

Abstract
OPA1 mutations are the major cause of dominant optic atrophy (DOA) and the syndromic form DOA plus, pathologies for which there is no established cure. We used a ‘drug repurposing’ approach to identify FDA-approved molecules able to rescue the mitochondrial dysfunctions induced by OPA1 mutations. We screened two different chemical libraries by using two yeast strains carrying the mgm1I322M and the chim3P646L mutations, identifying 26 drugs able to rescue their oxidative growth phenotype. Six of them, able to reduce the mitochondrial DNA instability in yeast, have been then tested in Opa1 deleted mouse embryonic fibroblasts expressing the human OPA1 isoform 1 bearing the R445H and D603H mutations. Some of these molecules were able to ameliorate the energetic functions and/or the mitochondrial network morphology, depending on the type of OPA1 mutation. The final validation has been performed in patients’ fibroblasts, allowing to select the most effective molecules. Our current results are instrumental to rapidly translating the findings of this drug repurposing approach into clinical trial for DOA and other neurodegenerations caused by OPA1 mutations.
Introduction
Mitochondria are extremely dynamic organelles that adjust their shape in response to the different energetic and metabolic requirements of the cell, through mitochondrial fusion and fission events. Highly conserved dynamin-like GTPases are involved in this process, Mitofusin 1 and Mitofusin 2 (MFN1 and MFN2) and dynamin-related protein 1 (DRP1), in charge of fusion and fission, respectively, at the outer membrane, and optic atrophy 1 (OPA1) possibly responsible for both the processes at the inner membrane (1–3). OPA1 is a multifunctional protein implicated, in addition to fusion, in structural organization of cristae, which in turn influences the assembly of the respiratory supercomplexes and the energetic efficiency (4,5), the mtDNA stability (6) and the apoptotic process by sequestering cytochrome c within the cristae (5). As a central player of mitochondrial dynamics OPA1 is also involved in the selective autophagy pathway termed mitophagy, a quality control system responsible for the clearance of damaged mitochondria (7–10). OPA1 is present in eight isoforms, anchored to the inner membrane as long forms and localized in the intermembrane space as soluble short forms, generated by a complex proteolytic maturation process (11,12). The multiplicity of OPA1 isoforms is likely essential to finely tune mitochondrial dynamics under different cellular conditions (13).
In humans, mutations in the OPA1 gene have been associated with dominant optic atrophy (DOA), a blinding disease characterized by selective degeneration of retinal ganglion cells and optic nerve atrophy (14,15). About 50% of the pathogenic mutations are predicted to produce a truncated protein, indicating haploinsufficiency as the genetic mechanism, whereas the missense mutations are mostly associated with a more severe multisystem disorder named DOA-plus, including DOA and mitochondrial myopathy with mtDNA multiple deletions (16–18). The DOA-plus associated missense mutations are most frequently clustered in the GTPase domain and are assumed to exert a dominant-negative effect (18,19). More recently, OPA1 mutations were shown to be causative of common neurodegenerative disorders, such as Parkinsonism and dementia and of multiple sclerosis (20–22).
Unfortunately, there is currently no established treatment for OPA1-linked diseases. It has been reported that idebenone treatment induces some degrees of visual improvement in OPA1-DOA patients (23,24), whereas it presents a limited therapeutic effect in a mouse model of OPA1-related DOA, strictly correlated to NQO1 expression (25). Even if there is an ongoing effort to genetically correct OPA1 mutations (26), gene therapy is still far from clinical use (12). Besides to some computational modeling of OPA1 protein structure (27), high-resolution structural features of mgm1 and s-OPA1 have been described only recently (28,29), making a drug-design approach still far from feasible. There is therefore a clear need to identifying molecules able at correcting the biochemical alterations associated with OPA1 mutations to advance therapeutic options for these diseases. To this end, the yeast model is a valuable tool in drug discovery, allowing to screening in a short time large molecular libraries, to identify chemical suppressors of pathological phenotypes (30). Saccharomyces cerevisiae possess an OPA1 orthologous, called MGM1, whose deletion makes the strain devoid of mtDNA (31–33) and causes abnormal mitochondrial morphology (34–36). Like OPA1, Mgm1 is also involved in mitochondrial fusion and maintenance of the cristae structure (35,37–39).
In this study, we report the results of a medium-size phenotypic screening of molecules by using a yeast mutant strains carrying the mgm1I322M mutation and the chim3P646L mutation, a chimeric protein composed by the N-terminal region of Mgm1 and the whole GTPase, middle and GED domains of OPA1 (40). We identified 26 drugs able to rescuing the oxidative growth phenotype of the chimeric mutant. Among these, six drugs were also able to restore the mtDNA stability perturbed by mgm1 mutations and have been then functionally tested in ad hoc generated Opa1 null mouse embryo fibroblasts (MEFs), expressing the human OPA1 isoform 1 bearing the R445H and D603H mutations, associated with DOA-plus and DOA, respectively. Some of these molecules effective in the yeast model were also able to ameliorate the pathological phenotype of MEFs under metabolic stress by specifically improving the energetic functions or the network dynamics, depending on the type of OPA1 mutation.
Results
Drugs screening of chemical libraries in yeast
Primary and secondary screening in the mgm1 mutant
To identify drugs able to rescue the respiratory defect due to mutation in MGM1, we took advantage of the thermal sensitive (ts) yeast mutant harboring mgm1I322M, equivalent to I382M mutation in OPA1 (27). This strain showed a limited growth in YP medium supplemented with oxidative carbon source (ethanol or glycerol) at 37°C (Fig. 1A). This growth defect is associated with both poor mtDNA maintenance at 37°C, which results in a petite frequency equal to 95%, and strong reduction of the respiratory activity (approximately 5% of residual respiration) (Fig. 1B and C). The chemical libraries screening was based on the capability of the drugs to partially restore the growth defect of such mutant on oxidative carbon source at high temperature. Two libraries were used. The first one is the Selleck FDA-approved chemical library, which contains 1018 molecules authorized for the treatment of several diseases by the FDA and can be used for ‘drug repurposing.’ In fact, this screening offers the advantage that the drugs, being already approved for human use, can be directly tested in clinical trials once the experimental evidence of their effects has been provided (41). The second library is the NCI BioServices Diversity Set IV (https://dtp.cancer.gov/organization/dscb/obtaining/available_plates.htm), which contains 1596 molecules chosen from almost 140 000 compounds for their structural diversity based on pharmacophoric centers and conformational isomers. The advantage of using this library is that the high degree of structural diversity among the molecules increases the probability of finding effective drugs, though, in this case, a direct clinical trial is not allowed.
Figure 1.
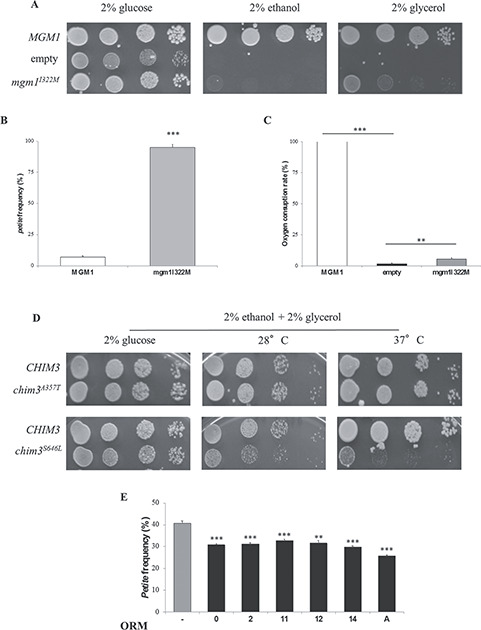
Yeast drugs screenings. (A) Spot assay of mgm1I322M mutant strain on YP medium supplemented with 2% ethanol or 2% glycerol at 37°C. 4 × 104, 4 × 103, 4 × 102 and 4 × 101 cells were present in each serial spot. (B) Petite frequency and (C) oxygen consumption rate normalized to the wild type MGM1 strain of the mgm1I322M mutant strain at 37°C. (D) Spot assay on YP medium supplemented with 2% ethanol and 2% glycerol of the chim3 mutant strains at 28 and 37°C. (E) Petite frequency of the mgm1I322M mutant strain treated with ORMs (32 μm ORM0, 420 nM ORM2, 16 μm ORM 11, 24 μm ORM12, 92 μm OMR14 and 1 μm ORMA) or with DMSO at 28°C. **Denotes P < 0.01; ***denotes P < 0.001.
Primary and secondary screenings were performed after a fine setup based on different oxidative carbon sources, different growth temperatures and different cell density aimed at identifying the best experimental conditions. Molecules were considered as positive hits if they produced a halo of enhanced growth around a soaked paper disk in both screenings. An example of a positive hit from the primary and secondary screening is reported in Supplementary Material, Figure S1. By such a screening, 22 drugs derived from the Selleck FDA-approved library and 21 drugs derived from the Diversity set IV chemical library were effective. Interestingly, benzbromarone, present in both libraries, resulted a positive hit in both cases, reinforcing the power of the screening. Molecules found in the Selleck library belong to different classes, such as anti-bacterial, anti-mycotic or anti-tumoral, and several share the primary target, i.e. the bacterial DNA gyrase A (nalidixic acid and piromidic acid); the squalene epoxidase (terbinafine and butenafine HCl); the topoisomerase II (doxorubicin, epirubicin HCl and daunorubicin HCl).
Construction and analysis of chim3 mutants for the tertiary screening
The 42 positive hits were then tested in a tertiary screening aimed at identify which of them was active not only in the rescue of the mgm1 mutation, but also of another OPA1 mutation, using a mutant strain harboring a MGM1/OPA1 chimeric gene, named CHIM3 in a previous study (40). Such a screening should be performed in a ‘leaky’ or ‘hypomorphic’ mutant with a significant growth impairment at 28°C or a ts phenotype. Unfortunately, the chim3I382M exhibited a slight growth defect, unsuitable for performing the screening either at 28°C or at 37°C. All the other chim3 mutant strains characterized so far were unable to grow on non-fermentable carbon sources due to total loss of mtDNA (27,40). To obtain chim3 mutant alleles suitable for the screening, we performed an in silico analysis in order to identify potential substitutions associated with a ts phenotype. The analysis is described in Material and Methods section. As reported at https://databases.lovd.nl/shared/genes/OPA1, 63 missense mutations were analyzed and five mutations, predicted to have a mild effect after analysis, were selected: A357T, R290Q, E487K, S646L and D438A. Chim3 mutant alleles containing such substitutions were constructed and introduced into the W303–1B Δmgm1 strain. Strains harboring R290Q, E487K and D438A mutations were unable to grow on medium supplemented with oxidative carbon sources (data not shown). Strain harboring the A357T mutation displayed a growth phenotype on non-fermentable carbon sources similar to that of strain harboring CHIM3 wt allele, both at 28°C and at 37°C (Fig. 1D). On the contrary, strain harboring the S646L allele displayed a ts phenotype, showing a slight growth reduction at 28°C and a strong growth impairment at 37°C compared to the CHIM3 strain (Fig. 1D).
After optimization of the growth conditions, the previously identified molecules were re-tested on chim3S646L mutant strain. Five drugs from the Selleck chemical library with anti-tumoral activity, strongly cytotoxic and unlikely suitable for a long treatment were excluded from this screening.
Fifteen drugs from the Selleck chemical library were able to partially restore the growth of the chim3P646L mutant strain, and were therefore named OPA1 rescuing molecules (ORMs) plus a number, as indicated in Table 1. These drugs belong to different therapeutic classes, mainly anti-bacterial, anti-inflammatory and anti-mycotic pharmaceuticals. A preliminary analysis of their chemical structures allows recognizing, among others, compounds endowed with aromatic and aliphatic amine groups but also with carboxylic acid moiety. In addition, some of the selected drugs share a common scaffold, which is an aromatic ring substituted with halogen atoms.
Table 1.
Selleck FDA-approved library screening in yeast
| Selleck FDA-approved library | |||||
|---|---|---|---|---|---|
| Drug | Pharmacological activity | Mechanism of action or primary target | ORM | Maximal concentration tested for petite analysis | Minimal concentration with rescue |
| Benzbromarone | Uricosuric | CYP2C9 inhibitor | ORM0 | 32 μm | 32 μm |
| Bromhexine HCl | Mucolytic | Medication for coughs | ORM1 | 16 μm | – |
| Chloroxine | Antibacterial | Treatment of dandruff and seborrheic dermatitis | ORM2 | 4 μm | 480 nM |
| Clofazimine | Antibacterial and anti-inflammatory | Intercalation with bacterial DNA and increasing levels of cellular phospholipase A2 | ORM3 | 125 μm | – |
| Diclazuril | Coccidiostat | anti-coccidial drug | ORM4 | 16 μm | – |
| Ethinyl Estradiol | Contraceptive | Orally bio-active estrogen (oral contraceptive pills) | ORM5 | 64 μm | – |
| Fesoterodine fumarate | Against urgent incontinency | Muscarinic receptor antagonist | ORM6 | 256 μm | – |
| Licofelone | Osteoarthritis | COX/LOX inhibitor (treatment for osteoarthritis) | ORM7 | 64 μm | – |
| Lithocholic acid | Cholagogue and choleretic | Bile acid (detergent to solubilize fats) | ORM8 | 32 μm | – |
| Nalidixic acid | Antibacterial | Inhibitor of the A subunit of bacterial DNA gyrase. | ORM9 | 64 μm | – |
| Piromidic acid | Antibacterial | Inhibitor of the A subunit of bacterial DNA gyrase | ORM10 | 16 μm | – |
| Retapamulin | Antibacterial | Bacterial protein synthesis inhibition | ORM11 | 256 μm | 16 μm |
| Salicylanilide | Antiviral, antibacterial and antimycotic | Hydrogen ionophore | ORM12 | 128 μm | 24 μm |
| Terbinafine hydrochloride | Antimycotic | Synthetic allylamine antifungal and a squalene epoxidase inhibitor | ORM13 | 2 μm | – |
| Tolfenamic acid | Anti-inflammatory | COX-2 inhibitor | ORM14 | 92 μm | 92 μm |
Among the drugs from the NCI Bioservices Diversity Set IV, 11 were positive, and were called ORMs plus a letter, as indicated in Table 2. With the exception of benzbromarone, found also in the Selleck collection, and ovalicin, the identified molecules were cytotoxic or predicted to be likely toxic on the basis of their structure, and/or unstable in aqueous solutions, or unavailable in sufficient quantity since not commercially sold. For this reason, only ovalicin was considered for the subsequent analysis.
Table 2.
Diversity set IV chemical library screening in yeast
| Drug | NSC code | CAS number | ORM |
|---|---|---|---|
| Ovalicin | 141358 | 19683-98-8 | ORMA |
| Pyridine-2-Azodimethylaniline | 9358 | 13103-75-8 | ORMB |
| 5-Fluoro-7-nitroquinolin-8-ol | 92207 | 18472-02-1 | ORMC |
| 4h-1,3-Thiazine-5,6-dihydro-4,4-dimethyl-2-(2-phenylethene | 150954 | no | ORMD |
| 1-Carbamimidoyl-3-(4-cholorophenyl) thiourea | 403374 | 7464-17-7 | ORME |
| 5-Benzyl-4-phenyldihydrofuran-2-3-dione | 17362 | 6362-70-5 | ORMF |
| 1,1,3-Triphenylpropargyl alcohol | 52241 | 1522-13-0 | ORMG |
| 4-Cholorophenylquinoline-6-sulphonate | 12544 | 5409-91-6 | ORMH |
| 5-(3-Chloroanilino)-1,2,4-dithiazol-3-ylidene]-dimethylammonium + hbr | 622689 | no | ORMI |
| Benzbromarone (3,5-dibromo-4-hydroxyphenyl-2-ethyl-3-benzofuranyl ketone), (methanone, (3,5-dibromo-4-hydroxyphenyl) (2-ethyl-3-benzofuranyl)-) | 85433 | 3562-84-3 | ORMK |
| Albacarcin V, chrysomycin A | 354844 | 82196-88-1 | ORMJ |
Effect of the ORMs on yeast mtDNA stability
We further investigated whether the rescue of growth defect of the mgmI322M and chim3S646L mutant strains by the 16 ORMs identified might be due to increased mtDNA stability. For this analysis, the strain mgm1I322M was chosen being its petite frequency approximately 40% at 28°C (Fig. 1E), thus indicating that the whole mtDNA is maintained in most cells. The chim3P646L mutant strain could not be utilized since its basal level of petite (80–85% at 28°C) is too high to detect a rescue, and after 48-h growth on glucose almost all cells lost their mtDNA.
For each ORM, four sub-lethal scalar concentrations were used to test their ability to rescue the defective mtDNA stability, identifying six ORMs able to decrease of the petite frequency at a specific concentration (Fig. 1E). These drugs were benzbromarone (ORM0), chloroxine (ORM2), retapamulin (ORM11), salicylanilide (ORM12), tolfenamic acid (ORM14), and ovalicin (ORMA) (Supplementary Material, Fig. S2).
Validation of ORMs efficacy in MEFs with OPA1 mutations
Effect of the ORMs on MEFs viability and ATP content
To assess the efficacy of the molecules identified in the yeast screening, we took advantage of the cell model we recently described (27), namely Opa1−/− MEFs expressing isoform 1 bearing two OPA1 mutations, the R445H and D603H, respectively localized in the GTPase and dynamin-like domains. We chose these two cell lines for their distinct biochemical features: the R445H MEFs, associated with DOA plus, present a severe energetic deficit, with loss of mtDNA and completely fragmented network, whereas the D603H MEFs, associated with non-syndromic DOA, show a significant mitochondrial network fragmentation, but normal mtDNA content and only a mild energetic defect (27). MEFs with wt OPA1 isoform 1 (ISO1) were used as control.
The three cell lines exhibited a similar growth in high glucose DMEM (Supplementary Material, Fig. S3A). However, when exposed to a metabolic stress, i.e. replacing glucose in the growth medium with galactose to force cells to rely on oxidative phosphorylation (OXPHOS) only to produce ATP (DMEM-galactose) (42), the R445H MEFs were unable to proliferate and, at longer times, also the growth of D603H MEFs was significantly reduced (Supplementary Material, Fig. S3B).
After determination of the optimal concentration of each ORM in DMEM (Table 3), MEFs were incubated for 24 h in DMEM-galactose in absence or presence of the drugs, to evaluate their capability to improve cell viability (Fig. 2A–C). As reported in Figure 2B and C, ORMs 0, 2, 12 and 14 increased the number of D603H viable cells, without reaching statistical relevance, whereas ORM12 only had a significant effect on R445H MEFs. No ORMs influenced the growth of ISO1 MEFs, except ORM12 that exhibited a weak effect only (Fig. 2A). ORM11 did not present any effect on MEFs viability (Supplementary Material, Fig. S4A).
Table 3.
Chemical names, CAS number and concentrations of functional ORMs in MEFs
| ORMs | Chemical name | CAS | Concentration (μm) |
|---|---|---|---|
| ORM0 | Benzbromarone | 3562-84-3 | 2 |
| ORM2 | Chloroxine | 773-76-2 | 1 |
| ORM11 | Retapamulin | 224452-66-8 | 1 |
| ORM12 | Salicylanilide | 87-17-2 | 10 |
| ORM14 | Tolfenamic acid | 13710-19-5 | 10 |
| ORMA | Ovalicin | 19683-98-8 | 1 |
Figure 2.
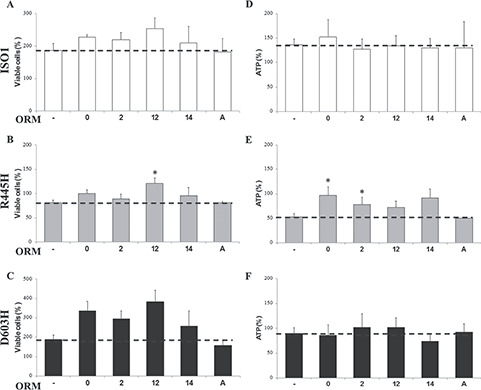
Viability and ATP content of MEFs bearing the indicated OPA1 mutations incubated with ORMs. (A–C) Number of viable cells of ISO1, R445H and D603H MEFs, after incubation for 24 h in DMEM-galactose in the absence or presence of ORMs. Data are expressed as percent of the SRB absorbance measured in DMEM-glucose at time = 0, considered as 100% value. Data are means ± SEM of 19 experiments in DMEM-galactose and 3–7 experiments in DMEM-galactose with ORMs treatment (ORMs 0, 2, 12: n = 3; ORM14, A: n = 7). (D–F) Cellular ATP levels of ISO1, R445H and D603H MEFs after incubation for 24 h in DMEM-galactose ± ORMs. Data are expressed as percentage of ATP content measured in DMEM-glucose at time = 0. Values are means ± SEM of 14 experiments in DMEM-galactose and 3–4 experiments in DMEM-galactose + ORMs (ORMs 0, 2, 12, 14 n = 4, ORMA n = 3). *Denotes P < 0.05.
We then evaluated the effects of the ORMs on ATP levels of MEFs incubated for 24 h in DMEM-galactose (Fig. 2D–F). We previously reported that under these conditions, the ATP content of ISO1 MEFs significantly increased as a consequence of the shift from glycolytic to oxidative metabolism. Differently, D603H MEFs were unable to enhance the ATP levels, whereas R445H MEFs exhibited a significant reduction (27). Figure 2E shows that ORMs 0, 2, 12 and 14 increased the ATP content of R445H MEFs, being the effect of ORMs 0 and 2 statistically significant. No ORMs enhanced the ATP content of both D603H and ISO1 MEFs (Fig. 2D and F). ORM11 did not present any effect on ATP content (Supplementary Material, Fig. S4B). It seems therefore that ORMs 0, 2 and, less efficiently, 12 and 14, specifically ameliorate the energetics of R445H MEFs, the only cell line suffering a reduction in cell viability and severe ATP depletion when grown in DMEM-galactose.
Effect of the ORMs on mitochondrial network morphology
The effect of ORMs on the shape of mitochondrial network was determined by fluorescence microscopy after cell loading with Mitotracker Red.
In DMEM, the mitochondria of ISO1 MEFs were mainly filamentous and intermediate, those of D603H MEFs were intermediate or fragmented and those of R445H were completely fragmented, as previously described (Fig. 3A–C and Supplementary Material, Fig. S5A and B) (27). Under this condition, ORMs 0 and 2 significantly recovered the mitochondrial fragmentation of both ISO1 and D603H MEFs, whereas ORMs 14 and A were selectively effective in ISO1 MEFs and D603H MEFs, respectively (Fig. 3B and C).
Figure 3.
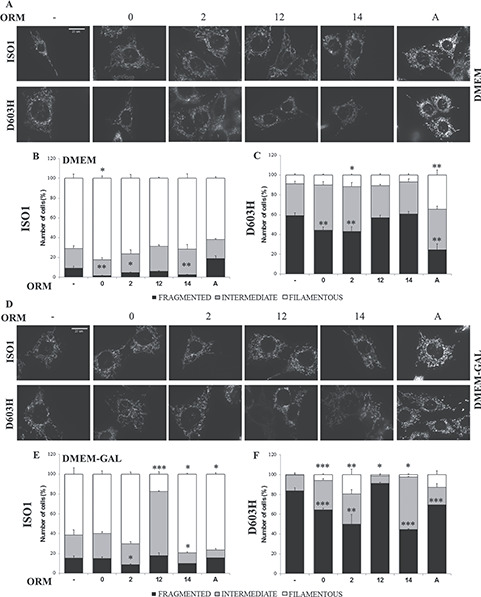
Mitochondrial network morphology of MEFs bearing the indicated OPA1 mutations incubated with ORMs. (A, D) Representative images of mitochondrial network of ISO1 and D603H MEFs after labeling with Mitotracker Red. MEFs lines were incubated for 24 h in DMEM (B, C) or in DMEM-galactose (E, F) in absence or presence of the ORMs. Cells were scored in three categories on the basis of mitochondrial network morphologies: cells with filamentous and interconnected network (filamentous), cells with short filamentous mitochondria (intermediate) and cells with fragmented mitochondria (fragmented). 100–120 cells were analyzed for ISO1 (B–E) and D603H (C–F) MEFs in each condition and for each experiment. Data are means ± SEM of 3–4 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
The incubation in DMEM-galactose did not alter the mitochondrial shape distribution in ISO1 MEFs; conversely it markedly increased intermediate and fragmented mitochondria in D603H MEFs (Fig. 3D–F). In R445H MEFs the mitochondrial network was completely fragmented also in DMEM-galactose (Supplementary Material, Fig. S5A and C). As reported in Figure 3E and F, ORMs 2, 14 and A were effective both in ISO1 and D603H MEFs, whereas ORM0 significantly reduced the mitochondrial fragmentation in D603H MEFs only. ORM12 statistically worsened the network morphology of ISO1 and D603H MEFs in DMEM-galactose. None of the ORMs was able to restore the mitochondrial morphology of R445H MEFs (Supplementary Material, Fig. S5B and C), in agreement with the previous finding that this mutation severely hampers the OPA1 GTPase activity and thus its fusogenic capacity (13,43), making the mitochondrial network of these cells similar to that of Opa1−/− MEFs (27). ORM11 did not present any effect on mitochondrial morphology (Supplementary Material, Fig. S4C and D).
Table 4 summarizes the biochemical effects of the ORMs in the MEF cell lines. From this set of data we can conclude that ORMs 0, 2 and 14 were efficient in ameliorating both energetics and mitochondrial morphology, whereas ORMs 12 and A had an effect only on energetics or restoring mitochondrial morphology, respectively. ORM11, although not having a toxic effect, did not present any effect in increasing cell viability, ATP content and filamentous mitochondrial network (Supplementary Material, Fig. S4) and was not included in the following screening.
Table 4.
Summary of the effect of different ORMs on mitochondrial functions in MEFs
| Phenotype | Cell lines | ORM0 | ORM2 | ORM12 | ORM14 | ORMA |
|---|---|---|---|---|---|---|
| Cell viability | ISO1 | |||||
| R445H | b | |||||
| D603H | ||||||
| ATP | ISO1 | |||||
| R445H | b | b | a | |||
| D603H | ||||||
| Mitochondrial morphology glucose | ISO1 | b | b | b | ||
| R445H | ||||||
| D603H | b | b | b | |||
| Mitochondrial morphology galactose | ISO1 | b | c(NEG) | b | b | |
| R445H | ||||||
| D603H | b | b | c(NEG) | b | b | |
| Efficacy |
−Energetics −Morphology |
−Energetics −Morphology |
−Cell viability | −Morphology | −Morphology |
aNot statistically significant trend.
bStatistically significant trend.
cStatistically significant trend in a negative way.
Identification of putative ORMs-targeted pathways
Mitochondrial shaping proteins
Being most of the ORMs able to ameliorate the mitochondrial network morphology, we decided to evaluate their effect on the expression levels of the main mitochondrial shaping proteins by western blot analysis. We previously reported that the levels of the OPA1, MFN2 and DRP1 were not influenced by the presence of the different OPA1 mutations (27). After 24 h in DMEM-galactose in presence of the ORMs, the OPA1 content was similar in all MEF lines (Fig. 4A and B). The ratio of OPA1 long- and short-forms was different in the three cell lines, since it tended to decrease in R445H MEFs and to increase in D604H compared to ISO1 MEFs, but it was not influenced by the different ORMs (Fig. 4A and C). The contents of MFN2, DRP1 and the activated DRP1-p-S616 were similar and not affected by any ORM (Fig. 4D–I). As the primary proteins shaping mitochondrial dynamics were not directly affected, it can be assumed that the ORMs’ targets are different, involving either their interactors or other pathways.
Figure 4.
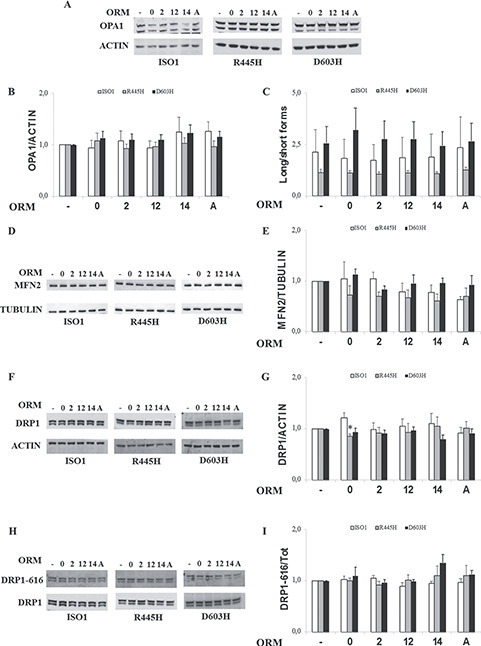
Levels of mitochondrial shaping proteins in MEFs bearing the indicated OPA1 mutations incubated with ORMs. MEFs were incubated for 24 h in DMEM-galactose in the absence or presence of ORMs. (A, D, F, H) Representative blots of OPA1, MFN2, DRP1 and DRP1-P-S616. ACTIN or TUBULIN were used as a loading control. (B, E, G) Densitometric analysis of OPA1, MFN2 AND DRP1 protein levels. Data are normalized to those of untreated cells in DMEM-galactose. Values are means ± SEM (n = 3–6). *P < 0.05, one simple t test. (C) Densitometric analysis of OPA1 long-/short-forms ratio. Values are means ± SEM (n = 4–6). (I) Densitometric analysis of DRP1-P-S616/DRP1 ratio. Values are means ± SEM (n = 3).
Mitochondrial mass, OXPHOS complexes and mtDNA content
The increased viability and the enhanced cellular ATP content induced by some ORMs in OPA1 mutated MEFs prompted us to investigate mitochondrial mass, mtDNA copy number and oxidative phosphorylation complexes content as possible ORMs targets.
To explore mitochondrial mass, we determined the levels of representative proteins of different mitochondrial sub-compartments, namely the TOM20 subunit of the translocase of the outer membrane, the TIM23 subunit of the translocase of the inner membrane, and the heat shock protein 60 (HSP60) and citrate synthase (CS) for the matrix. Figure 5A and B shows that TOM20 level did not change in ISO1 and R445H MEFs treated with ORMs, whereas it was slightly decreased by ORM12 in D603H MEFs. No significant difference was reported for TIM23, HSP60 and CS (Fig. 5A, C–E).
Figure 5.
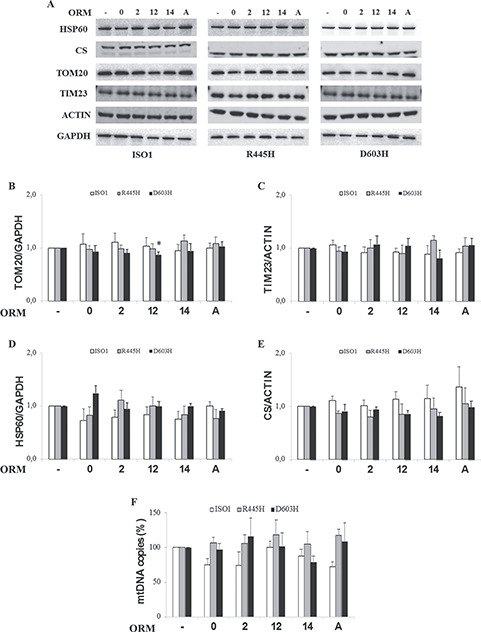
Mitochondrial protein content of MEFs bearing the indicated OPA1 mutations incubated with ORMs. MEFs were incubated for 24 h in DMEM-galactose in the absence or presence of ORMs. (A) Western blot of representative proteins of outer (TOM20) and inner (TIM23) mitochondrial membrane and of the matrix (citrate synthase and Hsp60). ACTIN or GAPDH were used as a loading control. (B–E) Densitometric analysis of the mitochondrial mass proteins; data are normalized to those of untreated cells in DMEM-galactose. Values are means ± SEM (n = 4–8). *P < 0.05, one simple t test. (F) mtDNA copy number. Data are means ± SEM (n = 3).
Similarly, the mtDNA copy number, measured by real time PCR, was also not significantly affected by any ORM (Fig. 5F).
Finally, we determined the level of representative subunit of the OXPHOS complexes, namely NDUFA9 for complex I, SDHA for complex II, UQCRC2 for complex III, COXIV for complex IV e ATP5a for complex V (Fig. 6A). Again, no significant difference was observed, although scattered tendency to increase was detected in D603H MEFs for ORM0 in complex IV, for ORM 2 in complexes II, III and IV, for ORM12 in complex II and for ORMA in complexes I, II and III (Fig. 6A–F). A similar trend was observed in R445H MEFs for ORMs 2 and 14 in complex IV (Fig. 6A and E).
Figure 6.
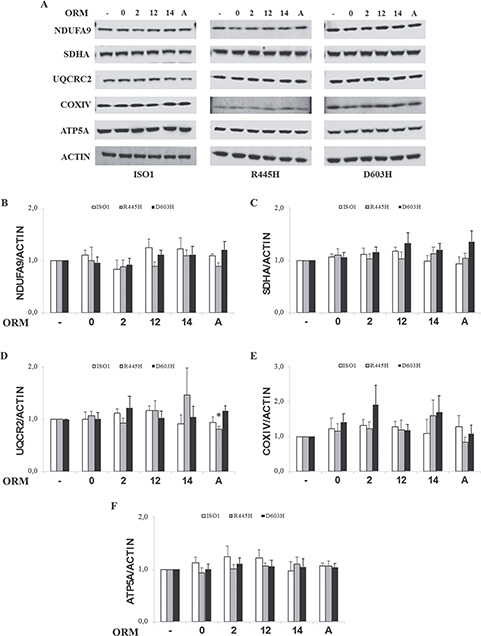
Levels of OXPHOS proteins of MEFs bearing the indicated OPA1 mutations incubated with ORMs. MEFs were incubated for 24 h in DMEM-galactose in the absence or presence of ORMs. (A) Western blot of representative subunits of complex I (NDUFA9), complex II (SDHA), complex III (UQCRC2), complex IV (COXIV) and complex V (ATP5A). ACTIN was used as a loading control. (B–F) Densitometric analysis of the OXPHOS subunits levels. Values are means ± SEM (n = 3–9) and are normalized to those of untreated cells in DMEM-galactose. *P < 0.05, one simple t test.
From these overall data, it can be concluded that the beneficial effect of ORMs on OPA1 mutant MEFs is not related with increased mitochondrial biogenesis.
Proteins involved in the autophagy process
OPA1 is involved in the autophagy/mitophagy processes (10) and missense and stop-codon OPA1 mutations have an opposite effect, increasing or decreasing this pathway, respectively (20,44). Thus, we went on to evaluate whether some ORMs may influence autophagy, whose efficiency is essential for clearance of damaged mitochondrial components. We monitored the level of the microtubule-associated proteins 1A/1B light chain 3B (here referred to as LC3), in particular of the cytoplasmic form (LC3-I) and its lipidated membrane-bound form (LC3-II). The amount of LC3-II correlates well with the number of autophagosomes. In DMEM, mutated MEFs showed a reduced ratio of LC3-II/I compared with ISO1 (Fig. 7A and B). Incubation in DMEM-galactose, an autophagy activator (20), increased proportionally this ratio in both ISO1 and OPA1 mutants, remaining lower in mutants, thus suggesting some impairment in the process (Fig. 7A and B).
Figure 7.
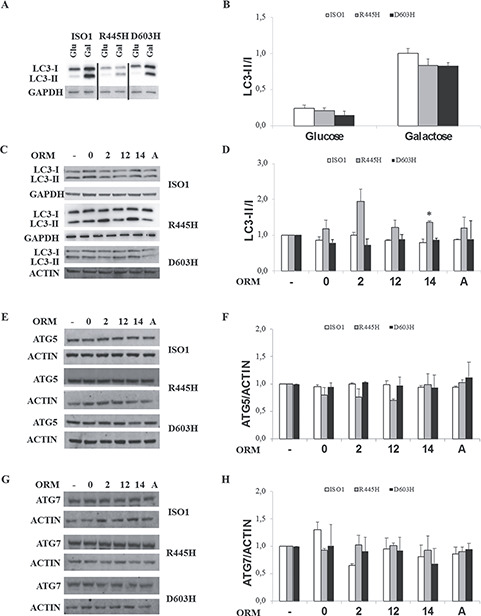
Levels of autophagy markers. Western blot of LC3 in DMEM and DMEM-galactose (A) and in DMEM-galactose in the absence or presence of ORMs (C) in MEFs. (B, D) Densitometric analysis of LC3II/I ratio. Values are means ± SEM (n = 3–4). *P < 0.05, student’s two tail t-test. (E, G) Representative blots of ATG5 and ATG7. (F, H) Densitometric analysis of ATG5 and AT7 protein levels. Values are means ± SEM (n = 3) and are normalized to those of untreated cells in DMEM-galactose. ACTIN or GAPDH were used as a loading control.
To verify this possibility, we analyzed the autophagic flux by treating MEFs with the mTOR inhibitor rapamycin, a strong autophagic inducer, and with chloroquine, which blocks lysosome-mediated proteolysis producing an increase of autophagosome numbers (45). We found that these treatments caused accumulation of the lipidated form of LC3, when compared to untreated conditions, and this occured equally in wt and OPA1 mutated MEFs (Supplementary Material, Fig. S6A–C), indicating that the autophagic flux is not significantly affected in our experimental conditions.
Incubations with ORMs induced an increase in LC3II/I ratio in R445H MEFs, being significant for ORM14 only, while they were ineffective on the ISO1 and D603H MEFs (Fig. 7C and D).
The first phase of the autophagy process is the phagophore formation. For this step, the processing of LC3 requires activation of ATG12 by ATG7 and then conjugation of ATG12 and ATG5 proteins (46,47). Figure 7E–H shows that the protein level of ATG5 and ATG7 was not significantly changed by any ORMs, except for ORM12 that reduced ATG5 in R445H MEFs, and for ORM2 in ISO1 MEFs, where ATG7 was decreased.
Taken together, these results indicate that, despite there was no evidence for an impairment in the autophagic process, the basal autophagic activity seems to be slowed down in the two OPA1 mutants compared to ISO1 in both DMEM and DMEM-galactose. This may be explained by the fact that only the mutated allele is present in MEFs. ORMs reverse this trend in R445H MEFs only, suggesting the possible use of autophagy as an alternative pathway to support energy production.
Validation of drugs efficacy in DOA patients’ fibroblasts
Finally, to consolidate these results, we decided to examine the ORMs efficacy in a more physiological cellular model: fibroblasts from DOA patients bearing the same OPA1 mutations as in MEFs, but in conjunction with the OPA1 wild-type allele. Despite being the most commonly used cell model to study the pathomechanism of DOA, fibroblasts suffer several limitations, the most relevant being the heterogeneity of OPA1-induced defect due to the individual nuclear and mitochondrial genomes variability. Accordingly, the OPA1 mutated fibroblasts present mild energetic dysfunction and different degree of mitochondrial network morphology impairment, mostly emerging only after metabolic stress, being this latter parameter the only one linearly correlated with the severity of patients’ clinical phenotype (27,48–51). For this reason, we considered the mitochondrial morphology as the best outcome measure for ORMs validation.
Representative images of mitochondrial network morphology of fibroblasts incubated in DMEM-galactose in absence or presence of ORMs are reported in Figure 8A and Supplementary Material, Figure S7A. Quantitative evaluation of mitochondrial morphology in DMEM-galactose showed that D603H fibroblasts had exclusively intermediate and filamentous mitochondria only (44 and 56% respectively), whereas R445H mutants presented also cells with fragmented mitochondria (27%) and a lower percentage of cells with filamentous mitochondria (27%), compared to D603H fibroblasts (Fig. 8B and C). All the ORMs, except ORM2, significantly ameliorated the mitochondrial network morphology of R445H fibroblasts, increasing the percentage of cells with filamentous mitochondria and reducing the cells with fragmented mito-network (Fig. 8B). ORMs 12, 14 and A were also effective in D603H fibroblasts (Fig. 8C). Thus, excluding ORM2, all other molecules exhibited a positive effect on the two OPA1 mutated fibroblasts. Since ORM2 was no effective on mitochondrial morphology, it was excluded from the subsequent analysis. ORM 0 was excluded only for D603H fibroblasts, since its effect on mitochondrial morphology was not statistically significant.
Figure 8.
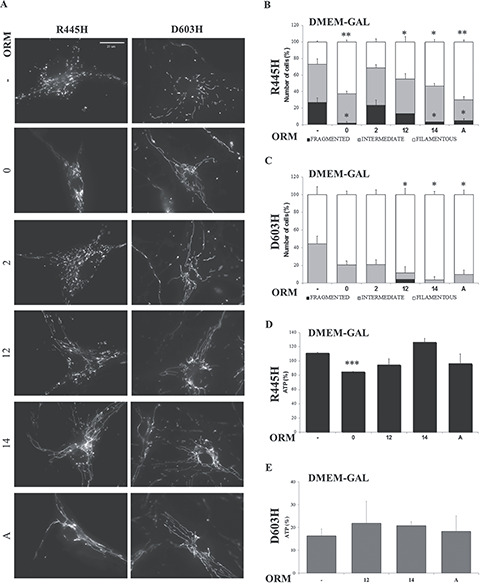
Effects of the ORMs in DOA patients’ fibroblasts. (A) Representative images of mitochondrial network of fibroblasts loaded with Mitotracker Red. (B, C) 60–80 cells were analyzed for R445H and D603H fibroblasts in each condition. Data are means ± SEM of three independent experiments. (D, E) Cellular ATP levels of R445H and D603H fibroblasts after 24 h incubation in DMEM-galactose in the absence or presence of ORMs. Data are expressed as percent of ATP content measured in DMEM-glucose at time = 0; values are means ± SEM of three experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
We then measured the energetic efficiency by determining the ATP levels after 24 h incubation in DMEM-galactose. We previously reported that under this condition the ATP levels of controls markedly increased, whereas fibroblasts bearing OPA1 mutations were unable to rise the ATP content (27). Only ORM14 increased the ATP levels in R445H fibroblasts without reaching statistical significance, whereas all other ORMs had no effect, being ORM0 apparently harmful (Fig. 8D). Conversely the ATP levels of D603H appear to be enhanced by the 3 ORMs tested (ORM12, 14 and A) without being statistically relevant (Fig. 8E).
All ORMs were effective in further improving the mitochondrial network morphology of control fibroblasts, and have null or positive effects, even if not statistically significant, also in further increasing cellular ATP in these cell lines (Supplementary Material, Fig. S7A–C).
All together, these results support the conclusions obtained in MEFs, suggesting that ORMs have a positive effect in OPA1 mutated fibroblasts mainly on morphology, which is the cellular parameter that better correlates with the severity of clinical phenotype. Considering that ORM14 also induces a slight increase in ATP levels, it seems reasonable to propose this molecule as the best candidate for therapeutic use in DOA patients.
Discussion
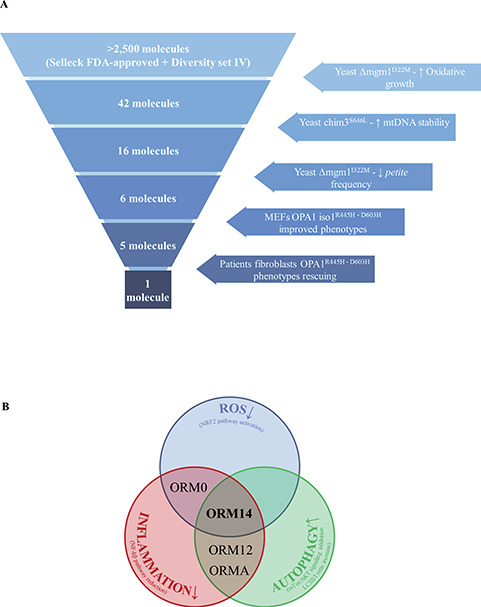
In this study, we have exploited a yeast-based screening of drugs, identifying pharmacological compounds able to rescue the defective yeast phenotype caused by mgm1/opa1 mutations. The efficacy of these ORMs has been then validated in the DOA cell model of MEFs and patients’ fibroblasts, bearing the R445H and D603H OPA1 mutations (Fig. 9A). These combined approaches have proved to be very efficient to isolate and validate compounds, most of which are FDA approved, and to investigate their molecular mechanisms. Indeed, the cellular analyses allowed us to rule out the involvement of mitochondrial biogenesis as major player of the rescue of defective cellular phenotype by ORMs. We did not observed alterations in the expression of the mitochondrial shaping proteins, suggesting alternative pathways for mitochondrial network morphology improvement. Finally, we present evidence suggesting that activation of autophagy is one of the intracellular mechanisms specifically targeted by ORMs.
Figure 9.
(A) Summary of the step-by-step experimental procedures leading to ORMs validation. (B) Hypothetical targets and mechanisms of action of the ORMs.
In the last two decades, yeast has emerged as a powerful screening tool, offering an affordable approach to study diseases and screen targets within a eukaryotic cellular context (30,52). Optimization of drug screening methods lead to the repurposing strategy, where ‘old’ molecules are tested to treat diseases, reducing development costs and timelines for the already de-risked compounds (53). We took advantage of both these approaches and, using our yeast models (mgm1 and chim3 mutants), we screened two different chemical libraries, identifying 6 molecules able to restore the growth impairment and improve mtDNA stability (Fig. 9A). Recently, Delerue et al. (54) described a similar screening, but using a different drug library, that identified 5 compounds able to efficiently prevent the lethality associated with loss of Msp1p, the ortholog of OPA1 in the fission yeast S. pombe, though no validation has been performed in mammalian cell models.
Among the 6 ORMs identified in yeast, 5 were effective in OPA1 mutated MEFs (Fig. 9A). Summarizing, ORMs 0, 2 and 14 improved both the mitochondrial morphology and ATP content, ORM12 ameliorated only cell viability, whereas ORMA recovered the mitochondrial network morphology. Considering both the two most representative dysfunctions, energetics and morphology, ORMs 0, 2 and 14 resulted the most effective. Moving on fibroblast cell model, ORMs 0, 12 and A were able to ameliorate the network morphology only, whereas ORM14 also slightly increased the cellular ATP amount. Having ruled out the direct involvement of ORMs in mitochondrial biogenesis and shaping proteins expression, to explain their beneficial effects on cells, we identified the activation of autophagy as a possible common mechanism of these ORMs.
ORM14 (tolfenamic acid), effective on the three cell models, is a non-steroidal anti-inflammatory drug (55), with inhibitory effects on cyclooxygenase-2 (COX-2) and NF-κB (55–57). Moreover, ORM14 has been already tested in various neurodegenerative disease models such as Alzheimer’s and Parkinson’s diseases, for which elevated levels of COX-2 have been reported (58,59). Treatment with ORM14 in Alzheimer’s disease mice reduces different pathological markers, including Cyclin Dependent Kinase 5 (CDK5) (60–63). Interestingly, it has been reported that CDK5 phosphorylates DRP1 at both serine 579 and 585 residues, inducing mitochondrial fission and apoptosis in neurons (64,65). Thus, the positive effect of ORM14 on mitochondrial morphology in OPA1 mutant cells may depend on a decrease in mitochondrial fission due to reduced CDK5 protein levels and, in turn, inhibition of DRP1. Recently, ORM14 treatment in a Huntington’s disease mouse model showed antioxidant properties, increasing the expression of NRF2, NQO1 and HO1, and also a pro-autophagic effect (66), as we also observed in R445H MEFs, suggesting that ORM14 may increase autophagic function in models of different diseases.
Intriguing, also ORMs 0, 12 and A identified by our screening seem to be involved in the same pathways or to be tested to treat similar diseases (Supplementary Material, Table S3). Indeed, the literature highlight three relevant common pathways associated with these ORMs: reduction of oxidative stress by NRF2 activation, increase of autophagy through inhibition of the mTOR/Akt pathway and lowering inflammation by downregulation of NF-kB signaling (Fig. 9B and Supplementary Material, Table S3). Furthermore, also ORM12 has been reported to have some positive effects on neurodegenerative diseases, such as Alzheimer (Supplementary Material, Table S3).
Disruption of critical mitochondrial pathways in neurones has deleterious effect on ATP production and elevated ROS level (67). The existence of an inverse relation between ROS production and mitochondrial filamentous network (68) may explain the results obtained on mitochondrial network morphology with ORMs in our OPA1 mutant cells. Indeed, both ORMs 0 and 14, which ameliorate mitochondrial network morphology in our models, have an antioxidant activity that involves NRF2, a master regulator of cellular redox homeostasis (69). NRF2 is involved also in autophagy, activating the transcription of p62 and other macroautophagy genes (70), a pathway that results weakened in mutated MEFs.
It is of interest that the substantial slowdown of autophagy in these cells is in line with the results of our metabolomics analysis evidencing also a significant fall in the cellular amino acids content, more severe in the R445H than in D603H MEFs (71). As stated above, ORMs 0, 2 and 14 were able to enhance the ATP content and, although ORM0 less efficiently, to induce the LC3-I cleavage in R445H MEFs, indicating re-activation of the autophagic process. It is reasonable to suggest that stimulation of autophagy in cells suffering a severe energetic failure may operate as a compensatory mechanism for cellular ATP supply (72). The lack of effect of ORMs on the LC3-I cleavage on D603H MEFs, which do not present a severe energetic impairment, may support this conclusion.
Remarkably, an intimate crosstalk occurs between autophagy and inflammasomes, multi-protein platforms assembled in responses to wide range of different stimuli, including damage-associated molecular patterns (DAMPs). By removing injured organelles as mitochondria, autophagy reduces the release of mitochondria-derived DAMPs and suppression of inflammasome activation (73). Inflammatory signaling and DAMPs have been reported to play a role in neurodegenerative diseases causation and progression (74,75). Indeed, Rodríguez-Nuevo and colleagues reported that a muscle-specific Opa1 ablation in mice resulted in mtDNA-mediated inflammation with activation of NF-kB and TLR9, and that this pathway was reduced by the NF-kB inhibitor sodium salicylate (76). The close link between OPA1 and NF-kB signaling is already known (77–79).
Remarkably, ORMs 0, 12 and 14 present anti-inflammatory activity, supporting the hypothesis of a role of inflammasome and NF-kB pathway in DOA pathogenesis (Fig. 9B).
Thus, the combined use of yeast, MEFs and human models, in addition to allowing the ‘deep phenotyping’ of OPA1 mutations useful for patient’s diagnosis (27), is a powerful tool for therapeutic intervention, taking advantage of the yeast model for high-throughput screening of molecules, whose efficiency is then further validated in the mouse and human cell models.
Finally, our results suggest the possibility that both ROS and inflammation are activated in mutated cells and that ORMs treatment induces both NRF2 and autophagy to counteract the dysregulation of these pathways. We cannot exclude a different involvement of these three pathways depending on the type of OPA1 mutation (Fig. 9A). Overall, comparing the ORMs effects on our DOA cell models, we suggest tolfenamic acid as the most promising compound to be used for drug repurposing in clinical trial for DOA or other neurodegeneration associated with OPA1 mutations.
Materials and Methods
Yeast studies
Yeast strains, plasmids and media
The strain used in this study is W303–1B Δmgm1 (MATα leu2–3 trp1–1 can1–100 ura3–1 ade 2–1 his3–11 mgm1::KanR) transformed with either MGM1 wild-type (wt) or mutant alleles cloned under their natural promoter in the plasmid pFL39 or with CHIM3 wt or mutant alleles cloned under the TETOFF promoter (40). Mutations were introduced in MGM1 or CHIM3 through the overlap PCR technique, using the oligonucleotides reported in Supplementary Material, Table S1. Plasmid were introduced in yeast through the quick LiAc/SS carrier DNA/PEG transformation protocol (80). YP medium contained 0.5% yeast extract (Formedium) and 1% peptone (Formedium); YPA medium was YP 2× supplemented with 40 mg/ml adenine base (Formedium). Carbon sources were added as indicated. Medium was solidified by adding 2% agar (Formedium).
Screening of the chemical libraries in yeast
The primary screening was performed with two chemical libraries: the NCI Bioservices Diversity Set IV, which contains 1596 different compounds (http://www.fisherbioservices.com/, catalog number Div4Oct2014), and the Selleck FDA-approved Drug Library, which contains 1018 compounds (http://www.selleckchem.com/, catalog number L1300, version 2014). The screening was carried out as reported by Lasserre et al. (81). Specifically, strain harboring mgm1I322M mutant allele was grown at 28°C in YP supplemented with 2% ethanol until the early stationary phase was reached. 6 × 105 cells were plated on 120 × 120 mm square Petri dishes (Greiner Bio-One) containing 90 ml of solid YP medium supplemented with 2% ethanol. 32 sterile 6-mm disks of 3MM paper (Whatman) were placed on each plate. Each disk was soaked with 2.5 μl of each drug at a concentration of 10 mm in DMSO, except for two disks, which were soaked with 2.5 μl of DMSO. Plates were incubated at 36.5°C and pictures were taken after 4–5 days. Molecules were considered as positive hits if they enhanced the growth around the paper disk.
The secondary screening was performed on positive hits from the primary screening in the same conditions, except that 4 disks were plated on each plate, among which one soaked with DMSO.
The tertiary screening was performed using W303-1B Δmgm1 strain transformed with chim3S646L. After growth in solid YP supplemented with 2% ethanol and 2% glycerol for three days, 2 × 105 cells were plated on the same square Petri dishes containing 90 ml of YP supplemented with 2% ethanol and 0.5% glycerol. For each positive hit deriving from the secondary screening, 3 disks were soaked with 2.5 μl of a drug dissolved in DMSO at different concentrations depending on the compound, and one disk was soaked with DMSO.
In silico analysis of OPA1 missense mutations and construction of the chim3 mutant alleles
The 63 missense mutations available at https://databases.lovd.nl/shared/genes/OPA1 were analyzed through 8 in silico prediction tools used to foresee their pathogenicity by different algorithms and parameters as reported in supplementary Material and Methods (Supplementary Material, Table S2). Based on the prediction of each tool, a ‘consensus’ score ranging from 0 to 16 was given to each of the missense mutations analyzed. Considering that all the mutations studied so far have a ‘consensus’ score ≥ 11, in most cases 15 or 16, and that all were found highly deleterious in yeast, we focused our attention on mutations having lower ‘consensus’ score, ranging from 5 to 11. Selected mutations were introduced in a YEPlac112-borne CHIM3 allele cloned under the TETOFF promoter through the overlap PCR technique using the oligonucleotides reported in Supplementary Material, Table S1, and the mutant plasmids were introduced in W303–1B Δmgm1 as previously reported (40).
Drug dilution and stock solutions
Positive drugs from the Selleck Chemical Library were purchased from different distributors as reported in Table 1, whereas drugs from the NCI Bioservices Diversity set IV (Table 2), most of which were not commercially available, were a kind gift of the National Cancer Institute. All drugs were diluted in DMSO at concentrations ranging from 5 to 100 mm depending on the maximal solubility. Maximal solubility in YP medium was defined as the concentration at which no pellet was observed after centrifugation of YP medium supplemented with the drug at 15 000g for 10 min.
Spot assay, respiratory activity, growth inhibition and petite frequency in yeast
Oxidative growth was evaluated by spotting serial cell dilutions (5 × 104, 5 × 103, 5 × 102 and 5 × 101 cell/spot), in a total volume of 5 μl, on solid YP medium, supplemented with 2% glucose, 2% glycerol or 2% ethanol.
To measure the respiratory activity, strains were pre-grown in YP supplemented with 2% ethanol to counter select petite cells. Cells were inoculated at a final concentration of 0.08 OD/ml in SC supplemented with 0.5% glucose and grown until glucose was exhausted for approximately 18 h. Petite frequency was lower than 10% in each case. The oxygen consumption rate was measured on whole cells at 30°C using a Clark-type oxygen electrode (Oxygraph System Hansatech Instruments England) as described previously (82).
Petite frequency without drugs was measured on liquid SC medium as previously reported (83). In the case of measurement with ORMs, a growth inhibition curve was performed by growing chim3S646L in YP supplemented with 2% glucose and each ORM at a concentration ranging from 256 μm (or the maximum concentration at which the drug did not precipitate in the medium) to 125 nM, or DMSO as control. 5 × 105 cells were inoculated, and cell density was measured after 24-h growth at 28°C. Petite frequency of strain harboring mgm1I322M allele was performed as previously reported (40), using, for each ORM, concentrations ranging from the concentration which inhibits the growth by 10–20% to concentration 2-, 4- and 8-fold lower. If a decrease of the petite frequency was observed, lower concentrations were tested.
Studies in mouse and human cells
Cells and culture conditions
MEFs stably expressing wt OPA1 isoforms 1 or OPA1 isoform 1 bearing the R445H or D603H mutations were previously described (27,84).
Skin fibroblasts were derived, after having obtained informed written consent, from five healthy donors and two DOA patients carrying the heterozygous R445H and D603H OPA1 mutations, respectively, as previously described in (27).
MEFs and fibroblasts were grown in DMEM (Gibco, Life Technologies, 21969035) containing 25 mm glucose supplemented with 10% fetal bovine serum (FBS, South America, Gibco, Life Technologies, 10270-106), 2 mm l-glutamine (Gibco, Life Technologies, 15140-122), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco, Life Technologies, 25030-024), in an incubator with a humidified atmosphere of 5% CO2 at 37°C. For some experiments, cells were grown in glucose-free DMEM (Gibco, Life Technologies, 11966025) supplemented with 5 mm galactose (Merck, 825V622860), 5 mm sodium pyruvate (Sigma, P2256) and 5% FBS (DMEM-galactose). For evaluation of autophagic flux, MEFs were treated with 1 μm rapamycin (Sigma, R0395) for 24 h and with 80 μm chloroquine diphosphate salt (Sigma, C6628) for 2 h.
MEFs and fibroblasts were grown on Falcon plastic ware (flasks, dishes, multiwells).
Cell viability
MEFs were seeded in 24-well plates and the day after incubated with DMEM-galactose for 24 h in the absence or presence of the ORMs. Cell viability was determined by the sulforhodamine B (SRB) assay (85). Data are expressed as percent of the SRB absorbance measured in DMEM before the incubation to DMEM-galactose (zero time).
Cellular ATP content
MEFs were incubated in DMEM for 24 h and then in DMEM-galactose for other 24 h, in the absence or presence of the ORMs. The ATP content was measured by using the luciferin/luciferase assay, as previously described (86). Data are expressed as percent of the ATP content measured in each cell line in DMEM before switching to DMEM-galactose (zero time).
Mitochondrial network morphology
Mitochondrial network morphology was analyzed as previously described (13).
mtDNA content
Quantification of mouse mtDNA copy number relative to nuclear DNA (nDNA) was carried out amplifying a mitochondrial gene (mt-Nd1) and a nuclear gene (β-Globin).
Quantification was performed using the ΔΔ C(t) method and mtDNA copy number was normalized on the reference nuclear gene β-Globin.
Western blotting
Cell lysates were prepared as previously described (49,87). Aliquots were separated on 8%, 10%, 12% or 15% SDS–PAGE, or on pre-cast NuPAGE 4–12% and 12% Bis-Tris Glycine gels (Life Technologies) and then transferred on nitrocellulose membranes (Bio-Rad). The chemiluminescence signals were measured with Gel Logic 1500 Imaging System, Biosense. The fluorescence signals were detected wtih Licor Odyssey instrument.
Reagents
Antibodies: DRP1(H00010059-M01) and MFN2 (H00009927-M01) from Abnova; OPA1 (612607) and TIM23 (611222) from BD Biosciences; HSP60 (SC-13966) from Santa Cruz; ATG5 (12994), ATG7 (8558), DRP1-phosphoS616 (3455), and TOM20 (42406) from Cell signaling; NDUFA9 (ab14713), SDHA (ab14715), UQCRC2 (ab14745), COX IV (ab14744), ATP5A (ab1801) and CS (ab129095) from Abcam; GAPDH (MAB374) from Millipore; LC3 (L7543), ACTIN (SAB00001) and TUBULIN (T6557) from Sigma-Aldrich; Mitotracker CMX Ros (M7512) from ThermoFisher Scientific. Horseradish peroxidase-conjugated secondary antibodies from Jackson Immuno Research (Mouse 115-035-146, Rabbit 111-035-144). ECL western blotting kit from Biorad or Thermo Scientific Pierce. Fluorescent secondary antibodies anti-rabbit (926-32210) or anti-mouse (926-68071) from Licor.
Statistical analysis
All numerical data are expressed as mean ± SD or SEM, as indicated. Number of biological replicates in independent experiments is detailed in each figure legend. For yeast data, statistical analyses were performed using a one-way ANOVA followed by a Bonferroni’s post-hoc test. For mammalian cell models data, the student’s t-test was used, unless otherwise indicated. Differences were considered statistically significant for P < 0.05.
Conflicts of Interest statement. The authors declare no conflict of interest.
Supplementary Material
Contributor Information
Serena J Aleo, Department of Pharmacy and Biotechnology (FABIT), University of Bologna, Bologna 40126, Italy.
Valentina Del Dotto, Unit of Neurology, Department of Biomedical and NeuroMotor Sciences (DIBINEM), University of Bologna, Bologna 40139, Italy.
Mario Fogazza, Department of Pharmacy and Biotechnology (FABIT), University of Bologna, Bologna 40126, Italy.
Alessandra Maresca, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna 40139, Italy.
Tiziana Lodi, Department of Chemistry, Life Science and Environmental Sustainability, University of Parma, Parma 43124, Italy.
Paola Goffrini, Department of Chemistry, Life Science and Environmental Sustainability, University of Parma, Parma 43124, Italy.
Anna Ghelli, Department of Pharmacy and Biotechnology (FABIT), University of Bologna, Bologna 40126, Italy.
Michela Rugolo, Department of Pharmacy and Biotechnology (FABIT), University of Bologna, Bologna 40126, Italy.
Valerio Carelli, Unit of Neurology, Department of Biomedical and NeuroMotor Sciences (DIBINEM), University of Bologna, Bologna 40139, Italy; IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna 40139, Italy.
Enrico Baruffini, Department of Chemistry, Life Science and Environmental Sustainability, University of Parma, Parma 43124, Italy.
Claudia Zanna, Department of Pharmacy and Biotechnology (FABIT), University of Bologna, Bologna 40126, Italy.
Funding
Futuro in Ricerca FIR2013 from the Ministero della Istruzione Università e Ricerca (MIUR) (RBFR131WDS-001 to C.Z. and RBFR131WDS-002 to E.B.); the Italian Ministry of Health (Ricerca Corrente to V.C.). C.Z. was supported by Fondazione Veronesi research fellowships 2019 and 2020.
References
- 1. Tilokani L., Nagashima S., Paupe V. and Prudent J. (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem., 62, 341–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ge Y., Shi X., Boopathy S., McDonald J., Smith A.W. and Chao L.H. (2020) Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. Elife, 9, e50973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ban T., Ishihara T., Kohno H., Saita S., Ichimura A., Maenaka K., Oka T., Mihara K. and Ishihara N. (2017) Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol., 19, 856–863. [DOI] [PubMed] [Google Scholar]
- 4. Cogliati S., Frezza C., Soriano M.E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L.C. et al. (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell, 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper B. et al. (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell, 14, 177–189. [DOI] [PubMed] [Google Scholar]
- 6. Elachouri G., Vidoni S., Zanna C., Pattyn A., Boukhaddaoui H., Gaget K., Yu-Wai-Man P., Gasparre G., Sarzi E., Delettre C. et al. (2011) OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res., 21, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Narendra D., Tanaka A., Suen D.-F. and Youle R.J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol., 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Twig G., Elorza A., Molina A.J.A., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G. et al. (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J., 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanna R.A., Quinsay M.N., Orogo A.M., Giang K., Rikka S. and Gustafsson A.B. (2012) Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem., 287, 19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu L., Feng D., Chen G., Chen M., Zheng Q., Song P., Ma Q., Zhu C., Wang R., Qi R. et al. (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol., 14, 177–185. [DOI] [PubMed] [Google Scholar]
- 11. Del Dotto V., Fogazza M., Carelli V., Rugolo M. and Zanna C. (2018) Eight human OPA1 isoforms, long and short: what are they for? Biochim. Biophys. Acta Bioenerg., 1859, 263–269. [DOI] [PubMed] [Google Scholar]
- 12. Del Dotto V., Fogazza M., Lenaers G., Rugolo M., Carelli V. and Zanna C. (2018) OPA1: how much do we know to approach therapy? Pharmacol. Res., 131, 199–210. [DOI] [PubMed] [Google Scholar]
- 13. Del Dotto V., Mishra P., Vidoni S., Fogazza M., Maresca A., Caporali L., McCaffery J.M., Cappelletti M., Baruffini E., Lenaers G. et al. (2017) OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep., 19, 2557–2571. [DOI] [PubMed] [Google Scholar]
- 14. Delettre C., Lenaers G., Griffoin J.M., Gigarel N., Lorenzo C., Belenguer P., Pelloquin L., Grosgeorge J., Turc-Carel C., Perret E. et al. (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet., 26, 207–210. [DOI] [PubMed] [Google Scholar]
- 15. Alexander C., Votruba M., Pesch U.E.A., Thiselton D.L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G. et al. (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet., 26, 211–215. [DOI] [PubMed] [Google Scholar]
- 16. Amati-Bonneau P., Valentino M.L., Reynier P., Gallardo M.E., Bornstein B., Boissière A., Campos Y., Rivera H., Aleja J.G., Carroccia R. et al. (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain, 131, 338–351. [DOI] [PubMed] [Google Scholar]
- 17. Hudson G., Amati-Bonneau P., Blakely E.L., Stewart J.D., He L., Schaefer A.M., Griffiths P.G., Ahlqvist K., Suomalainen A., Reynier P. et al. (2008) Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain, 131, 329–337. [DOI] [PubMed] [Google Scholar]
- 18. Yu-Wai-Man P., Griffiths P.G., Gorman G.S., Lourenco C.M., Wright A.F., Auer-Grumbach M., Toscano A., Musumeci O., Valentino M.L., Caporali L. et al. (2010) Multi-system neurological disease is common in patients with OPA1 mutations. Brain, 133, 771–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amati-Bonneau P., Milea D., Bonneau D., Chevrollier A., Ferré M., Guillet V., Gueguen N., Loiseau D., Pou de Crescenzo M.A., Verny C. et al. (2009) OPA1-associated disorders: phenotypes and pathophysiology. Int. J. Biochem. Cell Biol., 41, 1855–1865. [DOI] [PubMed] [Google Scholar]
- 20. Carelli V., Musumeci O., Caporali L., Zanna C., La Morgia C., Del Dotto V., Porcelli A.M., Rugolo M., Valentino M.L., Iommarini L. et al. (2015) Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol., 78, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lynch D.S., Loh S.H.Y., Harley J., Noyce A.J., Martins L.M., Wood N.W., Houlden H. and Plun-Favreau H. (2017) Clinical/scientific notes: nonsyndromic Parkinson disease in a family with autosomal dominant optic atrophy due to opa1 mutations. Neurol. Genet., 3, e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verny C., Loiseau D., Scherer C., Lejeune P., Chevrollier A., Gueguen N., Guillet V., Dubas F., Reynier P., Amati-Bonneau P. et al. (2008) Multiple sclerosis-like disorder in OPA1-related autosomal dominant optic atrophy. Neurology, 70, 1152–1153. [DOI] [PubMed] [Google Scholar]
- 23. Romagnoli M., La Morgia C., Carbonelli M., Di Vito L., Amore G., Zenesini C., Cascavilla M.L., Barboni P. and Carelli V. (2020) Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann. Clin. Transl. Neurol., 7, 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barboni P., Valentino M.L., La Morgia C., Carbonelli M., Savini G., De Negri A., Simonelli F., Sadun F., Caporali L., Maresca A. et al. (2013) Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain, 136, e231. [DOI] [PubMed] [Google Scholar]
- 25. Smith T.G., Seto S., Ganne P. and Votruba M. (2016) A randomized, placebo-controlled trial of the benzoquinone idebenone in a mouse model of OPA1-related dominant optic atrophy reveals a limited therapeutic effect on retinal ganglion cell dendropathy and visual function. Neuroscience, 319, 92–106. [DOI] [PubMed] [Google Scholar]
- 26. Sarzi E., Seveno M., Piro-Mégy C., Elzière L., Quilès M., Péquignot M., Müller A., Hamel C.P., Lenaers G. and Delettre C. (2018) OPA1 gene therapy prevents retinal ganglion cell loss in a dominant optic atrophy mouse model. Sci. Rep., 8, 2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Del Dotto V., Fogazza M., Musiani F., Maresca A., Aleo S.J., Caporali L., La Morgia C., Nolli C., Lodi T., Goffrini P. et al. (2018) Deciphering OPA1 mutations pathogenicity by combined analysis of human, mouse and yeast cell models. Biochim. Biophys. Acta Mol. Basis Dis., 1864, 3496–3514. [DOI] [PubMed] [Google Scholar]
- 28. Faelber K., Dietrich L., Noel J.K., Wollweber F., Pfitzner A.K., Mühleip A., Sánchez R., Kudryashev M., Chiaruttini N., Lilie H. et al. (2019) Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1. Nature, 571, 429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang D., Zhang Y., Ma J., Zhu C., Niu T., Chen W., Pang X., Zhai Y. and Sun F. (2020) Cryo-EM structures of S-OPA1 reveal its interactions with membrane and changes upon nucleotide binding. Elife, 9, e50294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Norcliffe J.L., Alvarez-Ruiz E., Martin-Plaza J.J., Steel P.J. and Denny P.W. (2014) The utility of yeast as a tool for cell-based, target-directed high-throughput screening. Parasitology, 141, 8–16. [DOI] [PubMed] [Google Scholar]
- 31. Jones B.A. and Fangman W.L. (1992) Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes Dev., 6, 380–389. [DOI] [PubMed] [Google Scholar]
- 32. Guan K., Farh L., Marshall T.K. and Deschenes R.J. (1993) Normal mitochondrial structure and genome maintenance in yeast requires the dynamin-like product of the MGM1 gene. Curr. Genet., 24, 141–148. [DOI] [PubMed] [Google Scholar]
- 33. Pelloquin L., Belenguer P., Menon Y. and Ducommun B. (1998) Identification of a fission yeast dynamin-related protein involved in mitochondrial DNA maintenance. Biochem. Biophys. Res. Commun., 251, 720–726. [DOI] [PubMed] [Google Scholar]
- 34. Shepard K.A. and Yaffe M.P. (1999) The yeast dynamin-like protein, Mgm1p, functions on the mitochondrial outer membrane to mediate mitochondrial inheritance. J. Cell Biol., 144, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong E.D., Wagner J.A., Gorsich S.W., McCaffery J.M., Shaw J.M. and Nunnari J. (2000) The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J. Cell Biol., 151, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wong E.D., Wagner J.A., Scott S.V., Okreglak V., Holewinske T.J., Cassidy-Stone A. and Nunnari J. (2003) The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J. Cell Biol., 160, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sesaki H., Southard S.M., Yaffe M.P. and Jensen R.E. (2003) Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol. Biol. Cell, 14, 2342–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amutha B., Gordon D.M., Gu Y. and Pain D. (2004) A novel role of Mgm1p, a dynamin-related GTPase, in ATP synthase assembly and cristae formation/maintenance. Biochem. J., 381, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meeusen S., DeVay R., Block J., Cassidy-Stone A., Wayson S., McCaffery J.M. and Nunnari J. (2006) Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell, 127, 383–395. [DOI] [PubMed] [Google Scholar]
- 40. Nolli C., Goffrini P., Lazzaretti M., Zanna C., Vitale R., Lodi T. and Baruffini E. (2015) Validation of a MGM1/OPA1 chimeric gene for functional analysis in yeast of mutations associated with dominant optic atrophy. Mitochondrion, 25, 38–48. [DOI] [PubMed] [Google Scholar]
- 41. Gns H.S., Gr S., Murahari M. and Krishnamurthy M. (2019) An update on drug repurposing: re-written saga of the drug’s fate. Biomed. Pharmacother., 110, 700–716. [DOI] [PubMed] [Google Scholar]
- 42. Ghelli A., Zanna C., Porcelli A.M., Schapira A.H.V., Martinuzzi A., Carelli V. and Rugolo M. (2003) Leber’s hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J. Biol. Chem., 278, 4145–5150. [DOI] [PubMed] [Google Scholar]
- 43. Ban T., Heymann J.A.W., Song Z., Hinshaw J.E. and Chan D.C. (2010) OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet., 19, 2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kane M.S., Alban J., Desquiret-Dumas V., Gueguen N., Ishak L., Ferre M., Amati-Bonneau P., Procaccio V., Bonneau D., Lenaers G. et al. (2017) Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J. Cell. Mol. Med., 21, 2284–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.D.J., Abeliovich H., Arozena A.A., Adachi H., Adams C.M., Adams P.D., Adeli k. et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy, 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wesselborg S. and Stork B. (2015) Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell. Mol. Life Sci., 72, 4721–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu L., Chen Y. and Tooze S.A. (2018) Autophagy pathway: cellular and molecular mechanisms. Autophagy, 14, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Amati-Bonneau P., Guichet A., Olichon A., Chevrollier A., Viala F., Miot S., Ayuso C., Odent S., Arrouet C., Verny C. et al. (2005) OPA1 R445H mutation in optic atrophy associated with sensorineural deafness. Ann. Neurol., 58, 958–963. [DOI] [PubMed] [Google Scholar]
- 49. Zanna C., Ghelli A., Porcelli A.M., Karbowski M., Youle R.J., Schimpf S., Wissinger B., Pinti M., Cossarizza A., Vidoni S. et al. (2008) OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain, 131, 352–367. [DOI] [PubMed] [Google Scholar]
- 50. Chevrollier A., Guillet V., Loiseau D., Gueguen N., Pou de Crescenzo M.A., Verny C., Ferre M., Dollfus H., Odent S., Milea D. et al. (2008) Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann. Neurol., 63, 794–798. [DOI] [PubMed] [Google Scholar]
- 51. Agier V., Oliviero P., Lainé J., L'Hermitte-Stead C., Girard S., Fillaut S., Jardel C., Bouillaud F., Bulteau A.L. and Lombès A. (2012) Defective mitochondrial fusion, altered respiratory function, and distorted cristae structure in skin fibroblasts with heterozygous OPA1 mutations. Biochim. Biophys. Acta, 1822, 1570–1580. [DOI] [PubMed] [Google Scholar]
- 52. Zimmermann A., Hofer S., Pendl T., Kainz K., Madeo F. and Carmona-Gutierrez D. (2018) Yeast as a tool to identify anti-aging compounds. FEMS Yeast Res., 18, foy020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C. et al. (2018) Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov., 18, 41–58. [DOI] [PubMed] [Google Scholar]
- 54. Delerue T., Tribouillard-Tanvier D., Daloyau M., Khosrobakhsh F., Emorine L.J., Friocourt G., Belenguer P., Blondel M. and Arnauné-Pelloquin L. (2019) A yeast-based screening assay identifies repurposed drugs that suppress mitochondrial fusion and mtDNA maintenance defects. DMM Dis. Model. Mech., 12, dmm036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shao H.J., Lou Z., Jeong J.B., Kim K.J., Lee J. and Lee S.H. (2015) Tolfenamic acid suppresses inflammatory stimuli-mediated activation of NF-κB signaling. Biomol. Ther., 23, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pathi S., Li X. and Safe S. (2014) Tolfenamic acid inhibits colon cancer cell and tumor growth and induces degradation of specificity protein (Sp) transcription factors. Mol. Carcinog., 53, E53–E61. [DOI] [PubMed] [Google Scholar]
- 57. Wilson W.J., Afzali M.F., Cummings J.E., Legare M.E., Tjalkens R.B., Allen C.P., Slayden R.A. and Hanneman W.H. (2016) Immune modulation as an effective adjunct post-exposure therapeutic for B. pseudomallei. PLoS Negl. Trop. Dis., 10, e0005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guan P.P. and Wang P. (2019) Integrated communications between cyclooxygenase-2 and Alzheimer’s disease. FASEB J., 33, 13–33. [DOI] [PubMed] [Google Scholar]
- 59. Kaur K., Gill J.S., Bansal P.K. and Deshmukh R. (2017) Neuroinflammation—a major cause for striatal dopaminergic degeneration in Parkinson’s disease. J. Neurol. Sci., 381, 308–314. [DOI] [PubMed] [Google Scholar]
- 60. Subaiea G.M., Adwan L.I., Ahmed A.H., Stevens K.E. and Zawia N.H. (2013) Short-term treatment with tolfenamic acid improves cognitive functions in Alzheimer’s disease mice. Neurobiol. Aging, 34, 2421–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Subaiea G.M., Ahmed A.H., Adwan L.I. and Zawia N.H. (2015) Reduction of amyloid-β deposition and attenuation of memory deficits by tolfenamic acid. J. Alzheimers Dis., 43, 425–433. [DOI] [PubMed] [Google Scholar]
- 62. Adwan L., Subaiea G.M., Basha R., Basha R. and Zawia N.H. (2015) Tolfenamic acid reduces tau and CDK5 levels: implications for dementia and tauopathies. J. Neurochem., 133, 266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chang J.K., Leso A., Subaiea G.M., Lahouel A., Masoud A., Mushtaq F., Deeb R., Eid A., Dash M., Bihaqiet S.W. et al. (2018) Tolfenamic acid: a modifier of the tau protein and its role in cognition and tauopathy. Curr. Alzheimer Res., 15, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jahani-Asl A., Huang E., Irrcher I., Rashidian J., Ishihara N., Lagace D.C., Slack R.S. and Park D.S. (2015) CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum. Mol. Genet., 24, 4573–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guo M.Y., Shang L., Hu Y.Y., Jiang L.P., Wan Y.Y., Zhou Q.Q., Zhang K., Liao H.F., Yi J.L. and Han X.J. (2018) The role of Cdk5-mediated Drp1 phosphorylation in Aβ 1-42 induced mitochondrial fission and neuronal apoptosis. J. Cell. Biochem., 119, 4815–4825. [DOI] [PubMed] [Google Scholar]
- 66. Liu P., Li Y., Yang W., Liu D., Ji X., Chi T., Guo Z., Li L. and Zou L. (2019) Prevention of Huntington’s disease-like behavioral deficits in R6/1 mouse by tolfenamic acid is associated with decreases in mutant huntingtin and oxidative stress. Oxidative Med. Cell. Longev., 2019, 4032428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yu-Wai-Man P., Votruba M., Burté F., La Morgia C., Barboni P. and Carelli V. (2016) A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol., 132, 789–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Willems P.H.G.M., Rossignol R., Dieteren C.E.J., Murphy M.P. and Koopman W.J.H. (2015) Redox homeostasis and mitochondrial dynamics. Cell Metab., 22, 207–218. [DOI] [PubMed] [Google Scholar]
- 69. Holmström K.M., Kostov R.V. and Dinkova-Kostova A.T. (2016) The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol., 1, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pajares M., Jiménez-Moreno N., García-Yagüe Á.J., Escoll M., Ceballos M.L., Van Leuven F., Rábano A., Yamamoto M., Rojo A.I. and Cuadrado A. (2016) Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy, 12, 1902–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chao de la Barca J.M., Fogazza M., Rugolo M., Chupin S., Del Dotto V., Ghelli A.M., Carelli V., Simard G., Procaccio V., Bonneau D. et al. (2020) Metabolomics hallmarks OPA1 variants correlating with their in-vitro phenotype and predicting clinical severity. Hum. Mol. Genet., 29, 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yang J., Zhou R. and Ma Z. (2019) Autophagy and energy metabolism. Adv. Exp. Med. Biol., 1206, 329–357. [DOI] [PubMed] [Google Scholar]
- 73. Sun Q., Fan J., Billiar T.R. and Scott M.J. (2017) Inflammasome and autophagy regulation: a two-way street. Mol. Med., 23, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chitnis T. and Weiner H.L. (2017) CNS inflammation and neurodegeneration. J. Clin. Invest., 127, 3577–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grazioli S. and Pugin J. (2018) Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front. Immunol., 9, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rodríguez-Nuevo A., Díaz-Ramos A., Noguera E., Díaz-Sáez F., Duran X., Muñoz J.P., Romero M., Plana N., Sebastián D., Tezze C. et al. (2018) Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J., 37, e96553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Laforge M., Rodrigues V., Silvestre R., Gautier C., Weil R., Corti O. and Estaquier J. (2016) NF-κB pathway controls mitochondrial dynamics. Cell Death Differ., 23, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Herkenne S., Ek O., Zamberlan M., Pellattiero A., Chergova M., Chivite I., Novotná E., Rigoni G., Branco Fonseca T., Samardzicet D. et al. (2020) Developmental and tumor angiogenesis requires the mitochondria-shaping protein Opa1. Cell Metab., 31, 987–1003. [DOI] [PubMed] [Google Scholar]
- 79. Albensi B.C. (2019) What is nuclear factor kappa B (NF-κB) doing in and to the mitochondrion? Front. Cell Dev. Biol., 7, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gietz R.D. (2014) Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol. Biol., 1205, 1–12. [DOI] [PubMed] [Google Scholar]
- 81. Lasserre J.-P., Dautant A., Aiyar R.S., Kucharczyk R., Glatigny A., Tribouillard-Tanvier D., Rytka J., Blondel M., Skoczen N., Reynier P. et al. (2015) Yeast as a system for modeling mitochondrial disease mechanisms and discovering therapies. Dis. Model. Mech., 8, 509–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Panizza E., Ercolino T., Mori L., Rapizzi E., Castellano M., Opocher G., Ferrero I., Neumann H.P.H., Mannelli M. and Goffrini P. (2013) Yeast model for evaluating the pathogenic significance of SDHB, SDHC and SDHD mutations in PHEO-PGL syndrome. Hum. Mol. Genet., 22, 804–815. [DOI] [PubMed] [Google Scholar]
- 83. Baruffini E., Ferrero I. and Foury F. (2010) In vivo analysis of mtDNA replication defects in yeast. Methods, 51, 426–436. [DOI] [PubMed] [Google Scholar]
- 84. Song Z., Chen H., Fiket M., Alexander C. and Chan D.C. (2007) OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol., 178, 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Porcelli A.M., Ghelli A., Iommarini L., Mariani E., Hoque M., Zanna C., Gasparre G. and Rugolo M. (2008) The antioxidant function of Bcl-2 preserves cytoskeletal stability of cells with defective respiratory complex I. Cell. Mol. Life Sci., 65, 2943–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zanna C., Ghelli A., Porcelli A.M., Martinuzzi A., Carelli V. and Rugolo M. (2005) Caspase-independent death of Leber’s hereditary optic neuropathy cybrids is driven by energetic failure and mediated by AIF and endonuclease G. Apoptosis, 10, 997–1007. [DOI] [PubMed] [Google Scholar]
- 87. Del Dotto V., Ullah F., Di Meo I., Magini P., Gusic M., Maresca M., Caporali L., Palombo F., Tagliavini F., Harris Baugh E. et al. (2020) SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. J. Clin. Invest., 130, 108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.