

Abstract
Glaucoma, a group of diseases characterized by progressive optic nerve degeneration that results in irreversible blindness, can be considered a neurodegenerative disorder of both the eye and the brain. Increasing evidence from human and animal studies have shown that glaucoma shares some common neurodegenerative pathways with Alzheimer’s disease (AD) and other tauopathies, such as chronic traumatic encephalopathy (CTE) and frontotemporal dementia. This hypothesis is based on the focal adhesion pathway hypothesis and the spreading hypothesis of tau. Not only has the Apolipoprotein E (APOE) gene been shown to be associated with AD, but also with primary open angle glaucoma (POAG). This review will highlight the relevant literature in the past 20 years from PubMed that show the pathogenic overlap between POAG and AD. Neurodegenerative pathways that contribute to transsynaptic neurodegeneration in AD and other tauopathies might also be similar to those in glaucomatous neurodegeneration.
Keywords: primary open-angle glaucoma, tauopathy, amyloid precursor protein, phosphorylated tau, Alzheimer’s disease
Introduction
Intraocular pressure (IOP) is the only known modifiable major risk factor for glaucoma, yet progression of visual loss remains inevitable in a subgroup of these patients even when conventional medical and surgical therapies optimize the IOP. The non-IOP factors contributing to visual loss include neuroinflammation, oxidative stress, dysregulation of calcium-dependent processes, defective autophagy, reactive gliosis, translaminar cribrosa pressure differences, and possibly the spreading of misfolded proteins.1 Some neurodegenerative pathways in primary open angle glaucoma (POAG) appear to overlap with those in Alzheimer’s disease (AD) and other tauopathies, such as chronic traumatic encephalopathy (CTE). Synaptic dysfunction and synaptic remodeling have recently been implicated as key pathogenic mechanisms in Alzheimer’s disease (AD).2 Two prevailing possible explanations for the synaptic dysfunction in AD and other tauopathies are the focal adhesion circular pathway hypothesis and the spreading hypothesis of tau.
Several genes involved in regulating the metabolic pathways of amyloid precursor protein (APP) and amyloid-beta (Aβ) also control the post-translational modification of tau protein species.2,3 Some AD genes, such as Apolipoprotein E (APOE), have been shown to increase the risk for POAG.4,5 Similar to the chronic insult of repetitive head trauma in chronic traumatic encephalopathy (CTE),3 recurrent IOP elevations might cause repetitive compressive compromise to the optic nerve head. These repetitive forces to the optic nerve head might potentially lead to tau accumulation, altered phosphorylation, and mis-sorting in glaucomatous neurodegeneration. Since transsynaptic neurodegeneration has been hypothesized to occur through the spreading of tau in AD, it might also explain the common neuropathologic involvement of the eye and brain in glaucomatous neurodegeneration.
The Focal Adhesion Circular Pathway Hypothesis in Synaptic Dysfunction
The classic amyloid cascade hypothesis proposed that defective APP metabolism leads to increased Aβ production, in which Aβ oligomers incite the spreading of tau from one neuron to another to cause synaptic and neuritic dysfunction. This linear model of the amyloid cascade hypothesis has recently taken a back seat to a vicious circle where dysfunctional metabolic pathways involving APP, tau, Aβ, and synaptic regulation revolve around the “focal adhesion core” (Figure 1).2,5 The focal adhesion pathway modulates the filamentous actin network by regulating actin-binding proteins, such as cofilin, that control dendritic spine morphology. This pathway depends on integrin to remodel the dendritic spine morphology for synaptic plasticity.2 This circular model takes into account the findings from genome-wide association studies, that genetic risk factors influence up to 80% of attributable risk in the more common forms of AD.6 The dysregulation of synaptic function and downstream cellular signaling through the focal adhesion circular pathway may be an important part of the pathogenesis of AD. The primary dysfunction in AD is located at the synapse where increased oligomeric amyloid-beta peptide causes N-methyl-D-aspartate (NMDA) receptor-dependent synaptic depression and loss of dendritic spines. Cell-surface APP is required for the regulation of normal synaptic function and is located in the focal adhesion complex where it recruits various intracellular proteins to modulate integrin signaling.7 APP interacts with integrin for the development of neurite outgrowth and serves as a synaptic adhesion molecule at the pre- and post-synaptic membranes for the proper connection with other synapses; and for the development of normal synaptic morphology and spine density.7 Therefore, cell-surface APP located in the focal adhesion complex serves a physiologic function in maintaining normal synaptic function, whereas the accumulated oligomeric amyloid-beta peptides in this complex can lead to synaptic loss.
Figure 1.

The focal adhesion complex regulates the actin cytoskeleton and downstream cell signaling pathways, such as in the spread of tau pathology. Integrin, amyloid precursor protein (APP), and receptor tyrosine kinase interact at the cell surface membrane to modulate cell adhesion. The genes for integrin, kindlin2, and CD2AP proteins regulate both APP metabolism and tau metabolism, as indicated by dashed outlines; whereas the genes for Cass4 and Pyk2 proteins are involved in only tau-related pathways and the gene for APP only in APP-related pathways. The genes for proteins inside the yellow-colored shapes are considered risk factors for AD. Adapted from Dourlen P, Kilinc D, Malmanche Net al The new genetic landscape of Alzheimer’s disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol 138, 221–236 (2019). Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).2
Any of the AD genes regulating APP metabolism, such as FERMT2, Cass4, CD2AP, and the ones regulating the actin cytoskeleton network, such as Kindlin-2, PUk2, CD2AP, and BIN1, can disrupt APP function and/or its interaction along the focal adhesion pathway.2 Amyloid-beta fibrils have been shown to activate integrin signaling to increase N-methyl-d-aspartate receptor (NMDAR) sensitivity to ultimately mediate Aß- induced neurotoxicity in hippocampal neurons.8 The scaffolding protein Ran BP9, that dephosphorylates cofilin, is a key regulator of actin filament dynamics, and it has been shown to lead to the formation of cofilin-actin rods in distal dendrites of synapses.9,10 Ran BP9 simultaneously increases APP and ß1-integrin endocytosis to cause elevated Aß production that then disrupts the focal adhesion pathway.11 This evidence supports the notion that cell surface APP plays an important role in synaptic function. Although microglia have been shown to form a protective barrier around amyloid deposits by compacting the fibrils into a less toxic form,12 microglial dysfunction can also influence the focal adhesion pathway through disrupted Aβ clearance, leading to increased toxicity in synapses and early synaptic loss/remodeling. The focal adhesion pathway does not preclude the spreading hypothesis, which might involve a downstream trigger of tau-related excitotoxicity causing synaptic failure. Because the focal adhesion pathway is reiterative, it appears to recapitulate the vicious cycle of neurodegeneration involving the dysregulation of APP and tau.
The Spreading Hypothesis of Tau in Synaptic Dysfunction
Tau is the major microtubule-associated protein expressed in neurons and retinal ganglion cells (RGCs) that is regulated by the microtubule-associated protein tau (MAPT) gene on the long arm of chromosome 17q21; it has 16 exons.13 Post-translational alternative splicing of its mRNA produces six tau isoforms in the adult human brain.13–15 The normal physiological function of tau is to promote the assembly of tubulin to form microtubules and to help stabilize microtubule polymers involved in intracellular axonal transport.16,17 The spreading hypothesis of tau in AD proposes that tau undergoes post-translational modifications, such as hyperphosphorylation, which is catalyzed by Pyk2 (tyrosine kinase), one of the proteins in the focal adhesion complex,2 to cause its detachment from microtubules that then disassemble. Within the focal adhesion circular pathway, it is postulated that APP interacts with phosphorylated tau (p-tau) to initiate the process of taumisfolding and aggregation that then leads to increased NMDAR sensitivity, followed by dendritic spine degradation.2 The p-tau is released from the dendrites into the extracellular space.18 The exact mechanism of abnormal tau propagation amongst neurons in the CNS is still not well understood at this time.19 Whether tau is spread from one neuron to another in POAG or in AD remains unproven.
The Role of Apolipoprotein E (APOE) Gene in Primary Open-Angle Glaucoma (POAG)
Genome-wide association studies (GWAS) have shown that the APOE gene ε2/ε3/ε4 polymorphism has been associated with the focal adhesion pathway in AD. Although not all the AD genes participating in the focal adhesion pathway model have been shown to increase the risk of POAG, the APOE ε4ε4 has been significantly associated with POAG. In a meta-analysis of fifteen studies including 2700 cases and 2365 controls from the literature since January 2014, the APOE ε4ε4 genotype was found to be significantly associated with an increased risk of POAG in Asians, but not in Caucasians (P = 0.003).5 Apolipoprotein E plays a key role in lipid metabolism, cholesterol transport, protein synthesis, tissue repair, cell growth and differentiation, and immune regulation.20–22 Apolipoprotein E binds to the low-density lipoprotein receptor to regulate the clearance of extracellular Aβ across the blood-brain barrier and the blood-ocular barrier. An increased Aß deposition in the cerebral blood vessels and plaques has been shown on histopathology in AD brains with the APOE ε4ε4 genotype.23 A more recent study showed the opposite finding, such that APOE ε4 decreased the risk of POAG, while it increased the risk of AD. These conflicting findings regarding APOE ε4 as a risk factor for POAG were thought to be attributed to the over-representation of normal tension glaucoma (NTG) in previous studies with mainly Asian subjects.24
Based on another meta-analysis of 108 case control studies with 35,389 POAG patients and 51,742 controls, the APOE gene was one of the 12 genes associated with an elevated risk of POAG,25 particularly APOE (−219G) with more optic nerve damage. APOE (−491T) also interacts with the myocilin (MYOC) promoter (−1000G) which is associated with an increased risk of ocular hypertension in POAG. Based on the pooled data, the rs449647 of the APOE gene had a significant association with POAG in the allelic, homozygote, heterozygote, and dominant comparisons. In a more recent GWAS study on 2320 patients of African ancestry with POAG and on 2121 without POAG,4 the Aß A4 precursor protein-binding family B member 2 (APBB2), located on chromosome 4 with the variant rs59892895T>C, was significantly associated with POAG (P = 2 x 10−8). Each copy of the rs59892895*C risk allele, most frequently found in individuals of African ancestry compared to those of European or Asian ancestry, was associated with an increased risk of POAG. The APBB2 gene is one of the genes regulating the processing of APP. Increased APBB2 gene expression was related to increased ß-amyloid plaque deposition in POAG human retinal and primary visual cortex tissues. Therefore, defective clearance of Aß in the optic nerve might contribute to the development of POAG.
Evidence for Abnormal Metabolism of Amyloid-Beta (Aß) and Tau in Glaucomatous Neurodegeneration
Recent epidemiologic studies have reported a potential association between glaucoma and AD.26–29 These studies support that glaucoma and AD might share similar pathophysiologic pathways in the metabolism of Aß and tau protein species. The histopathological defining features of AD are extracellular Aß plaques and intracellular neurofibrillary tangles consisting of phosphorylated tau (ptau).30 In glaucoma animal models, Aß has been shown to play a role in RGC apoptosis.31 Not only have ptau deposits been demonstrated in human glaucomatous retinas,32 but further evidence of Aß deposits in the retina of POAG patients have also been shown to be consistent with decreased vitreous Aß levels.33
Abnormal tau deposition in the retina has been found in 11 enucleated human eyes with a history of uncontrolled primary and secondary open-angle glaucoma compared to 10 age-matched controls.32 The immunofluorescent intensity for hyperphosphorylated tau, in the form of AT8 (phosphor-epitope at serine 202) was significantly higher in the outer region of the inner nuclear layer compared to controls. The optic nerve heads, however, did not show evidence of AT8 immunoreactivity. Normal tau immunoreactivity was absent in the glaucomatous retina, whereas it was present in all the control eyes. Normal tau was seen in the inner nuclear, inner plexiform layers, and less in RGCs and the retinal nerve fiber layer.32
Further evidence of abnormal beta-amyloid and abnormal tau accumulation in eyes have been demonstrated in animal models of ocular hypertension. In a rhesus monkey model of glaucoma,34 both eyes were induced to have elevated intraocular pressure by laser photocoagulation, and extracellular amyloid-beta 1–42 and intracellular abnormal phosphorylated tau (AT8) deposits were observed in the lateral genicular nucleus (LGN). More neurofibrillary tangles and amyloid plaques, like those in AD, were seen in the LGN than in the primary visual cortex, V1, which could imply that the disease progresses transsynaptically from the anterior to posterior visual pathways.34 In another study by Chiasseu, M. et al, tau accumulation, altered phosphorylation, and tau mis-sorting were shown in a rat glaucoma model.35 Like AD, glaucoma, and other tauopathies that progress with advancing age, tau accumulation in the rat retina increased with age and ocular hypertension,36,37 as compared with age-matched controls. It is known that different types of oxidative stress can lead to hyper-, hypo-, or de-phosphorylation of tau species.38 As shown in AD and other tauopathies, the ocular hypertension rat models demonstrate that phosphorylation of tau residues S396 and S404 is increased, but S199 phosphorylation is decreased, relative to the total tau levels in the retina.3 Chiasseu et al33 also showed that tau monomers likely converted to oligomers in the glaucomatous retinas because there was no evidence of transcriptional upregulation by increased mRNAs for these oligomers. Without any increase in retinal tau mRNA, they further hypothesized that tau accumulation in non-RGCs could be due to impaired autophagy or proteasomal degradation and/or spreading from one cell to another. Tau oligomers, rather than tau monomers and neurofibrillary tangles, were implicated as the main toxic forms that initiate and propagate glaucomatous degeneration.33,39,40 Like AD and other tauopathies, considerable tau mis-sorting was also observed in these glaucomatous rat retinas. The abnormal forms of tau were found in RGC dendrites and not in axons, suggesting that the abnormal tau was mis-sorted from axons to dendrites in the glaucomatous rat retinas, instead of anterogradely from RGC somas to the axons.41 It has also been hypothesized that impaired anterograde axonal transport could lead to tau accumulation in dendrites, where it could interact with postsynaptic protein Fyn and with A-beta to mediate excitotoxicity in the microtubules.42 These proposed neurodegenerative pathways are consistent with a previous study showing that elevated IOP can initiate damage to RGC dendrites via tau mis-sorting.43 Other studies also support the idea that remodeling of the RGC dendritic circuitry and axonal loss and synaptic dysfunction precedes RGC death in glaucoma.44,45 It, therefore, appears that tau dysregulation in these experimental glaucomatous models might recapitulate some of the key common pathological features in human glaucoma, AD, and other tauopathies.
Retinotopic Transsynaptic Neurodegeneration in Glaucoma
Elevated IOP is a major risk factor for the progression of glaucoma that leads to impaired axonal transport and synaptic degenerative changes that results in RGC apoptosis.46,47 In a mouse model of ocular hypertension, anterograde axonal transport deficits were observed to precede retrograde deficits located at the optic nerve head.48 Experimental glaucoma models have also demonstrated a significant correlation between mean IOP and the degree of LGN neuronal loss,49 and a higher mean IOP duration reduced the time to neuronal loss.50 CaMKII-α isoform (calcium/calmodulin-dependent kinase type II-alpha), an important post-synaptic density protein that regulates synaptic strength and the density of glutaminergic synapses, and its absence can lead to synaptic dysfunction and synaptic loss.51,52 Elevated IOP with or without optic nerve fiber loss was shown to reduce the expression of this protein in koniocellular neurons,49 leading to the theory of impaired transsynaptic changes in LGN in early glaucoma.
It has been demonstrated that atrophy of the posterior visual pathway is retinotopically consistent with the visual field defects and retinal nerve fiber layer loss in glaucoma. Examples of this anterograde transsynaptic degeneration has been demonstrated in several clinical, pathologic, and radiologic studies. In a clinicopathologic case study of a 79-year-old man with POAG, the clinical manifestations of glaucoma, which included his inferior neuroretinal rim atrophy in the optic disc and his superior visual field defect, correlated with the post-mortem pathological findings of neuronal atrophy in the intracranial optic nerves, the posterior lateral part of the LGN (Figure 2), and the primary visual cortex below the calcarine sulcus.53 Furthermore, magnetic resonance imaging (MRI) evidence of LGN atrophy was shown to correlate with neuropathologic findings showing neuronal shrinkage and loss associated with reactive astrogliosis in the LGN of experimental primate glaucoma models.50,54,55 Deafferentation of the larger nerve fibers in this glaucoma model was followed by atrophy in the relay neurons and their dendrites in both the magnocellular and parvocellular layers of the LGN.56,57 In another study of 18 patients with POAG compared with 18 age- and sex-matched healthy controls,58 ganglion cell-inner plexiform layer thinning significantly correlated with contralateral LGN volume reduction on 7-Tesla MRI.
Figure 2.
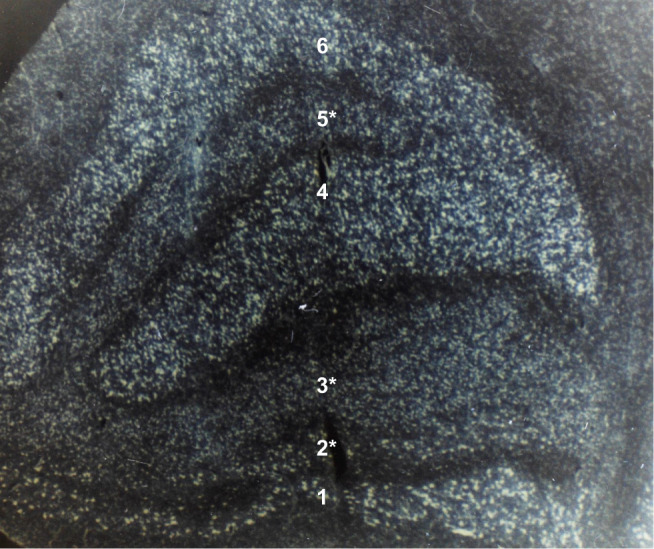
Coronal section of the left lateral geniculate nucleus in a 72-year-old patient after 8 years of optic atrophy from absolute glaucoma in the left eye. The smaller cells with less staining in ipsilateral layers 2, 3, and 5 (marked with an asterisk) represent transsynaptic atrophy. Layers 1 and 2 are the magnocellular layers. Layers 3 to 6 are the parvocellular layers. (Contrast-enhanced grayscale photo of a Nissl-stained specimen at 20x magnification; unpublished observation).
Further evidence for a transsynaptic degenerative process has been shown by the axonal degeneration of the visual pathway from the LGN to the primary visual cortex on neuroimaging studies.57 Compared to normal controls, significant atrophy of the optic tracts, optic radiations,58 and the occipital white matter59,60 in POAG patients was seen on 3-Tesla diffusion tensor imaging. The degree of visual cortical thinning correlated with the severity of RNFL thinning.61 In a more recent study using pixel-based analysis of the white matter visual pathway in 12 patients with POAG compared with 16 healthy controls, the optic tracts had significantly reduced fiber density (representing early microstructural axonal changes) and fiber-bundle cross-sectional area (representing later macrostructural axonal alterations) in the glaucomatous patients compared with controls.62 The more advanced white matter degeneration in the optic tracts of glaucomatous patients compared with that in the optic radiations implies that transsynaptic degeneration might account for the anterior to posterior visual pathway progression of glaucoma. Furthermore, functional MRI studies have also shown decreased activity in the primary visual cortex using blood-oxygen-level-dependent (BOLD) contrast.63,64
In addition to the transsynaptic degenerative changes in the brain and eye, other brain regions beyond the visual cortex have been shown to have reduced volumes. In 18 POAG patients, the left temporal and right nasal RNFL were found to be significantly thinner than the right temporal and left nasal RNFL.65 The voxel-based morphometry and diffusion tensor imaging in these patients showed significant volume reduction in the left visual cortex, the left LGN, and the intracranial portion of the optic nerves and chiasma. Fractional anisotropy was significantly decreased in the inferior frontal-occipital fasciculus, the longitudinal and inferior frontal fasciculi, the putamen, the caudate nucleus, the anterior and posterior thalamic radiations, and the anterior and posterior limbs of the internal capsule of the left hemisphere (P < 0.05).65 In 57 patients with POAG, ranging from the earliest stages to the end stages (absolute glaucoma), compared with 29 age-matched normal controls, the disruption of anatomical connectivity along the visual pathway and in the nonvisual white matter tracts was significantly correlated with the stages of POAG based on RNFL thinning and visual field defects.66 Functional connectivity was decreased in the visual and working memory networks, but it was increased in the default mode and subcortical networks. Both the anatomical connectivity and the functional connectivity were already affected in the early stage of POAG. Nonvisual regions of the brain, such as the hippocampus and the frontal cortex, also progressed with advancing POAG stage.66 These observations of concomitant non-visual pathway abnormalities in POAG patients could reflect pathology from comorbid conditions, such as AD or other types of dementias. Furthermore, the defective metabolism of APP secondary to the APOE gene mutation in POAG might account for the cognitive dysfunction that was shown to be significantly associated with POAG in African-Americans.67 Whether transsynaptic neurodegeneration in human glaucoma occurs via the focal adhesion pathway and/or the transsynaptic spreading of tau will require further detailed temporal and spatial transcriptomics and proteomic studies to decipher the regulatory pathways controlling beta-amyloid and tau metabolism in the retina and in the brain.
Conclusion
Lowering IOP remains to be the most modifiable risk factor for the successful treatment of POAG. However, future treatments will be developed to address the non-IOP-related factors of POAG to delay disease progression in the eye and in the brain. Structural and functional changes, such as inner retinal layer thinning, disc cupping, and reduced choline levels in the visual cortex from glaucomatous degeneration have been demonstrated to be already present in the brain before detectable visual field defects.68 The underlying genetic and metabolic pathways of transsynaptic neurodegeneration in glaucoma remains to be elucidated. If tauopathy is considered as a final common pathway for neurodegenerative diseases, then therapies should be aimed at these downstream pathways, in addition to the preventive therapies aimed at upstream ones. Future investigations are needed to search for more comprehensive therapeutic strategies that address not only the elevated IOP risk factors, but also the non-IOP-related genetic regulatory pathways contributing to glaucomatous neurodegeneration in the eye and in the brain. Currently, GAL-101, a novel small molecule that prevents and reverses the synaptotoxic effects of Aß 1–42, is being investigated as a promising therapy for glaucoma.69
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012;31(2):152–181. doi: 10.1016/j.preteyeres.2011.11.002 [DOI] [PubMed] [Google Scholar]
- 2.Dourlen P, Kilinc D, Malmanche N, Chapuis J, Lambert JC. The new genetic landscape of Alzheimer’s disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 2019;138(2):221–236. doi: 10.1007/s00401-019-02004-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68(7):709–735. doi: 10.1097/NEN.0b013e3181a9d503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hauser MA, Allingham RR, Aung T, Genetics of Glaucoma in People of African Descent C. Association of genetic variants with primary open-angle glaucoma among individuals with African ancestry. JAMA. 2019;322(17):1682–1691. doi: 10.1001/jama.2019.16161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Zhou YF, Zhao BY, Gu ZY, Li SL. Apolipoprotein E gene epsilon4epsilon4 is associated with elevated risk of primary open angle glaucoma in Asians: a meta-analysis. BMC Med Genet. 2014;15(1):60. doi: 10.1186/1471-2350-15-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–174. doi: 10.1001/archpsyc.63.2.168 [DOI] [PubMed] [Google Scholar]
- 7.Xu YX, Wang HQ, Yan J, Sun XB, Guo JC, Zhu CQ. Antibody binding to cell surface amyloid precursor protein induces neuronal injury by deregulating the phosphorylation of focal adhesion signaling related proteins. Neurosci Lett. 2009;465(3):276–281. doi: 10.1016/j.neulet.2009.09.022 [DOI] [PubMed] [Google Scholar]
- 8.Uhasz GJ, Barkoczi B, Vass G, et al. Fibrillar Abeta (1-42) enhances NMDA receptor sensitivity via the integrin signaling pathway. J Alzheimers Dis. 2010;19(3):1055–1067. doi: 10.3233/JAD-2010-1301 [DOI] [PubMed] [Google Scholar]
- 9.Woo JA, Jung AR, Lakshmana MK, et al. Pivotal role of the RanBP9-cofilin pathway in Abeta-induced apoptosis and neurodegeneration. Cell Death Differ. 2012;19(9):1413–1423. doi: 10.1038/cdd.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woo JA, Roh SE, Lakshmana MK, Kang DE. Pivotal role of RanBP9 in integrin-dependent focal adhesion signaling and assembly. FASEB J. 2012;26(4):1672–1681. doi: 10.1096/fj.11-194423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woo JA, Zhao X, Khan H, et al. Slingshot-cofilin activation mediates mitochondrial and synaptic dysfunction via Abeta ligation to beta1-integrin conformers. Cell Death Differ. 2015;22(6):1069–1070. doi: 10.1038/cdd.2015.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan P, Condello C, Keene CD, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;92(1):252–264. doi: 10.1016/j.neuron.2016.09.016 [DOI] [PubMed] [Google Scholar]
- 13.Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986;387(3):271–280. doi: 10.1016/0169-328x(86)90033-1 [DOI] [PubMed] [Google Scholar]
- 14.Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta. 2005;1739(2–3):91–103. doi: 10.1016/j.bbadis.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 15.Caillet-Boudin ML, Buee L, Sergeant N, Lefebvre B. Regulation of human MAPT gene expression. Mol Neurodegener. 2015;10(1):28. doi: 10.1186/s13024-015-0025-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343(6257):461–463. doi: 10.1038/343461a0 [DOI] [PubMed] [Google Scholar]
- 17.Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116(2):207–225. doi: 10.1016/0022-2836(77)90213-3 [DOI] [PubMed] [Google Scholar]
- 18.Lee S, Kim W, Li Z, Hall GF. Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. Int J Alzheimers Dis. 2012;2012:172837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14(7):399–415. doi: 10.1038/s41582-018-0013-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laws SM, Hone E, Gandy S, Martins RN. Expanding the association between the APOE gene and the risk of Alzheimer’s disease: possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J Neurochem. 2003;84(6):1215–1236. doi: 10.1046/j.1471-4159.2003.01615.x [DOI] [PubMed] [Google Scholar]
- 21.Saadat M. Apolipoprotein E (APOE) polymorphisms and susceptibility to breast cancer: a meta-analysis. Cancer Res Treat. 2012;44(2):121–126. doi: 10.4143/crt.2012.44.2.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- 23.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(20):9649–9653. doi: 10.1073/pnas.90.20.9649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margeta MA, Letcher SM, Igo RP Jr, et al. Association of APOE with primary open-angle glaucoma suggests a protective effect for APOE ε4. Invest Ophthalmol Vis Sci. 2020;61(8):3. doi: 10.1167/iovs.61.8.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen M, Yu X, Xu J, et al. Association of gene polymorphisms with primary open angle glaucoma: a systematic review and meta-analysis. Invest Ophthalmol Vis Sci. 2019;60(4):1105–1121. doi: 10.1167/iovs.18-25922 [DOI] [PubMed] [Google Scholar]
- 26.Bayer AU, Keller ON, Ferrari F, Maag KP. Association of glaucoma with neurodegenerative diseases with apoptotic cell death: alzheimer’s disease and Parkinson’s disease. Am J Ophthalmol. 2002;133(1):135–137. doi: 10.1016/S0002-9394(01)01196-5 [DOI] [PubMed] [Google Scholar]
- 27.Helmer C, Malet F, Rougier MB, et al. Is there a link between open-angle glaucoma and dementia? The three-city-alienor cohort. Ann Neurol. 2013;74(2):171–179. doi: 10.1002/ana.23926 [DOI] [PubMed] [Google Scholar]
- 28.Tamura H, Kawakami H, Kanamoto T, et al. High frequency of open-angle glaucoma in Japanese patients with Alzheimer’s disease. J Neurol Sci. 2006;246(1–2):79–83. doi: 10.1016/j.jns.2006.02.009 [DOI] [PubMed] [Google Scholar]
- 29.Kirby E, Bandelow S, Hogervorst E. Visual impairment in Alzheimer’s disease: a critical review. J Alzheimers Dis. 2010;21(1):15–34. doi: 10.3233/JAD-2010-080785 [DOI] [PubMed] [Google Scholar]
- 30.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(Suppl):S2–9. doi: 10.1038/nm1067 [DOI] [PubMed] [Google Scholar]
- 31.Zhu X, Zhou W, Cui Y, et al. Pilocarpine protects cobalt chloride-induced apoptosis of RGC-5 cells: involvement of muscarinic receptors and HIF-1 alpha pathway. Cell Mol Neurobiol. 2010;30(3):427–435. doi: 10.1007/s10571-009-9467-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta N, Fong J, Ang LC, Yucel YH. Retinal tau pathology in human glaucomas. Can J Ophthalmol. 2008;43(1):53–60. doi: 10.3129/i07-185 [DOI] [PubMed] [Google Scholar]
- 33.Yoneda S, Hara H, Hirata A, Fukushima M, Inomata Y, Tanihara H. Vitreous fluid levels of beta-amyloid((1-42)) and tau in patients with retinal diseases. Jpn J Ophthalmol. 2005;49(2):106–108. doi: 10.1007/s10384-004-0156-x [DOI] [PubMed] [Google Scholar]
- 34.Yan Z, Liao H, Chen H, et al. Elevated intraocular pressure induces amyloid-beta deposition and tauopathy in the lateral geniculate nucleus in a monkey model of glaucoma. Invest Ophthalmol Vis Sci. 2017;58(12):5434–5443. doi: 10.1167/iovs.17-22312 [DOI] [PubMed] [Google Scholar]
- 35.Chiasseu M, Cueva Vargas JL, Destroismaisons L, Vande Velde C, Leclerc N, Di Polo A. Tau accumulation, altered phosphorylation, and missorting promote neurodegeneration in glaucoma. J Neurosci. 2016;36:5785–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leger F, Fernagut PO, Canron MH, et al. Protein aggregation in the aging retina. J Neuropathol Exp Neurol. 2011;70(1):63–68. doi: 10.1097/NEN.0b013e31820376cc [DOI] [PubMed] [Google Scholar]
- 37.Ghoshal N, Garcia-Sierra F, Wuu J, et al. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp Neurol. 2002;177:475–493. [DOI] [PubMed] [Google Scholar]
- 38.Kuszczyk M, Gordon-Krajcer W, Lazarewicz JW. Homocysteine-induced acute excitotoxicity in cerebellar granule cells in vitro is accompanied by PP2A-mediated dephosphorylation of tau. Neurochem Int. 2009;55:174–180. [DOI] [PubMed] [Google Scholar]
- 39.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012;26(5):1946–1959. doi: 10.1096/fj.11-199851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patterson KR, Remmers C, Fu Y, et al. Characterization of prefibrillar tau oligomers in vitro and in alzheimer disease. J Biol Chem. 2011;286(26):23063–23076. doi: 10.1074/jbc.M111.237974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scholz T, Mandelkow E. Transport and diffusion of Tau protein in neurons. Cell Mol Life Sci. 2014;71(16):3139–3150. doi: 10.1007/s00018-014-1610-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nilsen LH, Rae C, Ittner LM, Gotz J, Sonnewald U. Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J Cereb Blood Flow Metab. 2013;33(5):684–691. doi: 10.1038/jcbfm.2012.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agostinone J, Di Polo A. Retinal ganglion cell dendrite pathology and synapse loss: implications for glaucoma. Prog Brain Res. 2015;220:199–216. [DOI] [PubMed] [Google Scholar]
- 44.Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171(2):313–325. doi: 10.1083/jcb.200506099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Della Santina L, Inman DM, Lupien CB, Horner PJ, Wong RO. Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J Neurosci. 2013;33(44):17444–17457. doi: 10.1523/JNEUROSCI.5461-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dandona L, Hendrickson A, Quigley HA. Selective effects of experimental glaucoma on axonal transport by retinal ganglion cells to the dorsal lateral geniculate nucleus. Invest Ophthalmol Vis Sci. 1991;32(5):1593–1599. [PubMed] [Google Scholar]
- 47.Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012;35(1):153–179. doi: 10.1146/annurev.neuro.051508.135728 [DOI] [PubMed] [Google Scholar]
- 48.Martin KR, Quigley HA, Valenta D, Kielczewski J, Pease ME. Optic nerve dynein motor protein distribution changes with intraocular pressure elevation in a rat model of glaucoma. Exp Eye Res. 2006;83:255–262. [DOI] [PubMed] [Google Scholar]
- 49.Yucel YH, Zhang Q, Weinreb RN, Kaufman PL, Gupta N. Atrophy of relay neurons in magno- and parvocellular layers in the lateral geniculate nucleus in experimental glaucoma. Invest Ophthalmol Vis Sci. 2001;42(13):3216–3222. [PubMed] [Google Scholar]
- 50.Weber AJ, Chen H, Hubbard WC, Kaufman PL. Experimental glaucoma and cell size, density, and number in the primate lateral geniculate nucleus. Invest Ophthalmol Vis Sci. 2000;41:1370–1379. [PubMed] [Google Scholar]
- 51.Hell JW. CaMKII: claiming center stage in postsynaptic function and organization. Neuron. 2014;81(2):249–265. doi: 10.1016/j.neuron.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamagata Y, Kobayashi S, Umeda T, et al. Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci. 2009;29(23):7607–7618. doi: 10.1523/JNEUROSCI.0707-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gupta N, Ang LC. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex. Br J Ophthalmol. 2006;90(6):674–678. doi: 10.1136/bjo.2005.086769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yucel YH, Gupta N, Zhang Q, Mizisin AP, Kalichman MW, Weinreb RN. Memantine protects neurons from shrinkage in the lateral geniculate nucleus in experimental glaucoma. Arch Ophthalmol. 2006;124(2):217–225. doi: 10.1001/archopht.124.2.217 [DOI] [PubMed] [Google Scholar]
- 55.Shimazawa M, Tomita G, Taniguchi T, et al. Morphometric evaluation of changes with time in optic disc structure and thickness of retinal nerve fibre layer in chronic ocular hypertensive monkeys. Exp Eye Res. 2006;82(3):427–440. doi: 10.1016/j.exer.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 56.Ly T, Gupta N, Weinreb RN, Kaufman PL, Yucel YH. Dendrite plasticity in the lateral geniculate nucleus in primate glaucoma. Vision Res. 2011;51(2):243–250. doi: 10.1016/j.visres.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yucel YH, Zhang Q, Weinreb RN, Kaufman PL, Gupta N. Effects of retinal ganglion cell loss on magno-, parvo-, koniocellular pathways in the lateral geniculate nucleus and visual cortex in glaucoma. Prog Retin Eye Res. 2003;22(4):465–481. doi: 10.1016/S1350-9462(03)00026-0 [DOI] [PubMed] [Google Scholar]
- 58.Chen Z, Lin F, Wang J, et al. Diffusion tensor magnetic resonance imaging reveals visual pathway damage that correlates with clinical severity in glaucoma. Clin Exp Ophthalmol. 2013;41(1):43–49. doi: 10.1111/j.1442-9071.2012.02832.x [DOI] [PubMed] [Google Scholar]
- 59.Lu P, Shi L, Du H, et al. Reduced white matter integrity in primary open-angle glaucoma: a DTI study using tract-based spatial statistics. J Neuroradiol. 2013;40(2):89–93. doi: 10.1016/j.neurad.2012.04.001 [DOI] [PubMed] [Google Scholar]
- 60.Bogorodzki P, Piatkowska-Janko E, Szaflik J, Szaflik JP, Gacek M, Grieb P. Mapping cortical thickness of the patients with unilateral end-stage open angle glaucoma on planar cerebral cortex maps. PLoS One. 2014;9(4):e93682. doi: 10.1371/journal.pone.0093682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu L, Xie B, Yin X, et al. Reduced cortical thickness in primary open-angle glaucoma and its relationship to the retinal nerve fiber layer thickness. PLoS One. 2013;8(9):e73208. doi: 10.1371/journal.pone.0073208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haykal S, Curcic-Blake B, Jansonius NM, Cornelissen FW. Fixel-based analysis of visual pathway white matter in primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2019;60(12):3803–3812. doi: 10.1167/iovs.19-27447 [DOI] [PubMed] [Google Scholar]
- 63.Duncan RO, Sample PA, Weinreb RN, Bowd C, Zangwill LM. Retinotopic organization of primary visual cortex in glaucoma: a method for comparing cortical function with damage to the optic disk. Invest Ophthalmol Vis Sci. 2007;48(2):733–744. doi: 10.1167/iovs.06-0773 [DOI] [PubMed] [Google Scholar]
- 64.Qing G, Zhang S, Wang B, Wang N. Functional MRI signal changes in primary visual cortex corresponding to the central normal visual field of patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2010;51(9):4627–4634. doi: 10.1167/iovs.09-4834 [DOI] [PubMed] [Google Scholar]
- 65.Zikou AK, Kitsos G, Tzarouchi LC, Astrakas L, Alexiou GA, Argyropoulou MI. Voxel-based morphometry and diffusion tensor imaging of the optic pathway in primary open-angle glaucoma: a preliminary study. AJNR Am J Neuroradiol. 2012;33(1):128–134. doi: 10.3174/ajnr.A2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frezzotti P, Giorgio A, Toto F, De Leucio A, De Stefano N. Early changes of brain connectivity in primary open angle glaucoma. Hum Brain Mapp. 2016;37:4581–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCoskey M, Addis V, Goodyear K, et al. Association between primary open-angle glaucoma and cognitive impairment as measured by the montreal cognitive assessment. Neurodegener Dis. 2018;18(5–6):315–322. doi: 10.1159/000496233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murphy MC, Conner IP, Teng CY, et al. Retinal structures and visual cortex activity are impaired prior to clinical vision loss in glaucoma. Sci Rep. 2016;6(1):31464. doi: 10.1038/srep31464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rammes G, Parsons CG. The Abeta aggregation modulator MRZ-99030 prevents and even reverses synaptotoxic effects of Abeta1-42 on LTP even following serial dilution to a 500:1 stoichiometric excess of Abeta1-42, suggesting a beneficial prion-like seeding mechanism. Neuropharmacology. 2020;179:108267. doi: 10.1016/j.neuropharm.2020.108267 [DOI] [PubMed] [Google Scholar]