

The obligate intracellular bacterium Chlamydia muridarum can colonize the mouse colon for a long period, but a gamma interferon (IFN-γ)-susceptible mutant clone fails to do so. Nevertheless, the mutant’s colonization is rescued in mice deficient in interleukin-7 receptor (IL-7R) (lacking both lymphocytes and innate lymphoid cells [ILCs]) or IFN-γ but not in mice lacking recombination-activated gene 1 (Rag1−/− mice) (lacking adaptive immunity lymphocytes), indicating a critical role of ILC-derived IFN-γ in regulating chlamydial colonization.
KEYWORDS: colon, ex-ILC3s, Chlamydia, spreading, IFN-γ, adoptive transfer, colon, innate lymphoid cells
ABSTRACT
The obligate intracellular bacterium Chlamydia muridarum can colonize the mouse colon for a long period, but a gamma interferon (IFN-γ)-susceptible mutant clone fails to do so. Nevertheless, the mutant’s colonization is rescued in mice deficient in interleukin-7 receptor (IL-7R) (lacking both lymphocytes and innate lymphoid cells [ILCs]) or IFN-γ but not in mice lacking recombination-activated gene 1 (Rag1−/− mice) (lacking adaptive immunity lymphocytes), indicating a critical role of ILC-derived IFN-γ in regulating chlamydial colonization. In the current study, we have used an adoptive transfer approach for further characterizing the responsible ILCs. First, intestinal ILCs isolated from Rag1−/− mice were able to rescue IL-7R-deficient mice to restrict the colonization of the IFN-γ-susceptible Chlamydia muridarum mutant. Second, the responsible ILCs were localized to the intestinal lamina propria since ILCs from the lamina propria but not the intraepithelial compartment conferred the restriction. Third, lamina propria ILCs enriched for RORγt expression but not those negative for RORγt rescued the IL-7R-deficient mice to restrict mutant colonization, indicating a critical role of group 3-like ILCs (ILC3s) since RORγt is a signature transcriptional factor of ILC3s. Fourth, a portion of the ILC3s expressed IFN-γ, thus defined as ex-ILC3s, and the transfer of the ex-ILC3s conferred colon resistance to mutant Chlamydia muridarum colonization in IFN-γ-deficient mice. Finally, genetically labeled RORγt-positive (RORγt+) ILCs were able to inhibit mutant colonization. Thus, we have demonstrated that ILC3s are sufficient for regulating chlamydial colonization, laying a foundation for further revealing the mechanisms by which an obligate intracellular bacterium activates colonic ILC3s.
INTRODUCTION
Chlamydia trachomatis, a leading cause of sexually transmitted bacterial infections (1), has been detected in both the genital and gastrointestinal (GI) tracts (2–6). However, the significance of GI C. trachomatis remains unknown. Nevertheless, there is no significant association between any significant GI pathology and infection with non-lymphogranuloma venereum (LGV) serovars of C. trachomatis (7–9). The mouse-adapted species Chlamydia muridarum has been used to investigate C. trachomatis infection biology because C. muridarum both causes pathological infection in the genital tract (10–13) and develops long-lasting colonization in the colon of mice (14–16). Mouse model-based studies have revealed that GI Chlamydia may either exacerbate or prevent chlamydial pathogenicity in the genital tract depending on the tissue order of first exposure to Chlamydia. When naive mice are first exposed to Chlamydia in the GI tract, GI Chlamydia may function as an oral vaccine to prevent subsequent chlamydial infections (17, 18). However, when Chlamydia is first inoculated into the mouse lower genital tract, chlamydial organisms may both ascend to the upper genital tract (19) and spread to the GI tract (20), possibly via blood circulation (21). Once arriving in the GI tract, chlamydial organisms may induce responses that exacerbate the pathogenicity of genital Chlamydia (22).
Bacteria can establish long-lasting colonization in the GI tract (23, 24) by interacting with gut mucosal innate lymphoid cells (ILCs) (25–28). The helper-like ILCs are grouped into group 1-like ILCs (ILC1s), ILC2s, and ILC3s based on their cell surface markers, cytokine profiles, and transcriptional requirements (29, 30). ILC3s are considered the most flexible group. They contain multiple subpopulations such as lymphoid tissue inducers (LTis) that express the chemokine receptor CCR6 and the subsets negative for CCR6 with or without natural cytotoxicity receptor (NCR) (31). ILC3s are highly adaptable to local tissue environments (32). For example, CCR6-negative ILC3s can be induced to express signature molecules of ILC1s such as T-bet and gamma interferon (IFN-γ) (33, 34). The RORγt-positive (RORγt+) ILC3s that coexpress T-bet are sometimes designated ex-ILC3s. The ILC3-to-ILC1 transition subpopulations (35) have been shown to contribute to both epithelial barrier function and pathology induction in response to infections with Salmonella (34) or Toxoplasma (36). It is not clear whether chlamydial colonization of the mouse colon can also induce ex-ILC3s.
Chlamydia is an obligate intracellular bacterium that replicates inside epithelial cells. Gaining insights into chlamydial interactions with gut mucosal ILCs may reveal unique mechanisms by which an obligate intracellular bacterium interacts with ILCs and shed new light on chlamydial pathogenesis. We recently identified a mutant Chlamydia muridarum strain that was no longer able to colonize the mouse colon stably (37). However, this mutant was rescued to colonize the colon of mice deficient in IFN-γ, which led us to define the mutant as an IFN-γ-susceptible mutant. Interestingly, the IFN-γ-susceptible mutant was rescued to colonize the colon of mice deficient in interleukin-7 receptor (IL-7R) (lacking both lymphocytes and ILCs) but not mice deficient in recombination-activated gene 1 (Rag1−/− mice) (lacking lymphocytes) (38), indicating a critical role of ILCs. However, it remains unclear whether the intestinal ILCs are sufficient for restricting the colonization of the IFN-γ-susceptible mutant and whether the responsible ILCs belong to ILC3s or ex-ILC3s.
In the current study, we have used an adoptive transfer approach to address the sufficiency issue and further characterize the responsible ILCs. IL-7R- or IFN-γ-deficient mice were used as the recipients since these mice allowed the IFN-γ-susceptible Chlamydia muridarum mutant to maintain long-lasting colonization in the colon, while the intestinal ILCs harvested from Rag1−/− mice were used as donor cells since Rag1−/− mice were still able to restrict the colonization of the mutant. We found that adoptive transfer of the donor cells completely inhibited the colonization of the Chlamydia mutant in the colon of the IL-7R-deficient mice. The responsible ILCs were further identified as ILC3s localized in the mouse intestinal lamina propria. Only when the lamina propria ILCs (LP-ILCs) enriched for RORγt expression were used as donor cells were the IL-7R-deficient mice rescued to inhibit mutant Chlamydia colonization. A portion of the RORγt+ ILC3s also expressed IFN-γ, and they were able to rescue IFN-γ-deficient mice to restrict mutant colonization, suggesting that the responsible ILCs are likely to be ex-ILC3s (35). Finally, genetically labeled RORγt-expressing ILCs were sufficient for inhibiting mutant Chlamydia colonization in IFN-γ-deficient mice. Thus, we have provided the first-ever experimental evidence demonstrating that ILC3s are sufficient for regulating chlamydial colonization in the mouse colon, which has laid a foundation for further revealing the molecular interactions between intestinal ILC3s and an obligate intracellular bacterium.
RESULTS
Intestinal lamina propria ILCs are sufficient for restoring ILC-deficient mice to inhibit the colonization of an IFN-γ-susceptible mutant Chlamydia strain.
As shown in Fig. 1, the mutant Chlamydia muridarum strain (designated G28.51.1) was no longer able to establish long-lasting colonization in wild-type C57BL/6J mice following intracolonic inoculation, but its colonization was restored in IL-7R-deficient mice, which is consistent with what we recently reported (37, 38). The IL-7R-deficient mice were then used as the recipient mice to evaluate the role of ILCs in regulating chlamydial colonization. The CD45/CD90 double-positive ILC populations were isolated from the intestinal tissues of Rag1−/− mice since Rag1−/− mice were still able to restrict the colonization of the mutant Chlamydia strain. When the ILCs were used as donor cells for adoptive transfer to the IL-7R-deficient mice, the mutant Chlamydia strain was prevented from colonization, indicating that intestinal ILCs are sufficient for regulating chlamydial colonization. We then used the adoptive transfer experimental approach to characterize the responsible ILCs by comparing the roles of the ILCs from Rag1−/− mouse intestinal intraepithelial versus lamina propria compartments (Fig. 2). Only the ILCs isolated from the lamina propria but not the intraepithelial compartment successfully conferred colon resistance to colonization by the mutant Chlamydia strain in IL-7R-deficient mice.
FIG 1.

Adoptive transfer of intestinal innate lymphoid cells for restoring IL-7R-deficient mice to gain colon resistance to colonization by an IFN-γ-susceptible mutant Chlamydia strain. (a) Live lin− (negative for CD3, Ly6G/Ly6C, CD11b, CD45R/B220, and TER-119) CD45+ CD90+ cells sorted from the total cellular extract of Rag1−/− mouse intestinal tissues were defined as innate lymphoid cells. These total innate lymphoid cells were used as donor cells in the adoptive transfer experiment. The transfer was carried out twice with 4 × 106 cells each and 1 day before and 1 day after infection, respectively, as indicated at the bottom. (b to g) The IFN-γ-susceptible mutant Chlamydia strain (clone G28.51.1) was used to infect groups of C57BL/6J mice (b and c) (n = 3) and IL-7R−/− mice (d to g) (n = 4 to 5) via intracolonic inoculation at a dose of 1 × 107 IFU (inclusion-forming units). IL-7R−/− mice were transferred without (d and e) or with (f and g) total intestinal innate lymphoid cells. On days 3 and 7 and weekly thereafter after inoculation, rectal swabs were taken (b, d, and f), or on day 28, mouse tissues were harvested (c, e, and g), as indicated along the x axis, for monitoring live chlamydial organisms. The mouse tissues include stomach (Sto); the small intestinal tissues (SI) duodenum (Duo), jejunum (Jej), and ileum (Ile); the large intestinal tissues (LI) cecum (Cec), colon (Col), and rectum (Rec); as well as the extragastrointestinal tissues (Extra-GI) spleen (Spl), liver (Liv), kidney (Kid), and mesenteric lymph nodes (mLN), as indicated along the x axis. The yields of chlamydial organisms from swabs or tissues are expressed as log10 IFU per swab or tissue, as shown along the y axis. Note that the mutant Chlamydia strain failed to colonize C57BL/6J mice, but IL-7R−/− mice restored the mutant’s colonization. More importantly, adoptive transfer of total intestinal innate lymphoid cells to IL-7R−/− mice rescued large intestinal resistance to mutant Chlamydia colonization. **, P < 0.01 (by a Wilcoxon rank sum test [areas under the curve between IL-7R−/− mice with and without adoptive transfer of intestinal innate lymphoid cells]). Data were acquired from two independent experiments.
FIG 2.

Adoptive transfer of innate lymphoid cells from the intestinal lamina propria for restoring IL-7R-deficient mice to gain colon resistance to colonization by the IFN-γ-susceptible mutant Chlamydia strain. (a and b) Live lin− (negative for CD3, Ly6G/Ly6C, CD11b, CD45R/B220, and TER-119) CD45+ CD90+ cells sorted from the intestinal intraepithelial cellular extract (defined as intraepithelial innate lymphoid cells [IE-ILCs]) (a) or the lamina propria cellular extract (defined as lamina propria innate lymphoid cells [LP-ILCs]) (b) of Rag1−/− mice were used as donor cells in the adoptive transfer experiment. The transfer was carried out twice with 1 × 106 cells each and 1 day before and 1 day after infection, respectively, as indicated at the bottom. (c to j) The IFN-γ-susceptible mutant Chlamydia strain (clone G28.51.1) was used to infect groups of C57BL/6J mice (c and d) (n = 3) and IL-7R−/− mice (e to j) (n = 3 to 5) via intracolonic inoculation at a dose of 1 × 107 IFU (inclusion-forming units). IL-7R−/− mice were transferred without (e and f) or with IE-ILCs (g and h) or LP-ILCs (i and j). On days 3 and 7 and weekly thereafter after inoculation, rectal swabs were taken (c, e, g, and i), or on day 28, mouse tissues were harvested (d, f, h, and j), as indicated along the x axis, for monitoring live chlamydial organisms (see the Fig. 1 legend for tissue name abbreviations). The yields of chlamydial organisms from swabs or tissues are expressed as log10 IFU per swab or tissue, as shown along the y axis. Note that adoptive transfer of LP-ILCs but not IE-ILCs conferred large intestinal resistance to mutant Chlamydia colonization in IL-7R−/− mice. *, P < 0.05; **, P < 0.01 (by a Wilcoxon rank sum test [areas under the curve between IL-7R−/− mice transferred with IE-ILCs and those transferred with LP-ILCs]). Data were acquired from two independent experiments.
Lamina propria ILCs enriched in RORγt expression are sufficient for rescuing IL-7R-deficient mice to inhibit mutant Chlamydia colonization.
As shown in Fig. 3, the CD45/CD90 double-positive cells isolated from the Rag1−/− intestinal lamina propria comprise two close clusters of cells after excluding dead and lineage-positive (lin+) cells. The minor cluster of cells with CD45high and CD90intermediate populations were mostly negative for the ILC3-specific transcriptional factor RORγt, thus defining non-ILC3s. The major cluster of cells with CD45intermediate and CD90high populations were mostly positive for RORγt, thus defined as ILC3s. We further compared the roles of the non-ILC3s versus ILC3-enriched clusters in conferring colon resistance to mutant Chlamydia colonization in IL-7R-deficient mice (Fig. 4). We found that only the ILC3-enriched cluster but not the non-ILC3s enabled the IL-7R-deficient mice to restrict mutant Chlamydia from colonizing the colon. Thus, we have demonstrated that the ILCs responsible for regulating chlamydial colonization are ILC3s.
FIG 3.

Identification of RORγt-enriched innate lymphoid cells (ILC3s) from Rag1−/− mouse intestinal lamina propria lymphoid cells. (a to c) The intestinal lamina propria lymphoid cell extract was labeled with vital dye and various antibodies as described in Materials and Methods and analyzed for live cells (a), lineage-negative cells (b), and CD45/CD90 double-positive cells (c). FSC, forward scatter. (d and e) Two adjacent cell clusters were identified from the CD45/CD90 double-positive cell population, and they were marked as non-ILC3s (representing 14.2% of the total lineage-negative lamina propria lymphoid cells) and ILC3s (30.1%), respectively, because the non-ILC3 cluster was 93.5% negative for the ILC3-specific transcriptional factor RORγt (d), while the ILC3 cluster was 97.2% positive for RORγt (e). Thus, RORγt-positive ILC3s can be purified from the CD45/CD90 double-positive cell population after excluding dead cells and lineage-positive cells using a flow cytometry cell sorter.
FIG 4.

Adoptive transfer of ILC3-enriched cells but not non-ILC3s sorted from the Rag1−/− mouse intestinal lamina propria for restoring IL-7R-deficient mice to gain colon resistance to colonization by the IFN-γ-susceptible mutant Chlamydia strain. (a) ILC3-enriched and non-ILC3 cells were sorted from CD45+ CD90+ double-positive cells, as described in the Fig. 3 legend, as donor cells for adoptive transfer experiments. The transfer was carried out twice with 1 × 105 cells each and 1 day before and 1 day after infection, respectively, as indicated at the bottom. (b to g) The IFN-γ-susceptible mutant Chlamydia strain (clone G28.51.1) was used to infect groups of IL-7R−/− mice without (b and c) or with the transfer of non-ILC3s (d and e) or ILC3s (f and g). On days 3 and 7 and weekly thereafter after inoculation, rectal swabs were taken (b, d, and f), or on day 28, mouse tissues were harvested (c, e, and g), as indicated along the x axis, for monitoring live chlamydial organisms (see the Fig. 1 legend for tissue name abbreviations). The yields of chlamydial organisms from swabs or tissues are expressed as log10 IFU per swab or tissue, as shown along the y axis. Note that adoptive transfer of ILC3s but not non-ILC3s conferred large intestinal resistance to mutant Chlamydia colonization in IL-7R−/− mice. *, P < 0.05; **, P < 0.01 (by a Wilcoxon rank sum test [areas under the curve between IL-7R−/− mice transferred with non-ILC3s and those transferred with ILC3s]). Data were acquired from two independent experiments.
RORγt-enriched ILC3s can produce IFN-γ and restore IFN-γ-deficient mice to restrict the colonization of mutant Chlamydia.
Having demonstrated that intestinal ILC3s can confer colon resistance to the colonization of an IFN-γ-susceptible mutant in IL-7R-deficient mice, we next evaluated whether the responsible ILC3s can produce IFN-γ (Fig. 5). When the ILC3-enriched cells were stimulated in vitro with mitogen, we found that a portion of these cells produced IFN-γ. This finding is consistent with the concept that some ILC3s can be induced to produce IFN-γ, becoming so-called ex-ILC3s (35, 39). We then further evaluated whether the IFN-γ-producing ILC3s could rescue IFN-γ-deficient mice to restrict the colonization of the IFN-γ-susceptible Chlamydia mutant (Fig. 6). As a control, the IFN-γ-susceptible Chlamydia mutant was able to colonize the colon of IFN-γ-deficient mice as expected (37, 38). Importantly, after adoptive transfer, the ILC3-enriched cluster but not the non-ILC3 cluster sorted from the CD45/CD90 double-positive cells (isolated from the Rag1−/− mouse intestinal lamina propria) fully restored colon resistance to the colonization of the IFN-γ-susceptible Chlamydia mutant in IFN-γ-deficient mice. Thus, we have demonstrated that ILC3 donor cells can complement IFN-γ-deficient mice with IFN-γ for inhibiting the colonization of the IFN-γ-susceptible Chlamydia mutant.
FIG 5.
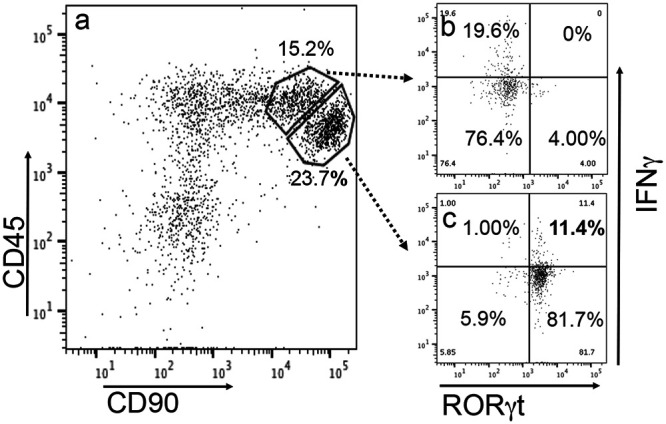
Detection of IFN-γ in RORγt-enriched ILC3s sorted from Rag1−/− intestinal lamina propria lymphoid cells. Intestinal lamina propria lymphoid cells in 96-well plates were stimulated with 50 ng/ml PMA and 1 μg/ml ionomycin for 4 h. Brefeldin A was added 2 h before cells were harvested for flow cytometry analysis. (a) CD45/CD90 double-positive cells were identified after excluding dead cells and lin+ (positive for CD3, Ly6G/Ly6C, CD11b, CD45R/B220, and TER-119) cells. (b and c) The non-ILC3 subpopulations (representing 15.2% of the total lineage-negative lamina propria lymphoid cells) and ILC3 subpopulations (23.7%) were detected for both RORγt and IFN-γ (b and c, respectively). RORγt+ IFN-γ+ cells (11.4%) were identified from the ILC3 subpopulation.
FIG 6.

Adoptive transfer of ILC3-enriched cells but not non-ILC3s sorted from the Rag1−/− mouse intestinal lamina propria for restoring IFN-γ-deficient mice to gain colon resistance to colonization by IFN-γ-susceptible mutant Chlamydia. (a) ILC3-enriched and non-ILC3 cells were sorted from live lin− (negative for CD3, Ly6G/Ly6C, CD11b, CD45R/B220, and TER-119) CD45+ CD90+ double-positive cells, as described in the Fig. 3 legend, as donor cells for adoptive transfer to IFN-γ-deficient mice. The transfer was carried out twice with 1 × 105 cells each and 1 day before and 1 day after infection, respectively, as indicated at the bottom. (b to g) The IFN-γ-susceptible mutant Chlamydia strain (clone G28.51.1) was used to infect groups of IFN-γ−/− mice without (b and c) (n = 4) or with the transfer of non-ILC3s (d and e) (n = 3) or ILC3s (f and g) (n = 3). On days 3 and 7 and weekly thereafter after inoculation, rectal swabs were taken (b, d, and f), or on day 28, mouse tissues were harvested (c, e, and g), as indicated along the x axis, for monitoring live chlamydial organisms (see the Fig. 1 legend for tissue name abbreviations). The yields of chlamydial organisms from swabs or tissues are expressed as log10 IFU per swab or tissue, as shown along the y axis. Note that adoptive transfer of ILC3s but not non-ILC3s conferred large intestinal resistance to mutant Chlamydia colonization in IFN-γ−/− mice. *, P < 0.05; **, P < 0.01 (by a Wilcoxon rank sum test [areas under the curve between IFN-γ−/− mice transferred with non-ILC3s and those transferred with ILC3s]). Data were acquired from two independent experiments.
RORγt-expressing ILC3s isolated from the intestinal lamina propria of RORγt-EGFP reporter mice can also rescue IFN-γ-deficient mice to inhibit the colonization of IFN-γ-susceptible mutant Chlamydia.
As described above, we were able to isolate ILCs highly enriched for expressing RORγt as donor cells to demonstrate the role of ILC3s in conferring colon resistance to mutant Chlamydia colonization in adoptive transfer experiments. However, the RORγt expression-enriched cells were sorted based on their unique distribution pattern in the CD45/CD90 plot but not based on their RORγt expression. It is not possible to obtain live cells with immune labeling of the ILC3-specific transcriptional factor RORγt. To overcome this difficulty and to further strengthen our conclusion on the role of RORγt+ ILC3s in mouse colon regulation of chlamydial colonization, we next isolated RORγt+ ILC3s from Rorc(γt)-EGFP reporter mice as donor cells. The reporter mice express EGFP (enhanced green fluorescent protein) under the control of the RORγt promoter (40). Thus, all RORγt-expressing cells are genetically marked with GFP. Heterozygous mice express both functional RORγt and GFP in RORγt+ cells. By taking advantage of the reporter mice, we sorted GFP-positive cells from the reporter mouse intestinal lamina propria ILCs as donor cells. Sorting was done after excluding dead cells and lineage-positive cells. As shown in Fig. 7, the IFN-γ-susceptible Chlamydia mutant failed to colonize C57BL/6J mice, while IFN-γ-deficient mice rescued mutant colonization. More importantly, adoptive transfer of GFP-positive ILCs but not GFP-negative ILCs sorted from the same reporter mouse led to restriction of IFN-γ-susceptible Chlamydia mutant colonization in IFN-γ-deficient mouse colons. Thus, we have used genetically labeled ILC3s to demonstrate that ILC3s are sufficient for regulating chlamydial colonization in the mouse colon.
FIG 7.

Adoptive transfer of RORγt GFP+ ILC3s but not RORγt GFP− non-ILC3s from the mouse intestinal lamina propria for restoring IFN-γ−/− mice to gain colon resistance to colonization by IFN-γ-susceptible mutant Chlamydia. (a) RORγt GFP+ ILC3s and RORγt GFP− non-ILC3s were sorted from intestinal lamina propria lymphoid cells after excluding dead and lin+ (positive for CD3, Ly6G/Ly6C, CD11b, CD45R/B220, and TER-119) cells as donor cells for adoptive transfer to IFN-γ-deficient mice. The transfer was carried out twice with 5 × 104 cells each and 1 day before and 1 day after infection, respectively, as indicated at the bottom. SSC, side scatter. (b to i) The IFN-γ-susceptible mutant Chlamydia strain (clone G28.51.1) was used to infect groups (n = 3) of C57BL/6J mice (b and c) or IFN-γ−/− mice without (d and e) or with transfer of RORγt GFP− non-ILC3s (f and g) or RORγt GFP+ ILC3s (h and i). On days 3 and 7 and weekly thereafter after inoculation, rectal swabs were taken (b, d, f, and h), or on day 28, mouse tissues were harvested (c, e, g, and i), as indicated along the x axis, for monitoring live chlamydial organisms (see the Fig. 1 legend for tissue name abbreviations). The yields of chlamydial organisms from swabs or tissues are expressed as log10 IFU per swab or tissue, as shown along the y axis. Note that adoptive transfer of ILC3s but not non-ILC3s conferred large intestinal resistance to mutant Chlamydia colonization in IFN-γ−/− mice. *, P < 0.05 (by a Wilcoxon rank sum test [areas under the curve between IFN-γ−/− mice transferred with RORγt GFP− non-ILC3s and those transferred with RORγt GFP+ ILC3s]). Data were acquired from two independent experiments.
DISCUSSION
In the present study, we have taken advantage of an IFN-γ-susceptible Chlamydia mutant (37, 38) for further characterizing the interactions of Chlamydia with intestinal ILCs. We have now presented evidence demonstrating that the intestinal ILCs are sufficient for inhibiting the Chlamydia mutant, and the responsible ILCs belong to ILC3s. First, adoptive transfer of total intestinal ILCs harvested from Rag1-deficient mice inhibited the colonization of the Chlamydia mutant in the colon of IL-7R-deficient mice. This observation demonstrated that the intestinal ILCs from Rag1-deficient mice are sufficient for reconstituting the colon inhibition of mutant Chlamydia and established an experimental platform for further characterizing the responsible ILCs. Second, the responsible ILCs were localized in the intestinal lamina propria but not the intraepithelial compartment. Third, only the lamina propria ILCs that express RORγt but not those that do not express RORγt were able to rescue the IL-7R-deficient mice to inhibit mutant colonization, indicating a critical role of group 3-like ILCs since RORγt is a critical transcriptional factor for ILC3s. Fourth, the RORγt+ ILC3s also enabled the IFN-γ-deficient mice to inhibit mutant Chlamydia colonization, consistent with the finding that a portion of the RORγt+ ILC3s were induced to express IFN-γ. These observations suggest that chlamydial colonization in the intestine may induce the ILC3-to-ILC1 transition, leading to the formation of ex-ILC3s. Finally, when RORγt+ ILC3s isolated from the RORγt reporter mice were transferred as donor cells to IFN-γ-deficient mice, the adoptive transfer fully restored colon resistance to mutant Chlamydia colonization. ILC3s are sufficient for regulating chlamydial colonization in the mouse colon, possibly via producing IFN-γ. However, many questions remain unanswered regarding gut ILC-Chlamydia interactions.
ILC3s have been shown to produce diverse cytokine profiles in response to local tissue environmental cues (32, 33, 35, 41). The IFN-γ-producing RORγt+ ILC3 cell subset can respond to microbial colonization (42, 43). Bacteria that can maintain long-lasting colonization in the gut become part of the gut microbiome known to confer colonization resistance to pathogenic infections (44–46). Maintaining a base level of IFN-γ in mucosal tissue is a colonization resistance mechanism (45). Since the mouse-adapted Chlamydia muridarum strain does not cause any significant pathology in the GI tract despite its long-lasting colonization in the gut (17), we hypothesize that C. muridarum may be a common commensal species in the mouse GI tract. This hypothesis is supported by observations that the IFN-γ-susceptible Chlamydia mutant is no longer able to maintain colonization in the mouse colon, while its parental wild-type Chlamydia strain can do so (37, 38). The wild-type Chlamydia strain may have fully adapted to the mouse colon environment by both inducing IFN-γ and evading the same IFN-γ. This hypothesis is consistent with the general concept that microbiota species can maintain colonization resistance and evade the same resistance. The constant level of IFN-γ may represent a critical component of colon mucosal colonization resistance to pathogens. Since the current study has demonstrated that ILC3s may represent the major cellular source of constant IFN-γ, we hypothesize that the ILC3s are a critical cellular component of intestinal colonization resistance that is strengthened during chlamydial colonization. Since the long-lasting colonization of the mouse colon by Chlamydia is nonpathological, it will be interesting to investigate whether and how chlamydial colonization may promote gut health.
We are aware of the limitations of the current study. Besides the IFN-γ-susceptible mutant used in the current study (47, 48), there are also other mutants that are susceptible to IFN-γ (49, 50). Interestingly, all IFN-γ-susceptible Chlamydia mutants identified so far carry mutations in various chromosomal genes rather than the plasmid-borne genes, suggesting that evading IFN-γ is probably a fundamental property of Chlamydia. It is worth noting that chlamydial plasmid-borne genes are also crucial for maintaining chlamydial colonization in the gut (37, 51–53). However, these plasmid gene mutants cannot be rescued to colonize the colon of IFN-γ-deficient mice, suggesting that Chlamydia may use the plasmid genes to evade IFN-γ-independent immunity in the gut. Interestingly, the plasmid gene mutants are more susceptible to antimicrobial activities in the stomach (54) and small intestine (55, 56), while the chromosomal gene mutants are more likely to be cleared in the large intestine (37, 38). We hypothesize that there may be IFN-γ-independent immunity for regulating chlamydial colonization in the small intestine, which may be evaded by chlamydial plasmid-borne genes.
MATERIALS AND METHODS
Chlamydia organisms.
The attenuated Chlamydia muridarum mutant clone G28.51.1 was used in the current study. G28.51.1 was produced previously (47, 48). Compared to the wild-type Chlamydia muridarum clone G13.32.1, G28.51.1 carries two mutations responsible for the attenuation phenotype in both genital tract pathogenicity (48, 52) and colon colonization (37, 38). These two mutations are a substitution mutation in gene tc0237, resulting in an amino acid substitution at codon 117 from glutamine (Q) to glutamic acid (E), designated mutation TC0237Q117E, and a 2nd mutation in gene tc0668, resulting in a stop codon at codon 216 originally coding for glycine (G), thus designated TC0668G216*. The chlamydial organisms were grown in HeLa cells (human cervical carcinoma epithelial cells; ATCC CCL-2) for density gradient purification into elementary bodies (EBs) and stored in aliquots at −80°C until use.
Mouse infection.
The mouse experiments were carried out according to the recommendations in the Guide for the Care and Use of Laboratory Animals endorsed by the National Institutes of Health (57). The protocol was approved by the Committee on the Ethics of Laboratory Animal Experiments of the University of Texas Health Science Center at San Antonio.
The following 5- to 7-week-old female mice used in the current study were all from Jackson Laboratories, Inc., Bar Harbor, ME: wild-type mice (C57BL/6J) (stock no. 000664), Rorc(γt)-EGFP heterozygote mice [B6.129P2(Cg)-Rorctm2Litt/J] (stock no. 007572) or mice deficient in IFN-γ (B6.129S7-Ifngtm1Ts/J) (IFN-γ−/−) (stock no. 002287), recombination-activating gene 1 (B6.129S7-Rag1tm1Mom/J) (Rag1−/−) (stock no. 002216), or the IL-7 receptor (B6.129S7-Il7rtm1Imx/J) (IL-7R−/−) (stock no. 002295). For recipient mice used for adoptive transfer experiments, all mice were inoculated with G28.51.1 EBs at 1 × 107 inclusion-forming units (IFU) per mouse via intracolonic inoculation as described previously (56). Briefly, EBs were diluted in 50 μl of SPG (220 mM sucrose, 12.5 mM phosphate, 4 mM l-glutamic acid [pH 7.5]) buffer and delivered to the colon using a straight ball-tipped needle designed for mouse oral gavage (catalog no. N-PK 020; Braintree Scientific, Inc., Braintree, MA). After inoculation, mice were monitored for live-organism shedding in rectal swabs or sacrificed for titrating live organisms in the designated organs/tissues.
Titrating live chlamydial organisms from mouse swabs and tissues.
To monitor live-organism shedding from GI tracts, rectal swabs were taken on day 3 postinoculation and weekly thereafter. Each swab was soaked in 0.5 ml of SPG buffer and vortexed with glass beads to release infectious EBs for quantitation. For titrating live chlamydial organisms from mouse tissues, various organs/tissues, as indicated for individual experiments, were harvested on the designated days after inoculation as specified for the individual experiments. Each organ or tissue segment was transferred to 0.5 ml (for each segment of the genital tract tissue) or 2 ml (for each remaining tissue/organ) of SPG buffer, followed by homogenization and brief sonication. After spinning down tissue debris, live C. muridarum organisms in the supernatants were titrated on HeLa cells in duplicate. The total number of IFU per swab or tissue was converted into log10 units for calculating the means and standard deviations across mice of the same group at each time point. The detection limits of the above-described titration method are 10 IFU per swab and 40 IFU per tissue sample. We always used 50 μl of the total volume (500 μl for swabs and 2,000 μl for tissue), starting with neat samples without dilution and then using serial dilutions. We counted the entire culture well for chlamydial inclusions when the inclusion density was low, allowing the detection of a single IFU per 50-μl sample.
Immunofluorescence assay.
An immunofluorescence assay was used to visualize and count chlamydial inclusions in the Chlamydia-infected HeLa culture, as described previously (58). Infected HeLa cells grown on coverslips were fixed with paraformaldehyde (Sigma, St. Louis, MO) and permeabilized with saponin (Sigma). After washing and blocking, the monolayers were labeled with a rabbit antichlamydial antibody (raised by immunization with C. muridarum EBs). The rabbit antibody was further probed with goat anti-rabbit IgG conjugated with Cy2 (green; Jackson ImmunoResearch Laboratories, Inc.) to visualize chlamydial inclusions, while Hoechst dye (blue; Sigma) was also added to the 2nd antibody solution for labeling nuclear DNA. The labeled cell samples were viewed under an Olympus (Melville, NY) IX-80 fluorescence microscope equipped with multiple filter sets.
Adoptive transfer.
Adoptive transfer of donor cells was applied to some groups of recipient mice. The transfer was carried out twice, each with the designated number of designated donor cells, as indicated for individual experiments. One transfer was done on the day before and the other was done on the day after infection via retro-orbital injection. After the recipient mice were intracolonically infected with Chlamydia, live chlamydial organisms were monitored in both rectal swabs and tissues, as described above.
To prepare innate lymphoid cells (ILCs) as donor cells, we used Rag1-deficient mice as donor mice since these mice lack lymphocytes. Before harvesting donor cells, the Rag1-deficient mice were intracolonically inoculated with 1 × 107 IFU of G28.51.1 for 7 days to enrich ILCs in the GI tract. The intestinal ILCs were isolated from both the intraepithelial (IE-ILCs) and lamina propria (LP-ILCs) compartments using a protocol described previously by Guo et al. (59). For the isolation of IE-ILCs, mice were sacrificed, and the intestine was taken out and placed in a 100- by 15-mm petri dish with ice-cold phosphate-buffered saline (PBS) for the removal of mesenteric materials, fat, and Peyer’s patches with dissecting scissors. The intestine was then cut open longitudinally and thoroughly washed in ice-cold PBS to remove all fecal matter. The cleaned intestine tissue was then cut into 0.5- to 1.0-cm pieces that were then transferred into a 50-ml tube with 15 ml prewarmed (at 37°C) washing buffer I (RPMI 1640 containing 3% FBS [fetal bovine serum] [catalog no. 900-108; Gemini Bio, West Sacramento, CA], 1 mM dithiothreitol [DTT], 5 mM EDTA, and 10 mM HEPES [the pH should be maintained between 7.2 and 7.4]). The tube was shaken at 250 rpm in a 37°C incubator for 30 min. It is worth pointing out that DTT should be added freshly into washing buffer I before each use, and all buffers used for isolating ILCs from now on should be kept at 37°C. The incubated tissue suspension was then filtered through a 70-μm cell strainer. The filterable solution contained IE-ILCs, which were collected while the remaining intestinal tissue pieces were shaken vigorously by vortexing (Analog vortex mixer, catalog no. 58816-121; VWR, Radnor, PA) in 7.5 ml fresh prewarmed washing buffer I for 20 s (remember that DTT should be added to buffer I only before use). The vortexed tissue mixture solution was passed through the same 70-μm cell strainer for collecting more IE-ILCs into the same IE-ILC tube. The washing buffer I vortexing-collection steps were repeated once to collect additional IE-ILCs. The filterable cell suspension in a 50-ml tube was kept on ice until further isolation and analysis.
After completing the collection of IE-ILCs, the remaining tissue pieces were washed twice each with 7.5 ml prewarmed washing buffer II (RPMI 1640 containing 10 mM HEPES) to remove EDTA. The tissue suspension was vortexed for 20 s during each wash. The supernatant from each wash was discarded. Note that the wash supernatants can also be collected into the IE-ILC tube. The washed tissue pieces were used for isolating LP-ILCs, which started with mincing the tissues into 1- to 2-mm pieces (individual tissue pieces were no longer identifiable) with dissecting scissors (it may take ∼2 min per intestine) in a sterile petri dish. The minced tissue mixture was digested in a 50-ml tube with 4 ml prewarmed digestion buffer containing RPMI 1640, 3% FBS, 10 mM HEPES, 45.9 U DNase I (catalog no. 18-047-019; Invitrogen, Carlsbad, CA), 150 μg/ml Liberase (catalog no. 5401020001; Sigma, St. Louis, MO), 10 μg/ml gentamicin (catalog no. 15710064; Gibco, Grand Island, NY), and 50 μg/ml vancomycin (catalog no. V06500-5.0; Research Products International, Mount Prospect, IL). The digestion tube was incubated at 37°C for 30 min with shaking at 200 rpm. Note that DNase I and Liberase should be added freshly immediately before use, and the optimal concentration for each of these two enzymes should be titrated in pilot experiments. After shaking incubation, the digested tissue mixture was passed through a 70-μm cell strainer, and the filterable solution that contained LP-ILCs was collected into a 50-ml tube, followed by using ∼5 ml washing buffer III (RPMI 1640 containing 3% FBS and 10 mM HEPES) to rinse the digestion tube and transferring the rinsing solution to the initial cell strainer. While the rinsing solution was passed through the cell strainer, the initial tissue sample retained in the cell strainer was gently homogenized with the flat end of a 1-ml syringe plunger. This rinsing/homogenizing/filtering cycle was repeated three more times. Note that as the cycle progresses, the tissue sample may disappear. All filterable solutions (∼24 ml in total) were collected and combined as LP-ILCs for further purification and analysis.
Both IE-ILC- and LP-ILC-containing suspensions were centrifuged at 1,500 rpm for 10 min at room temperature to pellet cells, and each pellet was resuspended in 5 ml RPMI 1640 containing 3% FBS and 10 mM HEPES. Next, 5 ml 80% Percoll was added to the cell suspension to make the final 40% Percoll solution. The 80% Percoll was diluted from 100% Percoll using sterile PBS, while 100% Percoll was prepared by adding 10 mM HEPES (using a 1 M stock solution), 10 μg/ml gentamicin (10-mg/ml stock), 50 μg/ml vancomycin (50-mg/ml stock), and 1× Hanks’ balanced salt solution (HBSS) (10× stock). The IE-ILC- and LP-ILC-containing 40% Percoll solutions were separately loaded into a 15-ml centrifuge tube containing 4 ml 80% Percoll. During sample loading, extreme care should be taken to avoid mixing the 40% Percoll layer with the 80% layer. The tubes were centrifuged at 2,500 rpm for 25 min at room temperature using a Sorvall ST 16R centrifuge (catalog no. 75004241; Thermo Fisher Scientific, Inc., Waltham, MA). Note that the centrifuge must be preset at “no break.” After centrifugation, a dense interphase that contains ILCs should be noticeable between the 40% and 80% Percoll layers if the separation is successful. The ILC-containing interphase was harvested into a new 15-ml centrifuge tube and washed using ∼10 ml RPMI 1640 containing 3% FBS and 10 mM HEPES. After centrifugation at 1,500 rpm for 10 min at room temperature, IE-ILC and LP-ILC pellets were resuspended in solutions suitable for subsequent experiments as indicated for the individual experiments. From a 7- to 8-week-old Rag1-deficient mouse, we were able to collect ∼1.5 × 107 total IE-ILCs and 3 × 106 LP-ILCs, respectively.
For experiments in which total intestinal ILCs were used as donor cells, IE-ILCs and LP-ILCs resuspended in sterile PBS were combined from the same mouse. For experiments in which sorted cells were used as donor cells, the cell pellets were resuspended in fluorescence-activated cell sorter (FACS) staining buffer. After excluding dead and lineage-positive cells (gating for live lineage-negative [lin−] cells), both the ILC3-enriched subset (CD45int CD90hi) and the non-ILC3 subset (CD45hi CD90hi) were sorted from Rag1-deficient mouse LP-ILCs (see below for flow cytometry protocols). We could typically obtain ∼5 × 106 live lin− IE-ILCs and ∼5 × 105 live lin− LP-ILCs from a single Rag1-deficient mouse. Since 20 to 30% of live lin− LP-ILCs were CD45int CD90hi, we could obtain ∼1 × 105 LP-ILC3s from a single donor mouse, which is sufficient for one adoptive transfer to one recipient mouse.
We also used genetically labeled RORγt-expressing cells as donor cells. These donor cells were harvested from Rorc(γt)-EGFP or RORγt-EGFP heterozygote mice. Although mice homozygous for the RORγt-EGFP mutant allele are defective in expressing the thymus-specific isoform RORγt, heterozygote mice maintain both functional RORγt and GFP expression in T cells. Thus, we can use GFP to sort for RORγt-expressing cells. To enrich for RORγt-GFP-positive cells in the mouse intestinal lamina propria, we inoculated G28.51.1 into RORγt-GFP mouse colon for 3 days before harvesting intestinal lamina propria lymphoid cells. Similarly, after excluding dead and lineage-positive cells, GFP+ cells were sorted from RORγt-GFP heterozygote mouse intestinal lamina propria lymphoid cells as donor cells.
Flow cytometry analysis and sorting.
For surface marker staining, each lamina propria lymphoid cell sample was resuspended in 100 μl FACS buffer containing the following antibodies: rat anti-mouse CD16/32 (to block nonspecific binding to Fc receptors) (clone 2.4G2, catalog no. BE0307; Bio X Cell, West Lebanon, NH); a rat anti-mouse lineage cocktail (conjugated with Pacific blue) (catalog no. 133310; BioLegend, Inc., San Diego, CA) consisting of rat anti-mouse CD3 (clone 17A2), rat anti-mouse Ly6G/Ly6C (clone RB6-8C5), rat anti-mouse CD11b (clone M1/70), rat anti-mouse B220 (clone RA3-6B2), and rat anti-mouse TER-119 (clone TER-119); rat anti-mouse CD45.2 (conjugated with allophycocyanin [APC]) (clone 104, catalog no. 17-0454-82; Thermo Fisher Scientific, Inc., Waltham, MA); and rat anti-mouse CD90.2 (conjugated with fluorescein isothiocyanate [FITC]) (clone 30-H12, catalog no. 105305; BioLegend, Inc.). Dead cells were excluded by staining with viability dye (conjugated with eFluor 506) (catalog no. 65-0866-14; Thermo Fisher Scientific). Antibody staining was permitted for 30 min at 4°C.
Cytokines were induced and accumulated inside cells for intracellular staining. Mouse intestinal lamina propria lymphoid cells (1 × 106 to 2 × 106) were resuspended in 200 μl medium (RPMI 1640 containing 10% FBS) per well (96-well plate) and stimulated with 50 ng/ml phorbol 12-myristate 13-acetate (PMA) (catalog no. P8139-1MG; Sigma) and 1 μg/ml ionomycin (catalog no. 10004974; Cayman Chemical Company, Inc., Ann Arbor, MI) for 4 h. Brefeldin A (catalog no. B7651; Sigma) was added at 10 μg/ml for 2 h before cells were harvested for analysis. After surface marker staining as described above, cells were fixed and permeabilized using a transcription factor staining buffer set (catalog no. 00-5523-00; Thermo Fisher Scientific). The cell samples were then stained with the following antibodies at 4°C in the dark for 30 min: mouse anti-mouse RORγt (conjugated with BV650) (clone Q31-378, catalog no. 564722; BD Bioscience, San Jose, CA) and rat anti-mouse IFN-γ (conjugated with phycoerythrin [PE]/Cy7) (clone XMG12, catalog no. 505826; BioLegend, Inc.). After cells were washed twice and resuspended with 200 μl FACS buffer, the cell samples were used for flow cytometry analysis on an LSRII instrument (BD Biosciences), and data were analyzed using FlowJo software (TreeStar).
A FACSAria II instrument (BD Biosciences) was used to sort the desired cell population as donor cells for adoptive transfer experiments. After excluding dead and lineage-positive cells (CD3+ Ly6C+ CD11b+ B220+ TER-119+), the ILC3-enriched subset (CD45int CD90hi) and the non-ILC3 subset (CD45hi CD90hi) were sorted from Rag1-deficient mouse LP-ILCs into ILC3-enriched and non-ILC3 subsets. GFP+ cells were sorted from RORγt-GFP reporter mice.
Statistics.
The numbers of live organisms as IFU at individual data points or over a time course were compared using the Wilcoxon rank sum test. The area under the curve (AUC) was used for comparing the time course or clusters of tissue sample data. When multiple groups were included in a given experiment, analysis of variance (ANOVA) was first used to determine whether there was an overall significant difference among all groups. Only when the P value was <0.05 (ANOVA) were the differences between every two groups further analyzed using a Wilcoxon test.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2017. Sexually transmitted disease surveillance, 2016. Centers for Disease Control and Prevention, Atlanta, GA: https://www.cdc.gov/std/stats16/default.htm. [Google Scholar]
- 2.Dukers-Muijrers NH, Schachter J, van Liere GA, Wolffs PF, Hoebe CJ. 2015. What is needed to guide testing for anorectal and pharyngeal Chlamydia trachomatis and Neisseria gonorrhoeae in women and men? Evidence and opinion. BMC Infect Dis 15:533. doi: 10.1186/s12879-015-1280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gratrix J, Singh AE, Bergman J, Egan C, Plitt SS, McGinnis J, Bell CA, Drews SJ, Read R. 2015. Evidence for increased Chlamydia case finding after the introduction of rectal screening among women attending 2 Canadian sexually transmitted infection clinics. Clin Infect Dis 60:398–404. doi: 10.1093/cid/ciu831. [DOI] [PubMed] [Google Scholar]
- 4.Musil K, Currie M, Sherley M, Martin S. 2016. Rectal chlamydia infection in women at high risk of chlamydia attending Canberra Sexual Health Centre. Int J STD AIDS 27:526–530. doi: 10.1177/0956462415586317. [DOI] [PubMed] [Google Scholar]
- 5.Pabbaraju K, Wong S, Gill K, Severini A, Roy F, Gratrix J, Singh AE, Naidu P, Read R, Drews SJ. 2017. Use of the APTIMA Combo 2 assay and a secondary algorithm to detect and confirm Chlamydia trachomatis in rectal-only infections. Sex Transm Dis 44:118–119. doi: 10.1097/OLQ.0000000000000552. [DOI] [PubMed] [Google Scholar]
- 6.Peters RP, Dubbink JH, van der Eem L, Verweij SP, Bos ML, Ouburg S, Lewis DA, Struthers H, McIntyre JA, Morre SA. 2014. Cross-sectional study of genital, rectal, and pharyngeal Chlamydia and gonorrhea in women in rural South Africa. Sex Transm Dis 41:564–569. doi: 10.1097/OLQ.0000000000000175. [DOI] [PubMed] [Google Scholar]
- 7.Mardh PA, Ursing B, Sandgren E. 1980. Lack of evidence for an association between infection with Chlamydia trachomatis and Crohn’s disease, as indicated by micro-immunofluorescence antibody tests. Acta Pathol Microbiol Scand B 88:57–59. doi: 10.1111/j.1699-0463.1980.tb02604.x. [DOI] [PubMed] [Google Scholar]
- 8.McGarity BH, Robertson DA, Clarke IN, Wright R. 1991. Deoxyribonucleic acid amplification and hybridisation in Crohn’s disease using a chlamydial plasmid probe. Gut 32:1011–1015. doi: 10.1136/gut.32.9.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munro J, Mayberry JF, Matthews N, Rhodes J. 1979. Chlamydia and Crohn’s disease. Lancet ii:45–46. doi: 10.1016/S0140-6736(79)90213-7. [DOI] [PubMed] [Google Scholar]
- 10.de la Maza LM, Pal S, Khamesipour A, Peterson EM. 1994. Intravaginal inoculation of mice with the Chlamydia trachomatis mouse pneumonitis biovar results in infertility. Infect Immun 62:2094–2097. doi: 10.1128/IAI.62.5.2094-2097.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison RP, Caldwell HD. 2002. Immunity to murine chlamydial genital infection. Infect Immun 70:2741–2751. doi: 10.1128/iai.70.6.2741-2751.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Zhang H, Zhou Z, Yang Z, Ding Y, Zhou Z, Zhong E, Arulanandam B, Baseman J, Zhong G. 2014. Chlamydial induction of hydrosalpinx in 11 strains of mice reveals multiple host mechanisms for preventing upper genital tract pathology. PLoS One 9:e95076. doi: 10.1371/journal.pone.0095076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shekhar S, Peng Y, Wang S, Yang X. 2018. CD103+ lung dendritic cells (LDCs) induce stronger Th1/Th17 immunity to a bacterial lung infection than CD11b(hi) LDCs. Cell Mol Immunol 15:377–387. doi: 10.1038/cmi.2016.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Igietseme JU, Portis JL, Perry LL. 2001. Inflammation and clearance of Chlamydia trachomatis in enteric and nonenteric mucosae. Infect Immun 69:1832–1840. doi: 10.1128/IAI.69.3.1832-1840.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeruva L, Spencer N, Bowlin AK, Wang Y, Rank RG. 2013. Chlamydial infection of the gastrointestinal tract: a reservoir for persistent infection. Pathog Dis 68:88–95. doi: 10.1111/2049-632X.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Zhang Q, Zhang T, Zhang Y, Zhu C, Sun X, Zhang N, Xue M, Zhong G. 2016. The Chlamydia muridarum organisms fail to auto-inoculate the mouse genital tract after colonization in the gastrointestinal tract for 70 days. PLoS One 11:e0155880. doi: 10.1371/journal.pone.0155880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Zhu C, Zhang T, Tian Q, Zhang N, Morrison S, Morrison R, Xue M, Zhong G. 2018. Nonpathogenic colonization with Chlamydia in the gastrointestinal tract as oral vaccination for inducing transmucosal protection. Infect Immun 86:e00630-17. doi: 10.1128/IAI.00630-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu C, Lin H, Tang L, Chen J, Wu Y, Zhong G. 2018. Oral Chlamydia vaccination induces transmucosal protection in the airway. Vaccine 36:2061–2068. doi: 10.1016/j.vaccine.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 19.Campbell J, Huang Y, Liu Y, Schenken R, Arulanandam B, Zhong G. 2014. Bioluminescence imaging of Chlamydia muridarum ascending infection in mice. PLoS One 9:e101634. doi: 10.1371/journal.pone.0101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q, Huang Y, Gong S, Yang Z, Sun X, Schenken R, Zhong G. 2015. In vivo and ex vivo imaging reveals a long-lasting chlamydial infection in the mouse gastrointestinal tract following genital tract inoculation. Infect Immun 83:3568–3577. doi: 10.1128/IAI.00673-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai J, Zhang T, Wang L, Shao L, Zhu C, Zhang Y, Failor C, Schenken R, Baseman J, He C, Zhong G. 2016. Intravenous inoculation with Chlamydia muridarum leads to a long-lasting infection restricted to the gastrointestinal tract. Infect Immun 84:2382–2388. doi: 10.1128/IAI.00432-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong G. 2018. Chlamydia spreading from the genital tract to the gastrointestinal tract—a two-hit hypothesis. Trends Microbiol 26:611–623. doi: 10.1016/j.tim.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atarashi K, Umesaki Y, Honda K. 2011. Microbiotal influence on T cell subset development. Semin Immunol 23:146–153. doi: 10.1016/j.smim.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 24.Belkaid Y, Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonnenberg GF, Artis D. 2012. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37:601–610. doi: 10.1016/j.immuni.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Britanova L, Diefenbach A. 2017. Interplay of innate lymphoid cells and the microbiota. Immunol Rev 279:36–51. doi: 10.1111/imr.12580. [DOI] [PubMed] [Google Scholar]
- 27.Castellanos JG, Longman RS. 2020. Innate lymphoid cells link gut microbes with mucosal T cell immunity. Gut Microbes 11:231–236. doi: 10.1080/19490976.2019.1638725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim M, Kim CH. 2016. Colonization and effector functions of innate lymphoid cells in mucosal tissues. Microbes Infect 18:604–614. doi: 10.1016/j.micinf.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eberl G, Colonna M, Di Santo JP, McKenzie AN. 2015. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H. 2018. Innate lymphoid cells: 10 years on. Cell 174:1054–1066. doi: 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 31.Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F. 2019. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis 10:315. doi: 10.1038/s41419-019-1540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nussbaum K, Burkhard SH, Ohs I, Mair F, Klose CSN, Arnold SJ, Diefenbach A, Tugues S, Becher B. 2017. Tissue microenvironment dictates the fate and tumor-suppressive function of type 3 ILCs. J Exp Med 214:2331–2347. doi: 10.1084/jem.20162031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, Honig M, Pannicke U, Schwarz K, Ware CF, Finke D, Diefenbach A. 2010. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, Goppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A. 2013. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 494:261–265. doi: 10.1038/nature11813. [DOI] [PubMed] [Google Scholar]
- 35.Cella M, Gamini R, Secca C, Collins PL, Zhao S, Peng V, Robinette ML, Schettini J, Zaitsev K, Gordon W, Bando JK, Yomogida K, Cortez V, Fronick C, Fulton R, Lin LL, Gilfillan S, Flavell RA, Shan L, Artyomov MN, Bowman M, Oltz EM, Jelinsky SA, Colonna M. 2019. Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat Immunol 20:980–991. doi: 10.1038/s41590-019-0425-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulthess J, Meresse B, Ramiro-Puig E, Montcuquet N, Darche S, Begue B, Ruemmele F, Combadiere C, Di Santo JP, Buzoni-Gatel D, Cerf-Bensussan N. 2012. Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity 37:108–121. doi: 10.1016/j.immuni.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 37.Koprivsek JJ, Zhang T, Tian Q, He Y, Xu H, Xu Z, Zhong G. 2019. Distinct roles of chromosome- versus plasmid-encoded genital tract virulence factors in promoting Chlamydia muridarum colonization in the gastrointestinal tract. Infect Immun 87:e00265-19. doi: 10.1128/IAI.00265-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koprivsek JJ, He Y, Song C, Zhang N, Tumanov A, Zhong G. 2020. Evasion of innate lymphoid cell-regulated gamma interferon responses by Chlamydia muridarum to achieve long-lasting colonization in mouse colon. Infect Immun 88:e00798-19. doi: 10.1128/IAI.00798-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montaldo E, Juelke K, Romagnani C. 2015. Group 3 innate lymphoid cells (ILC3s): origin, differentiation, and plasticity in humans and mice. Eur J Immunol 45:2171–2182. doi: 10.1002/eji.201545598. [DOI] [PubMed] [Google Scholar]
- 40.Eberl G, Littman DR. 2003. The role of the nuclear hormone receptor RORgammat in the development of lymph nodes and Peyer’s patches. Immunol Rev 195:81–90. doi: 10.1034/j.1600-065x.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- 41.Sciume G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, Spencer SP, Wilhelm C, Poholek AC, Vahedi G, Kanno Y, Belkaid Y, O’Shea JJ. 2012. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med 209:2331–2338. doi: 10.1084/jem.20122097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Withers DR, Hepworth MR. 2017. Group 3 innate lymphoid cells: communications hubs of the intestinal immune system. Front Immunol 8:1298. doi: 10.3389/fimmu.2017.01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cording S, Medvedovic J, Lecuyer E, Aychek T, Dejardin F, Eberl G. 2018. Mouse models for the study of fate and function of innate lymphoid cells. Eur J Immunol 48:1271–1280. doi: 10.1002/eji.201747388. [DOI] [PubMed] [Google Scholar]
- 44.Litvak Y, Mon KKZ, Nguyen H, Chanthavixay G, Liou M, Velazquez EM, Kutter L, Alcantara MA, Byndloss MX, Tiffany CR, Walker GT, Faber F, Zhu Y, Bronner DN, Byndloss AJ, Tsolis RM, Zhou H, Baumler AJ. 2019. Commensal Enterobacteriaceae protect against Salmonella colonization through oxygen competition. Cell Host Microbe 25:128–139.e5. doi: 10.1016/j.chom.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiemann S, Smit N, Roy U, Lesker TR, Galvez EJC, Helmecke J, Basic M, Bleich A, Goodman AL, Kalinke U, Flavell RA, Erhardt M, Strowig T. 2017. Enhancement of IFNgamma production by distinct commensals ameliorates Salmonella-induced disease. Cell Host Microbe 21:682–694.e5. doi: 10.1016/j.chom.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 46.Caballero S, Kim S, Carter RA, Leiner IM, Susac B, Miller L, Kim GJ, Ling L, Pamer EG. 2017. Cooperating commensals restore colonization resistance to vancomycin-resistant Enterococcus faecium. Cell Host Microbe 21:592–602.e4. doi: 10.1016/j.chom.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen C, Zhou Z, Conrad T, Yang Z, Dai J, Li Z, Wu Y, Zhong G. 2015. In vitro passage selects for Chlamydia muridarum with enhanced infectivity in cultured cells but attenuated pathogenicity in mouse upper genital tract. Infect Immun 83:1881–1892. doi: 10.1128/IAI.03158-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conrad TA, Gong S, Yang Z, Matulich P, Keck J, Beltrami N, Chen C, Zhou Z, Dai J, Zhong G. 2016. The chromosome-encoded hypothetical protein TC0668 is an upper genital tract pathogenicity factor of Chlamydia muridarum. Infect Immun 84:467–479. doi: 10.1128/IAI.01171-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morrison SG, Giebel AM, Toh EC, Spencer HJ III, Nelson DE, Morrison RP. 2018. Chlamydia muridarum genital and gastrointestinal infection tropism is mediated by distinct chromosomal factors. Infect Immun 86:e00141-18. doi: 10.1128/IAI.00141-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giebel AM, Hu S, Rajaram K, Finethy R, Toh E, Brothwell JA, Morrison SG, Suchland RJ, Stein BD, Coers J, Morrison RP, Nelson DE. 2019. Genetic screen in Chlamydia muridarum reveals role for an interferon-induced host cell death program in antimicrobial inclusion rupture. mBio 10:e00385-19. doi: 10.1128/mBio.00385-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shao L, Melero J, Zhang N, Arulanandam B, Baseman J, Liu Q, Zhong G. 2017. The cryptic plasmid is more important for Chlamydia muridarum to colonize the mouse gastrointestinal tract than to infect the genital tract. PLoS One 12:e0177691. doi: 10.1371/journal.pone.0177691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shao L, Zhang T, Liu Q, Wang J, Zhong G. 2017. Chlamydia muridarum with mutations in chromosomal genes tc0237 and/or tc0668 is deficient in colonizing the mouse gastrointestinal tract. Infect Immun 85:e00321-17. doi: 10.1128/IAI.00321-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shao L, Zhang T, Melero J, Huang Y, Liu Y, Liu Q, He C, Nelson DE, Zhong G. 2018. The genital tract virulence factor pGP3 is essential for Chlamydia muridarum colonization in the gastrointestinal tract. Infect Immun 86:e00429-17. doi: 10.1128/IAI.00429-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang T, Huo Z, Ma J, He C, Zhong G. 2019. The plasmid-encoded pGP3 promotes Chlamydia evasion of acidic barriers in both stomach and vagina. Infect Immun 87:e00844-18. doi: 10.1128/IAI.00844-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma J, He C, Huo Z, Xu Y, Arulanandam B, Liu Q, Zhong G. 2020. The cryptic plasmid improves Chlamydia fitness in different regions of the gastrointestinal tract. Infect Immun 88:e00860-19. doi: 10.1128/IAI.00860-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huo Z, He C, Xu Y, Jia T, Wang J, Zhong G. 2020. Chlamydia deficient in plasmid-encoded pGP3 is prevented from spreading to large intestine. Infect Immun 88:e00120-20. doi: 10.1128/IAI.00120-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 58.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo X, Muite K, Wroblewska J, Fu Y-X. 2016. Purification and adoptive transfer of group 3 gut innate lymphoid cells. Methods Mol Biol 1422:189–196. doi: 10.1007/978-1-4939-3603-8_18. [DOI] [PubMed] [Google Scholar]