

Abstract
Adolescent idiopathic scoliosis (AIS), a sideways curvature of the spine, is the most common pediatric musculoskeletal disorder, affecting ~3% of the population worldwide. However, its genetic bases and tissues of origin remain largely unknown. Several genome-wide association studies (GWAS) have implicated nucleotide variants in non-coding sequences that control genes with important roles in cartilage, muscle, bone, connective tissue and intervertebral disks (IVDs) as drivers of AIS susceptibility. Here, we set out to define the expression of AIS-associated genes and active regulatory elements by performing RNA-seq and chromatin immunoprecipitation-sequencing against H3 lysine 27 acetylation in these tissues in mouse and human. Our study highlights genetic pathways involving AIS-associated loci that regulate chondrogenesis, IVD development and connective tissue maintenance and homeostasis. In addition, we identify thousands of putative AIS-associated regulatory elements which may orchestrate tissue-specific expression in musculoskeletal tissues of the spine. Quantification of enhancer activity of several candidate regulatory elements from our study identifies three functional enhancers carrying AIS-associated GWAS SNPs at the ADGRG6 and BNC2 loci. Our findings provide a novel genome-wide catalog of AIS-relevant genes and regulatory elements and aid in the identification of novel targets for AIS causality and treatment.
Introduction
Adolescent idiopathic scoliosis (AIS) is a lateral spinal curvature with no obvious etiology, which manifests during the adolescent growth spurt (1,2). This condition affects around 3% of school-aged children worldwide. The only treatments to prevent curve progression are restrictive bracing or corrective surgery, the latter often requiring extensive postoperative pain management. Hospital costs of operative treatment alone exceed 1 billion USD annually (3). Currently, the genetic basis, the underlying biological mechanisms and the tissues of origin of AIS remain largely unknown. Thus, there are no reliable methods to predict susceptibility to AIS or medications to prevent curve progression. Therefore, it is crucial to gain insight into disease mechanisms and pathogenesis in order to devise new methods to diagnose and treat AIS.
Genome-wide association studies (GWAS) identified at least 13 genetic loci underlying AIS susceptibility (4–12), all of which reside within the non-coding regions of the genome. These results suggest that nucleotide changes in gene regulatory sequences such as enhancers could be the potential drivers of AIS susceptibility (8,9). These gene regulatory sequences are thought to control genes with roles in chondrocytes: ADGRG6 (also called GPR126) (5,13), SOX6, CDH13 (4), BNC2 (8) and SOX9 (7); intervertebral disks (IVDs): ADGRG6, PAX1 (9) and SOX9; muscle: LBX1 (6,11), PAX1, BNC2, SOX6 and PAX3 (12); bone: BNC2 and connective tissue: FBN1 and FBN2 (14), COL11A2 (15). It is worth noting that AIS-associated variants are not necessarily causal but rather point to disease candidates that are in linkage disequilibrium (LD). For the majority of tissues implicated in AIS susceptibility, we currently do not have publicly available (e.g. Encyclopedia of DNA Elements (ENCODE)) genome-wide maps of candidate regulatory elements and also lack transcriptomic data of the genes they could be regulating. Although multiple risk factors are now understood to affect the AIS susceptibility, the interplay of these factors in genetic networks and potential common pathways that could be affected has not been defined.
To obtain a genomic understanding of the AIS-associated genetic networks and the regulatory elements that control them, we performed RNA-seq and chromatin immunoprecipitation-sequencing (ChIP-seq) for the active chromatin mark histone H3 lysine 27 acetylation (H3K27ac) (16,17) on six relevant tissues/cell types: human cartilage, bone and muscle and mouse IVDs, chondrocytes and connective tissue. Our systematic genome-wide analysis establishes an extensive catalog of genes, genetic pathways and active regulatory elements in AIS-relevant tissues. To demonstrate the power of these datasets, we identified gene expression profiles and active regulatory elements for all known AIS GWAS loci in the six examined tissues. We identified putative regulatory elements overlapping the AIS-associated SNPs as well as genetic networks around the AIS-associated genes. By performing enhancer assays in chondrocytes, we functionally validated three novel regulatory elements harboring AIS-associated SNPs, two at the ADGRG6 locus and one at the BNC2 locus. Our transcriptional and epigenetic profiling of AIS-relevant tissues provides a basis for the additional functional follow-up studies to investigate the pathogenic mechanisms underlying AIS susceptibility.
Results
Expression profiling of AIS-relevant tissues identifies AIS-associated gene regulatory networks
To systematically identify genes expressed in the AIS-relevant tissues, we carried out RNA-seq on human cartilage, muscle and bone and on mouse IVDs, connective tissue and chondrocytes (as we were not able to obtain equivalent human tissues) (Fig. 1). The above-mentioned tissues were chosen as they are implicated in AIS-susceptibility through GWAS and animal studies (4–13). RNA was collected from the three biological replicates of each tissue, reverse-transcribed and sequenced. These analyses revealed a list of genes expressed in each of the six tissues (we identified over 4300 genes per tissue) as well as genes that are expressed in multiple tissues (Fig. 2A and Table 1).
Figure 1.
Schematic outline of the study. Cartilage, bone and muscle were isolated from three human individuals, and intervertebral disks, chondrocytes and connective tissue were isolated from mice. All tissues were subjected to RNA-seq and H3K27ac ChIP-seq to identify gene expression profiles and tissue-specific regulatory elements.
Figure 2.
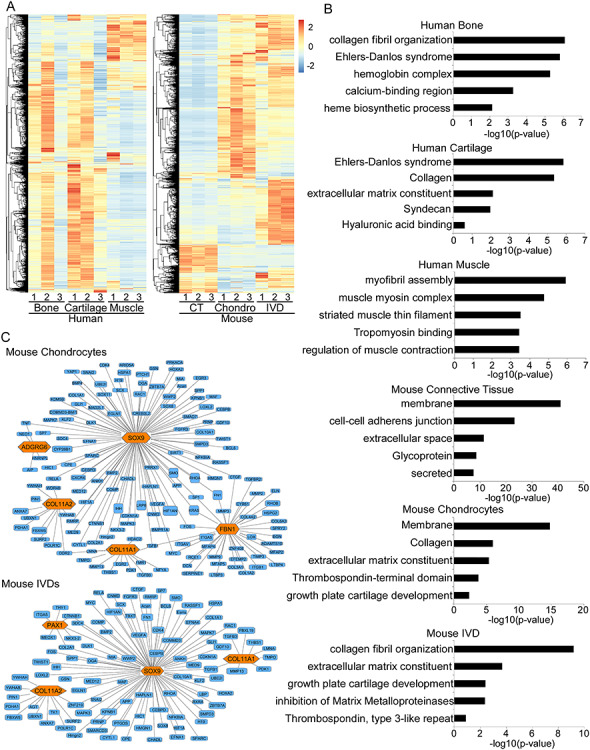
RNA-seq gene expression profiling of AIS-relevant tissues and pathway analysis of AIS-associated genes. (A) Heat maps of gene expression profiles of the six examined tissues. (B) Top gene ontology terms as determined by DAVID (19). (C) Gene regulatory networks for AIS-associated loci (shown in orange) as identified using Ingenuity Pathway Analysis. CT, connective tissue; Chondro, chondrocytes; IVD, intervertebral disk.
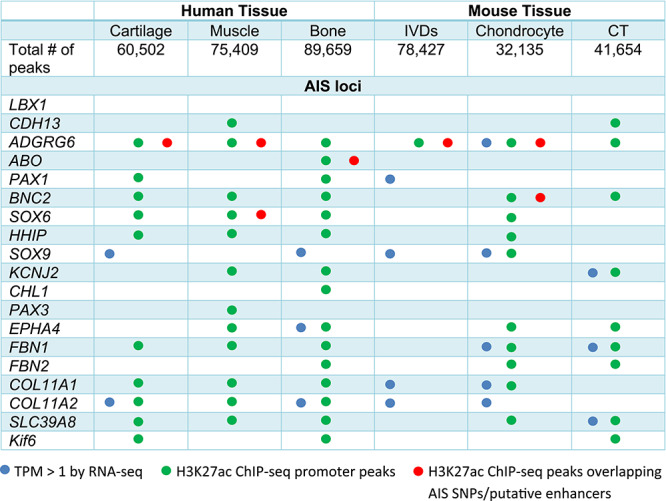
Table 1.
Overview of RNA-seq and ChIP-seq results for AIS GWAS loci
|
We generated unique RNA-seq datasets for AIS-relevant tissues, as we did not identify similar datasets in publicly available datasets such as ENCODE (18). The closest human RNA-seq dataset we found on ENCODE is for skeletal muscle myoblasts (HSMM). When comparing expression profiles, we see that the profile of HSMM cells is different from our RNA-seq datasets obtained from human spinal cartilage, bone and muscle, but it is overall more similar to the human muscle than to cartilage and bone (Supplementary Material, Fig. S1). As we could not identify similar mouse RNA-seq datasets in ENCODE, we compared it to the mouse forebrain RNA-seq data and found that the expression profiles of mouse forebrain are substantially different from our mouse RNA-seq profiles for chondrocytes, connective tissue and IVD (Supplementary Material, Fig. S2).
Analysis of AIS-associated genes via GWAS found many of these genes to be expressed in at least one of the examined tissues (Table 1). For example, Adgrg6 expression was detected in mouse chondrocytes, Kcnj2 in mouse connective tissue and EPHA4 in human bone. We also identified AIS-associated transcription factors that are known to be critical regulators of chondrogenesis in each of the tissues, such as SOX9 and PAX1 in cartilage and IVDs.
When comparing gene expression profiles between individual patients, we noticed significant differences in the bone samples between two of the patients and the third one (Fig. 2A). While all three patients were non-Hispanic white females without remarkable histories, the two patients whose bone RNA-seq profiles clustered closer together were 11 and 13 years of age and the bone samples were taken at the thoracic level T10, while the third patient was 16-years old and the sample was taken at level T10–11 (Supplementary Material, Table S2). These differences in expression profiles might be due to the differences in the skeletal maturity of the patients and/or the thoracic level of the spine. We next set out to characterize enriched pathways and genetic networks around AIS-associated genes in each tissue. Functional annotation clustering using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (19) found an enrichment of gene ontology (GO) categories for each tissue. For example, chondrocytes are enriched for GO categories related to collagen (P-value = 9.8E-7), extracellular matrix structural constituent (P-value = 4.3E-6) and growth plate cartilage development (P-value = 4.6E-3) (Fig. 2B). Further analysis of our tissue-specific datasets using the Ingenuity Pathway Analysis (IPA) (20) revealed novel genetic networks up- and down-stream of AIS GWAS genes as well as interactions between multiple AIS genes in specific tissues (Fig. 2C). For example, we identified common genetic networks between the following AIS-associated genes in chondrocytes: ADGRG6, SOX9, FBN1, COL11A1 and COL11A2. Tumor necrosis factor-alpha (TNF-alpha) for example, was shown to be a potent inhibitor of SOX9 in cartilage (21), and ADGRG6 expression is decreased when SOX9 is deleted (22). In IVD, we identified common genetic networks between SOX9, PAX1, COL11A1 and COL11A2. Our pathway analyses identified active tissue-specific genetic networks around AIS-associated genes and thereby putative regulators, downstream effectors or interaction partners of AIS-associated genes.
ChIP-seq identifies AIS-associated regulatory elements
To identify active regulatory elements in AIS-relevant tissues, we performed ChIP-seq for H3K27ac on all six tissues (Fig. 1). Each tissue was processed using two biological replicates, chromatin was crosslinked, immunoprecipitated and DNA sequenced. We found over 30 000 peaks in each tissue (Table 1). We also identified numerous peaks that are shared between two or more tissues. For example, 5152 peaks were shared between mouse chondrocytes and IVDs (Fig. 3A). Using the Genomic Regions Enrichment of Annotations (GREAT) tool (23), we observed tissue-specific enrichment of peak regions nearby genes belonging to several GO categories. For example, in chondrocytes, we observed a significant association with genes involved in chondrodystrophy (P-value = 1.8E-18), decreased skin tensile strength (P-value = 1.5E-16) and absent somites (P-value = 1.3E-14). In connective tissue, we saw an enrichment with genes involved in joint laxity (P-value = 8.9E-26), vertebral compression fractures (P-value = 1.3183E-8) and premature skin wrinkling (P-value = 1.2589E-7). These results highlight putative mechanisms that might contribute to the pathogenesis of AIS.
Figure 3.
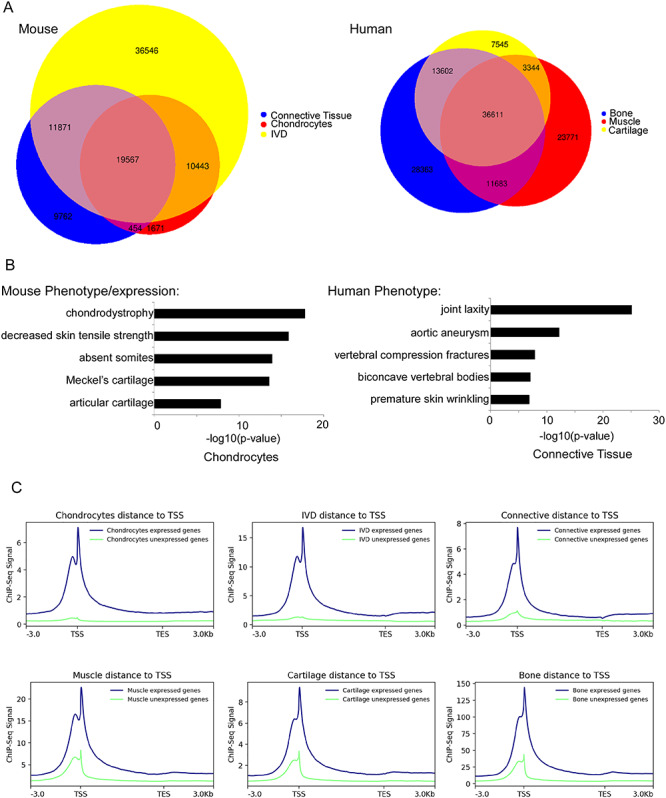
ChIP-seq identifies active regulatory elements in AIS-associated tissues. (A) Venn diagram showing the overlap between H3K27ac peaks in mouse and human tissues. (B) Top mouse phenotype and gene expression according to the Mouse Genome Informatics, and human phenotype term enrichment for mouse chondrocytes and human connective tissue, respectively, according to GREAT (23). (C) H3K27ac peaks show higher correlation around the TSS of genes that are expressed in the RNA-seq from the same tissue.
In order to test for the correlation between the H3K27ac peaks and actively expressed genes in each tissue, as determined by ChIP-seq and RNA-seq, respectively, we grouped genes in each tissue into either expressed [transcripts per kilobase million (TPM) ≥ 1] or not expressed, and we determined for each group the correlation to ChIP-seq signal and the distance to the transcriptional start site (TSS). We found a positive correlation between the actively expressed genes and regions having H3K27ac signals for each tissue (Fig. 3C). These findings corroborate the overlapping GO term enrichments found for both our RNA-seq and ChIP-seq genes and peaks (Figs 2B and 3B). GO terms related to cartilage development and extracellular matrix constituent were found for both active genes and H3K27ac peaks. We also examined the correlation between our RNA-seq and ChIP-seq data specifically for the AIS-associated loci and found that many associated genes that are expressed (as determined by RNA-seq) have H3K27ac peaks around the TSS (Table 1).
Identification and functional characterization of novel AIS-associated enhancers
One major challenge in the functional follow-up of GWAS is to assign function to non-coding variants in particular as the lead SNP is not necessarily pathogenic. We thus set out to determine whether the AIS-associated SNPs identified in previous GWAS overlap with the putative regulatory elements identified in our ChIP-seq experiments. For these analyses, we identified all SNPs in LD (r2 ≥ 0.8) with lead AIS GWAS SNPs and overlaid them with H3K27ac peaks for each of the six tissues. We identified tissue-specific H3K27ac peaks that overlap GWAS SNPs for several of the AIS-associated loci (Table 1). These sequences represent putative regulatory elements that could be affected by the disease-associated SNPs.
To functionally validate some of these sequences, we focused our analysis on the H3K27ac peaks identified in mouse chondrocytes, as chondrocytes were implicated as a major cell type underlying AIS susceptibility (13,23,24) and the existing cell lines can be easily transfected for in vitro validation experiments. We identified chondrocyte-specific H3K27ac peaks overlapping AIS-associated SNPs at the ADGRG6 (Fig. 4A) and BNC2 loci. We selected six sequences at the ADGRG6 locus and three at the BNC2 locus to test for enhancer activity using luciferase reporter assays. These sequences were around 3 kb in length and were chosen based on the chondrocyte-specific H3K27ac peaks, their overlap with the AIS-associated SNPs, conservation among vertebrates and overlap with DNaseI hypersensitivity sites and transcription factor ChIP-seq from ENCODE as previously described (25–28). Sequences were cloned into an enhancer assay vector (pGL4.23; Promega), which contains a minimal promoter followed by a luciferase reporter gene. As a positive control, we used pGL4.13 (Promega) with an SV40 early enhancer (pGL4.13; Promega) and the pGL4.23 empty vector as a negative control. Out of the nine assayed sequences, three showed significant luciferase activity in a human chondrocyte cell line (SW1353) (Fig. 4B and Supplementary Material, Fig. S3). Two of these functional enhancers are located in the ADGRG6 locus, with one sequence (ADGRG6_4) residing in the fourth intron and the second (ADGRG6_6) in the 3′UTR of this gene (Fig. 4B). The third sequence is located in the third intron of BNC2 (BNC2_2) (Supplementary Material, Fig. S3). We examined available chromatin conformation capture datasets using the 3D Genome Browser (29) for H1-MSC cells (human mesenchymal stem cell line) (30) and human GM12878 (human lymphoblastoid cell line) (31) and found that ADGRG6_4 is predicted to interact with the ADGRG6 promoter and the BNC2_2 sequence is predicted to interact with the BNC2 promoter. In sum, we discovered three novel AIS-associated regulatory elements that could affect gene expression levels and contribute to disease susceptibility.
Figure 4.
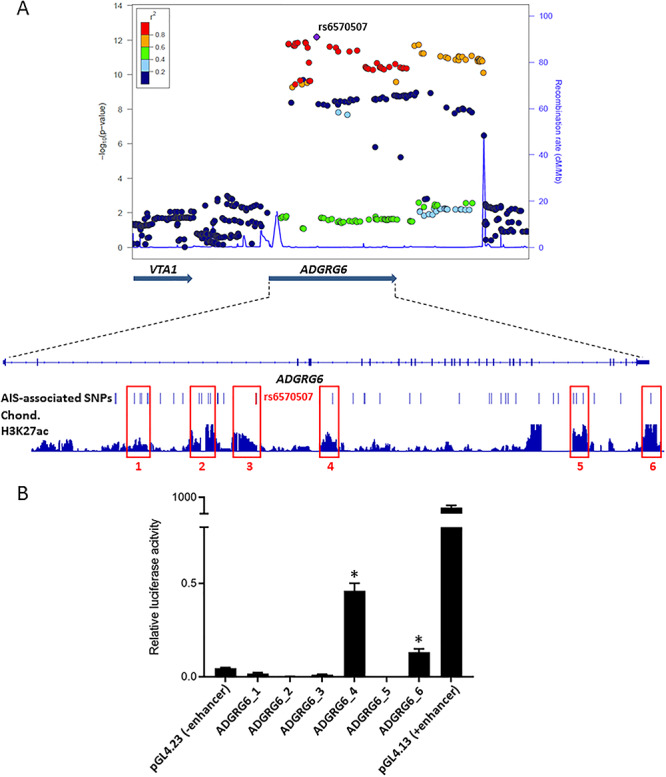
Identification and characterization of AIS-associated enhancers at the ADGRG6 locus. (A) Integrative genomic viewer snapshot and Locuszoom plot for the ADGRG6 locus, highlighting the overlap of AIS-associated SNPs with H3K27ac ChIP-seq peaks. The Locuszoom plot is indexed by the lead SNP rs6570507(4). The colors of the spots reflect the degree of LD, with the index SNP measured by r2. (B) Luciferase assays in human SW1353 cells identify two novel enhancers at the ADGRG6 locus (*P < 0.02).
Discussion
In this study, we set out to identify genes, genetic networks and regulatory elements underlying AIS susceptibility in a genome-wide manner. We performed RNA-seq and ChIP-seq on six AIS-relevant tissues: IVDs, chondrocytes, connective tissue, cartilage, bone and muscle. Our RNA-seq results allowed us to define enriched pathways and biological processes in these tissues. We also identified transcriptional profiles and novel genetic networks of important AIS genes. As AIS is a complex disease, multiple risk factors and tissues/cell types are thought to affect AIS susceptibility. Genome-wide meta-analysis determined that the vast majority of AIS risk loci remain to be discovered (4). Our analyses provide novel candidate loci that can be selectively probed for their association with AIS. In addition, it will be important to determine whether multiple AIS loci act in common pathways and how this affects disease risk. Our pathway analysis determined that some of the previously identified AIS loci interact in pathways in specific tissues. For example, the genetic network surrounding ADGRG6, an important AIS susceptibility gene (4,5,13), showed interactions with several other genes that have been implicated in AIS, with one of these genes being SOX9, a master regulator of chondrogenesis (32). This interaction is supported by direct observation that loss of Adgrg6 in the IVD is associated with reduced SOX9 expression in cartilaginous endplate and annulus fibrosus regions of the IVD in mouse (22). Thus, our analyses allowed us to identify genetic networks and predict interactions between several AIS genes, which will assist future studies on the intricate interplay of these loci in driving susceptibly to this complex disease. Our H3K27ac ChIP-seq datasets revealed thousands of novel putative enhancers in the six tissues and provided exceptional candidate sequences for subsequent studies to probe the functional relevance of AIS-associated non-coding SNPs. Using GREAT, we observed tissue-specific enrichment and a significant association with genes involved in human and mouse phenotypes such as chondrodystrophy and vertebral compression fractures, which might contribute to the pathogenesis of scoliosis. We also found a strong overlap between active genes as shown by RNA-seq and active ChIP-seq chromatin marks by both genome-wide comparisons as well as by examining specific AIS loci, demonstrating the correlation between our datasets.
One major focus of post-GWAS studies is to assign function to disease-associated non-coding SNPs. As a first step in this direction, we generated a database of tissue-specific regulatory regions in the AIS-relevant tissues. We used these data to identify novel putative enhancers overlapping with associated SNPs at several AIS-associated GWAS loci: LBX1, CDH13, GPR126, ABO, PAX1, BNC2, SOX6, HHIP, SOX9/KCNJ2, CHL1, PAX3, AJAP1/EPHA4 and BCL2 (4–13). These putative AIS-associated enhancers are a valuable resource for future functional and mechanistic follow-up studies. As a great majority of risk factors underlying AIS remain undiscovered, future GWAS, whole-genome sequencing and family studies will uncover many additional AIS loci, which can then also be analyzed using our genomic datasets.
As chondrocytes were shown to be an important cell type underlying AIS susceptibility (13,22,24), we functionally tested putative chondrocyte enhancers that overlap AIS-associated SNPs using enhancer assays. We identified three novel functional enhancer sequences that encompass SNPs in LD with AIS GWAS SNPs. One enhancer is located in the fourth intron and one in the 3′UTR of ADGRG6 and a third enhancer is located in the third intron of BNC2 overlapping an alternative promoter. The ADGRG6 and BNC2 loci were found to be associated with AIS both via GWAS and animal studies (4,5,8,13). Additional work on how these loci could affect their expression and subsequent function would be of interest. ADGRG6 in particular represents an exciting locus as it encodes for a G-protein-coupled receptor and is therefore a potential ‘druggable’ target. Further investigation is needed to dissect the regulation and interactions of ADGRG6 and other AIS-associated loci as well as shared mechanisms that lead to AIS susceptibility. Our study provides the first comprehensive genome-wide survey of the transcriptional and epigenetic profiles of AIS-relevant tissues and highlights novel AIS-associated regulatory elements and genetic networks, enabling the identification of new candidates for the diagnosis and treatment of AIS.
Materials and Methods
Human and mouse tissue samples
All human tissue samples were collected at the Texas Scottish Rite Hospital for Children in Dallas and were approved by an Institutional Review Board (data were analyzed anonymously). Facets and muscle were collected from the thoracolumbar spine of AIS patients aged 11–17 during spinal surgery, and cartilage was dissected off the bone. As facets were specifically removed during the spinal fusion surgery, which is performed to treat AIS, it was not possible for us to acquire equivalent tissues from the patients without AIS. Tissues were snap-frozen for RNA-seq or fixed in 1% formaldehyde when used for ChIP-seq (see in the following). All mouse work was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California and the University of Texas, Austin. We isolated, IVDs from the thoracic and lumbar spine of postnatal day (P) 20 mice, costal chondrocytes from the anterior rib cage and sternum of P 2–4 mice and connective tissue from 8 to 10-week-old CD1 mice (Charles River) from the kidney capsule, as it is the cleanest connective tissue that can be isolated from the mouse without contamination from other tissues. Chondrocytes were isolated as previously described (13). Briefly, rib cages were dissected, soft tissues were removed and rib cages were digested at 37°C for 1 h in 2 mg/ml pronase and then washed in phosphate-buffered saline (PBS). Rib cages were then further digested for 1 h using 3 mg/ml collagenase D in Dulbecco’s Modified Eagle Medium at 37°C and 5% CO2 followed by three PBS washes to remove the digested tissue. Remaining cartilage was then further digested for 4–6 h in 3 mg/ml collagenase D and then filtered through a 45 μm cell strainer to obtain chondrocytes.
RNA isolation, sequencing and analysis
Each tissue was processed using three biological replicates. Each human replicate consisted of a tissue from one patient. For mouse chondrocytes, we pooled tissue from 6 to 10 mice per replicates, for connective tissue we pooled 4 mice and for IVDs we isolated 20–24 IVDs from 2 mice as 1 replicate. RNA was isolated from chondrocytes using the RNeasy Mini Kit and from connective tissue using the RNeasy Fibrous Tissue Mini Kit (Qiagen). All other tissues were first homogenized using Lysing Matrix S, 50 × 2 ml Tubes (Fisher, 116925050) in TRIzol Reagent (Invitrogen, 15596026) on the Tissue Lyser II. Total RNA was then cleaned up with the Direct-zol RNA miniprep kit (Zymo Research, Z2070). RNA quantity and purity were analyzed on a Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent). Total RNA was subjected to isolate poly(A) mRNA with poly-T oligo attached magnetic beads (Invitrogen). RNA fragments were reverse-transcribed to create the final cDNA libraries following the NEBNext® Ultra™ RNA Library Prep Kit (Illumina), and paired-end sequencing 150 bp was performed on a Novaseq 6000. Sequence quality was verified using FastQC (33). Raw sequencing reads were mapped to the mouse (GRCm38/m10) or human (GRCh37/hg19) genome (hg19) using STAR (34). Transcripts were quantified and transcripts per million (TPM) values were used for expression normalization. For each tissue, a list of genes with TPM ≥ 1 was generated and used for downstream analysis. Heatmaps of gene expression profiles of the six examined tissues were generated using the R heatmap package. Functional annotation clustering was carried out using the DAVID (19). IPA (20) was used to carry out pathway analysis for AIS-associated genes expressed in each tissue.
Chromatin immunoprecipitation-sequencing
Each tissue was processed using two biological replicates and the samples were pooled in the same manner described before for RNA sequencing. Tissues were crosslinked using 1% formaldehyde by standard techniques (35). ChIP was performed using an antibody against H3K27ac (Millipore, Burlington, MA 05-1334) using the LowCell# ChIP kit (Diagenode). Illumina sequencing libraries were generated using the Accel-NGS 2S Plus DNA Library Kit (Swift Biosciences). Single-end 50 bp sequencing was done on a HiSeq 4000 and computational analyses were performed using BWA (36) and MACS2 (37) following the ENCODE guidelines for ChIP-seq analyses (38). Replicated peaks are called from the pooled replicates that are observed in both replicates.
Luciferase reporter assays
Enhancer candidate sequences (Supplementary Material, Table S1) were selected based on having an H3K27ac peak in mouse chondrocytes and overlapping SNPs in LD with an AIS-associated GWAS lead SNP. The SNPs in LD were determined using ldlink (39) on data from the 1000 Genomes Project (40,41) in the HapMap Caucasian (CEU) population. Enhancer candidate sequences were PCR-amplified from human genomic DNA, cloned into a pGL4.23 enhancer assay vector (Promega) and sequence-verified. Empty pGL4.23 was used as negative control and pGL4.13 (Promega) with an SV40 early enhancer as a positive control. Luciferase assays for enhancer candidates were carried out in chondrocyte cell lines [human SW1353 cells (ATCC® HTB-94™)]. SW1353 cells were grown in DMEM (Invitrogen). SW1353 media was supplemented with 1% penicillin/streptomycin and 10 and 5% FBS, respectively. Forty-eight hours before transfection, 60 000 cells were plated out in 96 well plates and were grown up to 90% confluency. Cells were transfected with 130 ng of the assayed plasmid and 16 ng of pGL4.73 (hRluc/SV40) (Promega) containing Renilla luciferase to correct for transfection efficiency, using X-tremeGENE (Roche) according to the manufacturer’s protocol. Transfections were performed in triplicates. Forty-eight hours after transfection, cells were lysed, and luciferase activity was measured using the Dual-Luciferase Reporter Assay Kit (Promega). Measurements were performed on a GloMax 96-microplate luminometer (Promega).
Supplementary Material
{kind=link}
Acknowledgements
We would like to thank Fumitaka Inoue for his help with sequence sample submission and Anna Williams and Nandina Paria for their help with human tissue collection. Funding Section Research in this publication was supported by the National Institutes of Child and Human Development (grant number 1P01HD084387 to N.A. and C.A.W.) and the Arthritis and Musculoskeletal and Skin Diseases (R01AR072009-01 to R.S.G.) and (F32AR073648 to Z.L.).
Conflict of Interest statement. There is no conflict of interest.
Author’s contribution
N.M. and N.A. conceived the experiments. N.M. performed the experiments. J.Z., N.M., W.L.E. and J.W. performed RNA-seq and ChIP-seq analysis. A.U. helped with ChIP-seq samples. A.M.K. compiled AIS SNPs. Z.L., R.S.G., J.R. and C.A.W. provided tissues, expertise and feedback. N.M. and N.A. wrote the manuscript.
References
- 1. Hresko M.T. (2013) Idiopathic scoliosis in adolescents. N. Engl. J. Med., 368, 834–841. [DOI] [PubMed] [Google Scholar]
- 2. Wise C.A., Gao X., Shoemaker S., Gordon D. and Herring J.A. (2008) Understanding genetic factors in idiopathic scoliosis, a complex disease of childhood. Curr. Genomics, 9, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martin C.T., Pugely A.J., Gao Y., Mendoza-lattes S.A., Ilgenfritz R.M., Callaghan J.J. and Weinstein S.L. (2014) Increasing hospital charges for adolescent idiopathic scoliosis in the United States. Spine, 39, 1676–1682. [DOI] [PubMed] [Google Scholar]
- 4. Khanshour A.M., Kou I., Fan Y., Einarsdottir E., Makki N., Kidane Y.H., Kere J., Grauers A., Johnson T.A., Paria N. et al. (2018) Genome-wide meta-analysis and replication studies in multiple ethnicities identify novel adolescent idiopathic scoliosis susceptibility loci. Hum. Mol. Genet., 27, 3986–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kou I., Takahashi Y., Johnson T.A., Takahashi A., Guo L., Dai J., Qiu X., Sharma S., Takimoto A., Ogura Y. et al. (2013) Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat. Genet., 45, 676–679. [DOI] [PubMed] [Google Scholar]
- 6. Londono D., Kou I., Johnson T.A., Sharma S., Ogura Y., Tsunoda T., Takahashi A., Matsumoto M., Herring J.A., Lam T.-P. et al. (2014) A meta-analysis identifies adolescent idiopathic scoliosis association with LBX1 locus in multiple ethnic groups. J. Med. Genet., 51, 401–406. [DOI] [PubMed] [Google Scholar]
- 7. Miyake A., Kou I., Takahashi Y., Johnson T.A., Ogura Y., Dai J., Qiu X., Takahashi A., Jiang H., Yan H. et al. (2013) Identification of a susceptibility locus for severe adolescent idiopathic scoliosis on chromosome 17q24.3. PLoS One, 8, e72802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ogura Y., Kou I., Miura S., Takahashi A., Xu L., Takeda K., Takahashi Y., Kono K., Kawakami N., Uno K. et al. (2015) A functional SNP in BNC2 is associated with adolescent idiopathic scoliosis. Am. J. Hum. Genet., 97, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharma S., Londono D., Eckalbar W.L., Gao X., Zhang D., Mauldin K., Kou I., Takahashi A., Matsumoto M., Kamiya N. et al. (2015) A PAX1 enhancer locus is associated with susceptibility to idiopathic scoliosis in females. Nat. Commun., 6, 6452–6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharma S., Gao X., Londono D., Devroy S.E., Mauldin K.N., Frankel J.T., Brandon J.M., Zhang D., Li Q.-Z., Dobbs M.B. et al. (2011) Genome-wide association studies of adolescent idiopathic scoliosis suggest candidate susceptibility genes. Hum. Mol. Genet., 20, 1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takahashi Y., Kou I., Takahashi A., Johnson T.A., Kono K., Kawakami N., Uno K., Ito M., Minami S., Yanagida H. et al. (2011) A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet., 43, 1237–1240. [DOI] [PubMed] [Google Scholar]
- 12. Zhu Z., Tang N.L.-S., Xu L., Qin X., Mao S., Song Y., Liu L., Li F., Liu P., Yi L. et al. (2015) Genome-wide association study identifies new susceptibility loci for adolescent idiopathic scoliosis in Chinese girls. Nat. Commun., 6:8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karner C.M., Long F., Solnica-Krezel L., Monk K.R. and Gray R.S. (2015) Gpr126/Adgrg6 deletion in cartilage models idiopathic scoliosis and pectus excavatum in mice. Hum. Mol. Genet., 24, 4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buchan J.G., Alvarado D.M., Haller G.E., Cruchaga C., Harms M.B., Zhang T., Willing M.C., Grange D.K., Braverman A.C., Miller N.H. et al. (2014) Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum. Mol. Genet., 23, 5271–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haller G., Alvarado D., Mccall K., Yang P., Cruchaga C., Harms M., Goate A., Willing M., Morcuende J.A., Baschal E. et al. (2016) A polygenic burden of rare variants across extracellular matrix genes among individuals with adolescent idiopathic scoliosis. Hum. Mol. Genet., 25, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U S A, 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rada-Iglesias A., Bajpai R., Prescott S., Brugmann S.A., Swigut T. and Wysocka J. (2012) Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell, 11, 633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Consortium E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang D.W., Sherman B.T., Tan Q., Collins J.R., Alvord W.G., Roayaei J., Stephens R., Baseler M.W., Lane H.C. and Lempicki R.A. (2007) The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol., 8, R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krämer A., Green J., Pollard J. and Tugendreich S. (2014) Causal analysis approaches in ingenuity pathway analysis. Bioinformatics, 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murakami S., Lefebvre V. and Crombrugghe B. (2000) Potent inhibition of the master chondrogenic factor Sox9 gene by interleukin-1 and tumor necrosis factor-alpha. J. Biol. Chem., 275, 3687–3692. [DOI] [PubMed] [Google Scholar]
- 22. Liu Z., Easson G.W.D., Zhao J., Makki N., Ahituv N., Hilton M.J., Tang S.Y. and Gray R.S. (2019) Dysregulation of STAT3 signaling is associated with endplate-oriented herniations of the intervertebral disc in Adgrg6 mutant mice. PLoS Genet., 15, e1008096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M. and Bejerano G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wise C.A., Sepich D., Ushiki A., Khanshour A.M., Kidane Y.H., Makki N., Gurnett Ca., Gray R.S., Rios J.J., Ahituv N. Solnica-Krezel L. (2020) The cartilage matrisome in adolescent idiopathic scoliosis. Bone Res. 8, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huo Y., Li S., Liu J., Li X. and Luo X.-J. (2019) Functional genomics reveal gene regulatory mechanisms underlying schizophrenia risk. Nat. Commun., 10, 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Inoue F., Kircher M., Martin B., Cooper G.M., Witten D.M., McManus M.T., Ahituv N. and Shendure J. (2017) A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res., 27, 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luizon M.R., Eckalbar W.L., Wang Y., Jones S.L., Smith R.P., Laurance M., Lin L., Gallins P.J., Etheridge A.S., Wright F. et al. (2016) Genomic characterization of metformin hepatic response. PLoS Genet., 12, e1006449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu H., Leslie E.J., Carlson J.C., Beaty T.H., Marazita M.L., Lidral A.C. and Cornell R.A. (2017) Identification of common non-coding variants at 1p22 that are functional for non-syndromic orofacial clefting. Nat. Commun., 8, 14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y., Song F., Zhang B., Zhang L., Xu J., Kuang D., Li D., Choudhary M.N.K., Li Y., Hu M. et al. (2018) The 3D genome browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol., 19, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W. et al. (2015) Chromatin architecture reorganization during stem cell differentiation. Nature, 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dy P., Wang W., Bhattaram P., Wang Q., Wang L., Ballock R.T. and Lefebvre V. (2012) Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell, 22, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wingett S.W. and Andrews S. (2018) FastQ screen: a tool for multi-genome mapping and quality control. F1000Research, 7, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dobin A. and Gingeras T.R. (2015) Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics, 51, 11.14.11–11.14.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. VanderMeer J.E., Smith R.P., Jones S.L. and Ahituv N. (2014) Genome-wide identification of signaling center enhancers in the developing limb. Development, 141, 4194–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li H. and Durbin R. (2009) Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res., 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Machiela M.J. and Chanock S.J. (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants: Fig. 1. Bioinformatics, 31, 3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Hsi-Yang Fritz M. et al. (2015) An integrated map of structural variation in 2,504 human genomes. Nature, 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Genomes Project C., Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.