|
| Cpd. | R | X | IC50 [nM] 1 | |
| E. coli | ||||
| DNA Gyrase | Topoisomerase IV | |||
| 3a | H |
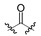
|
450 ± 180 | 1000 ± 150 |
| 3b | OBn |
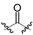
|
3300 ± 1500 | >10,000 |
| 7a | H |
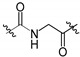
|
92 ± 10 | 2500 ± 1300 |
| 7b | H |
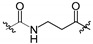
|
130 ± 10 | 3100 ± 0 |
| 11a 2 | H |
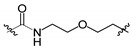
|
130 ± 30 | >10,000 |
| 11b 3 | OBn |
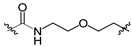
|
14 ± 9 | 2500 ± 1100 |
| 1a 4 | 13 ± 0 | 500 ± 280 | ||
| 1b 5 | <10 | 350 ± 50 | ||
| CP 6 | 120 ± 20 | 5400 ± 2100 | ||
| NB 7 | 170 ± 20 | 11,000 ± 2000 | ||
1 Concentration of compound (mean ±SD, in nM) that inhibits the enzyme activity by 50%; 2, 3 Compounds 11a and 11b were obtained as hydrochloride salts; 4, 5 1a and 1b, GyrB and ParE inhibitors; 6 CP, ciprofloxacin; 7 NB, novobiocin.
Table 2.
Minimum inhibitory concentration (MIC) values of the new hybrid compounds against the indicated Gram-negative bacterial strains.
| Cpd. | MIC [µg/mL] 1 | ||||
|---|---|---|---|---|---|
| A. baumannii ATCC 17978 | P. aeruginosa ATCC 27863 | K. pneumoniae ATCC 10031 | E. cloacae spp. cloacae ATCC 13047 | E. coli ATCC 25922 | |
| 3a | 16 | 64 | 0.5 | 4 | 2 |
| (23.4 µM) | (93.6 µM) | (0.73 µM) | (5.85 µM) | (2.93 µM) | |
| 3b | >64 | >64 | >64 | >64 | >64 |
| (>81.0 µM) | (>81.0 µM) | (>81.0 µM) | (>81.0 µM) | (>81.0 µM) | |
| 7a | >64 | >64 | 4 | >64 | >64 |
| (>86.4 µM) | (>86.4 µM) | (5.40 µM) | (>86.4 µM) | (>86.4 µM) | |
| 7b | >64 | >64 | 8 | >64 | >64 |
| (>84.8 µM) | (>84.8 µM) | (10.6 µM) | (>84.8 µM) | (>84.8 µM) | |
| 11a | >64 | >64 | 4 | 64 | 32 |
| (>82.9 µM) | (>82.9 µM) | (5.18 µM) | (82.9 µM) | (41.5 µM) | |
| 11b | 64 | >64 | 1 | 16 | 4 |
| (72.9 µM) | (>72.9 µM) | (1.14 µM) | (18.2 µM) | (4.56 µM) | |
| 1a 2 | 4 | 8 | 1 | >64 | 4 |
| (10.8 µM) | (21.6 µM) | (2.70 µM) | (>173 µM) | (10.8 µM) | |
| 1b 3 | 2 | 2 | 4 | >64 | 16 |
| (4.20 µM) | (4.20 µM) | (8.40 µM) | (>134 µM) | (33.6 µM) | |
| CP 4 | 0.25 | 0.25 | <0.03125 | <0.03125 | <0.03125 |
| (0.75 µM) | (0.75 µM) | (<0.0943 µM) | (<0.0943 µM) | (<0.0943 µM) | |
1 MIC, minimum inhibitory concentration; 2, 3 1a and 1b, GyrB and ParE inhibitors; 4 CP, ciprofloxacin. Measurements were performed according to the Clinical and Laboratory Standards Institute guidelines, with three independent measurements.
Four hybrids (7a, 7b, 11a, 11b) showed potent nanomolar inhibitory activities against DNA gyrase from E. coli (IC50 range, 14–130 nM; Table 1), which were comparable or superior to the activity of the GyrA inhibitor ciprofloxacin (IC50, 120 nM; Table 1). Compared to the GyrB inhibitors 1a and 1b, the hybrids showed weaker inhibition, with the exception of 11b, which showed very promising activity, with an IC50 of 14 nM. Inhibition of topoisomerase IV was weaker for all of these tested compounds, with low micromolar activities for 3a, 7a, 7b, and 11b, and lack of activity for 3b and 11a. Ciprofloxacin also inhibited topoisomerase IV in the low micromolar range, with an IC50 of 5.4 µM. These data demonstrate that the addition of these linkers that give flexibility to the molecules was beneficial for the inhibitory activities: here, the fused molecules 3a and 3b showed weaker inhibition of DNA gyrase compared to the linked molecules 7a, 7b, 11a, and 11b. On the contrary, the best antibacterial activity was obtained for the fused hybrid 3a. Compound 3a showed potent MICs against K. pneumoniae (0.5 µg/mL), E. cloacae (4 µg/mL), and E. coli (2 µg/mL) (Table 2). Compared to the other five hybrids, 3a is the smallest molecule, so one hypothesis for its superior activity is that this size difference contributes to better cellular uptake. However, because of experiments detailed below we came to doubt that differences in cellular uptake account for its superior activity. Compounds 7a and 7b, with the glycine and β-alanine linkers, showed no significant antibacterial activities, except against K. pneumoniae, while the ether compounds 11a and 11b were active against E. coli in addition to K. pneumoniae (Table 2). The activities of 11a and 11b against these two bacterial strains were comparable to the GyrB inhibitors 1a and 1b, while they were lower than the antibacterial activity of ciprofloxacin. In addition, the dual-targeting ligands 3a and 11b showed promising antibacterial activities also against E. cloacae (MICs, 4, 16 µg/mL, respectively), against which the GyrB inhibitors 1a and 1b were inactive. Compound 3b showed only weak micromolar enzyme inhibition, and was therefore almost inactive against all of these bacterial strains.
To study the factors that influence the antibacterial activities of these compounds, the hybrids were also tested on E. coli BW25113 wild-type and mutant strains (Table 3). The first three of the mutated genes for dapF, mrcB, and surA are involved in peptidoglycan biosynthesis and maturation of outer membrane proteins [31]; this leads to mutants with disturbed cell walls, which are thus more permeable. The other two mutants were for the acrB and tolC genes that encode efflux pump proteins [32]. When the hybrids were tested against the ΔdapF, ΔmrcB, and ΔsurA mutants with impaired cell walls, there were no differences in the activities compared to the wild-type strain. Therefore, poor penetration does not appear to be the main reason for the weak activities of these GyrA/GyrB inhibitor hybrids. In contrast, when tested against the ΔtolC mutant with an impaired efflux pump, the activities were improved for all of these tested hybrids. For 3a and 11b, the MICs showed changes of 4-fold or 8-fold, while for 7a, 7b, and 11a the changes were above 32-fold or 64-fold. From these data, we can conclude that the hybrids 7a, 7b, and 11a underwent strong efflux from the bacterial cell, which might be the reason for their weak activity or inactivity against the wild-type strains (Table 2). Although 3a and 11b also showed improved activities here, the MIC changes were not as pronounced, and their good activities on the wild-type strain show that the efflux was not detrimental to their antibacterial activities.
Table 3.
Minimum inhibitory concentrations of these new hybrid compounds against E. coli BW25113 wild-type and mutant strains with impaired cell-wall structure (ΔdapF, ΔmrcB, ΔsurA) and efflux pumps (ΔacrB, ΔtolC).
| Cpd. | Minimum Inhibitory Concentration [µg/mL] | |||||
|---|---|---|---|---|---|---|
| E. coli BW25113 WT 1 | E. coli BW25113 ΔdapF | E. coli BW25113 ΔmrcB | E. coli BW25113 ΔsurA | E. coli BW25113 ΔacrB | E. coli BW25113 ΔtolC | |
| 3a | 4 | 4 | 4 | 4 | 4 | 1 |
| (5.85 µM) | (5.85 µM) | (5.85 µM) | (5.85 µM) | (5.85 µM) | (1.46 µM) | |
| 3b | >64 | >64 | >64 | >64 | >64 | 16 |
| (>81.0 µM) | (>81.0 µM) | (>81.0 µM) | (>81.0 µM) | (>81.0 µM) | (20.3 µM) | |
| 7a | >64 | >64 | >64 | >64 | >64 | 2 |
| (>86.4 µM) | (>86.4 µM) | (>86.4 µM) | (>86.4 µM) | (>86.4 µM) | (2.70 µM) | |
| 7b | >64 | >64 | >64 | >64 | >64 | 2 |
| (>84.8 µM) | (>84.8 µM) | (>84.8 µM) | (>84.8 µM) | (>84.8 µM) | (2.65 µM) | |
| 11a | 32 | 32 | 32 | 16 | 32 | 0.5 |
| (41.5 µM) | (41.5 µM) | (41.5 µM) | (20.7 µM) | (41.5 µM) | (0.65 µM) | |
| 11b | 8 | 8 | 8 | 8 | 8 | 1 |
| (9.11 µM) | (9.11 µM) | (9.11 µM) | (9.11 µM) | (9.11 µM) | (1.14 µM) | |
| CP 2 | <0.03125 | <0.03125 | <0.03125 | <0.03125 | <0.03125 | <0.03125 |
| (<0.0943 µM) | (<0.0943 µM) | (<0.0943 µM) | (<0.0943 µM) | (<0.0943 µM) | (<0.0943 µM) | |
1 WT, wild-type; 2 CP, ciprofloxacin.
To investigate whether the observed antibacterial activities of these hybrids is due to their interactions with the catalytic or the ATP binding sites of DNA gyrase, or both, we tested them against three E. coli MG1655 strains in the presence of an efflux-pump substrate phenylalanine-arginine β-naphthylamide (PAβN): wild-type; a GyrB R136C mutant carrying a mutation at the ATP binding site; and the GyrA S83L, D87N and ParC S80I, E84G mutants, with the fluoroquinolone-binding site mutated (Table 4). These mutated amino acids are the most common sites for target-based resistance in E. coli. The results for these mutant strains confirm the hypothesis that these compounds can interact with both subunits; i.e., GyrA (and/or ParC) and GyrB. Compound 7b showed equipotent antibacterial activities against the strain with a mutated GyrB binding site and against the strain with the mutated fluoroquinolone binding site (GyrA and ParC). The activities against the two mutants were also comparable for compounds 7a and 11b, which suggests that 7a, 7b, and 11b have a balanced interaction with both of the binding sites. For 3a and 11a, the mutation in the GyrB binding site did not result in weaker activities compared to the wild type, while the mutation at the fluoroquinolone binding site did; this indicated that these two compounds interacted more strongly with the GyrA and ParC subunits. Nevertheless, the activities for this mutant were not completely lost. From these data, it can be concluded that our new hybrids can interact with both the GyrA and GyrB binding sites.
Table 4.
Minimum inhibitory concentrations of these new hybrid compounds against the three indicated E. coli MG1655 strains. The assays were carried out in the presence of 50 µg/mL PAβN.
| Cpd. | Minimum Inhibitory Concentration [µg/mL] | ||
|---|---|---|---|
| E. coli MG1655 WT 1 | E. coli MG1655 GyrB R136C | E. coli MG1655 GyrA S83L, D87N ParC S80I, E84G | |
| 3a | 0.5 | 0.5 | 16 |
| (0.73 µM) | (0.73 µM) | (23.4 µM) | |
| 3b | 64 | 32 | >64 |
| (81.0 µM) | (40.5 µM) | (>81.0 µM) | |
| 7a | 1 | 2 | 0.5 |
| (1.35 µM) | (2.70 µM) | (0.675 µM) | |
| 7b | 1 | 1 | 1 |
| (1.325 µM) | (1.325 µM) | (1.325 µM) | |
| 11a | 0.5 | 0.5 | 8 |
| (0.65 µM) | (0.65 µM) | (10.4 µM) | |
| 11b | 1 | 2 | 4 |
| (1.14 µM) | (2.28 µM) | (4.56 µM) | |
| CP 2 | <0.03125 | <0.03125 | 64 |
| (<0.0943 µM) | (<0.0943 µM) | (193 µM) | |
1 WT, wild type; 2 CP, ciprofloxacin.
4. Conclusions
In summary, we have designed and synthesized new hybrid ligands that can interact with both the catalytic and the ATP binding sites of DNA gyrase. These new hybrids show low nanomolar inhibitory activities on E. coli DNA gyrase, and good antibacterial activities, especially against K. pneumoniae and E. coli, two pathogens from the problematic ESKAPE group. Additionally, compounds 3a and 11b do not undergo extensive efflux. Testing of these compounds on mutant E. coli strains confirmed that they can bind to both the GyrA (and/or ParC) and GyrB binding sites. Targeting both of the binding sites results in potent antibacterial activities against Gram-negative pathogens, and should also have a strong advantage in terms of delayed or prevented emergence of bacterial resistance.
Acknowledgments
The authors thank Chris Berrie for editing the manuscript.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4923/13/1/6/s1: Figure S1. Dose-response curves for compounds 3a, 3b, 7a, 7b, 11a, and 11b for E. coli DNA gyrase, Figure S2. Dose-response curves for compounds 3a, 7a, 7b, and 11b for E. coli topoisomerase IV, Figure S3. 1H NMR spectra of compound 3a, Figure S4. HPLC chromatogram of compound 3a, Figure S5. 1H NMR spectra of compound 3b, Figure S6. HPLC chromatogram of compound 3b, Figure S7. 1H NMR spectra of compound 7a, Figure S8. HPLC chromatogram of compound 7a, Figure S9. 1H NMR spectra of compound 7b, Figure S10. HPLC chromatogram of compound 7b, Figure S11. 1H NMR spectra of compound 11a, Figure S12. HPLC chromatogram of compound 11a, Figure S13. 1H NMR spectra of compound 11b, Figure S14. HPLC chromatogram of compound 11b.
Appendix A
Synthetic Procedures and Analytical Data
General procedure A. Synthesis of compounds 3a-b, 5a-b, 7a-b, and 11a-b (with 3a as the example). A solution of 2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzo[d]thiazole-6-carboxylic acid (1a, 75 mg, 0.203 mmol) in N,N-dimethylformamide (8 mL) was cooled to 0 °C, and then EDC (47 mg, 0.244 mmol), HOBt (36 mg, 0.264 mmol), and N-methylmorpholine (45 µL, 0.406 mmol) were added, and the reaction mixture was stirred for 30 min at 0 °C. Ciprofloxacin (2, 67 mg, 0.203 mmol) was then added, and the reaction mixture was stirred at room temperature overnight. The solvent was evaporated in vacuo, with ethyl acetate (30 mL) and 10% citric acid (20 mL) added to the residue. The precipitate obtained was filtered off, washed with ethyl acetate, and dried.
1-Cyclopropyl-7-(4-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzo[d]thiazole-6-carbonyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3a). Yield: 91 mg (65.7%); dark purple solid; mp > 300 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.15–1.23 (m, 2H, cyclopropyl-CH2), 1.30–1.37 (m, 2H, cyclopropyl-CH2), 2.28 (s, 3H, pyrr-CH3), 3.30–3.45 (signal is overlapped with the signal for water, 4H, 2 × piperazine-CH2), 3.65–3.90 (m, 5H, 2 × piperazine-CH2, CH), 7.58 (dd, 1H, J = 8.4, 1.6 Hz, Ar-H′-5), 7.62 (d, 1H, J = 7.6 Hz, Ar-H′-4/Ar-H-8), 7.85 (s, 1H, Ar-H′-4/Ar-H-8), 7.96 (d, 1H, J = 13.1 Hz, Ar-H-5), 8.19 (s, 1H, Ar-H′-7), 8.69 (s, 1H, Ar-H-2), 11.86 (s, 1H, pyrr-NH/CONH), 12.39 (s, 1H, pyrr-NH/CONH), 15.21 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3368 (NH), 3173 (NH), 3066 (OH), 1713 (CO), 1663 (CO), 1603, 1530, 1491, 1331, 1256, 1162, 1085, 1020, 944, 900, 830, 809, 771, 742, 706, 624, 552, 517. HRMS (ESI-) m/z for C31H24Cl2FN6O5S ([M-H]-): calculated: 681.0885, found: 681.0909, delta: 3.66 ppm. HPLC: tr 6.86 min (95.8% at 254 nm).
7-(4-(4-(Benzyloxy)-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzo[d]thiazole-6-carbonyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3b). Synthesized according to General procedure A using 1b (60 mg, 0.162 mmol) as reactant instead of 1a. The crude product was purified with flash column chromatography using dichloromethane/methanol (15:1) as eluent. Yield: 20 mg (20.1%); white solid; mp > 300 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.19 (s, 2H, cyclopropyl-CH2), 1.28–1.36 (m, 2H, cyclopropyl-CH2), 2.26 (s, 3H, pyrr-CH3), 3.39 (s, 4H, 2 × piperazine-CH2), 3.58–3.94 (m, 5H, 2 × piperazine-CH2, CH), 5.32 (s, 2H, CH2Bn), 7.19 (s, 1H, Ar-H′-5), 7.29–7.48 (m, 3H, 3 × Ar-H″ (Bn)), 7.48–7.57 (m, 2H, 2 × Ar-H″ (Bn)), 7.61 (d, J = 7.4 Hz, 1H, Ar-H-8), 7.74 (s, 1H, Ar-H′-7), 7.95 (d, J = 13.1 Hz, 1H, Ar-H-5), 8.68 (s, 1H, Ar-H-2), 12.06 (s, 1H, pyrr-NH/CONH), 12.22 (s, 1H, pyrr-NH/CONH), 15.19 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 2915 (NH), 2843 (OH), 1723 (CO), 1658 (CO), 1626, 1531, 1471, 1402, 1331, 1258, 1131, 1029, 902, 743, 617, 551. HRMS (ESI+) m/z for C38H32Cl2FN6O6S ([M + H]+): calculated 789.1460, found 789.1401, delta: −7.42 ppm. HPLC: tr 7.48 min (98.9% at 254 nm).
7-(4-((tert-Butoxycarbonyl)glycyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5a). Synthesized according to General procedure A using (tert-butoxycarbonyl)glycine (185 mg, 1.06 mmol) as reactant. During isolation, the phases of the mother liquor after the filtration of the precipitated product were separated, where the water phase was extracted with ethyl acetate (2 × 20 mL), and the organic phases were combined and washed with 10% citric acid (2 × 10 mL) and brine (20 mL). The organic phase was dried over Na2SO4 and filtered, and the solvent was removed in vacuo. The crude product was purified first by crystallization from methanol, where the mother liquor was evaporated, and the residue was purified again by crystallization from a methanol/diethyl ether mixture. All three precipitates were combined to give 148 mg of pure product. Yield: 148 mg (28.7%); pale yellow solid; mp 218–220 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.15–1.23 (m, 2H, cyclopropyl-CH2), 1.28–1.35 (m, 2H, cyclopropyl-CH2), 1.39 (s, 9H, 3 × CH3 (t-Bu)), 3.28–3.40 (signal is overlapped with the signal for water, 4H, 2 × piperazine-CH2), 3.67 (br s, 4H, 2 × piperazine-CH2), 3.79–3.89 (m, 3H, CH2CO, CH), 6.84 (t, 1H, J = 5.6 Hz, NH), 7.59 (d, 1H, J = 7.4 Hz, Ar-H-8), 7.95 (d, 1H, J = 13.2 Hz, Ar-H-5), 8.68 (s, 1H, Ar-H-2), 15.20 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3406 (NH), 3001 (OH), 1729 (CO), 1702 (CO), 1650 (CO), 1622, 1496, 1468, 1439, 1387, 1341, 1298, 1243, 1217, 1164, 1098, 1054, 1017, 917, 931, 871, 833, 781, 750, 709, 618, 582. MS (ESI+) m/z = 489.3 ([M + H]+). HRMS (ESI+) m/z for C24H30FN4O6 ([M + H]+): calculated: 489.2144, found: 489.2130, delta: −2.80 ppm.
7-(4-(3-((tert-Butoxycarbonyl)amino)propanoyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5b). Synthesized according to General procedure A using 3-((tert-butoxycarbonyl)amino)propanoic acid (171 mg, 0.91 mmol) as reactant. During isolation, the phases of the mother liquor after filtration of precipitated product were separated, where water phase was extracted with ethyl acetate (2 × 20 mL), and the organic phases were combined and washed with 10% citric acid (2 × 10 mL) and brine (20 mL). The organic phase was dried over Na2SO4 and filtered, and the solvent was removed in vacuo. The crude product was purified by crystallization from methanol. The two precipitates were combined to give 243 mg of pure product. Yield: 243 mg (53.4%); pale yellow crystals; mp 185–187 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.17–1.23 (m, 2H, cyclopropyl-CH2), 1.29–1.36 (m, 2H, cyclopropyl-CH2), 1.38 (s, 9H, 3 × CH3 (t-Bu)), 3.18 (q, 2H, J = 6.8 Hz, NHCH2CH2), 3.29–3.42 (signal is overlapped with the signal for water, 6H, 2 × piperazine-CH2, NHCH2CH2), 3.64–3.73 (m, 4H, 2 × piperazine-CH2), 3.83 (sept, 1H, J = 4.1 Hz, CH), 6.77 (t, 1H, J = 5.6 Hz, CONHCH2), 7.59 (d, 1H, J = 7.5 Hz, Ar-H-8), 7.95 (d, 1H, J = 13.2 Hz, Ar-H-5), 8.68 (s, 1H, Ar-H-2), 15.21 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3272 (NH), 2985 (OH), 1732 (CO), 1700 (CO), 1623 (CO), 1503, 1470, 1382, 1343, 1260, 1237, 1169, 1107, 1056, 1020, 966, 933, 876, 833, 803, 780, 747, 699, 666, 636, 564. MS (ESI+) m/z = 503.3 ([M + H]+). HRMS (ESI+) m/z for C25H32FN4O6 ([M + H]+): calculated: 503.2300, found: 503.2290, delta: −2.16 ppm.
General procedure B. Synthesis of compounds 6a-b and 10 (with 6a as the example). Compound 5a (171 mg, 0.350 mmol, 1 equiv.) was suspended in 1,4-dioxane (5 mL) and stirred at room temperature for 5 min. Then, 4 M HCl in 1,4-dioxane (6 mL) was added, and the reaction mixture was stirred at room temperature for 3 h. The solvent was removed in vacuo, and the crude product was purified with crystallization from a methanol/diethyl ether mixture, and dried.
2-(4-(3-Carboxy-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinolin-7-yl)piperazin-1-yl)-2-oxoethan-1-aminium chloride (6a). Yield: 95 mg (63.9%); yellow powder; mp 178–180 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.17–1.23 (m, 2H, cyclopropyl-CH2), 1.27–1.35 (m, 2H, cyclopropyl-CH2), 3.22–3.54 (signal is overlapped with the signal for water, 4H, 2 × piperazine-CH2), 3.59–3.65 (m, 2H, piperazine-CH2), 3.72–3.78 (m, 2H, piperazine-CH2), 3.83 (sept, 1H, J = 4.0 Hz, CH), 3.92–3.40 (m, 2H, CH2NH3+), 7.59 (d, 1H, J = 7.5 Hz, Ar-H-8), 7.97 (d, 1H, J = 13.1 Hz, Ar-H-5), 8.10 (s, 3H, NH3+), 8.69 (s, 1H, Ar-H-2), 15.19 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3395 (NH), 2921 (OH), 2863 (CH), 2631 (CH), 1719 (CO), 1659 (CO), 1624 (CO), 1493, 1452, 1386, 1336, 1304, 1245, 1146, 1056, 1018, 949, 923, 886, 830, 805, 747, 705, 645, 615, 530. MS (ESI+) m/z = 389.2 ([M + H]+). HRMS (ESI+) m/z for C19H22FN4O4 ([M + H]+): calculated: 389.1620, found: 389.1612, delta: −1.93 ppm.
3-(4-(3-Carboxy-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinolin-7-yl)piperazin-1-yl)-3-oxopropan-1-aminium chloride (6b). Synthesized according to General procedure B. Yield: 142 mg (77.1%); pale yellow powder; mp 207–209 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.16–1.24 (m, 2H, cyclopropyl-CH2), 1.28–1.35 (m, 2H, cyclopropyl-CH2), 2.75 (t, 2H, J = 6.4 Hz, NH3+CH2CH2), 2.98–3.10 (m, 2H, NH3+CH2CH2), 3.29–3.50 (signal is overlapped with the signal for water, 4H, 2 × piperazine-CH2), 3.63–3.70 (m, 2H, piperazine-CH2), 3.70–3.76 (m, 2H, piperazine-CH2), 3.83 (sept, 1H, J = 4.0 Hz, CH), 7.58 (d, 1H, J = 7.5 Hz, Ar-H-8), 7.77 (br s, 3H, NH3+), 7.96 (d, 1H, J = 13.2 Hz, Ar-H-5), 8.69 (s, 1H, Ar-H-2), 15.19 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3459 (NH), 3233 (NH), 3058 (OH), 2953, 2849 (CH), 1700 (CO), 1621 (CO), 1477, 1443, 1332, 1246, 1216, 1188, 1143, 1107, 1044, 1023, 988, 947, 930, 873, 833, 808, 750, 626, 539. MS (ESI+) m/z = 403.1 ([M + H]+). HRMS (ESI+) m/z for C20H24FN4O4 ([M + H]+): calculated: 403.1776, found: 403.1769, delta: −1.71 ppm.
1-Cyclopropyl-7-(4-((2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzo[d]thiazole-6-carbonyl)glycyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7a). Synthesized according to General procedure A with 1a (61 mg, 0.165 mmol) and 6a (70 mg, 0.165 mmol) as reactants and 3 equiv. of N-methylmorpholine (54 µL, 0.495 mmol) instead of 2 equiv. The precipitate after filtration was resuspended in methanol, sonicated, heated, and filtered off. The procedure was repeated twice. Yield: 29.5 mg (24.2%); light grey powder; mp > 300 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.16–1.23 (m, 2H, cyclopropyl-CH2), 1.28–1.36 (m, 2H, cyclopropyl-CH2), 2.28 (s, 3H, pyrr-CH3), 3.42 (s, 4H, 2 × piperazine-CH2), 3.69–3.78 (m, 4H, 2 × piperazine-CH2), 3.84 (s, 1H, CH), 4.25 (d, 2H, J = 5.4 Hz, NHCH2CO), 7.61 (d, 1H, J = 7.4 Hz, Ar-H′-4/Ar-H-8), 7.82 (s, 1H, Ar-H), 7.93–8.02 (m, 2H, 2 × Ar-H), 8.53 (s, 1H, Ar-H), 8.68 (s, 1H, Ar-H-2), 8.69–8.72 (m, 1H, ArCONH), 11.93 (s, 1H, pyrr-NH/pyrr-CONH), 12.35 (s, 1H, pyrr-NH/pyrr-CONH), 15.21 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3361 (NH), 3232 (NH), 2989 (OH), 1712 (CO), 1660 (CO), 1625 (CO), 1504, 1449, 1331, 1253, 1024, 938, 888, 831, 765, 746, 701, 619. HRMS (ESI+) m/z for C33H29Cl2FN7O6S ([M + H]+): calculated: 740.1256, found: 740.1226, delta: −3.98 ppm. HPLC: tr 6.44 min (95.0% at 254 nm).
1-Cyclopropyl-7-(4-(3-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzo[d]thiazole-6-carboxamido)propanoyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7b). Synthesized according to General procedure A with 1a (50 mg, 0.135 mmol) and 6b (59 mg, 0.135 mmol) as reactants and 3 equiv. of N-methylmorpholine (45 µL, 0.405 mmol) instead of 2 equiv. The precipitate after filtration was suspended in water, sonicated, and filtered off, washed with hot tetrahydrofuran and dried. Yield: 63 mg (61.5%); brown solid; mp > 300 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.14–1.22 (m, 2H, cyclopropyl-CH2), 1.24–1.36 (m, 2H, cyclopropyl-CH2), 2.28 (s, 3H, pyrr-CH3), 2.72 (t, 2H, J = 7.1 Hz, NHCH2CH2CO), 3.32 (signal is overlapped with the signal for water, 4H, 2 × piperazine-CH2), 3.53–3.58 (m, 2H, NHCH2CH2 CO), 3.72 (s, 4H, 2 × piperazine-CH2), 3.80 (s, 1H, CH), 7.54 (d, 1H, J = 7.4 Hz, Ar-H′-4/Ar-H-8), 7.79 (s, 1H, Ar-H), 7.89–7.99 (m, 2H, 2 × Ar-H), 8.48 (s, 1H, Ar-H), 8.63 (t, 1H, J = 4.0 Hz, ArCONH), 8.67 (s, 1H, Ar-H-2), 11.90 (s, 1H, pyrr-NH/pyrr-CONH), 12.33 (s, 1H, pyrr-NH/pyrr-CONH), 15.20 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3382 (NH), 3205 (NH), 3002 (OH), 1721 (CO), 1625 (CO), 1508, 1448, 1408, 1372, 1260, 1089, 1025, 887, 807, 767, 743, 699, 621, 573, 521. HRMS (ESI+) m/z for C34H31Cl2FN7O6S ([M + H]+): calculated: 754.1412, found: 754.1386, delta: −3.53 ppm. HPLC: tr 6.25 min (95.1% at 254 nm).
7-(4-(2-(2-((tert-Butoxycarbonyl)amino)ethoxy)ethyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (9). To a solution of ciprofloxacin (2, 500 mg, 1.51 mmol) in DMF (33 mL), Et3N (694 µL, 4.98 mmol) and 2-(2-((tert-butoxycarbonyl)amino)ethoxy)ethyl methanesulfonate (855 mg, 3.02 mmol) dissolved in 3 mL of DMF were added, and the reaction mixture was stirred at 80 °C. Then, 5 drops of water were added, and the reaction mixture was stirred at 80 °C for 2 days. An additional 1 equiv. of 2-(2-((tert-butoxycarbonyl)amino)ethoxy)ethyl methanesulfonate (428 mg, 1.51 mmol) dissolved in 3 mL of DMF and 5 drops of water were added, and the reaction mixture was stirred at 80 °C overnight. The solvent was removed in vacuo, and ethyl acetate (200 mL) and water (100 mL) were added to the residue. The phases were separated, with the organic phase dried over Na2SO4 and filtered, and the solvent removed in vacuo. The crude product was purified with crystallization from methanol, and then additionally purified by addition of water (25 mL), with sonication and filtration of the product. The procedure was repeated twice. Yield: 243 mg (31.0%); pale yellow solid; mp 147–150 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.15–1.23 (m, 2H, cyclopropyl-CH2), 1.29–1.35 (m, 2H, cyclopropyl-CH2), 1.38 (s, 9H, 3 × CH3 (t-Bu)), 2.57 (t, 2H, J = 5.6 Hz, OCH2CH2N), 2.66-2.69 (m, 4H, 2 × piperazine-CH2), 3.09 (q, 2H, J = 5.6 Hz, NHCH2CH2O), 3.30–3.43 (signal is overlapped with the signal for water, 6H, 2 × piperazine-CH2, OCH2CH2N), 3.56 (t, 2H, J = 5.6 Hz, NHCH2CH2O), 3.83 (sept, 1H, J = 3.6 Hz, CH), 8.82 (t, 1H, J = 5.5 Hz, t-BuOCONH), 7.57 (d, 1H, J = 7.5 Hz, Ar-H-8), 7.92 (d, 1H, J = 13.4 Hz, Ar-H-5), 8.67 (s, 1H, Ar-H-2), 15.25 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3231 (NH), 2969 (OH), 1727 (CO), 1687 (CO), 1628 (CO), 1503, 1451, 1388, 1359, 1338, 1268, 1249, 1166, 1121, 1041, 1027, 1001, 942, 891, 868, 830, 802, 746, 696, 637, 577, 549. MS (ESI+) m/z = 519.4 ([M + H]+). HRMS (ESI+) m/z for C26H36FN4O6 ([M + H]+): calculated: 519.2613, found: 519.2606, delta: −1.48 ppm.
1-(2-(2-Ammonioethoxy)ethyl)-4-(3-carboxy-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinolin-7-yl)piperazin-1-ium chloride (10). Synthesized according to General procedure B. Instead of removing the solvent in vacuo, the formed precipitate was filtered off, washed with 1,4-dioxane (10 mL) and dried. Yield: 230 mg (100.0%); grey powder; mp 176–180 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.13–1.26 (m, 2H, cyclopropyl-CH2), 1.28–1.40 (m, 2H, cyclopropyl-CH2), 2.97–3.10 (m, 2H, CH2), 3.32–3.92 (signal is overlapped with the signal for water, 15H, 7 × CH2, CH), 7.63 (d, 1H, J = 7.6 Hz, Ar-H-8), 7.99 (d, 1H, J = 13.1 Hz, Ar-H-5), 8.25 (s, 3H, NH3+), 8.71 (s, 1H, Ar-H-2), 11.12 (s, 1H, NH+), signal for COOH group is not seen. IR (ATR) ν [cm−¹] = 3422 (NH), 3343 (NH), 2971 (OH), 1717 (CO), 1628 (CO), 1612, 1505, 1485, 1453, 1431, 1376, 1335, 1278, 1111, 1066, 1014, 943, 902, 884, 809, 786, 704, 627, 549. MS (ESI+) m/z = 419.2 ([M + H]+). HRMS (ESI+) m/z for C21H28FN4O4 ([M + H]+): calculated: 419.2089, found: 419.2082, delta: −1.65 ppm.
4-(3-Carboxy-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinolin-7-yl)-1-(2-(2-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzo[d]thiazole-6-carboxamido)ethoxy)ethyl)piperazin-1-ium chloride (11a). Synthesized according to General procedure A with 1a (69 mg, 0.186 mmol) and 10 (92 mg, 0.186 mmol) as reactants. After isolation the product was additionally purified by crystallization from methanol. Yield: 36 mg (25.3%); light purple solid; mp 187–190 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.20–1.27 (m, 2H, cyclopropyl-CH2), 1.28–1.35 (m, 2H, cyclopropyl-CH2), 2.28 (s, 3H, pyrr-CH3), 3.12–3.31 (m, 4H, 2 × CH2), 3.39–3.84 (s, 4H, 2 × CH2) 3.52–3.61 (m, 4H, 2 × CH2), 3.67 (t, 2H, J = 4.8 Hz, CH2), 3.78–3.92 (m, 3H, CH2, CH), 7.39 (d, 1H, J = 7.4 Hz, Ar-H), 7.70–7.83 (m, 2H, 2 × Ar-H), 7.98 (dd, 1H, J = 1.7, 8.5 Hz, Ar-H), 8.57 (s, 2H, 2 × Ar-H), 8.70 (t, 1H, J = 4.9 Hz, ArCONH), 10.73 (s, 1H, NH+), 11.68 (s, 1H, pyrr-NH/pyrr-CONH), 12.23 (s, 1H, pyrr-NH/pyrr-CONH), 14.91 (s, 1H, COOH). IR (ATR) ν [cm−¹] = 3365 (NH), 3223 (NH), 2925 (OH), 1719 (CO), 1657 (CO), 1627 (CO), 1525, 1455, 1400, 1331, 1264, 1167, 1104, 1036, 944, 913, 892, 833, 748, 702, 621, 551, 519. HRMS (ESI+) m/z for C35H35Cl2FN7O6S ([M + H]+): calculated: 770.1725, found: 770.1725, delta: −0.03 ppm. HPLC: tr 4.82 min (95.1% at 254 nm).
1-(2-(2-(4-(Benzyloxy)-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzo[d]thiazole-6-carboxamido)ethoxy)ethyl)-4-(3-carboxy-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinolin-7-yl)piperazin-1-ium chloride (11b). Synthesized according to General procedure A with 1b (40 mg, 0.084 mmol) and 10 (41 mg, 0.084 mmol) as reactants. After isolation, the product was additionally purified by washing with hot water three times and by crystallization from methanol. Yield: 3.42 mg (4.5%); light beige solid; mp > 300 °C. 1H NMR (400 MHz, DMSO-d6) δ [ppm] 1.13–1.22 (m, 4H, 2 × cyclopropyl-CH2), 2.25 (s, 3H, pyrr-CH3), 3.03 (t, 2H, J = 12.1 Hz, CH2), 3.24–3.29 (m, 2H, CH2), 3.39–3.48 (m, 4H, 2 × CH2), 3.52–3.61 (m, 4H, 2 × CH2), 3.63–3.70 (m, 2H, CH2), 3.74–3.79 (m, 1H, CH), 3.84 (s, 2H, CH2), 5.29 (s, 2H, Bn-CH2), 7.26 (d, 1H, J = 7.2 Hz, Ar-H), 7.35–7.47 (m, 3H, 3 × Ar-H), 7.48–7.55 (m, 2H, 2 × Ar-H), 7.64 (d, J = 1.5 Hz, Ar-H), 7.75 (d, 1H, J = 12.9 Hz, Ar-H), 8.12 (s, 1H, Ar-H), 8.54 (s, 1H, Ar-H), 8.64 (s, 1H, ArCONH), 10.12 (s, 1H, NH+), 11.80 (s, 1H, pyrr-NH/pyrr-CONH), 11.92 (s, 1H, pyrr-NH/pyrr-CONH), 14.87 (s, 1H, COOH). HRMS (ESI+) m/z for C42H41Cl2FN7O7S ([M + H]+): calculated: 876.2144, found: 876.2135, delta: −1.00 ppm. HPLC: tr 5.54 min (93.1% at 254 nm).
Author Contributions
Conceptualization, L.P.M. and T.T.; methodology, M.D., Ž.S., J.I., N.Z., A.Z., P.É.S., A.N., G.D., T.R., C.P., D.K., L.P.M. and T.T.; software, T.T.; validation, M.D., P.É.S., A.N., G.D., T.R. and T.T.; investigation, M.D., Ž.S., P.É.S., G.D. and T.R.; writing—original draft preparation, M.D. and T.T.; writing—review and editing, M.D., T.T. and L.P.M.; visualization, M.D. and T.T.; supervision, L.P.M. and T.T.; funding acquisition, D.K. and C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Slovenian Research Agency (Grant No. P1-0208) and supported by the following research grants: European Research Council H2020-ERC-2014-CoG 648364—Resistance Evolution (C.P.); ‘Célzott Lendület’ Programme of the Hungarian Academy of Sciences LP-2017–10/2017 (C.P.); ‘Élvonal’ KKP 126506 (C.P.), GINOP-2.3.2–15–2016–00014 (EVOMER, for C.P.), GINOP-2.3.2–15–2016–00020 (MolMedEx TUMORDNS) and GINOP-2.3.3–15–2016–00001, EFOP 3.6.3-VEKOP-16-2017-00009 (P.Sz, T.R.), UNKP-20-3 New National Excellence Programme of the Ministry for Innovation and Technology (P.Sz.), and a PhD Fellowship from the Boehringer Ingelheim Fonds (A.N.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available at www.mdpi.com/xxx/s1.
Conflicts of Interest
The authors declare the following financial interests/personal relationships which might be considered as potential competing interests: a PCT patent application (New class of DNA gyrase and/or topoisomerase IV inhibitors with activity against gram-positive and gram-negative bacteria: PCT/EP2019/073412073412073412) has been filed by Tomašič, N.T., Zidar, M., Durcik, J., Ilaš, A., Zega, C., Cruz, PD., Tammela, C., Pál, A., Nyerges, D., Kikelj, L., Mašič, P. The funders had no role in the design of the study, in the collection, analyses, or interpretation of the data, in the writing of the manuscript, or in the decision to publish the results.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Silver L.L. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 2007;6:41–55. doi: 10.1038/nrd2202. [DOI] [PubMed] [Google Scholar]
- 2.Mayer C., Janin Y.L. Non-quinolone inhibitors of bacterial type IIA topoisomerases: A feat of bioisosterism. Chem. Rev. 2014;114:2313–2342. doi: 10.1021/cr4003984. [DOI] [PubMed] [Google Scholar]
- 3.Tomašić T., Mašič L.P. Prospects for developing new antibacterials targeting bacterial type IIA topoisomerases. Curr. Top. Med. Chem. 2014;14:130–151. doi: 10.2174/1568026613666131113153251. [DOI] [PubMed] [Google Scholar]
- 4.Champoux J.J. DNA Topoisomerases: Structure, function, and mechanism. Ann. Rev. Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 5.Hooper D.C., Jacoby G.A. Topoisomerase inhibitors: Fluoroquinolone mechanisms of action and resistance. Cold Spring Harb. Perspect. Med. 2016;6 doi: 10.1101/cshperspect.a025320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pham T.D.M., Ziora Z.M., Blaskovich M.A.T. Quinolone antibiotics. Med. Chem. Commun. 2019;10:1719–1739. doi: 10.1039/C9MD00120D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bisacchi G.S., Manchester J.I. A new-class antibacterial—Almost. Lessons in drug discovery and development: A critical analysis of more than 50 years of effort toward ATPase Inhibitors of DNA gyrase and topoisomerase IV. ACS Infect. Dis. 2015;1:4–41. doi: 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- 8.Gross C.H., Parsons J.D., Grossman T.H., Charifson P.S., Bellon S., Jernee J., Dwyer M., Chambers S.P., Markland W., Botfield M., et al. Active-site residues of Escherichia coli DNA gyrase required in coupling ATP hydrolysis to DNA supercoiling and amino acid substitutions leading to novobiocin resistance. Antimicrob. Agents Chemother. 2003;47:1037–1046. doi: 10.1128/AAC.47.3.1037-1046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Redgrave L.S., Sutton S.B., Webber M.A., Piddock L.J.V. Fluoroquinolone resistance: Mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014;22:438–445. doi: 10.1016/j.tim.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Fluoroquinolone and Quinolone Antibiotics: PRAC Recommends Restrictions on Use. [(accessed on 5 October 2019)]; Available online: https://www.ema.europa.eu/en/documents/press-release/fluoroquinolone-quinolone-antibiotics-prac-recommends-restrictions-use_en.pdf.
- 11.Kim E.S., Hooper D.C. Clinical importance and epidemiology of quinolone resistance. Infect. Chemother. 2014;46:226–238. doi: 10.3947/ic.2014.46.4.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aldred K.J., Kerns R.J., Osheroff N. Mechanism of quinolone action and resistance. Biochem. 2014;53:1565–1574. doi: 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Correia S., Poeta P., Hébraud M., Capelo J.L., Igrejas G. Mechanisms of quinolone action and resistance: Where do we stand? J. Med. Microbiol. 2017;66:551–559. doi: 10.1099/jmm.0.000475. [DOI] [PubMed] [Google Scholar]
- 14.Collin F., Karkare S., Maxwell A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011;92:479–497. doi: 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomašič T., Katsamakas S., Hodnik Ž., Ilaš J., Brvar M., Solmajer T., Montalvão S., Tammela P., Banjanac M., Ergović G., et al. Discovery of 4,5,6,7-Tetrahydrobenzo[1,2- d ]thiazoles as Novel DNA gyrase inhibitors targeting the ATP-binding site. J. Med. Chem. 2015;58:5501–5521. doi: 10.1021/acs.jmedchem.5b00489. [DOI] [PubMed] [Google Scholar]
- 16.Zidar N., Macut H., Tomašič T., Brvar M., Montalvão S., Tammela P., Solmajer T., Peterlin Mašič L., Ilaš J., Kikelj D. N -Phenyl-4,5-dibromopyrrolamides and N -Phenylindolamides as ATP competitive DNA gyrase B inhibitors: Design, synthesis, and evaluation. J. Med. Chem. 2015;58:6179–6194. doi: 10.1021/acs.jmedchem.5b00775. [DOI] [PubMed] [Google Scholar]
- 17.Gjorgjieva M., Tomašič T., Barančokova M., Katsamakas S., Ilaš J., Tammela P., Peterlin Mašič L., Kikelj D. Discovery of benzothiazole scaffold-based DNA gyrase B inhibitors. J. Med. Chem. 2016;59:8941–8954. doi: 10.1021/acs.jmedchem.6b00864. [DOI] [PubMed] [Google Scholar]
- 18.Durcik M., Lovison D., Skok Ž., Durante Cruz C., Tammela P., Tomašič T., Benedetto Tiz D., Draskovits G., Nyerges Á., Pál C., et al. New N-phenylpyrrolamide DNA gyrase B inhibitors: Optimization of efficacy and antibacterial activity. Eur. J. Med. Chem. 2018;154:117–132. doi: 10.1016/j.ejmech.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomašič T., Barančoková M., Zidar N., Ilaš J., Tammela P., Kikelj D. Design, synthesis, and biological evaluation of 1-ethyl-3-(thiazol-2-yl)urea derivatives as Escherichia coli DNA gyrase inhibitors. Arch. Pharm. 2018;351:1700333. doi: 10.1002/ardp.201700333. [DOI] [PubMed] [Google Scholar]
- 20.Fois B., Skok Ž., Tomašič T., Ilaš J., Zidar N., Zega A., Peterlin Mašič L., Szili P., Draskovits G., Nyerges Á. Dual Escherichia coli DNA Gyrase A and B Inhibitors with Antibacterial Activity. Chem. Med. Chem. 2020;15:265–269. doi: 10.1002/cmdc.201900607. [DOI] [PubMed] [Google Scholar]
- 21.Nyerges A., Tomašič T., Durcik M., Revesz T., Szili P., Draskovits G., Bogar F., Skok Ž., Zidar N., Ilaš J. Rational design of balanced dual-targeting antibiotics with limited resistance. PLoS Biol. 2020;18:e3000819. doi: 10.1371/journal.pbio.3000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rice L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008;197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 23.Vanden Broeck A., Lotz C., Ortiz J., Lamour V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019;10:4935. doi: 10.1038/s41467-019-12914-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan P.F., Srikannathasan V., Huang J., Cui H., Fosberry A.P., Gu M., Hann M.M., Hibbs M., Homes P., Ingraham K., et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015;6:10048. doi: 10.1038/ncomms10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The PyMOL Molecular Graphics System, Version 1.8. Schrödinger, LLC.; New York, NY, USA: 2014. [Google Scholar]
- 26.Durcik M., Tammela P., Barančoková M., Tomašič T., Ilaš J., Kikelj D., Zidar N. Synthesis and Evaluation of N-Phenylpyrrolamides as DNA Gyrase B Inhibitors. Chem. Med. Chem. 2018;13:186–198. doi: 10.1002/cmdc.201700549. [DOI] [PubMed] [Google Scholar]
- 27.Nyerges Á., Csörgő B., Nagy I., Bálint B., Bihari P., Lázár V., Apjok G., Umenhoffer K., Bogos B., Pósfai G. A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. Proc. Natl. Acad. Sci. USA. 2016;113:2502–2507. doi: 10.1073/pnas.1520040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szili P., Draskovits G., Révész T., Bogár F., Balogh D., Martinek T., Daruka L., Spohn R., Vásárhelyi B.M., Czikkely M., et al. Rapid evolution of reduced susceptibility against a balanced dual-targeting antibiotic through stepping-stone mutations. Antimicrob. Agents Chemother. 2019;63 doi: 10.1128/AAC.00207-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.CLSI . Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria. Clinical and Laboratory Standards Institute; Wayne, PA, USA: 2012. Approved Standard—Eighth Edition. CLSI document M11-A8. [Google Scholar]
- 31.Baker K.R., Sigurðardóttir H.H., Jana B., Guardabassi L. Cephem potentiation by inactivation of nonessential genes involved in cell wall biogenesis of β-lactamase-producing Escherichia coli. Antimicrob. Agents Chemother. 2017;61 doi: 10.1128/AAC.01773-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pourahmad Jaktaji R., Jazayeri N. Expression of acrA and acrB Genes in Esherichia coli Mutants with or without marR or acrR Mutations. Iran. J. Basic Med. Sci. 2013;16:1254–1258. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available at www.mdpi.com/xxx/s1.