

Abstract
Mutations in retinitis pigmentosa GTPase regulator (RPGR) cause severe retinal ciliopathy, X-linked retinitis pigmentosa. Although two major alternatively spliced isoforms, RPGRex1-19 and RPGRORF15, are expressed, the relative importance of these isoforms in disease pathogenesis is unclear. Here, we analyzed fibroblast samples from eight patients and found that all of them form longer cilia than normal controls, albeit to different degrees. Although all mutant RPGRORF15 messenger RNAs (mRNAs) are unstable, their steady-state levels were similar or higher than those in the control cells, suggesting there may be increased transcription. Three of the fibroblasts that had higher levels of mutant RPGRORF15 mRNA also exhibited significantly higher levels of RPGRex1-19 mRNA. Four samples with unaltered RPGRex1-19 levels carried mutations in RPGRORF15 that resulted in this isoform being relatively less stable. Thus, in all cases, the RPGRex1-19/RPGRORF15 isoform ratio was increased, and this was highly correlative to the cilia extension defect. Moreover, overexpression of RPGRex1-19 (mimicking the increase in RPGRex1-19 to RPGRORF15 isoform ratio) or RPGRORF15 (mimicking reduction of the ratio) resulted in significantly longer or shorter cilia, respectively. Notably, the cilia length defect appears to be attributable to both the loss of the wild-type RPGRORF15 protein and to the higher levels of the RPGRex1-19 isoform, indicating that the observed defect is due to the altered isoform ratios. These results suggest that maintaining the optimal RPGRex1-9 to RPGRORF15 ratio is critical for cilia growth and that designing strategies that focus on the best ways to restore the RPGRex1-19/RPGRORF15 ratio may lead to better therapeutic outcomes.
Introduction
Primary cilia are evolutionarily conserved microtubule-based non-motile protrusions of the plasma membrane of most vertebrate cell types (1). They are nucleated from the basal bodies (mother centrioles) that dock at the apical plasma membrane in quiescent cells. The proximal region of the cilia, also called transition zone, acts as a gate. The microtubules further extend to form a distal axoneme surrounded by the ciliary membrane. The primary cilium regulates critical developmental and homeostatic signaling cascades by targeting several integral and peripheral membrane proteins as well as soluble moieties along the axoneme via a conserved mechanism termed intraflagellar transport (2,3). The trafficking is also regulated by macromolecular complexes of several proteins including retinitis pigmentosa GTPase regulator (RPGR), centrosomal protein 290 kDa (CEP290), centrosomal protein 110 kDa (CP110), Usher Syndrome proteins and Bardet Biedl Syndrome proteins (4,5). Mutations in these proteins are associated with severe developmental and homeostatic disorders collectively termed ciliopathies (6). These include Meckel–Gruber Syndrome, Joubert Syndrome, Senior–Loken Syndrome, Usher Syndrome and some forms of non-syndromic photoreceptor degenerations.
Photoreceptors (rods and cones) have a modified sensory cilium in the form of the light-sensing outer segment. The photoreceptors are one of the highly metabolically active cell types in the body. They depend upon stringently controlled proteins and lipid trafficking from their site of synthesis in the inner segment to the outer segment for light detection (7). Perturbations in the trafficking machinery due to functional deficits in ciliary proteins lead to severe photoreceptor degeneration (2,3,8). One such ciliary trafficking regulator is RPGR.
Mutations in RPGR are associated with X-linked forms of retinitis pigmentosa (RP), a debilitating blindness characterized by severe and progressive photoreceptor degeneration (9–12). RPGR mutations account for the majority (~70%) of X-linked retinitis pigmentosa (XLRP) cases and 12–15% of simplex RP, with no family history (12–14). In addition, RPGR mutations are associated with cone–rod degeneration, macular dystrophy and extraocular presentations, such as sperm abnormalities, hearing defects and primary cilia dyskinesia (15–19). Overall, RPGR mutations are a leading cause of blindness accounting for almost 20% of all RP cases.
The RPGR gene encodes two major alternatively spliced isoforms: RPGRex1-19 (19 exons) and RPGRORF15 (terminating in intron 15; exon ORF15) (20–23). The terminal exon 19 of the RPGRex1-19 isoform carries an isoprenylation signal involved in its localization to cilia (24,25). Exon ORF15 of the RPGRORF15 isoform contains purine-rich repeats that encode for a glutamic acid and glycine-rich domain (26). As majority of RPGR mutations are located in exon ORF15, the RPGRORF15 isoform has been considered the major disease-associated isoform in humans (26). The great majority of exon ORF15 mutations are predicted to prematurely truncate the RPGRORF15 protein.
Both RPGR isoforms are expressed throughout the body, but are particularly abundant in the retina. Previous studies have identified mouse and canine models of RPGR mutations and have suggested the involvement of RPGR in regulating ciliary trafficking by acting as a gatekeeper at the transition zone (27–31). Loss of RPGR alters protein trafficking to the photoreceptor outer segment, but permits its initial generation. RPGR depletion alters cilia length in cultured cell lines and fibroblasts (32). However, the effect on the ciliary defects of different RPGRORF15 mutations is unclear.
To investigate the effect of the human disease-causing mutations in RPGRORF15 on its expression and on cilia formation, we analyzed RPGR isoform expression, cilia formation and ciliary protein trafficking in patient-derived cells. Our studies unravel a new potential pathogenic mechanism of RPGRORF15 disease, which may assist in developing new therapies for these conditions.
Results
Ciliary defects in RPGRORF15-mutant fibroblasts
Given that RPGR regulates cilia length (32), we examined cilia formation in the patient and control fibroblasts. We generated primary fibroblasts from skin biopsy specimens from eight male RPGRORF15 patients (RPGR-1, RPGR-2, RPGR-3, RPGR-4, RPGR-5, RPGR-6, RPGR-7 and RPGR-8) with frameshift and truncation mutations in exon ORF15 (Fig. 1A and B). The patients were all from families previously confirmed to have X-linked RP due to RPGRORF15 mutations (33,34); their ages at skin biopsy ranged from 21 to 66 years. The patients had ophthalmoscopic features of pigmentary retinopathy, abnormal visual fields and reduced or non-detectable electroretinograms (33,34). As the control samples from the same family members could not be obtained, we included six random asymptomatic control samples without RPGR mutations (CTL-1, CTL-2, CTL-3, CTL-4, CTL-5 and CTL-6).
Figure 1.
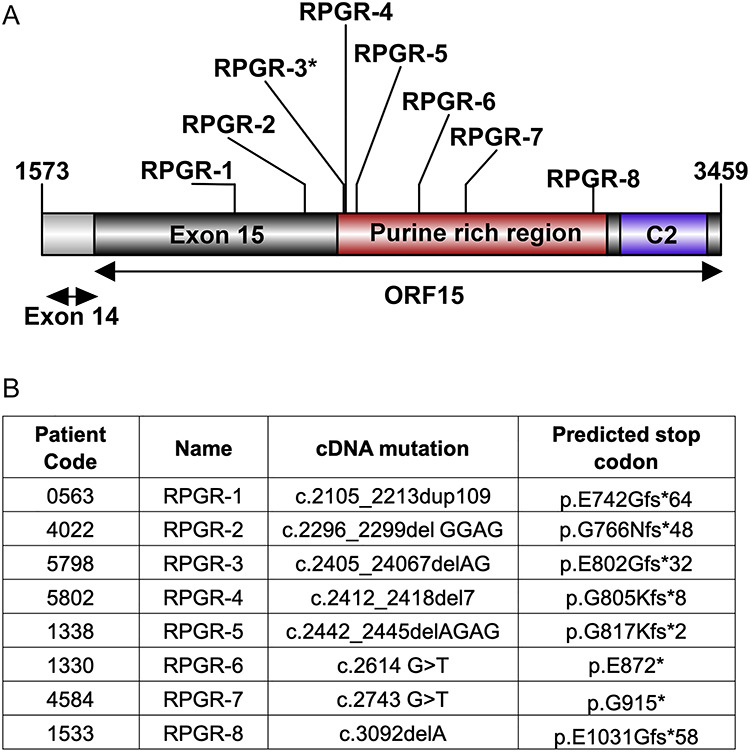
RPGRORF15 mutations. Schematic representation (A) and predicted effect of RPGRORF15 mutations (B) in XLRP patients. *: optic cups of RPGR-3 were generated.
Although the control and RPGR-2 fibroblasts grew cilia in the range of 1–1.5 μm, we detected two groups of mutations with longer cilia: cilia were elongated to a range of 2.5–3.5 μm range in the RPGR-1, RPGR-3, RPGR-4 and RPGR-5 fibroblasts and to a lesser extent (1.8–2.5 μm) in RPGR-6, RPGR-7 and RPGR-8 (Fig. 2A and B). As predicted, we did not detect an effect on the number of ciliated cells.
Figure 2.
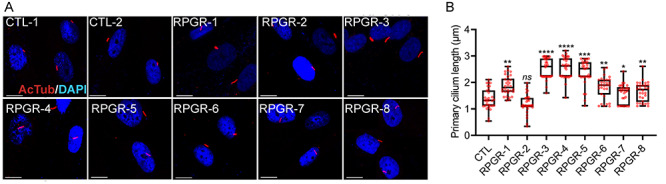
Ciliary defects in fibroblasts. (A) Control (CTL-1 and CTL-2) and mutant fibroblasts were stained for acetylated α-tubulin (red, ciliary marker) and 4′,6-diamidino-2-phenylindole (DAPI) (blue, nucleus). Scale: 5 μm. (B) The cilia length was quantified using the ImageJ software (n > 100). The CTL data represents the average cilia length of all control fibroblasts. ****P < 0.0001; ***P < 0.002; **P < 0.01; *P < 0.05.
Correlation of cilia length to RPGR isoform levels in fibroblasts
We next asked whether the observed ciliary phenotype was associated with an effect of exon ORF15 mutations on RPGR expression. To this end, we performed quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) analysis of the two major isoforms. We found significantly higher levels (8–10-fold) of the RPGRex1-19 isoform in RPGR-3, RPGR-4 and RPGR-5 fibroblasts samples when compared with the other RPGR mutants or the control fibroblasts (Fig. 3A). We also detected higher levels (2–3-fold) of the mutant RPGRORF15 isoform in RPGR-3, RPGR-4 and RPGR-5. Interestingly, the level of the mutant RPGRORF15 in RPGR-1 was significantly reduced when compared with the controls (Fig. 3B).
Figure 3.
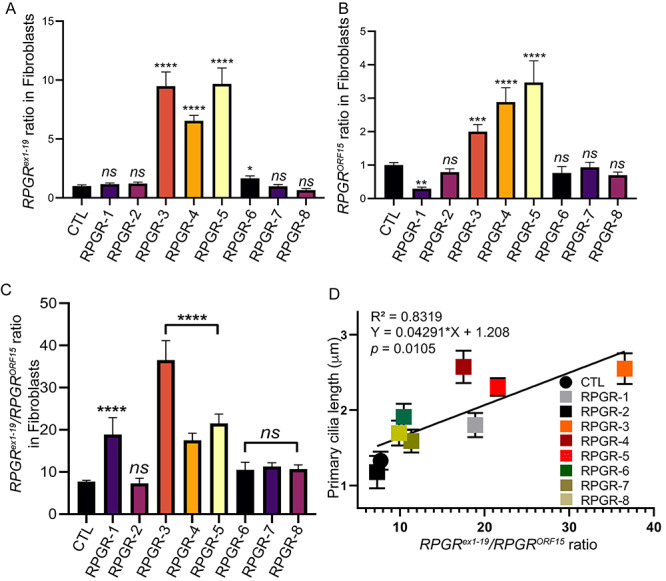
RPGR isoform levels in patient-derived fibroblasts. (A, B) RPGRex1-19 (A) or RPGRORF15 levels in the mutant fibroblasts relative to controls (CTL) were analyzed. (C) The ratio of RPGRex1-19 to RPGRORF15 mRNA levels was determined. RPGR-1, RPGR-3, RPGR-4 and RPGR-5 showed significantly higher ratio when compared with control (CTL) cells. ns, not significant. Data are mean ± SD from five independent experiments. (D) Positive correlation of the cilium length with the RPGRex1-19/RPGRORF15 ratio was observed. Each dot represents average cilium length of >100 cilia compared with the RPGRex1-19/RPGRORF15 ratio from three independent experiments. Black line shows a linear fit through the data. Inset displays the coefficient of determination (R2) and significance (P-value).
Given that RPGR-1, RPGR-3, RPGR-4 and RPGR-5 also formed relatively longer cilia, we tested whether there was a correlation between the RPGRex1-19/RPGRORF15 ratio and the cilia length. Although the RPGR-2, RPGR-6, RPGR-7, RPGR-8 and control fibroblasts showed an RPGRex1-19/RPGRORF15 ratio of ~10-fold, in RPGR-1, RPGR-3, RPGR-4 and RPGR-5 fibroblasts, there was an increased ratio of between 18- and 35-fold (Fig. 3C). Furthermore, we found that the length of the cilia was highly correlative to RPGRex1-19/RPGRORF15 ratio, with the coefficient of determination (R2) of ~83% (Fig. 3D). Using RPGR isoform-specific antibodies (Supplementary Fig. S1A), we also detected a significant increase (18–20-fold) in the ratio of the RPGRex1-19/RPGRORF15 protein levels in RPGR-1, RPGR-3, RPGR-4 and RPGR-5 when compared with other RPGR mutations or controls (Supplementary Fig. S1B and C). We then checked whether such phenomenon of increased RPGRex1-19/RPGRORF15 ratio is also conserved in retinal microenvironment. Using retinal organoids derived from reprogrammed control and RPGR-3 human-induced pluripotent stem cells (iPSCs; Supplementary Fig. S2A and B) (West et al. 2020, manuscript submitted), we found significant (~2.8-fold) increase in the RPGRex1-19/RPGRORF15 ratio in the mutant sample as compared with the controls (Supplementary Fig. S2C).
These results suggest that RPGRORF15 mutations tested in this study affect not only mutant RPGRORF15 expression levels but also the RPGRex1-19 isoform levels, albeit to different extents. Although in fibroblast samples from all RPGR patients there was a positive correlation between the RPGRex1-19/RPGRORF15 ratio and cilia length, in RPGR-1, RPGR-3, RPGR-4 and RPGR-5 samples, there were distinct effects on the RPGR messenger RNA (mRNA) levels. The RPGR-1 mutation results in significantly reduced mutant RPGRORF15 expression, but does not affect the RPGRex1-19 levels. On the other hand, although the mutant RPGRORF15 is expressed at relatively higher levels in RPGR-3, RPGR-4 and RPGR-5 fibroblast samples, they exhibit significantly higher levels of RPGRex1-19. To further understand the ciliary defects, we focused on RPGR-1, RPGR-3, RPGR-4 and RPGR-5 samples. We also used RPGR-2 as it showed similar RPGR expression as the control fibroblasts.
Localization of cilia length regulators in RPGRORF15-mutant fibroblasts
To investigate the ciliary defect in the fibroblasts with elongated cilia, we assessed the distribution of ciliary length regulators CEP290 and distribution of glutamylated tubulin. CEP290 mutations are a frequent cause of Leber congenital amaurosis and syndromic ciliopathies (35–37). Immunofluorescence staining of RPGR-1, RPGR-3, RPGR-4 and RPGR-5 with a previously reported CEP290 antibody demonstrated ~50% reduction in the levels of CEP290 at the basal body (determined by the co-localization with basal body marker γ-tubulin) and concomitant increase in the cytosolic CEP290 distribution when compared with RPGR-2 or control fibroblasts (Fig. 4A and B). However, the number of CEP290-positive cells was not affected in the mutant as compared with the control fibroblasts (Fig. 4C). These results are consistent with previous studies that demonstrated longer cilia in CEP290-mutant patient fibroblasts and in Cep290-mutant mice (38,39) and suggest an association between the observed cilia length differences and CEP290 levels in the RPGR-mutant fibroblasts.
Figure 4.
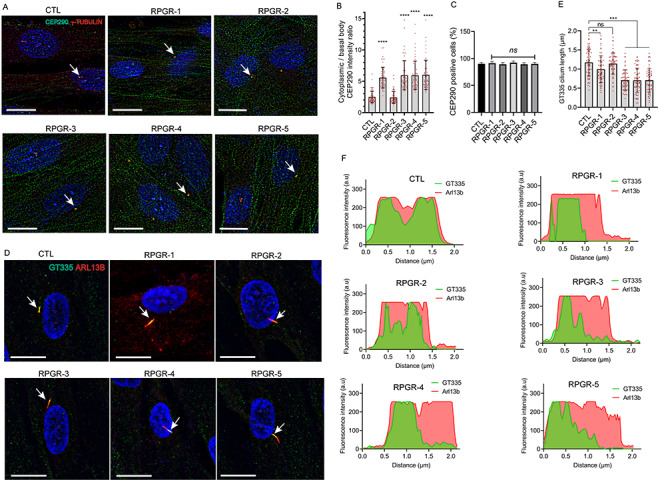
Distribution of ciliary markers. CEP290 (A–C) and GT335 (D–F) staining in the indicated fibroblasts was assessed. (A) The cells were stained with CEP290 (green) and γ-tubulin (basal body marker, red) antibodies and the ratio of the CEP290-positive signal in the cytoplasm to that at the basal body (B) and the number of CEP290-positive cells (C) were quantified using ImageJ (n > 100). (D) The cells were stained with GT335 (green, glutamylated tubulin) and ARL13B (red, cilia marker). (E) The distribution of GT335 (green signal intensity) along the length of the cilium when compared with ARL13B (red intensity) (distance, μm) was quantified using IMARIS software (Oxford Instruments). a.u., arbitrary units.
Tubulin polyglutamylation is a modification of the stable ciliary microtubules, which can be detected using the GT335 antibody (40,41). We found that the distribution of GT335 was altered in the RPGRORF15-mutant fibroblasts. Immunofluorescence analysis revealed that GT335 immunoreactivity was restricted to the proximal region of the cilia in RPGR-1, RPGR-3, RPGR-4 and RPGR-5 (within ~1.0 μm) (Fig. 4D and E) when compared with RPGR-2 and the controls, in which GT335 staining is distributed across the whole cilium (Fig. 4F).
Expression of wild-type RPGRORF15 in mutant fibroblasts
We then sought to determine whether the observed cilia length defects are due to the lack of the wild-type RPGRORF15 protein. Transfection of the wild-type human green fluorescent protein (GFP)-RPGRORF15-encoding complementary DNA (cDNA) into RPGR-3, RPGR-4 and RPGR-5 did not completely reverse the elongated cilia phenotype when compared with cells transfected with only GFP. Although the RPGR-3, RPGR-4 and RPGR-5 fibroblasts transfected with GFP alone formed longer cilia than the CTL (control) fibroblasts, the cilia length reduced from 2.2–2.4 μm to 1.3–1.5 μm after transfection with GFP-RPGRORF15 (Fig. 5A and B). RPGR-1, which had relatively modest (~1.8 μm) increase in the cilia length and RPGRex1-19/RPGRORF15 ratio showed almost complete reversal (~1 μm) of the elongated cilia phenotype upon GFP-RPGRORF15 expression. These data strongly suggest that the normal RPGRORF15 protein function partly contributes to the observed cilia length changes. Samples with relatively higher RPGRex1-19/RPGRORF15 ratio did not show complete reversal of the cilia length phenotype. Further support of this hypothesis comes from the observation that expressing RPGRORF15 in the control or RPGR-2 (~20% longer cilia) cells significantly reduced the cilia length 10–20% less than the average lengths of the GFP-expressing control cells. All cells transfected with RPGRORF15-encoding cDNA overexpressed RPGRORF15 to similar levels (4–5-fold higher) when compared with GFP-expressing control cells (Supplementary Fig. S3A) and did not alter the RPGRex1-19 levels (Supplementary Fig. S3B).
Figure 5.
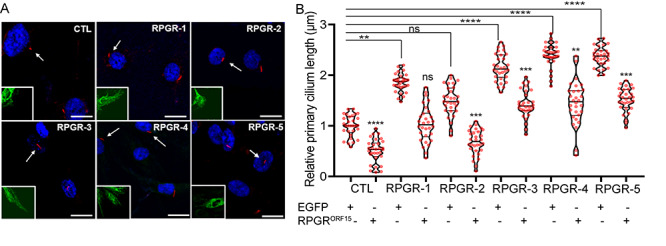
Effect of wild-type RPGRORF15 overexpression on the cilia defect. (A) The fibroblasts were transiently transfected with plasmids-encoding GFP or GFP-RPGRORF15 and processed for staining with the GFP (green) or acetylated α-tubulin (red, ciliary marker). Arrows point to the ciliated cells marked in the inset in green channel. The nuclei are stained with DAPI (blue). (B) The cilia length was quantified using ImageJ (n > 100). The graph shows the cilia length relative to the length of the control (CTL) fibroblasts transfected with only GFP-encoding cDNA. The statistical significance of the cilia length in the GFP or GFP-RPGRORF15-transfected mutant fibroblasts cells was determined relative to the GFP-transfected control fibroblasts. **P < 0.01; ***P < 0.0002; ****P < 0.0001; ns, not significant. Note that the GFP-RPGRORF15-transfected RPGR-2 and CTL cells have significantly shorter cilia when compared with the GFP-transfected counterparts.
Some RPGRORF15-mutant isoforms are less stable
Varying mRNA levels of the RPGR isoforms could be due to the mutations affecting the purine-rich repeat region of the genomic DNA or RPGR mRNA stability. As some RPGRORF15 mutations (RPGR-2, RPGR-6, RPGR-7 and RPGR-8) did not affect the isoform ratio, we concluded that the DNA architecture in the region of those mutations is not sufficient to cause the observed changes. We then tested whether the effect on RPGR isoform ratio is due to changes in its stability. We performed transcription inhibition assays using actinomycin D to assess the half-lives of the RPGR isoforms. Our studies revealed that the mutant RPGRORF15 levels rapidly decrease to ~10% of the total levels within the first hour of actinomycin D treatment and are at minimal levels by 4 h in RPGR-3, RPGR-4 and RPGR-5 samples. Their levels also remained significantly lower than the control at all time points. However, the rate of decline of the mutant RPGRORF15 levels in RPGR-1 and RPGR-2 samples was overall similar to that of the control RPGRORF15 mRNA. There was, however, a slightly faster decline of the mutant RPGRORF15 levels in RPGR-1 and RPGR-2 at 6 and 8 h of actinomycin D treatment when compared with the control (Fig. 6A). As predicted, we did not detect a significant difference in the decay of the RPGRex1-19 mRNA in the patient fibroblasts relative to the controls (Fig. 6B). Based on these studies, we found that the half-life of the mutant RPGRORF15 in RPGR-1 and RPGR-2 after actinomycin D treatment relative to vehicle treatment was similar to that in control fibroblasts (~2.3 and 2.2 h for RPGR-1 and RPGR-2, respectively, and 1.6 h for control) (Fig. 4C), whereas the half-life of the mutant RPGRORF15 in RPGR-3, RPGR-4 and RPGR-5 was 0.67, 0.51 and 0.28 h, respectively. Linear regression analysis further validated that the decay kinetics of mutant RPGRORF15 in RPGR-3, RPGR-4 and RPGR-5 does not follow a linear correlation (Fig. 6C).
Figure 6.
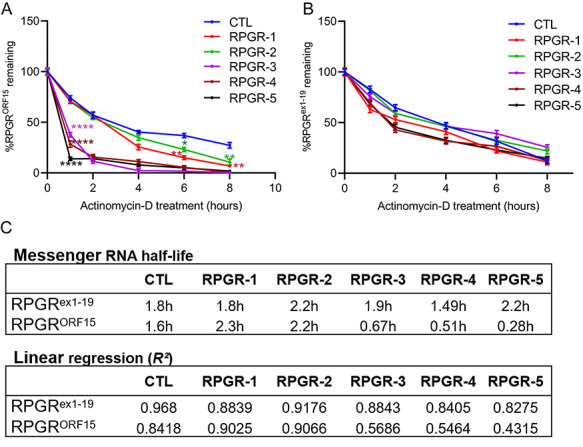
Mutant mRNA decay in RPGRORF15 fibroblasts. Quantitative PCR analysis of RPGRORF15 (A) or RPGRex1-19 (B) isoform was performed in fibroblasts treated with actinomycin D for indicated time. The levels of the mutant isoforms were calculated relative to the levels in the control (CTL) fibroblasts at the same time points. Statistical significance was determined by assessing the mRNA levels at each time point relative to that of the control (CTL) at the same time point. ****P < 0.0001; **P < 0.01; *P < 0.05. No significant changes in the degradation of RPGRex-19 mRNA were detected at the same time points (B). (C) The half-life and linear regression of the mutant RPGRORF15 and RPGRex1-19 mRNAs was calculated by determining the time (in hours) taken by the isoforms to reduce to half the original levels.
RPGRex1-19 knockdown in mutant fibroblasts
If RPGRORF15 only partly reversed the cilia length, the increase in the RPGRex1-19 levels could also be relevant to the observed cilia elongation in the RPGRORF15-mutant fibroblasts. To test this, we overexpressed RPGRex1-19 in human telomerase reverse transcriptase-retinal pigmented epithelium (hTERT-RPE1) cells by transient transfection. Our analysis revealed 3–4-fold longer cilia in the RPGRex1-19 overexpressing cells as compared with the control cells (Supplementary Fig. S4A). Thus, we hypothesized that reducing the RPGRex1-19 levels will alleviate the elongated cilia phenotype. To this end, we performed small interfering ribonucleic acid (siRNA)-mediated depletion of RPGRex1-19. We designed three siRNAs (siRNA-1, siRNA-2 and siRNA-3) against RPGRex1-19. All siRNAs reduced the expression of RPGRex1-19, with maximal inhibition observed with siRNA-3 (Supplementary Fig. S4B), when compared with the levels expressed in cells transfected with a scrambled sequence control. Moreover, the siRNAs did not affect RPGRORF15 levels in the fibroblasts (Supplementary Fig. S4C). Transfection of RPGRex1-19-siRNAs followed by immunofluorescence analysis in RPGR-3 and RPGR-5 revealed an almost complete reversal of the cilia elongation phenotype with the cilia length comparable with that of the scrambled siRNA-treated control fibroblasts. RPGR-4 showed ~90% reduction in cilia length compared with scrambled siRNA-treated cells (Fig. 7A and B).
Figure 7.
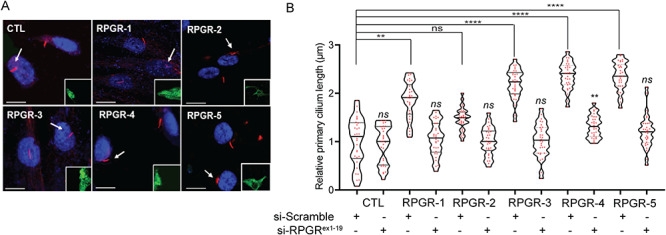
Effect of RPGRex1-19 knockdown on the cilia defect. (A) The fibroblasts were transiently transfected with RPGR-siRNA-3 or scrambled siRNA followed by GFP (green, inset) and acetylated α-tubulin staining. (B) The cilia length was quantified represented as the length relative to the scrambled siRNA-treated control (CTL) cells. Scale: 5 μm; ns, not significant.
Discussion
Although RPGRORF15 gene augmentation therapy has been successful in naturally occurring canine mutations and murine models (42,43) and is currently in human clinical trials, efficacy of the treatment across the large spectrum of human exon ORF15 mutations is currently not known. We show that all eight RPGRORF15 mutations tested here cause cilia length defects of varying extents, lead to mutant mRNA instability and four mutations lead to a significant increase in the RPGRex1-19/RPGRORF15 ratio. Our results indicate that three out of four exon ORF15 mutations that result in an increase in the RPGRex1-19/RPGRORF15 mRNA ratio have shorter half-life due to relatively faster mutant mRNA decay, yet have relatively higher steady-state levels of the mutant RPGRORF15 mRNA and protein. The precise mechanism of the transcriptional complexity observed in this study is not clear. Nonetheless, these results point to transcriptional adaptation in which the unstable mRNA transcripts can result in a feedback response to increase the levels of the protein. RPGR transcriptional adaptation could increase the RPGRORF15 protein levels; however, at the same time also increase RPGRex1-19 levels, as observed in this study. Such transcriptional adaptation may result from the need to compensate for the decaying mutant RPGRORF15 transcripts (44–46); however, its relevance to retinal disease remains to be determined. We also found that the expression of wild-type RPGRORF15 did not affect the elevated RPGRex1-19 levels in fibroblasts. This suggests that the mechanism of increase in the RPGRex1-19 levels is not due to the absence of the wild-type RPGRORF15 and implicates the expression of the mutant ORF15 isoforms. The observed effects on cilia growth, on the other hand, seem to be due in part to the lack of the RPGRORF15 protein and to the relatively increased expression of RPGRex1-19. This is because RPGRORF15 overexpression or RPGRex1-19 knockdown were able to partly reverse the cilia defect, with RPGRex1-19 knockdown showing better reversal of the cilia length. These results suggest that the relative levels of both RPGR isoforms are critical for optimal cilia growth. Further support of this hypothesis comes from our observation that the overexpression of RPGRex1-19 leads to longer cilia while that of RPGRORF15 results in shorter cilia. In addition, previous studies showed that overexpression of Rpgrex1-19 or RPGRORF15 in mice results in severe retinal degeneration (47,48).
What is the mechanism of cilia elongation in RPGRORF15-mutant samples? We found that the imbalanced RPGRex1-19/RPGRORF15 ratio subsequently results in higher overall levels of the RPGR proteins. This could lead to increased context-dependent function of RPGR-containing protein complexes at the cilia and alteration of the gatekeeping function at the transition zone. We previously showed that RPGR regulates the composition of the ciliary outer segments of photoreceptors by affecting its ability to maintain the barrier at the transition zone (30). RPGRex1-19 is also involved in regulating ciliary cargo trafficking in cultured cells as well as mouse photoreceptors (24). Moreover, both RPGR isoforms interact genetically and physically with CEP290 and this interaction is critical for photoreceptor outer segment viability and function (24). Hence, RPGR–CEP290 interaction could take place in overlapping as well as distinct complexes consisting of the different isoforms. We, therefore, propose that RPGRORF15 mutations that perturb ratio of the two isoforms and subsequently the integrity or recruitment of the multiprotein complexes result in altered ciliary trafficking. Further studies are underway to delineate the mechanism of such effects.
Mutations in retinal ciliary proteins have been mostly associated with reduced cilia length and photoreceptor outer segment degeneration. Knockdown of rpgr in zebrafish or in cultured human RPE cell lines results in shorter cilia (49–52). Although the fibroblasts derived from RPGRORF15 patients exhibited longer cilia, longer photoreceptor outer segments were not observed in iPSC-derived optic cups or in RPGR patients. These results could reflect cell-type specific differences in the utilization of distinct ciliary regulation pathways. Previous clinical findings and animal model studies also showed that RPGR mutations do not prevent the initial generation of the photoreceptor outer segments, but that they undergo progressive degeneration (28,29,31). Of note, context-dependent cilia length regulation and outer segment defects have been reported with CEP290-patient fibroblasts and CP110-mouse fibroblasts (39,53).
The RPGR-2, RPGR-3, RPGR-4 and RPGR-5 are similar in nature (small frameshifts), they produce profoundly different effects. These could partly be due to the location of these mutations: while the RPGR-2 mutation is located towards the 3′-region of the non-repetitive exon 15, the RPGR-3, RPGR-4 and RPGR-5 mutations lie in the 5′-region of the repetitive purine-rich region. The repetitive region of these mutations may be involved in the recognition or recruitment of mRNA decay factors or splicing effectors. On the other hand, the RPGR-1 mutation, which lies within exon 15 does not show increased RPGRex1-19 levels, yet the steady-state levels of the mutant RPGRORF15 mRNA are significantly reduced. As the mRNA stability profile of this mutant is similar to that of the control, there may be a different mechanism underlying reduced mutant RPGRORF15 levels. However, the resultant increase in the RPGRex1-19/RPGRORF15 ratio indicates the similar cilia extension defect as RPGR-3, RPGR-4 and RPGR-5.
The partial effect of overexpression of RPGRORF15 in the mutant fibroblasts on the rescue of the ciliary phenotype potentially also raises questions that they may be relevant to the ongoing clinical trials that utilize RPGRORF15 gene augmentation strategy. These studies are based upon the rationale that the RPGRORF15 mutations result in a complete or partial loss of function. Although our study was in progress, first results of a phase 1/2 study reported encouraging results with 7 out of 18 patients showing early improvement in visual function after RPGRORF15 gene augmentation therapy (54). However, at least one of the patients with the mutation c.2405_2406delAG (RPGR-3) did not reveal reliable clinical improvement. Our results predict that this mutation would cause a significant disruption in the RPGR isoform ratio and abnormally longer cilia that might not be completely reversed by RPGRORF15 overexpression. This study underscores the complexity of RPGR gene defects and suggests that gene therapy strategies may need to focus on ways to restore the appropriate RPGRex1-19/RPGRORF15 ratio, taking into account the consequences of different ORF15 mutations; ORF15 overexpression may be a less effective therapeutic strategy for patients with certain RPGRORF15 mutations.
Materials and Methods
Study approval
The skin biopsies were obtained from previously published XLRP patients and unaffected individuals after written informed consent. All procedures followed the Declaration of Helsinki and approved by the Institutional Review Board of the University of Pennsylvania and UMass Medical School.
Plasmid DNA constructs and antibodies
The plasmids-encoding human RPGRORF15 or RPGRex1-19 have been described (50,51). Antibody details are provided in Supplementary Table S1.
Fibroblast isolation
The biopsies were dissociated as previously described to obtain primary fibroblasts (39). Briefly, skin biopsies were washed in iodine followed by three washes in phosphate-buffered saline (Invitrogen). The sample was minced with a sharp scalpel in a culture plate and incubated with Dulbecco’s-modified Eagle’s medium (DMEM)/F12 (Invitrogen) supplemented with 20% fetal bovine serum (FBS) (Sigma-Aldrich, Saint-Louis, MO, USA), penicillin and streptomycin at 37°C in a humidified 5% CO2 incubator. Ten days after harvesting, the skin was removed, and fibroblasts were passaged for storage and analysis. After dissociation, cells were grown in DMEM/F12 media containing 10% FBS at 37°C and 5% CO2 incubator. All analyses were done in the fibroblasts at the same passage stages.
Fibroblast transfection assays
Plasmid DNA transfections was performed using FuGENE 6 (Promega) according to the manufacturer’s protocol. All Dicer substrate siRNAs (RPGRex1-19, positive and negative controls, see Supplementary Table S2) were designed and purchased from Integrated DNA Technology and transfected at 20 nM using Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific), as per manufacturer’s instructions. Cells were plated and transfected 24 h later at 50–70% confluency and induced to form primary cilia 24 h post-transfection by serum starvation for 24 h.
RNA extraction and quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) analysis
Total RNAs were isolated with QIAzol Lysis Reagent (Qiagen) and precipitated according to the manufacturer’s instructions. Reverse transcription was performed with 1 μg of total RNAs with the SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen). The resulting cDNA was used to perform gene expression analysis using the BioRad CFX96 qPCR instrument and Power SYBR Green PCR Master Mix (Thermo Fisher Scientific). The isoform-specific quantitative PCR (qPCR) primers are described in Supplementary Table S2.
RNA stability assay
Messenger RNA decay after transcriptional inhibition with actinomycin D treatment was performed as previously described (55). Briefly, the fibroblasts were plated at 50% confluency in six-well plates; 24 h later, cells were treated with 10 μg/ml actinomycin D (Sigma-Aldrich) in DMEM/F12 media for 1, 2, 4, 6 and 8 h. Total mRNA was extracted and subjected to RT-qPCR. Experiments were conducted in triplicates and mRNA decay rate was determined by non-linear regression curve fitting (one-phase decay) using GraphPad Prism Software.
Immunoblotting
The cells were lyzed on ice in Pierce RIPA Buffer (Thermo Scientific) containing Halt™ Protease Inhibitor Cocktail (100X, Thermo Fisher). Lysates were sonicated and centrifugated at 12 000g for 15 min. Supernatants were quantified and 20 μg of total protein lysate was incubated for 5 min at 95°C with Laemmli sample buffer. Proteins were separated on a 4–20% Mini-PROTEAN® TGX™ Precast Protein Gel (BioRad) and transferred to a nitrocellulose membrane (BioRad). The blots were processed using LI-COR western blotting instructions and blocking, and primary and secondary antibodies solutions were prepared in Intercept Blocking buffer (LI-COR). Primary antibodies were incubated overnight at 4°C and secondary antibodies were incubated at room temperature for 2 h (see supplementary table for antibodies details and dilution information). Protein expression was detected using LI-COR Odyssey Fc detection system and quantified with Image Studio Lite quantification software (LI-COR).
Immunofluorescence, microscopy and co-localization analysis
The primary fibroblasts were plated on coverslips (AmScope CS-R18-100) and fixed in pre-chilled 100% methanol at −20°C for 5 min or in 4% paraformaldehyde/1X Dulbecco’s phosphate-buffered saline (DPBS; potassium chloride 0.2 g/L, potassium dihydrogen phosphate 0.2 g/L, sodium chloride 8 g/L and disodium hydrogen phosphate 1.15 g/L) for 10 min, washed three times in DPBS and blocked in DPBS containing 5% normal goat serum and 0.5% Triton X-100 for 1 h. Primary antibody solution was prepared in blocking solution and incubated overnight at 4°C. After washing in DPBS, cells were incubated in blocking solution containing Alexa-488-conjugated and Alexa-546-conjugated (Invitrogen) for 1 h. Finally, cells were washed in DPBS and incubated for 5 min with DAPI for nuclear staining. Coverslips were mounted with Antifade Mounting Media (Vectashield) and all images were visualized over 63× objective using a Leica DM6 Thunder microscope with a 16 bit monochrome camera.
Cilia length was measured on single z-stacks using the ImageJ software and line and measure tool. An average of 15 cilia were measured in at least five different coverslip area using acetylated tubulin or ADP-ribosylation factor like 13B (ARL13B) ciliary markers. The IMARIS platform was used to process the two channel intensities simultaneously and to measure the degree of overlap of the two channels. After setting intensity threshold, the basal body marker γ-tubulin was used to define a region of interest and a co-localization channel was built on different z-stacks to obtain the percentage of co-localized signal and intensity. When needed, adjacent optical sections were merged in the z-axis to form a projected image (ImageJ). Co-localization value was plotted using the GraphPad Prism software.
Statistics
All data are presented as means ± standard derivation from the mean. Two groups comparison was analyzed by Student’s t-tests using the Prism GraphPad software. Multiple groups comparison was performed with one-way analysis of variance. Differences between groups were considered statistically significant if P < 0.05. The statistical significance is denoted with asterisks (*P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001).
Funding
National Institutes of Health (EY022372 and EY029050 to H.K., EY028273 and P30EY01583 to V.R.M.C. and EY017549 to S.G.J. and A.V.C.); Medical Research Council UK (MR/T002735/1, MR/J004553/1); European Research Council (323147-STEMRD); Retina UK (GR576 to R.R.A.); Research to Prevent Blindness Unrestricted Grant Funds to Scheie Eye Institute.
Supplementary Material
Acknowledgement
The authors thank the patients and families who participated in these studies. The authors also thank Dr Gregory Pazour (UMMS) for providing the fibroblasts of control individuals and Dr Michael Volkert (UMMS) for critical inputs and suggestions on this project.
Conflict of Interest statement. None declared.
Contributor Information
Laura Moreno-Leon, Department of Ophthalmology & Visual Sciences, UMass Medical School, Worcester, MA 01655, USA.
Emma L West, Division of Molecular Therapy, UCL Institute of Ophthalmology, London EC1V 9El, UK.
Michelle O’Hara-Wright, Division of Molecular Therapy, UCL Institute of Ophthalmology, London EC1V 9El, UK.
Linjing Li, Department of Ophthalmology & Visual Sciences, UMass Medical School, Worcester, MA 01655, USA.
Rohini Nair, Department of Ophthalmology, Scheie Eye Institute, University of Pennsylvania, Philadelphia, PA 19104, USA.
Jie He, Department of Ophthalmology, Scheie Eye Institute, University of Pennsylvania, Philadelphia, PA 19104, USA.
Manisha Anand, Department of Ophthalmology & Visual Sciences, UMass Medical School, Worcester, MA 01655, USA.
Bhubanananda Sahu, Department of Ophthalmology & Visual Sciences, UMass Medical School, Worcester, MA 01655, USA.
Venkat Ramana Murthy Chavali, Department of Ophthalmology, Scheie Eye Institute, University of Pennsylvania, Philadelphia, PA 19104, USA.
Alexander J Smith, Division of Molecular Therapy, UCL Institute of Ophthalmology, London EC1V 9El, UK.
Robin R Ali, Division of Molecular Therapy, UCL Institute of Ophthalmology, London EC1V 9El, UK.
Samuel G Jacobson, Department of Ophthalmology, Scheie Eye Institute, University of Pennsylvania, Philadelphia, PA 19104, USA.
Artur V Cideciyan, Department of Ophthalmology, Scheie Eye Institute, University of Pennsylvania, Philadelphia, PA 19104, USA.
Hemant Khanna, Department of Ophthalmology & Visual Sciences, UMass Medical School, Worcester, MA 01655, USA.
References
- 1. Singla V. and Reiter J.F. (2006) The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science, 313, 629–633. [DOI] [PubMed] [Google Scholar]
- 2. Pazour G.J. and Rosenbaum J.L. (2002) Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol., 12, 551–555. [DOI] [PubMed] [Google Scholar]
- 3. Baker S.A., Freeman K., Luby-Phelps K., Pazour G.J. and Besharse J.C. (2003) IFT20 links kinesin II with a mammalian intraflagellar transport complex that is conserved in motile flagella and sensory cilia. J. Biol. Chem., 278, 34211–34218. [DOI] [PubMed] [Google Scholar]
- 4. Khanna H. (2015) Photoreceptor sensory cilium: traversing the ciliary gate. Cell, 4, 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen H.Y., Kelley R.A., Li T. and Swaroop A. (2020) Primary cilia biogenesis and associated retinal ciliopathies. Semin. Cell Dev. Biol. 19, 30167–30173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Badano J.L., Leitch C.C., Ansley S.J., May-Simera H., Lawson S., Lewis R.A., Beales P.L., Dietz H.C., Fisher S. and Katsanis N. (2006) Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature, 439, 326–330. [DOI] [PubMed] [Google Scholar]
- 7. Besharse J.C. (1986) The Retina: A Model for Cell Biological Studies Part I. Academic, New York. [Google Scholar]
- 8. Arts H.H., Doherty D., Beersum S.E., Parisi M.A., Letteboer S.J., Gorden N.T., Peters T.A., Marker T., Voesenek K., Kartono A. et al. (2007) Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet., 39, 882–888. [DOI] [PubMed] [Google Scholar]
- 9. Berson E.L. (1996) Retinitis pigmentosa: unfolding its mystery. Proc. Natl. Acad. Sci. U. S. A., 93, 4526–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bird A.C. (1987) Clinical investigation of retinitis pigmentosa. Prog. Clin. Biol. Res., 247, 3–20. [PubMed] [Google Scholar]
- 11. Hanany M., Rivolta C. and Sharon D. (2020) Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. U. S. A., 117, 2710–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Churchill J.D., Bowne S.J., Sullivan L.S., Lewis R.A., Wheaton D.K., Birch D.G., Branham K.E., Heckenlively J.R. and Daiger S.P. (2013) Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci., 54, 1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Breuer D.K., Yashar B.M., Filippova E., Hiriyanna S., Lyons R.H., Mears A.J., Asaye B., Acar C., Vervoort R., Wright A.F. et al. (2002) A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet., 70, 1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharon D., Sandberg M.A., Rabe V.W., Stillberger M., Dryja T.P. and Berson E.L. (2003) RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet., 73, 1131–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ayyagari R., Demirci F.Y., Liu J., Bingham E.L., Stringham H., Kakuk L.E., Boehnke M., Gorin M.B., Richards J.E. and Sieving P.A. (2002) X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics, 80, 166–171. [DOI] [PubMed] [Google Scholar]
- 16. Hunter D.G., Fishman G.A. and Kretzer F.L. (1988) Abnormal axonemes in X-linked retinitis pigmentosa. Arch. Ophthalmol., 106, 362–368. [DOI] [PubMed] [Google Scholar]
- 17. Iannaccone A., Breuer D.K., Wang X.F., Kuo S.F., Normando E.M., Filippova E., Baldi A., Hiriyanna S., MacDonald C.B., Baldi F. et al. (2003) Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J. Med. Genet., 40, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koenekoop R.K., Loyer M., Hand C.K., Al Mahdi H., Dembinska O., Beneish R., Racine J. and Rouleau G.A. (2003) Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am. J. Ophthalmol., 136, 678–687. [DOI] [PubMed] [Google Scholar]
- 19. Bukowy-Bieryllo Z., Zietkiewicz E., Loges N.T., Wittmer M., Geremek M., Olbrich H., Fliegauf M., Voelkel K., Rutkiewicz E., Rutland J. et al. (2013) RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr. Pulmonol., 48, 352–363. [DOI] [PubMed] [Google Scholar]
- 20. Meindl A., Dry K., Herrmann K., Manson F., Ciccodicola A., Edgar A., Carvalho M.R., Achatz H., Hellebrand H., Lennon A. et al. (1996) A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat. Genet., 13, 35–42. [DOI] [PubMed] [Google Scholar]
- 21. Roepman R., Duijnhoven G., Rosenberg T., Pinckers A.J., Bleeker-Wagemakers L.M., Bergen A.A., Post J., Beck A., Reinhardt R., Ropers H.H. et al. (1996) Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum. Mol. Genet., 5, 1035–1041. [DOI] [PubMed] [Google Scholar]
- 22. Mears A.J., Hiriyanna S., Vervoort R., Yashar B., Gieser L., Fahrner S., Daiger S.P., Heckenlively J.R., Sieving P.A., Wright A.F. and Swaroop A. (2000) Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am. J. Hum. Genet., 67, 1000–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He S., Parapuram S.K., Hurd T.W., Behnam B., Margolis B., Swaroop A. and Khanna H. (2008) Retinitis pigmentosa GTPase regulator (RPGR) protein isoforms in mammalian retina: insights into X-linked retinitis pigmentosa and associated ciliopathies. Vision Res., 48, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rao K.N., Zhang W., Li L., Ronquillo C., Baehr W. and Khanna H. (2016) Ciliopathy-associated protein CEP290 modifies the severity of retinal degeneration due to loss of RPGR. Hum. Mol. Genet., 25, 2005–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee J.J. and Seo S. (2015) PDE6D binds to the C-terminus of RPGR in a prenylation-dependent manner. EMBO Rep. 16, 1581–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vervoort R. and Wright A.F. (2002) Mutations of RPGR in X-linked retinitis pigmentosa (RP3). Hum. Mutat., 19, 486–500. [DOI] [PubMed] [Google Scholar]
- 27. Hosch J., Lorenz B. and Stieger K. (2011) RPGR: role in the photoreceptor cilium, human retinal disease, and gene therapy. Ophthalmic Genet., 32, 1–11. [DOI] [PubMed] [Google Scholar]
- 28. Schlegel J., Hoffmann J., Roll D., Muller B., Gunther S., Zhang W., Janise A., Vossing C., Fuhler B., Neidhardt J. et al. (2019) Toward genome editing in X-linked RP-development of a mouse model with specific treatment relevant features. Transl. Res., 203, 57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Q., Acland G.M., Wu W.X., Johnson J.L., Pearce-Kelling S., Tulloch B., Vervoort R., Wright A.F. and Aguirre G.D. (2002) Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum. Mol. Genet., 11, 993–1003. [DOI] [PubMed] [Google Scholar]
- 30. Rao K.N., Li L., Anand M. and Khanna H. (2015) Ablation of retinal ciliopathy protein RPGR results in altered photoreceptor ciliary composition. Sci. Rep., 5, 11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hong D.H., Pawlyk B.S., Shang J., Sandberg M.A., Berson E.L. and Li T. (2000) A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc. Natl. Acad. Sci. U. S. A., 97, 3649–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rao K.N., Zhang W., Li L., Anand M. and Khanna H. (2016) Prenylated retinal ciliopathy protein RPGR interacts with PDE6delta and regulates ciliary localization of Joubert syndrome-associated protein INPP5E. Hum. Mol. Genet., 25, 4533–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Charng J., Cideciyan A.V., Jacobson S.G., Sumaroka A., Schwartz S.B., Swider M., Roman A.J., Sheplock R., Anand M., Peden M.C. et al. (2016) Variegated yet non-random rod and cone photoreceptor disease patterns in RPGR-ORF15-associated retinal degeneration. Hum. Mol. Genet., 25, 5444–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang W.C., Wright A.F., Roman A.J., Cideciyan A.V., Manson F.D., Gewaily D.Y., Schwartz S.B., Sadigh S., Limberis M.P., Bell P. et al. (2012) RPGR-associated retinal degeneration in human X-linked RP and a murine model. Invest. Ophthalmol. Vis. Sci., 53, 5594–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hollander A.I., Koenekoop R.K., Yzer S., Lopez I., Arends M.L., Voesenek K.E., Zonneveld M.N., Strom T.M., Meitinger T., Brunner H.G. et al. (2006) Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet., 79, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hollander A.I., Roepman R., Koenekoop R.K. and Cremers F.P. (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res., 27, 391–419. [DOI] [PubMed] [Google Scholar]
- 37. Sayer J.A., Otto E.A., O'Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V. et al. (2006) The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet., 38, 674–681. [DOI] [PubMed] [Google Scholar]
- 38. Rachel R.A., Yamamoto E.A., Dewanjee M.K., May-Simera H.L., Sergeev Y.V., Hackett A.N., Pohida K., Munasinghe J., Gotoh N., Wickstead B. et al. (2015) CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum. Mol. Genet., 24, 3775–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shimada H., Lu Q., Insinna-Kettenhofen C., Nagashima K., English M.A., Semler E.M., Mahgerefteh J., Cideciyan A.V., Li T., Brooks B.P. et al. (2017) In vitro modeling using ciliopathy-patient-derived cells reveals distinct cilia dysfunctions caused by CEP290 mutations. Cell Rep., 20, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dijk J., Miro J., Strub J.M., Lacroix B., Dorsselaer A., Edde B. and Janke C. (2008) Polyglutamylation is a post-translational modification with a broad range of substrates. J. Biol. Chem., 283, 3915–3922. [DOI] [PubMed] [Google Scholar]
- 41. Rao K.N., Anand M. and Khanna H. (2016) The carboxyl terminal mutational hotspot of the ciliary disease protein RPGRORF15 (retinitis pigmentosa GTPase regulator) is glutamylated in vivo. Biol. Open, 5, 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beltran W.A., Cideciyan A.V., Lewin A.S., Iwabe S., Khanna H., Sumaroka A., Chiodo V.A., Fajardo D.S., Roman A.J., Deng W.T. et al. (2012) Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A., 109, 2132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pawlyk B.S., Bulgakov O.V., Sun X., Adamian M., Shu X., Smith A.J., Berson E.L., Ali R.R., Khani S., Wright A.F. et al. (2015) Photoreceptor rescue by an abbreviated human RPGR gene in a murine model of X-linked retinitis pigmentosa. Gene Ther. 23, 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. El-Brolosy M.A., Kontarakis Z., Rossi A., Kuenne C., Gunther S., Fukuda N., Kikhi K., Boezio G.L.M., Takacs C.M., Lai S.L. et al. (2019) Genetic compensation triggered by mutant mRNA degradation. Nature, 568, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rossi A., Kontarakis Z., Gerri C., Nolte H., Holper S., Kruger M. and Stainier D.Y. (2015) Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature, 524, 230–233. [DOI] [PubMed] [Google Scholar]
- 46. Tautz D. (2000) A genetic uncertainty problem. Trends Genet., 16, 475–477. [DOI] [PubMed] [Google Scholar]
- 47. Hong D.H., Pawlyk B.S., Adamian M. and Li T. (2004) Dominant, gain-of-function mutant produced by truncation of RPGR. Invest. Ophthalmol. Vis. Sci., 45, 36–41. [DOI] [PubMed] [Google Scholar]
- 48. Wright R.N., Hong D.H. and Perkins B. (2011) Misexpression of the constitutive Rpgr(ex1-19) variant leads to severe photoreceptor degeneration. Invest. Ophthalmol. Vis. Sci., 52, 5189–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gakovic M., Shu X., Kasioulis I., Carpanini S., Moraga I. and Wright A.F. (2011) The role of RPGR in cilia formation and actin stability. Hum. Mol. Genet., 20, 4840–4850. [DOI] [PubMed] [Google Scholar]
- 50. Ghosh A.K., Murga-Zamalloa C.A., Chan L., Hitchcock P.F., Swaroop A. and Khanna H. (2010) Human retinopathy-associated ciliary protein retinitis pigmentosa GTPase regulator mediates cilia-dependent vertebrate development. Hum. Mol. Genet., 19, 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murga-Zamalloa C.A., Atkins S.J., Peranen J., Swaroop A. and Khanna H. (2010) Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum. Mol. Genet., 19, 3591–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shu X., Zeng Z., Gautier P., Lennon A., Gakovic M., Patton E.E. and Wright A.F. (2010) Zebrafish Rpgr is required for normal retinal development and plays a role in dynein-based retrograde transport processes. Hum. Mol. Genet., 19, 657–670. [DOI] [PubMed] [Google Scholar]
- 53. Yadav S.P., Sharma N.K., Liu C., Dong L., Li T. and Swaroop A. (2016) Centrosomal protein CP110 controls maturation of the mother centriole during cilia biogenesis. Development, 143, 1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cehajic-Kapetanovic J., Xue K., Martinez-Fernandez de la Camara C., Nanda A., Davies A., Wood L.J., Salvetti A.P., Fischer M.D., Aylward J.W., Barnard A.R. et al. (2020) Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med., 26, 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ratnadiwakara M. and Änkö M.-L. (2018) mRNA stability assay using transcription inhibition by actinomycin D in mouse pluripotent stem cells. Bio-Protocol, 8, e3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.