

ABSTRACT
The Myostatin/Activin branch of the TGF-β superfamily acts as a negative regulator of vertebrate skeletal muscle size, in part, through downregulation of insulin/insulin-like growth factor 1 (IGF-1) signaling. Surprisingly, recent studies in Drosophila indicate that motoneuron-derived Activin signaling acts as a positive regulator of muscle size. Here we demonstrate that Drosophila Activin signaling promotes the growth of muscle cells along all three axes: width, thickness and length. Activin signaling positively regulates the insulin receptor (InR)/TORC1 pathway and the level of Myosin heavy chain (Mhc), an essential sarcomeric protein, via increased Pdk1 and Akt1 expression. Enhancing InR/TORC1 signaling in the muscle of Activin pathway mutants restores Mhc levels close to those of the wild type, but only increases muscle width. In contrast, hyperactivation of the Activin pathway in muscles increases overall larval body and muscle fiber length, even when Mhc levels are lowered by suppression of TORC1. Together, these results indicate that the Drosophila Activin pathway regulates larval muscle geometry and body size via promoting InR/TORC1-dependent Mhc production and the differential assembly of sarcomeric components into either pre-existing or new sarcomeric units depending on the balance of InR/TORC1 and Activin signals.
KEY WORDS: Activin, Drosophila, Insulin, Muscle
Summary: The Activin branch of the TGF-β superfamily regulates Drosophila larval body wall muscle size in all three spatial dimensions through differential control of sarcomeric building block production and assembly.
INTRODUCTION
Skeletal muscle accounts for a large portion of the body mass in various species including mammals (Gunn, 1989) and flying insects (Marden, 2000). It is essential not only for mobility but also for organismal energy balance and metabolism as it is a primary tissue for insulin-stimulated glucose consumption (reviewed by Stump et al., 2006). Skeletal muscle is also proposed to be an endocrine organ that secretes a plethora of bioactive molecules, known as myokines, many of which depend on muscle contraction for production and secretion. Current evidence suggests that myokines exert substantial influence on the physiology and activity of their various target tissues (reviewed by Pedersen and Febbraio, 2012). Therefore, achieving and maintaining an appropriate skeletal muscle mass and cellular function is likely to be essential for good health and quality of life.
Multiple signaling pathways are known to act in concert to achieve and maintain proper muscle mass (reviewed by Piccirillo et al., 2014; Schiaffino et al., 2013). Among these, Myostatin (Mstn), a member of the TGF-β superfamily of growth and differentiation factors, has proven to be a prominent player. Loss-of-function mutations in mstn have been identified or induced in a large variety of vertebrates including humans (Kambadur et al., 1997; McPherron et al., 1997; Bhattacharya et al., 2019; Gao et al., 2016; Schuelke et al., 2004). In all these species, loss of mstn results in larger skeletal muscles, leading to the conclusion that Mstn is a negative regulator of muscle mass. Mechanistically, the increase in skeletal muscle mass caused by disruption of the mstn gene has been attributed to excess proliferation of muscle progenitors (hyperplasia) that is manifested by a larger number of fibers, as well as to hypertrophy of each muscle fiber causing bigger cross-sectional area (Amthor et al., 2009; McPherron et al., 1997). However, several more recent studies suggest that the hypertrophy of individual muscle fibers may be the predominant mechanism for increasing muscle mass, with a minimal contribution from hyperplasia (Amthor et al., 2009; Lee et al., 2012; Sartori et al., 2009). In addition, satellite cells (muscle stem cells) do not appear to contribute to the muscle hypertrophy (Amthor et al., 2009; Sartori et al., 2009). Taken together, these results indicate that the hypertrophy of individual myofibers, rather than increases in myofiber and myonuclei number, is the chief mechanism for enhanced muscle growth in mstn mutants.
In addition to Mstn, activins, which are also members of the TGF-β superfamily, have also been shown to negatively affect muscle mass (Chen et al., 2017, 2014). The Mstn and activins appear to synergize in suppressing muscle growth, as co-inhibition of both factors results in greater increases in muscle mass than inhibition of the activity of individual factors (Chen et al., 2017). Moreover, expression of a dominant negative form of ActRIIB, a high affinity activin type 2 receptor for Mstn, activins and several other ligands in the TGF-β/Activin subfamily (Lee and McPherron, 2001; Souza et al., 2008), leads to muscle hypertrophy (Lee and McPherron, 2001). Conversely, overexpression of these factors promotes the loss of muscle weight in both rats (Amirouche et al., 2009) and mice (Chen et al., 2014; Zimmers et al., 2002).
Several different muscle pathologies, including cancer cachexia (Lokireddy et al., 2012; Loumaye et al., 2015; Marino et al., 2015) and muscle disuse due to paralytic injury (Gustafsson et al., 2010; Reardon et al., 2001; Wehling et al., 2000), also show a correlation between muscle disuse and increased expression of Mstn or Activin. In the case of cancer cachexia and dystrophic muscle wasting, muscle loss can be partially reversed by inhibition of Mstn or Activin signaling (Bogdanovich et al., 2002; Chen et al., 2017; Zhou et al., 2010). Therefore, Mstn- and Activin-induced signaling pathways may provide potent therapeutic targets for the treatment of muscle atrophy in multiple settings, including age-related sarcopenia (Bergen et al., 2015; White and LeBrasseur, 2014).
A well-documented consequence of Mstn- or Activin-induced signaling in skeletal muscle is inhibition of the insulin-like growth factor 1/phosphoinositide 3-kinase/protein kinase B (IGF-1/PI3K/AKT) pathway. Specifically, overexpression of mstn via in vivo transfection in the adult muscle leads to attenuated phosphorylation of AKT, S6 and 4E-BP (Amirouche et al., 2009). Conversely, inhibition of Mstn and Activin by prodomain-derived antagonists or a soluble decoy receptor of ActRIIB leads to increased phosphorylation of mTOR and S6RP or increased phosphorylation of AKT and FOXO3a in skeletal muscle, respectively (Chen et al., 2017; Zhou et al., 2010). These are all consistent with the idea that Mstn- or Activin-induced signals lead to inhibition of the IGF-1/AKT/mTOR function. Because the IGF-1/AKT/mTOR pathway is the most important anabolic stimulus for muscle growth (Egerman and Glass, 2014), much of the influence of Mstn and activins on muscle mass can most probably be attributed to inhibition of the IGF-1/AKT/mTOR pathway.
Drosophila skeletal muscles exhibit tremendous (around 50-fold) growth during larval stages (Piccirillo et al., 2014). This growth occurs through an increase in individual cell size without contribution from muscle stem cells (Demontis and Perrimon, 2009; Piccirillo et al., 2014). The process is mechanistically similar to the mammalian muscle hypertrophy shown by mstn mutants that largely depends on growth of individual myofibers, making Drosophila a good model for exploring the role of Activin signaling in regulating muscle fiber growth. In addition, the Activin signaling network is significantly simpler in Drosophila than in vertebrates and consists of only three ligands: Activin beta (Actβ), Dawdle (Daw) and Myoglianin (Myo), a close homolog of vertebrate myostatin. These ligands signal through a single type I receptor, Baboon, and a single R-Smad known as Smad2 or Smox (reviewed by Upadhyay et al., 2017).
Intriguingly, we have recently shown that, unlike vertebrates, the Drosophila Activin-like ligand Actβ is a positive regulator of larval muscle mass (Moss-Taylor et al., 2019). To distinguish whether this positive growth function is a general feature of the entire Activin signaling network or represents an aberration resulting from loss of only one ligand, we analyzed muscle growth in babo and Smad2 (Smox) null mutants that eliminate signaling by the entire pathway. We found that when Activin signaling is completely compromised, larval muscle reductions in length, width and thickness are similar to those observed for Actβ loss alone. Hyperactivation of the Activin pathway through expression of constitutively activated Babo produces larger muscles and bodies in which length is disproportionally increased relative to width. We present evidence that Drosophila Activin signaling differentially controls larval muscle growth in three dimensions using both insulin-dependent (width) and insulin-independent (length and thickness) pathways depending on the strength of the Activin signal.
RESULTS
Removal of the entire Activin signal pathway results in smaller larval muscles
To examine how muscle size is altered in Activin pathway mutants, we counted Z-discs from larval skeletal muscle 6 of abdominal segment 2 stained with an α-Actinin (α-Actn) antibody. The Z-disc number is a proxy for sarcomere number and reflects the anterior-posterior length of the muscle cell. We also measured the lateral width of the muscle. We utilized a heteroallelic combination of baobfd4 and babodf(babofd4/d.f.), as well as Smad2F4/Y, as TGF-β/Activin pathway mutants in which canonical signaling was completely abolished (Fig. S1A). The α-Actn antibody labeled Z-discs with a similar intensity in wild-type (wt) and Activin pathway mutant muscles (Fig. 1A), consistent with the results from immunoblot analysis (see Fig. 5A). Notably, however, the surface area of each muscle cell was smaller in the babo and Smad2 mutants than in the wild type (Fig. 1A). Quantitatively, the babo and Smad2 muscles exhibited ∼35% reduction in Z-disc number (56.16±1.39 for wt versus 36.58±0.78 for babo and 38.58±0.71 for Smad2) and ∼25% decrease in muscle width (1.02±0.02 for wt versus 0.79±0.02 for babo and 0.73±0.03 for Smad2) (Fig. 1B), demonstrating that Activin signaling plays an essential role in new sarcomere addition, leading to muscle lengthening, as well as lateral expansion of sarcomeres, which adds to muscle width. In addition to the decrease in Z-disc number, the size of sarcomeres (as measured by Z-disc interval) was also decreased in babo but not Smad2 mutants (Fig. S1D). Therefore, there appears to be an additional effect on muscle length in the babo mutant. Why the sarcomere size is affected by babo but not Smad2 mutation needs further study.2
Fig. 1.

Activin signaling is necessary for proper muscle growth. (A) Schematic showing muscle geometry and representative images of muscles 6 and 7 of abdominal segment 2 of wild-type (wt) and Activin/TGF-β pathway mutants stained with α-Actn antibody. (B) Assessment of muscle length by counting Z-discs and measurement of relative width of the muscle 6 of abdominal segment 2. Both the Z-disc number and muscle width are decreased in babo and Smad2 mutants. (C) Restoring Activin signaling in Smad2 muscle by expressing a wild-type Smad2 transgene rescues the reduced Z-disc number and muscle width. (D) Orthogonal views from z-stack of optical sections representing muscle thickness. (E) Quantification of muscle thickness. Expressing a wild-type Smad2 transgene rescues the reduced thickness of Smad2 muscle. (F) Only Actβ mutant displays a reduction in Z-disc number and muscle width. (G,H) Expression of Actβ in the motor neurons of Actβ mutant rescues the reduced Z-disc number and thickness (G) as well as thickness (H) of the muscle. Values are mean±s.e.m. *P<0.05 and ***P<0.001 from one-way ANOVA followed by Dunnett's test in which each genotype was compared with wt (B,C,E,F) or to Actβ/+;Actβed80 control (G,H). Additionally, unpaired t-tests were performed in C and E as indicated by lines; ##P<0.01 and ###P<0.001. Scale bar: 50 μm.
Fig. 5.
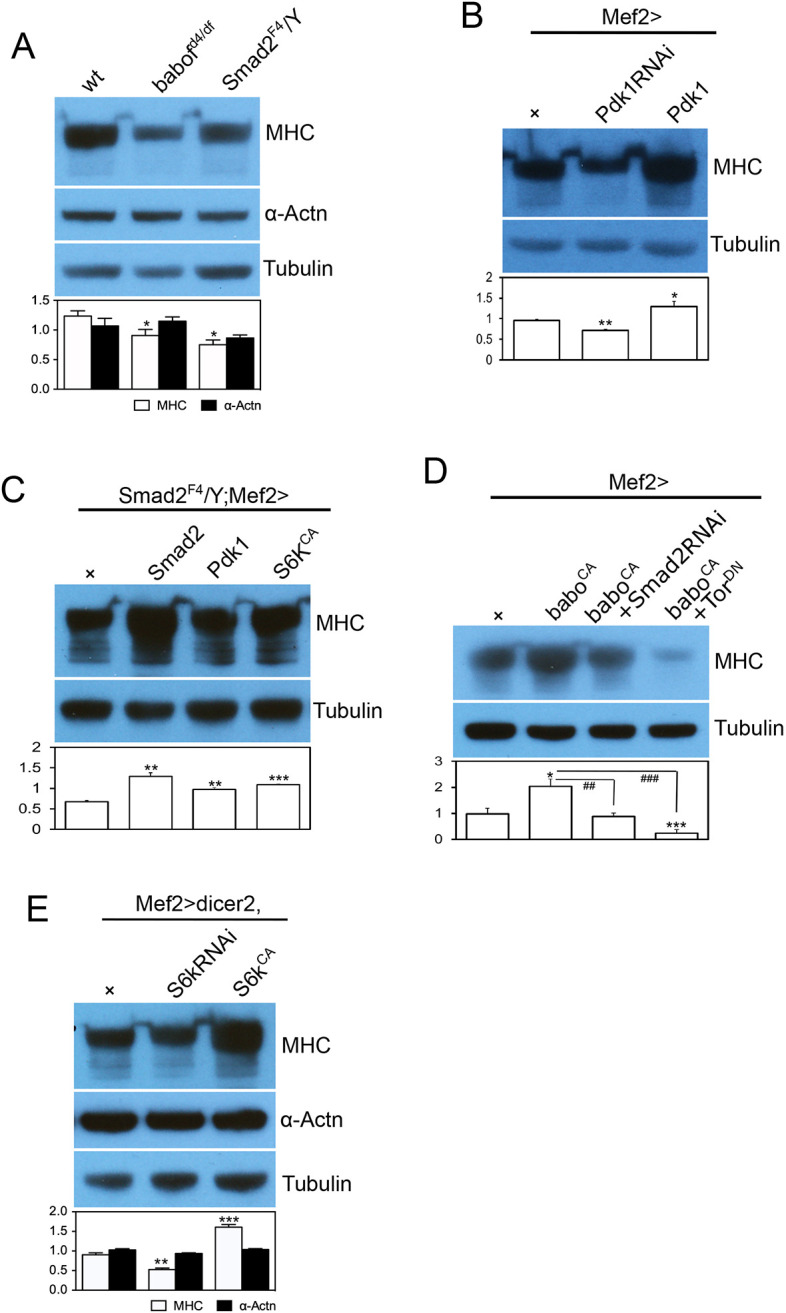
Activin signaling positively regulates Mhc production through its effect on InR/TOR1 activity. Representative immunoblot images and quantification of sarcomeric proteins. (A) The amount of Mhc, a key sarcomeric protein, is decreased in the larval body walls of babo and Smad2 mutants, whereas α-Actn, another sarcomeric protein that localizes to Z-discs, is not affected by these mutations. (B) Muscle expression of Pdk1RNAi decreases Mhc abundance whereas expression of wild-type Pdk1 in the muscle increases it, indicating a positive correlation between Pdk1 expression level and the amount of Mhc. (C) Expressing Smad2, Pdk1 or S6kCA transgenes in Smad2 mutant muscle rescues the decreased Mhc level. (D) Expressing baboCA causes hyperproduction of Mhc, which is suppressed by coexpression of Smad2RNAi or TorDN. (E) Expression of S6kRNAi and S6kCA in wild-type muscle reduces and increases the Mhc level, respectively, without affecting the α-Actn level. Values are mean±s.e.m. *P<0.05, **P<0.01 and ***P<0.001 from one-way ANOVA followed by Dunnett's test in which each genotype was compared with wt (A), Mef2-Gal4/+ (B,D), UAS-dicer2/+;Mef2-Gal4/+ (E) or Mef2-Gal4/+ control in Smad2 mutant background (C). Additionally, unpaired t-tests were performed in D as indicated by lines; ##P<0.01 and ###P<0.001.
We also performed the same assay using muscle 7 of abdominal segment 2 (Fig. S1B) and muscle 6 of abdominal segment 3 (Fig. S1G), from which we obtained similar results. Therefore, it appears that the positive role of Activin signaling in muscle growth is not limited to certain muscle(s) but might be general to all the muscles in the body wall. The effect of Activin signaling on muscle length and width was found to be cell-autonomous, as expression of a Smad2 transgene using a muscle driver restored the decreased Z-disc number and muscle width of the Smad2 mutant (Fig. 1C). To complete the assessment of muscle size along the three axes, we also measured the muscle thickness and found that it decreased by ∼25% in Smad2 muscle (10.58 μm for wt versus 7.81 μm for Smad2F4/Y;Mef2>+; Fig. 1D,E). In addition, expression of Smad2 transgene in the Smad2 muscle completely rescued the muscle thickness (7.81 μm for Smad2F4/Y;Mef2>+ versus 13.6 μm for Smad2F4/Y;Mef2>Smad2; Fig. 1E), indicating that Activin signaling cell-autonomously and positively regulates the growth of muscle cells along the height as well as length and width. Notably, myonuclei number was not changed in babo and Smad2 mutants (Fig. S1F), indicating that myoblast fusion occurs properly in these mutants, but the growth of individual muscle fibers is affected.
A similar reduction in the width and length of muscle cells was observed in Actβ mutants (Fig. 1F) (Moss-Taylor et al., 2019), but not in myo or daw mutants. Furthermore, overexpression of Actβ in motor neurons rescued the decreased Z-disc number and width (Fig. 1G) as well as thickness (Fig. 1H) of Actβ mutant muscle, consistent with the idea that motor neuron-derived Actβ is the major Activin-like ligand that regulates larval muscle growth in Drosophila. Interestingly, the skeletal muscle was disproportionately smaller than other organs in Actβ mutants (Moss-Taylor et al., 2019). Together with the finding that Activin signaling regulates muscle size in a cell-autonomous manner (Fig. 1C,E), these results indicate that Activin signaling influences primarily the muscle growth in many organs. It has also been shown that myonuclei of Actβ mutants are smaller than those of the wild type (Moss-Taylor et al., 2019). To see whether the reduction in myonculei size, which is related to endoreplication, is responsible for the smaller muscle phenotype of Activin pathway mutants, we overexpressed Myc in Smad2 muscle to increase the nuclei size. As presented in Fig. S4A,B, Myc overexpression greatly increased the myonuclei size but failed to increase the muscle size of Smad2 mutants. Furthermore, the ratio between myonuclei and muscle area was not significantly changed in Smad2 mutants (Fig. S4C). Taken together, these results demonstrate that Activin signaling controls muscle growth via a mechanism other than one that affects myonuclei size and/or endoreplication.
Influence of the Activin pathway on InR/TOR signaling in larval skeletal muscle
In mammalian skeletal muscle, Mstn signaling is known to inhibit the IGF-1/PI3K/mTOR pathway (Amirouche et al., 2009; Chen et al., 2017; Zhou et al., 2010). To determine whether the two pathways interact similarly in non-mammalian muscle, we investigated phosphorylation of AKT1 and S6K in Drosophila larval skeletal muscle-epidermis complexes of wild-type and Activin pathway mutants. The pAKT1 antibody used in this study detects phosphorylation of AKT1 at Ser505. This site corresponds to Ser473 of mammalian AKT and is phosphorylated by TORC2 (Hietakangas and Cohen, 2007; Sarbassov et al., 2005; Yang et al., 2006). The pS6K antibody detects phosphorylation at Thr398, which corresponds to Thr389 of mammalian S6K, and is phosphorylated by TORC1 (Kockel et al., 2010; Lindquist et al., 2011; Sarbassov et al., 2005; Yang et al., 2006).
We first confirmed that phosphorylation at these sites is indeed dependent on InR activity in the larval skeletal muscle by overexpressing wild-type InR or by RNA intereference (InR-RNAi, using the Mef2-Gal4 driver) to increase or suppress InR activity, respectively. As shown in Fig. 2A, phosphorylation of AKT1 was greatly reduced by InR-RNAi and increased by InR overexpression in the muscle-epidermis complexes, confirming that the phosphorylation level at Ser505 of AKT1 faithfully reflects InR activity. The results also indicate that TORC2 activation is one of the downstream events of InR activation, because the Ser505 of AKT1 is exclusively phosphorylated by TORC2 (Hietakangas and Cohen, 2007; Sarbassov et al., 2005). The phosphorylation at Thr398 of S6K was similarly regulated by InR activity, that is, decreased by InR-RNAi and increased by InR overexpression (Fig. 2A), suggesting that TORC1 is also positively regulated by InR signaling in Drosophila larval skeletal muscle.
Fig. 2.
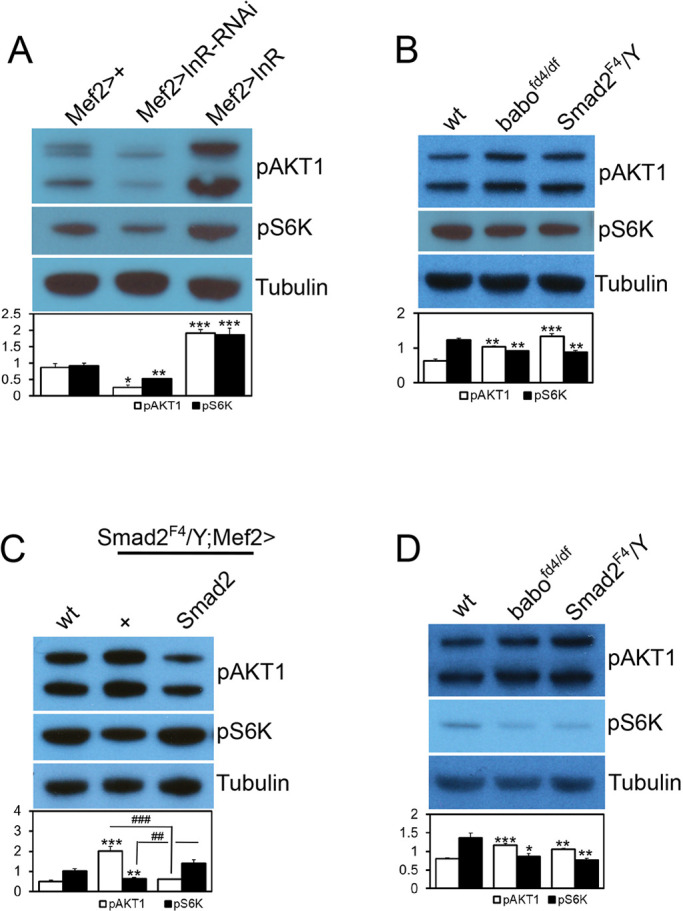
Activin pathway regulates InR/TOR signaling in the body wall. Representative immunoblot images and quantification of pAKT1 and pS6K. (A) Phosphorylation of AKT1 at S505 and S6K at T398 sites are down- and upregulated by muscular expression of InR-RNAi and InR, respectively, suggesting that InR signaling positively regulates the phosphorylation at these sites. (B) Phosphorylation of AKT1 is increased, whereas the pS6K level is decreased in the larval body walls of babo and Smad2 mutants. (C) Resupply of Activin signaling in Smad2 muscle restores pAKT1 and pS6K levels close to those of the wild type. (D) Wandering larvae display the same pattern of alteration in the AKT1 and S6K phosphorylation in the larval body wall as foraging larvae. Values are mean±s.e.m. *P<0.05, **P<0.01 and ***P<0.001 from one-way ANOVA followed by Dunnett's test in which each genotype was compared with Mef2-Gal4/+ control (A) or wt (B-D). Additionally, unpaired t-tests were performed in C as indicated by lines; ##P<0.01 and ###P<0.001.
We next examined the effect of loss of babo and Smad2 on phosphorylation of AKT1 and S6K. In mammalian skeletal muscle and myoblast culture, phosphorylation of AKT at Ser473 is negatively regulated by Myostatin-induced Activin/TGF-β signaling (Lokireddy et al., 2011; Tan et al., 2011; Trendelenburg et al., 2009). We found increased phosphorylation at the corresponding site of Drosophila AKT1 (Ser505) in the babo and Smad2 mutants (Fig. 2B), which implies a negative effect of Activin/TGF-β signaling on AKT1 phosphorylation. The result also suggests that TORC2 activity is elevated in Drosophila Activin/TGF-β pathway mutants. In contrast to AKT1, phosphorylation of S6K was mildly decreased in babo and Smad2 mutants (Fig. 2B), indicating a decreased TORC1 activity. The influence of TGF-β/Activin signaling on phosphorylation of AKT1 and S6K appears to be muscle-specific and cell-autonomous in the muscle-epidermis complexes, because expression of a wild type Smad2 transgene in the muscle of a Smad2 mutant resulted in restoration of pAKT1 and pS6K levels toward those of the wild type (Fig. 2C). In addition to the continuously feeding, foraging larvae that are used for all the immunoblot analyses in this study, we also examined wandering larvae that have ceased feeding, in order to determine whether the alterations in pAKT1 and pS6K levels are dependent on feeding status. As in the foraging stage, the wandering larvae of babo and Smad2 mutants also exhibited elevated pAKT1 and decreased pS6K levels (Fig. 2D). Taken together, these results indicate that TORC1 activity is downregulated in Activin/TGF-β pathway mutants, leading to decreased phosphorylation of S6K (Thr398), whereas TORC2 activity is upregulated, resulting in elevated phosphorylation of AKT1 (Ser505). Furthermore, the regulatory effects of Activin signaling on TORC1 and TORC2 activities appear to be independent of the feeding status.
Negative feedback loop by S6K
Our findings suggest that the TORC1 and TORC2 activities are differentially affected by the loss of canonical Activin signaling, whereas they are both similarly regulated by InR activity (Fig. 2A). To uncover why the phosphorylation statuses of AKT1 and S6K changes in opposite directions, we examined whether a negative feedback loop involving S6K might play a role. It has been shown that mTORC1 negatively regulates the IGF-1/PI3K/AKT pathway by inducing S6K-mediated phosphorylation and degradation of insulin receptor substrate (Harrington et al., 2004; Um et al., 2004). The inhibitory effect of S6K activation on AKT1 phosphorylation at Ser505 has also been demonstrated in Drosophila (Kockel et al., 2010; Sarbassov et al., 2005). Because S6K activity is probably decreased in Activin pathway mutants, as judged by reduced phosphorylation at T398 (Fig. 2B) and the fact that there is lower protein synthesis capacity in these mutants (see below), it is possible that the elevated pAKT1 level in babo and Smad2 muscles is an indirect result of weakened inhibitory feedback of S6K on the InR-AKT1 axis. To test this possibility, we overexpressed an activated form of S6K (S6KCA) in Smad2 muscle to compensate for the decreased S6K activity and found that the pAKT1 level decreased towards that of the wild type (Fig. 3A). In addition, knockdown of S6k in wild-type muscle increased the pAKT1 level, whereas overexpression of S6kCA decreased it (Fig. 3B). To further demonstrate the importance of negative feedback on the phosphorylation status of AKT1 at Ser505, we suppressed TORC1 activity by knocking down raptor, a key component of TORC1, and observed an increase in the pAKT1 level (Fig. 3C). Knocking down rictor, a crucial component of the TORC2 complex, on the other hand, led to decreased phosphorylation of pAKT1, further confirming that TORC2 is the primary player in phosphorylating AKT1 at this site (Fig. 3C). Taken together, these results demonstrate that the elevated AKT1 phosphorylation in babo and Smad2 muscles is a consequence of decreased activity of TORC1 and S6K.
Fig. 3.

Increased phosphorylation of AKT1 is an indirect result of reduced inhibitory feedback by S6K. (A) Overexpressing a constitutively active form of S6k (S6kCA) suppresses the hyperphosphorylation of AKT1 in Smad2 mutant muscle. (B) Overexpression of S6kRNAi and S6kCA in wild-type muscle causes hyper-phosphorylation and hypo-phosphorylation of AKT1, respectively, indicating that the negative feedback loop from S6K to the InR-AKT1 axis functions efficiently in larval body wall muscle. (C) Overexpression of raptorRNAi to inhibit TORC1 activity results in an elevated pAKT1 level, whereas rictorRNAi inhibits TORC2 activity leading to decreased phosphorylation of AKT1. (D) The larval body wall tissue of Activin pathway mutants exhibits lower protein synthesis capacity, as assayed by the SUnSET method. (E) Rescue of decreased protein synthesis capacity by muscle-specific expression of Smad2 transgene. Values are mean±s.e.m. *P<0.05 from one-way ANOVA followed by Dunnett's test in which each genotype was compared with wt (A) or UAS-dicer2/+;Mef2-Gal4/+ control (B,C). Additionally, an unpaired t-test was performed in A as indicated by lines; ##P<0.01.
Finally, we investigated the protein synthesis capacity of wild-type and TGF-β/Activin pathway mutants, which is known to be controlled by TORC1 and S6K activities. By adopting the SUnSET method (Schmidt et al., 2009), we found that protein synthesis capacity was reduced in the body wall tissue of babo and Smad2 mutants (Fig. 3D). Furthermore, the decreased capacity in protein synthesis was rescued by muscle-specific expression of wild-type Smad2 in Smad2 mutants (Fig. 3E). These results further demonstrate that the activities of TORC1 and S6K, both key regulators of protein synthesis, are downregulated in the muscle of TGF-β/Activin pathway mutants.
Transcriptional regulation of InR/TOR pathway components by Activin signaling
Considering that Smad2, the R-Smad of the Activin/TGF-β pathway, is a transcription factor, one possibility is that the Drosophila Activin/TGF-β pathway influences the InR/TOR pathway via transcriptional control of one or more of its signal transduction component(s). To gain insight into the transcriptional influence of the Activin/TGF-β pathway on InR/TOR signaling, we performed RNA sequencing (RNA-seq) using wild-type and Smad2 mutant muscles as well as Mhc-Gal4;tub-Gal80ts controls and baboCA gain of function-expressing samples. The heat map using FPKM values of genes encoding InR/TOR signaling components showed that transcripts of Pdk1 and Akt1 were significantly decreased in the Smad2 mutant, whereas transcription of the rest of the genes was unaffected, suggesting a positive role of the Activin/TGF-β pathway in the transcription of specific sets of genes in the InR/TOR signaling pathway (Fig. 4A). In addition, temporal expression of activated Babo led to an increase in the transcripts of Pdk1 and Akt1, further demonstrating the positive role of the Activin/TGF-β pathway (Fig. 4A). The RNA-seq results were then validated by qPCR analysis, which also demonstrated a decrease in the transcripts of Pdk1 and Akt1 (Fig. 4B). Consistent with the findings from RNA-seq and qPCR analyses, the total protein level of AKT1 was found to be decreased in babo and Smad2 mutants (Fig. 4C), even though the pAKT1 level was elevated in these mutants (Fig. 2B). Taken together, these results suggest that the Activin pathway impinges on the InR/TOR pathway via control of the transcription of some of its signal transduction components.
Fig. 4.

Activin signaling promotes transcription of the InR/TOR pathway components Pdk1 and Akt1. (A) Heat map shows the effects of Smad2 loss and temporal overexpression of baboCA on the transcript levels of InR/TOR pathway components. Although most of the components are not affected, the expression of Pdk1 and Akt1, highlighted by a red rectangle in the heat map, are downregulated by Smad2 mutation and upregulated by temporal expression of baboCA. (B) Verification of the RNA-seq results by qPCR. (C) Consistent with the RNA-seq and qPCR results, the total protein level of AKT1 is decreased in babo and Smad2 mutants. (D) Overexpression of Pdk1 in Smad2 muscle restores the increased pAKT1 level towards that of wild type. (E) Expression of Pdk1RNAi in the skeletal muscle produces a similar phenotype in pAKT1 level as loss of Activin signaling, whereas Pdk1 overexpression suppresses AKT1 phosphorylation. (F) A heteroallelic combination of Pdk1 mutations causes hyperphosphorylation of AKT1. Values are mean±s.e.m. *P<0.05, **P<0.01 and ***P<0.001 from one-way ANOVA followed by Dunnett's test in which each genotype was compared with wt (B-D,F) or Mef2-Gal4/+ control (E). Additionally, unpaired t-tests were performed in D and F as indicated by lines; ###P<0.001.
To determine whether the decrease in the transcripts of InR/TOR signaling components is responsible for any of the phenotypes exhibited by Activin pathway mutants, we sought to restore the expression level of Pdk1 in the Smad2 mutant muscle and examined pAKT1 (Ser505) levels, which were inversely correlated with TOCR1 and S6K activities (Fig. 3). As shown in Fig. 4D, overexpressing Pdk1 in Smad2 muscle reduced the elevated pAKT1 level toward that of the wild type, suggesting that the decrease in expression of Pdk1 is, at least partly, responsible for the elevated phosphorylation of AKT1. In a converse experiment, Pdk1 was either knocked down or overexpressed in wild-type muscles and the phosphorylation of AKT1 was examined. In line with the idea that the Pdk1 expression level negatively correlates with AKT1 phosphorylation, Pdk1 knockdown increased the pAKT1 level whereas Pdk1 overexpression decreased it (Fig. 4E). Because the pAKT1 level also negatively correlated with S6K activity (Fig. 3B) and S6K activity was enhanced upon phosphorylation at the active site (Thr238) by PDK1, as inferred from mammalian results (Pullen et al., 1998), we propose that the effect of changes in Pdk1 expression on pAKT1 phosphorylation can be ascribed to the alterations in S6K activity. Moreover, AKT1 phosphorylation was also found to be increased in Pdk1 mutants (Fig. 3F), further supporting the idea of negative correlation between PDK1 activity and pAKT1 level. Together, these results suggest that an alteration in the expression of downstream signal transduction components can significantly affect the output of the InR/TOR signaling pathway, even in the absence of a change in ligand availability or activity and this appears to be the mechanism by which the Activin pathway influences InR/TOR signaling.
Effect of Activin/TGF-β pathway on sarcomeric protein levels
In some cases, the Mstn/Activin pathway has been shown to negatively regulate Myosin heavy chain (Mhc) levels, which in turn correlate with the change in muscle size in mammalian myoblast culture and in skeletal muscle (Hulmi et al., 2013; Lokireddy et al., 2011). We examined whether the Activin pathway similarly affects Mhc levels in Drosophila muscle. In immunoblot analysis using muscle-epidermis tissue, babo and Smad2 mutants exhibited reduced Mhc abundance (Fig. 5A), even though the transcript level of Mhc was not changed (Fig. S2A). Furthermore, expressing a wild-type Smad2 transgene in Smad2 muscle rescued the decrease in Mhc level, indicating a tissue-autonomous effect of the Activin pathway (Fig. 5C). In contrast, the total amount of α-Actn, another sarcomeric protein, was not altered in babo and Smad2 mutants (Fig. 5A) despite the increase in its transcript level (Fig. S2A). Therefore, it appears that the Activin pathway has variable effects on protein and transcript levels of different sarcomeric proteins. Specifically, the Activin pathway either positively regulates the translation or suppresses the degradation of Mhc in Drosophila larval skeletal muscle.
As shown above, a decrease in Pdk1 expression gives rise to altered InR/TOR signaling in babo and Smad2 mutants. Because InR/TOR signaling has a role in protein synthesis and degradation, we investigated whether the decrease in Pdk1 expression also contributes to the change in Mhc level found in Activin pathway mutants. As illustrated in Fig. 5B, knockdown or overexpression of Pdk1 resulted in decreased or increased levels of Mhc, respectively, demonstrating a positive relationship between Pdk1 expression and Mhc protein levels. We then overexpressed Pdk1 in Smad2 muscle and found that it rescued the decreased Mhc level (Fig. 5C). From these results, we conclude that a decrease in Pdk1 expression is, at least in part, responsible for the reduced Mhc level shown by Activin pathway mutants.
We found that decreased Pdk1 expression led to diminished TORC1 activity in Activin pathway mutants, as illustrated by inversely correlating the pAKT1 level (Fig. 4D,E). To investigate the contribution of TORC1 in the regulation of Mhc level by the Activin pathway, we expressed dominant negative Tor (TorDN) together with activated babo (baboCA). Activated Babo alone increased Mhc production twofold, which was suppressed by coexpression of Smad2RNAi (Fig. 5D), meaning that BaboCA promoted Mhc production primarily through canonical Smad2-dependent signaling. Interestingly, coexpression of TorDN resulted in an even stronger suppression of the hyper-Mhc production induced by BaboCA (Fig. 5D). Furthermore, a similar result was obtained by coexpression of baboCA and raptorRNAi (Fig. S2B), suggesting that TORC1 activity mediates almost all of the effect of BaboCA on the Mhc level. Furthermore, we overexpressed S6kCA in Smad2 mutant muscle to assess the contribution of S6K, a key downstream effector of TORC1, and found that it rescued the Mhc level (Fig. 5C). In line with the essential role of S6K in regulating Mhc production, expression of S6kRNAi caused a significant decrease in Mhc levels and S6KCA a significant increase (Fig. 5E). As in babo and Smad2 mutants, the α-Actn level was not affected by alterations in S6K activity (Fig. 5E). These results emphasize the importance of the TORC1-S6K axis in mediating the effect of the Activin pathway on Mhc abundance.
Activin signaling promotes muscle growth through both InR/TORC1-dependent and -independent mechanisms
The finding that TORC1 signaling and Mhc levels are downregulated in Activin pathway mutants led us to hypothesize that the reduction in Mhc via reduced TORC1 signaling is primarily responsible for the decreased muscle growth observed in Activin pathway mutants. As shown above, the decreased Mhc level of Smad2 muscle was rescued by overexpression of Pdk1 and S6kCA (Fig. 5C). Because Mhc is an essential building block of sarcomeres, we reasoned that overexpression of Pdk1 or S6kCA would rescue the sarcomere number of Smad2 muscle. Surprisingly, however, overexpression of either of these transgenes failed to rescue the sarcomere number, as assayed by counting the Z-discs in Smad2 muscle (Fig. 6A). Moreover, S6kCA even further decreased the sarcomere number from that of control Smad2 mutant muscle (Fig. 6A). Therefore, these results indicate that sarcomere formation can be decoupled from sarcomeric protein production and also suggest that the Activin pathway promotes sarcomere formation independently of its influence on InR/TORC1 signaling and Mhc production. Interestingly, overexpression of Pdk1 or S6kCA increased the width (Fig. 6A) but not the thickness (Fig. 6B) of Smad2 muscle. From these results, we suggest that if Mhc is overproduced in the absence of canonical Activin signaling, it is primarily used for lateral expansion of the muscle probably through addition to existing sarcomeres. To further test the role of Pdk1 in lateral growth of the muscle, we overexpressed Pdk1RNAi to deplete PDK1 in wild-type muscle and found a decrease in muscle width, but no change in Z-disc number (Fig. 6C). We also altered S6K activity by overexpressing S6kRNAi or S6kCA. Despite profoundly affecting Mhc abundance (Fig. 5E), neither S6kRNAi nor S6kCA had much effect on the Z-disc number (Fig. 6D), further demonstrating the lack of correlation between Mhc production and formation of new sarcomeres.
Fig. 6.

Z-disc number and muscle thickness are decoupled from Mhc level. (A) Overexpression of Pdk1 or S6kCA in Smad2 muscle rescues the muscle width but not the Z-disc number. (B) Overexpression of Pdk1 or S6kCA fails to restore the reduced thickness of Smad2 muscle. (C) Pdk1RNAi expression decreases the muscle width but not the Z-disc number. (D) Although the S6kRNAi and S6kCA profoundly affect the Mhc level in larval body wall tissue (Fig. 5E), they have no or little effect on Z-disc number of the muscle. However, the muscle width is significantly reduced by S6kRNAi and increased by S6kCA expression. (E) Representative images of muscles 6 and 7 of abdominal segment 2 stained with α-Actn antibody. (F) Z-disc number and relative width of the muscle expressing baboCA alone and together with Smad2RNAi or TorDN. (G) Representative wandering stage larvae expressing Smad2RNAi or baboCA in the muscle and a control larva. Values are mean±s.e.m. *P<0.05, **P<0.01 and ***P<0.001 from one-way ANOVA followed by Dunnett's test in which each genotype was compared with UAS-dicer2/+;Mef2-Gal4/+ control (D), Mef2-Gal4/+ control (F) or Mef2-Gal4/+ control in Smad2 mutant background (A,B). Unpaired t-tests were performed in C and F (indicated by lines); #P<0.05, ###P<0.001. Scale bar: 120 μm.
We have shown that overexpressing baboCA alone caused a twofold increase in the Mhc level, which was suppressed by coexpressing Smad2RNAi or TorDN (Fig. 5D). We next investigated the effects of these manipulations on muscle size. As presented in Fig. 6E, overexpression of baboCA greatly increased muscle length, which was reverted by coexpression of Smad2RNAi. By contrast, TorDN failed to suppress the increase in muscle length caused by baboCA. In the Z-disc counting assay, baboCA-expressing muscles exhibited, on average, 20 more sarcomeres than the Mef-Gal4 controls (70.9±1.43 for Mef2>+ versus 89.45±0.91 for Mef2>baboCA; Fig. 6F). As expected, Smad2RNAi completely blocked the increase in sarcomere number caused by baboCA overexpression and even further reduced the sarcomere number from that of controls (89.45±0.91 for Mef2>baboCA versus 61±0.72 for Mef2>baboCA+Smad2RNAi versus 70.9±1.43 for Mef2>+; Fig. 6F). In contrast, TorDN only mildly suppressed the effect of baboCA so that the sarcomere number was still higher than that of controls (89.45±0.91 for Mef2>baboCA versus 79.63±2.13 for Mef2>baboCA+TorDN versus 70.9±1.43 for Mef2>+; Fig. 6F). Considering that the Mef2>baboCA+TorDN muscle probably had a higher level of Activin signaling but a lesser amount of Mhc (Fig. 5D) than the Mef2>baboCA+Smad2RNAi and Mef2>+ control muscles, we conclude that the sarcomere number better correlates with the level of Activin signaling than with sarcomeric protein abundance. In contrast to the sarcomere number, muscle width was reduced by baboCA overexpression and rescued by Smad2RNAi (Fig. 6F). We rationalize that the muscle width is smaller in baboCA-overexpressing muscle to accommodate the large increase in sarcomere number. In other words, sarcomeric subunits are assembled into new sarcomeres, expanding the muscle length at the expense of muscle widening, through addition of sarcomeric proteins into existing Z-discs.
Finally, we examined the relationship between muscle size and whole larval body size. As illustrated in Fig. 6G, blocking Activin signaling in the muscle via Smad2RNAi expression resulted in short larvae, whereas BaboCA expression in the muscle produced very elongated larvae. Considering that the babo and Smad2 mutants have shorter muscles with fewer number of Z-discs (Fig. 1A,B) and baboCA-expressing larvae bear elongated muscles (Fig. 6E,F), there is a positive correlation between muscle and larval body length.
DISCUSSION
In this study, we assess the effect of canonical Drosophila Activin signaling on InR/TOR pathway activity and its relation to larval body-wall muscle growth. Our findings reveal an unexpected and striking difference in the way that Activin signaling regulates muscle size in Drosophila larvae compared to mammals. In Drosophila, Activin signaling promotes muscle growth, whereas in developing vertebrates it limits muscle mass. As in vertebrates, we find that the InR/TORC1 pathway is a core conserved target that mediates muscle size control in response to Activin, but the activity of the IGF-1/TORC1 pathway is regulated in opposing directions in the two systems. We also find that stimulation of InR/TORC1 signaling in Drosophila in the absence of Activin leads to upregulation of Mhc, which is incorporated into existing sarcomeres to increase their width and thickness. However, in the presence of Activin signaling both the width and length of muscle fibers are enhanced. The combinatorial effect of these two sarcomeric assembly processes is the formation of larger larval body wall muscles with an increased volume.
Differential modulation of the IGF-1/TORC1 pathway accounts for the opposing effects of Activin signaling on mammalian versus Drosophila somatic muscles size.
The present study demonstrates that the Activin pathway in Drosophila controls the output of the InR pathway by regulating the expression levels of PDK1 and AKT1, two downstream InR signal transduction components. Because the steady-state levels of these two transcripts are lower in the body walls of Smad2 and babo mutants and are increased by expression of an activated Babo in muscle, it seems likely that these two genes are direct transcriptionally regulated targets of Smad2 and that Activin signaling boosts InR/TORC1 activity, resulting in enhanced S6K activity, higher general levels of protein synthesis and increased levels of Mhc. When reception of Activin signaling is compromised, then the opposite occurs. This mechanism is quite different from that proposed for how Mstn/Activin signaling impinges on the insulin/IGF-1 activity in mammals. In general, it has been reported that AKT phosphorylation is upregulated in the absence of Mstn/Activin signaling (Hitachi et al., 2014; Tan et al., 2011). Other points of intersection between the pathways have also been reported, including several studies in mice suggesting that Mstn/Activin signaling suppresses expression of miRNAs that inhibit PTEN translation (Goodman et al., 2013; Hitachi et al., 2014). This leads to lower levels of AKT phosphorylation, decreased mTORC1 activation and smaller muscles in the presence of Mstn/Activin signals.
The IGF-1/TORC1 pathway is only one point of intersection from which to understand muscle size control. In general, muscle homeostasis is thought to be regulated by balancing the activities of protein synthesis and degradation pathways (Bonaldo and Sandri, 2013; Schiaffino and Mammucari, 2011). IGF-1/TORC1 signaling clearly interfaces with both these modes of protein homeostasis control; however, in most cases it is not clear whether Smad directly regulates expression of specific components in either the synthesis or degradation pathways or whether most of its effects can be attributed to regulation of IGF-1/TORC1.
Why mammals and Drosophila are wired in opposite directions in terms of the influence of Activin signaling on IGF-1/TORC1 activity and muscle size control is unclear. The Mstn/Activin branch of the TGF-β family is very ancient and is present in some pre-Bilateria groups, including several cnidarian species (Watanabe et al., 2014). Clearly, many additional phyla, classes and species of animals need to be examined to more fully understand how and why these contrasting roles in muscle size control have evolved.
IGF-1/TORC1 negative feedback and muscle homeostasis
It is well documented in mammals that there is a negative feedback loop formed by S6K toward insulin receptor substrate, which profoundly diminishes the efficacy of signaling from insulin/IGF-1 to the PI3K-AKT axis (Harrington et al., 2004; Shah et al., 2004; Zhang et al., 2008). A similar negative feedback loop has also been demonstrated in Drosophila cell culture (Sarbassov et al., 2005) and wing imaginal discs (Kockel et al., 2010), but has never been studied in skeletal muscle. Here we demonstrate that this negative feedback loop does indeed work efficiently in Drosophila skeletal muscle. As the inhibitory feedback has a profound effect on insulin responsiveness, it will be interesting to determine how the peripheral tissues in Activin pathway mutants react to Drosophila insulin-like peptides. Considering the fact that the absolute expression levels of PDK1 and AKT1 are downregulated in the Activin pathway mutants, the response to insulin-like peptides may be compromised even though the inhibitory feedback is alleviated. Consistent with this idea, we previously reported that both Smad2 and daw mutants display increased hemolymph sugar levels (Ghosh and O'Connor, 2014), suggesting an impairment in the regulation of blood sugar level. In addition, it might indicate that the decrease in PDK1 and AKT1 expression overrides the effect of relieved negative feedback. Further study is required to unveil how these competing effects are summed by tissues to determine their responsiveness to insulin or other growth factors.
Which TGF-β superfamily members control muscle size?
In mammals, the TGF-β superfamily consists of at least 30 ligands that are broadly classified into the bone morphogenetic proteins (BMPs) that transduce signals through Smads1,5,8 and members of the TGF-β/Activin subgroup (including Mstn) that signal through Smads2 and 3 (Sartori et al., 2014). The Drosophila system is much simpler with only six clear family members, three of which are classified as BMPs and three that belong to the Activin subgroup including Myoglianin, the homolog of vertebrate Mstn (Upadhyay et al., 2017). In vertebrates, the full complement of ligands that participate in muscle size control in not known. Mstn is by far the best-studied family member in terms of post-myogenic muscle growth control; however, several lines of evidence suggest that other activins, as well as BMP family members, also participate in muscle size homeostasis. Overexpression of various types of ligand-binding proteins with diverse specificity for the Activin family members produced more extreme muscle hypertrophy in mice than mstn knockout alone, implying that other Activin/TGF-β factors probably contribute to muscle growth control (Chen et al., 2015, 2017, 2014; Lee and McPherron, 2001; Lee et al., 2005; Winbanks et al., 2012).
Although the BMP arm of the superfamily has not received as much attention, overexpression of BMP-7 or its activated type I receptor in muscle resulted in enhanced Smad1,5,8 phosphorylation and hypertrophic muscle growth (Sartori et al., 2013; Stantzou et al., 2017; Winbanks et al., 2013). Intriguingly, this appears to be accomplished, in part, through mTORC1 activation, increased protein synthesis and reduced protein turnover, very similar to our findings for the Activin pathway in Drosophila. At present, no specific studies addressing the role of BMPs in muscle growth control have been reported in Drosophila, although it is worth noting that the BMP-7 homolog, Gbb, is expressed in larval muscle and strongly affects neuromuscular junction size and function (McCabe et al., 2003). In terms of three Drosophila Activin-like ligands, muscle size regulation appears to be primarily accomplished by motoneuron delivery of Actβ to the muscle during larval growth (Moss-Taylor et al., 2019). Loss of Actβ results in reduction of larval muscle size to a similar extent as reported here for Babo and Smad2 loss, whereas genetic null mutations in either myo or daw, the other two Activin-like ligands, produce no change in muscle size (Fig. 1D) (Upadhyay et al., 2020). Furthermore, loss of Actβ also results in similar electrophysiological defects at the neuromuscular junction as found for Babo and Smad2, whereas loss of Daw or Myo have little effect (Kim and O'Connor, 2014). These data all support Actβ as the primary Drosophila TGF-β-like ligand involved in muscle size control.
Differential assembly of sarcomeres controls muscle geometry
A major unanswered question concerns how the formation of new sarcomeres versus expansion of existing sarcomeres is differentially regulated. Although autonomous mechanisms within muscles probably contribute to the dimensional assembly of sarcomeres, it is important to recognize the probable role that external signals play in this process. Because skeletal muscles are attached to the epidermis, which is tightly associated with the cuticle, epidermis-cuticle size is probably able to restrict or shape the way that skeletal muscle cells grow. For example, it has been shown that attachment of flight muscle to the tendon during metamorphosis generates tension at the muscle-tendon junction which, in turn, triggers simultaneous formation of sarcomeres that become aligned along the tendon-tendon axis, resulting in myofibrillogenesis (Weitkunat et al., 2014). Later, the muscle grows and new sarcomeres are added somewhere along the long axis, although the location has not yet been determined (Spletter et al., 2018). It is not clear whether a similar mechanism is utilized during larval skeletal muscle fiber assembly because the larval exoskeleton is soft cuticle that keeps changing in length as larvae move, making the myotendinous system more dynamic than that of pupae and adults, which have a more ridged cuticle.
In addition to the mechanical tension, the epidermis may also set a limit on muscle growth and influence sarcomere addition through a ‘coordination signal’. For instance, if the epidermis-cuticle grows at a slower rate than muscle, a signal may be produced, perhaps through mechanical tension, that will restrict the lengthwise growth of the muscle but still accommodates some lateral growth. Our finding that manipulating Activin signal reception only in muscles induced corresponding changes in the size of the whole body (Fig. 6E,G) could be explained by coordinated intertissue signaling whereby the muscle sends an instructive growth signal to the epidermis-cuticle, which then signals back to the muscle so that growth is coordinated between these two functionally entwined tissues.
In summary, we envision a multistep mechanism for Activin control of Drosophila muscle fiber growth (Fig. 7). First, it regulates production of muscle fiber structural subunits such as Mhc through enhancement of Akt1 and Pdk1 transcript levels. In a second step, these subunits are assembled, under guidance by Activin signaling, into either existing sarcomeres to build muscle fiber width and thickness or into new sarcomeres, leading to elongation of the muscle. We propose that elongation of muscles in particular requires coordination between muscle and epidermal growth and that Activin signaling, either directly or indirectly, manages this communication.
Fig. 7.

Control of InR/TOR signaling network and muscle growth by Activin signaling pathway. Activin signaling positively regulates InR/TOR signaling by promoting the transcription of Pdk1 and Akt1. Activation of InR increases PI3K-dependent PIP3 generation, leading to increased activity of PDK1. PDK1 then phosphorylates AKT1 at Thr342 and S6K at Thr238. The PI3K-generated PIP3 is also necessary for activation of the TORC2 complex, which phosphorylates Ser505 of AKT1. When phosphorylated at Thr342 and Ser505 sites, AKT1 initiates a cascade of inhibition leading to activation of TOCR1 complex that phosphorylates S6K at Thr398. The sequential phosphorylations at Thr398 and Thr238 sites fully activate S6K. Mammalian homologous sites of phosphorylation are presented in parentheses. The activated S6K then promotes the production of certain sarcomeric proteins as well as inhibits signal transduction from InR to PI3K. InR/TORC1 signaling increases the steady-state level of Mhc, which can be preferentially added to the lateral side of existing sarcomeres when Activin signaling is low or absent. However, when activin signaling is high in the muscle this promotes epidermal growth, which enables lengthwise addition of sarcomeres, perhaps as a consequence of reduced muscle tension and/or the expression of putative factor(s) whose function is to shift the mode of Mhc addition toward formation of new sarcomeres and vertical expansion of existing sarcomeres.
MATERIALS AND METHODS
Drosophila strains and husbandry
Fly lines were kept on standard cornmeal-yeast-agar medium at 25°C. For experiments involving babo, Smad2 and daw mutants, larvae were raised on yeast paste placed on apple juice-agar plates because these mutants do not grow well on standard medium. The w1118 strain was used as a wild-type (wt) control for babo, Smad2 and ligand mutants. Smad2F4, babofd4, babodf, Actβed80, daw1, daw11, myo1, UAS-Smad2 and UAS-baboCA (constitutively active) lines have been described previously (Brummel et al., 1999; Kim and O'Connor, 2014; Ting et al., 2007). myoCR2 was generated by BestGene by CRISPR-mediated mutagenesis using 5′-CTTCGACTATTCACCGCGCTATTA-3′ as a guide RNA. The resulting line contains a 1 bp deletion, resulting in frameshift and stop prior to the ligand domain, and is a putative null mutant (Fig. S1E). The Mef2-Gal4 (BL27390) line was used as a muscle driver throughout the study except in the RNA-seq analysis. To ensure that the results observed in this study were the consequence of muscle-specific acts of the Mef2-Gal4 driver, we repeated some of the key experiments using the Mhc-Gal4 (Demontis et al., 2014) driver, which is considered to be more specific to skeletal muscle, and obtained similar results (Fig. S3). Other stocks used were Pdk13 (Rintelen et al., 2001), Pdk133 (Cheng et al., 2011), UAS-dicer2 (BL24650), UAS-S6kRNAi (NIG 10539R-2), UAS-S6KCA (BL6914), UAS-raptorRNAi (BL31528-JF01087), UAS-rictorRNAi (BL31527-JF01086), UAS-Pdk1 (Cheng et al., 2011), UAS-Pdk1RNAi (BL27725-JF02807), UAS-TorDN (BL7013), UAS-InR (BL8284), UAS-InR-RNAi (VDRC 992), UAS-Myc (BL9675).
Immunoblot analysis
Late foraging third instar larvae were used for immunoblots unless otherwise noted. Four to six larval body wall tissues containing muscle-epidermis complex were homogenized in 21 μl of RIPA buffer (Sigma, #R0278) supplemented with a cocktail of protease inhibitors (Complete mini, Roche) and incubated at 4°C for 40 min with agitation. After centrifugation, 13 μl of supernatant from each sample was transferred into a new tube, mixed with 7 μl of 3× loading buffer and denatured for 5 min at 95°C. Equal volumes from each sample were run on 4-12% Bis-Tris gel (Novex, #NP0322BOX) and transferred onto PVDF membrane (Millipore, #IPFL00010). The membranes were then blocked with Casein-containing buffer (Bio-Rad, #1610783) and incubated with primary antibody at 4°C overnight. The primary antibodies used were rabbit anti-AKT1 (1:1000; Cell Signaling Technology, #4691), rabbit anti-pAKT1 (1:1000; Cell Signaling Technology, #4054), rabbit anti-pS6K (1:250; Cell Signaling Technology, #9209), rabbit anti-pSmad2 (1:500; Cell Signaling Technology, #3108), mouse anti-α-Actn (1:50; DSHB, 2G3-3D7), anti-β-Tubulin (1:1000; DSHB, E7). Secondary antibodies were HRP-conjugated anti-rabbit or mouse IgG (1:10,000; Cell Signaling Technology, #7074 and #7076, respectively). Bands were visualized using Pierce ECL Western Blotting Substrate (Thermo Scientific, #32209) and band intensities were quantified using ImageJ (NIH) software. The quantification graphs are presented beneath the representative immunoblot images and show mean±s.e.m. from at least three independent samplings. The title of the y-axis of each graph is ‘Relative protein level’ and has been omitted for simplification.
Muscle size assessment
Wandering larvae were rinsed in double distilled H2O and dissected in Ca2+-free HL3 medium as described previously (Kim and O'Connor, 2014). The larval fillets were then fixed in 3.7% paraformaldehyde solution (Electron Microscopy Sciences) for 15 min at room temperature (RT). After washing in 1× PBS and permeabilization in 1× PBT (0.5% BSA plus 0.2% Triton X-100 in 1× PBS), the fillets were then incubated with mouse α-Actn antibody (1:100; DSHB, 2G3-3D7) overnight at 4°C and Alexa Fluor 555-conjugated secondary antibody (1:200; Molecular Probes) at RT for 2 h.
Images were taken using a Zeiss LSM 710 confocal microscope. From the α-Actn staining, the Z-disc numbers were counted using the PeakFinder macro in ImageJ (NIH) followed by manual adjustment (Fig. S1C). The PeakFinder macro was also used for finding Z-disc intervals (Fig. S1C). The muscle width was measured using ImageJ software and normalized to wild-type or control values. Muscle thickness was obtained from the orthogonal view of the z-stack of 1 μm optical sections.
Protein synthesis assay
The surface sensing of translation (SUnSET) method (Schmidt et al., 2009) was adopted to monitor the protein synthesis capacity of the skeletal muscle with little modification. The SUnSET assay takes advantage of the fact that puromycin, a structural analog of aminoacyl-tRNA, can be incorporated into elongating polypeptide chains and then immunologically detected. In the assay, two or three fillets of late foraging larvae were incubated in M3 insect medium (Sigma-Aldrich) supplemented with 2 mM of trehalose (Sigma-Aldrich), 15 μg/ml of puromycin (Sigma-Aldrich) and 10 μg/ml of insulin (Sigma-Aldrich) for 20 min at RT. The fillets were washed three times in 1× PBS and then sampled for immunoblotting. Anti-puromycin antibody (Millipore, #MABE343) was used at 1:10,000 dilution. Representative images from triplicate assays are shown.
qRT-PCR
Seven to ten larvae were dissected in 1× PBS to remove the internal organs. Total RNAs were prepared from the remaining muscle-epidermis complexes using TRIzol reagent (Invitrogen), followed by cleanup with RNAeasy Mini kit (Qiagen). The Superscript III first-strand synthesis kit (Invitrogen) was used to synthesize cDNA, and qRT-PCR reactions were performed on the LightCycler 480 (Roche) using a SYBR Green kit. Each sample was triplicated per reaction. Rpl23 was used as a normalization control. The fold changes were calculated based on values obtained using the second derivative maximum method. Data are mean±s.e.m. from at least three independent mRNA preparations.
RNA sequencing analysis
Total RNAs were extracted using TRIzol reagent (Invitrogen) and further cleaned using the RNAeasy kit (Qiagen) from 30 muscle-epidermis complexes of wt and Smad2 mutants, as well as tub-Gal80ts/+;Mhc-Gal4/+ and tub-Gal80ts/+;Mhc-Gal4/baboCA animals that were heat-shocked for 12 h at 30°C for temporal expression of baboCA. Total RNA (3 μg per genotype) was submitted to the University of Minnesota Genomics Center (UMGC) for quality assessment and Illumina next-generation sequencing. In the UMGC, the integrity of RNAs was assessed using capillary electrophoresis (Agilent Bioanalyzer 2100). The sequencing libraries were constructed using TruSeq RNA preparation kit v2 after the mRNAs were enriched by oligo-dT-mediated purification. The libraries were then sequenced on a 100 bp paired-end run on the Illumina HiSeq 2500. Over 10 million reads were generated per library. The RNA-seq reads were mapped to the Drosophila genome using TopHat. After mapping, the reads were assembled into transcripts using Cufflinks, which generated fragments per kilo base of transcript per million mapped reads (FPKM) values. Gene differential expression tests were performed using Cuffdiff. Finally, heat maps were drawn using R with the ggplot2 package.
Statistical analysis
Statistical analyses were performed using Prism software (version 6.0, GraphPad Software). Data are presented as mean±s.e.m. One-way ANOVA followed by Dunnett's test was used for comparisons among multiple groups and asterisks are used to denote the significance. Comparisons between two groups were performed by unpaired t-test and significances are denoted by pound signs. Graphs were drawn using either Prism or Exel software.
Supplementary Material
Acknowledgements
We thank Graeme W. Davis and Ernst Hafen for providing Pdk1 mutants. We are grateful to the Bloomington Drosophila Stock Center for providing fly lines and Developmental Studies Hybridoma Bank for α-Actn antibody. We also thank Aidan Peterson, Thomas Neufeld and Hiroshi Nakato for comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.K., M.B.O.; Methodology: M.K.; Validation: M.K.; Formal analysis: M.K.; Investigation: M.K.; Resources: M.B.O.; Data curation: M.K.; Writing - original draft: M.K.; Writing - review & editing: M.K., M.B.O.; Supervision: M.B.O.; Project administration: M.B.O.; Funding acquisition: M.B.O.
Funding
This work was supported by the National Institutes of Health (1R35 GM-118029 to M.B.O.). Deposited in PMC for release after 12 months.
Data availability
The RNA-seq data have been deposited in GEO under accession number GSE162810.
Supplementary information
Supplementary information available online at https://dev.biologists.org/lookup/doi/10.1242/dev.190868.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.190868.reviewer-comments.pdf
References
- Amirouche A., Durieux A.-C., Banzet S., Koulmann N., Bonnefoy R., Mouret C., Bigard X., Peinnequin A. and Freyssenet D. (2009). Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology 150, 286-294. 10.1210/en.2008-0959 [DOI] [PubMed] [Google Scholar]
- Amthor H., Otto A., Vulin A., Rochat A., Dumonceaux J., Garcia L., Mouisel E., Hourdé C., Macharia R., Friedrichs M. et al. (2009). Muscle hypertrophy driven by myostatin blockade does not require stem/precursor-cell activity. Proc. Natl. Acad. Sci. USA 106, 7479-7484. 10.1073/pnas.0811129106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen H. R., Farr J. N., Vanderboom P. M., Atkinson E. J., White T. A., Singh R. J., Khosla S. and LeBrasseur N. K. (2015). Myostatin as a mediator of sarcopenia versus homeostatic regulator of muscle mass: insights using a new mass spectrometry-based assay. Skelet. Muscle 5, 21 10.1186/s13395-015-0047-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya T. K., Shukla R., Chatterjee R. N. and Bhanja S. K. (2019). Comparative analysis of silencing expression of myostatin (MSTN) and its two receptors (ACVR2A and ACVR2B) genes affecting growth traits in knock down chicken. Sci. Rep. 9, 7789 10.1038/s41598-019-44217-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovich S., Krag T. O. B., Barton E. R., Morris L. D., Whittemore L.-A., Ahima R. S. and Khurana T. S. (2002). Functional improvement of dystrophic muscle by myostatin blockade. Nature 420, 418-421. 10.1038/nature01154 [DOI] [PubMed] [Google Scholar]
- Bonaldo P. and Sandri M. (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 6, 25-39. 10.1242/dmm.010389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummel T., Abdollah S., Haerry T. E., Shimell M. J., Merriam J., Raftery L., Wrana J. L. and O'Connor M. B. (1999). The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev. 13, 98-111. 10.1101/gad.13.1.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. L., Walton K. L., Winbanks C. E., Murphy K. T., Thomson R. E., Makanji Y., Qian H., Lynch G. S., Harrison C. A. and Gregorevic P. (2014). Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 28, 1711-1723. 10.1096/fj.13-245894 [DOI] [PubMed] [Google Scholar]
- Chen J. L., Walton K. L., Al-Musawi S. L., Kelly E. K., Qian H., La M., Lu L., Lovrecz G., Ziemann M., Lazarus R. et al. (2015). Development of novel activin-targeted therapeutics. Mol. Ther. 23, 434-444. 10.1038/mt.2014.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. L., Walton K. L., Hagg A., Colgan T. D., Johnson K., Qian H., Gregorevic P. and Harrison C. A. (2017). Specific targeting of TGF-β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. USA 114, E5266-E5275. 10.1073/pnas.1620013114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L., Locke C. and Davis G. W. (2011). S6 kinase localizes to the presynaptic active zone and functions with PDK1 to control synapse development. J. Cell Biol. 194, 921-935. 10.1083/jcb.201101042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F. and Perrimon N. (2009). Integration of Insulin receptor/Foxo signaling and dMyc activity during muscle growth regulates body size in Drosophila. Development 136, 983-993. 10.1242/dev.027466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F., Patel V. K., Swindell W. R. and Perrimon N. (2014). Intertissue control of the nucleolus via a myokine-dependent longevity pathway. Cell Rep 7, 1481-1494. 10.1016/j.celrep.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerman M. A. and Glass D. J. (2014). Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 49, 59-68. 10.3109/10409238.2013.857291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Dai Z., Shi C., Zhai G., Jin X., He J., Lou Q. and Yin Z. (2016). Depletion of Myostatin b promotes somatic growth and lipid metabolism in zebrafish. Front. Endocrinol. 7, 88 10.3389/fendo.2016.00088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. C. and O'Connor M. B. (2014). Systemic Activin signaling independently regulates sugar homeostasis, cellular metabolism, and pH balance in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 111, 5729-5734. 10.1073/pnas.1319116111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman C. A., McNally R. M., Hoffmann F. M. and Hornberger T. A. (2013). Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol. Endocrinol. 27, 1946-1957. 10.1210/me.2013-1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn H. M. (1989). Heart weight and running ability. J. Anat. 167, 225-233. [PMC free article] [PubMed] [Google Scholar]
- Gustafsson T., Osterlund T., Flanagan J. N., von Waldén F., Trappe T. A., Linnehan R. M. and Tesch P. A. (2010). Effects of 3 days unloading on molecular regulators of muscle size in humans. J. Appl. Physiol. 109, 721-727. 10.1152/japplphysiol.00110.2009 [DOI] [PubMed] [Google Scholar]
- Harrington L. S., Findlay G. M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N. R., Cheng S., Shepherd P. R. et al. (2004). The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213-223. 10.1083/jcb.200403069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietakangas V. and Cohen S. M. (2007). Re-evaluating AKT regulation: role of TOR complex 2 in tissue growth. Genes Dev. 21, 632-637. 10.1101/gad.416307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitachi K., Nakatani M. and Tsuchida K. (2014). Myostatin signaling regulates Akt activity via the regulation of miR-486 expression. Int. J. Biochem. Cell Biol. 47, 93-103. 10.1016/j.biocel.2013.12.003 [DOI] [PubMed] [Google Scholar]
- Hulmi J. J., Oliveira B. M., Silvennoinen M., Hoogaars W. M. H., Ma H., Pierre P., Pasternack A., Kainulainen H. and Ritvos O. (2013). Muscle protein synthesis, mTORC1/MAPK/Hippo signaling, and capillary density are altered by blocking of myostatin and activins. Am. J. Physiol. Endocrinol. Metab. 304, E41-E50. 10.1152/ajpendo.00389.2012 [DOI] [PubMed] [Google Scholar]
- Kambadur R., Sharma M., Smith T. P. L. and Bass J. J. (1997). Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 7, 910-916. 10.1101/gr.7.9.910 [DOI] [PubMed] [Google Scholar]
- Kim M.-J. and O'Connor M. B. (2014). Anterograde Activin signaling regulates postsynaptic membrane potential and GluRIIA/B abundance at the Drosophila neuromuscular junction. PLoS ONE 9, e107443 10.1371/journal.pone.0107443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockel L., Kerr K. S., Melnick M., Brückner K., Hebrok M. and Perrimon N. (2010). Dynamic switch of negative feedback regulation in Drosophila Akt-TOR signaling. PLoS Genet. 6, e1000990 10.1371/journal.pgen.1000990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-J. and McPherron A. C. (2001). Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 98, 9306-9311. 10.1073/pnas.151270098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-J., Reed L. A., Davies M. V., Girgenrath S., Goad M. E. P., Tomkinson K. N., Wright J. F., Barker C., Ehrmantraut G., Holmstrom J. et al. (2005). Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc. Natl. Acad. Sci. USA 102, 18117-18122. 10.1073/pnas.0505996102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-J., Huynh T. V., Lee Y.-S., Sebald S. M., Wilcox-Adelman S. A., Iwamori N., Lepper C., Matzuk M. M. and Fan C.-M. (2012). Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathway. Proc. Natl. Acad. Sci. USA 109, E2353-E2360. 10.1073/pnas.1206410109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist R. A., Ottina K. A., Wheeler D. B., Hsu P. P., Thoreen C. C., Guertin D. A., Ali S. M., Sengupta S., Shaul Y. D., Lamprecht M. R. et al. (2011). Genome-scale RNAi on living-cell microarrays identifies novel regulators of Drosophila melanogaster TORC1-S6K pathway signaling. Genome Res. 21, 433-446. 10.1101/gr.111492.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokireddy S., Mouly V., Butler-Browne G., Gluckman P. D., Sharma M., Kambadur R. and McFarlane C. (2011). Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am. J. Physiol. Cell Physiol. 301, C1316-C1324. 10.1152/ajpcell.00114.2011 [DOI] [PubMed] [Google Scholar]
- Lokireddy S., Wijesoma I. W., Bonala S., Wei M., Sze S. K., McFarlane C., Kambadur R. and Sharma M. (2012). Myostatin is a novel tumoral factor that induces cancer cachexia. Biochem. J. 446, 23-36. 10.1042/BJ20112024 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Loumaye A., de Barsy M., Nachit M., Lause P., Frateur L., van Maanen A., Trefois P., Gruson D. and Thissen J.-P. (2015). Role of Activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab. 100, 2030-2038. 10.1210/jc.2014-4318 [DOI] [PubMed] [Google Scholar]
- Marden J. H. (2000). Variability in the size, composition, and function of insect flight muscles. Annu. Rev. Physiol. 62, 157-178. 10.1146/annurev.physiol.62.1.157 [DOI] [PubMed] [Google Scholar]
- Marino F. E., Risbridger G. and Gold E. (2015). Activin-βC modulates cachexia by repressing the ubiquitin-proteasome and autophagic degradation pathways. J. Cachexia Sarcopenia Muscle 6, 365-380. 10.1002/jcsm.12031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe B. D., Marqués G., Haghighi A. P., Fetter R. D., Crotty M. L., Haerry T. E., Goodman C. S. and O'Connor M. B. (2003). The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39, 241-254. 10.1016/S0896-6273(03)00426-4 [DOI] [PubMed] [Google Scholar]
- McPherron A. C., Lawler A. M. and Lee S.-J. (1997). Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387, 83-90. 10.1038/387083a0 [DOI] [PubMed] [Google Scholar]
- Moss-Taylor L., Upadhyay A., Pan X., Kim M.-J. and O'Connor M. B. (2019). Body size and tissue-scaling is regulated by motoneuron-derived Activinß in Drosophila melanogaster. Genetics 213, 1447-1464. 10.1534/genetics.119.302394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen B. K. and Febbraio M. A. (2012). Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 8, 457-465. 10.1038/nrendo.2012.49 [DOI] [PubMed] [Google Scholar]
- Piccirillo R., Demontis F., Perrimon N. and Goldberg A. L. (2014). Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev. Dyn. 243, 201-215. 10.1002/dvdy.24036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N., Dennis P. B., Andjelkovic M., Dufner A., Kozma S. C., Hemmings B. A. and Thomas G. (1998). Phosphorylation and activation of p70s6k by PDK1. Science 279, 707-710. 10.1126/science.279.5351.707 [DOI] [PubMed] [Google Scholar]
- Reardon K. A., Davis J., Kapsa R. M. I., Choong P. and Byrne E. (2001). Myostatin, insulin-like growth factor-1, and leukemia inhibitory factor mRNAs are upregulated in chronic human disuse muscle atrophy. Muscle Nerve 24, 893-899. 10.1002/mus.1086 [DOI] [PubMed] [Google Scholar]
- Rintelen F., Stocker H., Thomas G. and Hafen E. (2001). PDK1 regulates growth through Akt and S6K in Drosophila. Proc. Natl. Acad. Sci. USA 98, 15020-15025. 10.1073/pnas.011318098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov D. D., Guertin D. A., Ali S. M. and Sabatini D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098-1101. 10.1126/science.1106148 [DOI] [PubMed] [Google Scholar]
- Sartori R., Milan G., Patron M., Mammucari C., Blaauw B., Abraham R. and Sandri M. (2009). Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 296, C1248-C1257. 10.1152/ajpcell.00104.2009 [DOI] [PubMed] [Google Scholar]
- Sartori R., Schirwis E., Blaauw B., Bortolanza S., Zhao J., Enzo E., Stantzou A., Mouisel E., Toniolo L., Ferry A. et al. (2013). BMP signaling controls muscle mass. Nat. Genet. 45, 1309-1318. 10.1038/ng.2772 [DOI] [PubMed] [Google Scholar]
- Sartori R., Gregorevic P. and Sandri M. (2014). TGFβ and BMP signaling in skeletal muscle: potential significance for muscle-related disease. Trends Endocrinol. Metab. 25, 464-471. 10.1016/j.tem.2014.06.002 [DOI] [PubMed] [Google Scholar]
- Schiaffino S. and Mammucari C. (2011). Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle 1, 4 10.1186/2044-5040-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S., Dyar K. A., Ciciliot S., Blaauw B. and Sandri M. (2013). Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 280, 4294-4314. 10.1111/febs.12253 [DOI] [PubMed] [Google Scholar]
- Schmidt E. K., Clavarino G., Ceppi M. and Pierre P. (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275-277. 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- Schuelke M., Wagner K. R., Stolz L. E., Hübner C., Riebel T., Kömen W., Braun T., Tobin J. F. and Lee S.-J. (2004). Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 350, 2682-2688. 10.1056/NEJMoa040933 [DOI] [PubMed] [Google Scholar]
- Shah O. J., Wang Z. and Hunter T. (2004). Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14, 1650-1656. 10.1016/j.cub.2004.08.026 [DOI] [PubMed] [Google Scholar]
- Souza T. A., Chen X., Guo Y., Sava P., Zhang J., Hill J. J., Yaworsky P. J. and Qiu Y. (2008). Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol. Endocrinol. 22, 2689-2702. 10.1210/me.2008-0290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spletter M. L., Barz C., Yeroslaviz A., Zhang X., Lemke S. B., Bonnard A., Brunner E., Cardone G., Basler K., Habermann B. H. et al. (2018). A transcriptomics resource reveals a transcriptional transition during ordered sarcomere morphogenesis in flight muscle. eLife 7, e34058 10.7554/eLife.34058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stantzou A., Schirwis E., Swist S., Alonso-Martin S., Polydorou I., Zarrouki F., Mouisel E., Beley C., Julien A., Le Grand F. et al. (2017). BMP signaling regulates satellite cell-dependent postnatal muscle growth. Development 144, 2737-2747. 10.1242/dev.144089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump C. S., Henriksen E. J., Wei Y. and Sowers J. R. (2006). The metabolic syndrome: role of skeletal muscle metabolism. Ann. Med. 38, 389-402. 10.1080/07853890600888413 [DOI] [PubMed] [Google Scholar]
- Tan C. K., Leuenberger N., Tan M. J., Yan Y. W., Chen Y., Kambadur R., Wahli W. and Tan N. S. (2011). Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 60, 464-476. 10.2337/db10-0801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting C.-Y., Herman T., Yonekura S., Gao S., Wang J., Serpe M., O'Connor M. B., Zipursky S. L. and Lee C.-H. (2007). Tiling of r7 axons in the Drosophila visual system is mediated both by transduction of an activin signal to the nucleus and by mutual repulsion. Neuron 56, 793-806. 10.1016/j.neuron.2007.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg A. U., Meyer A., Rohner D., Boyle J., Hatakeyama S. and Glass D. J. (2009). Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 296, C1258-C1270. 10.1152/ajpcell.00105.2009 [DOI] [PubMed] [Google Scholar]
- Um S. H., Frigerio F., Watanabe M., Picard F., Joaquin M., Sticker M., Fumagalli S., Allegrini P. R., Kozma S. C., Auwerx J. et al. (2004). Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200-205. 10.1038/nature02866 [DOI] [PubMed] [Google Scholar]
- Upadhyay A., Moss-Taylor L., Kim M.-J., Ghosh A. C. and O'Connor M. B. (2017). TGF-β family signaling in Drosophila. Cold Spring Harb. Perspect. Biol. 9, a022152 10.1101/cshperspect.a022152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay A., Peterson A. J., Kim M. J. and O'Connor M. B. (2020). Muscle-derived myoglianin regulates Drosophila imaginal disc growth. eLife9, e51710. 10.7554/eLife.51710 [DOI]
- Watanabe H., Schmidt H. A., Kuhn A., Höger S. K., Kocagöz Y., Laumann-Lipp N., Özbek S. and Holstein T. W. (2014). Nodal signalling determines biradial asymmetry in Hydra. Nature 515, 112-115. 10.1038/nature13666 [DOI] [PubMed] [Google Scholar]
- Wehling M., Cai B. and Tidball J. G. (2000). Modulation of myostatin expression during modified muscle use. FASEB J. 14, 103-110. 10.1096/fasebj.14.1.103 [DOI] [PubMed] [Google Scholar]
- Weitkunat M., Kaya-Çopur A., Grill S. W. and Schnorrer F. (2014). Tension and force-resistant attachment are essential for myofibrillogenesis in Drosophila flight muscle. Curr. Biol. 24, 705-716. 10.1016/j.cub.2014.02.032 [DOI] [PubMed] [Google Scholar]
- White T. A. and LeBrasseur N. K. (2014). Myostatin and sarcopenia: opportunities and challenges - a mini-review. Gerontology 60, 289-293. 10.1159/000356740 [DOI] [PubMed] [Google Scholar]
- Winbanks C. E., Weeks K. L., Thomson R. E., Sepulveda P. V., Beyer C., Qian H., Chen J. L., Allen J. M., Lancaster G. I., Febbraio M. A. et al. (2012). Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J. Cell Biol. 197, 997-1008. 10.1083/jcb.201109091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winbanks C. E., Chen J. L., Qian H., Liu Y., Bernardo B. C., Beyer C., Watt K. I., Thomson R. E., Connor T., Turner B. J. et al. (2013). The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. J. Cell Biol. 203, 345-357. 10.1083/jcb.201211134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Inoki K., Kim E. and Guan K.-L. (2006). TSC1/TSC2 and Rheb have different effects on TORC1 and TORC2 activity. Proc. Natl. Acad. Sci. USA 103, 6811-6816. 10.1073/pnas.0602282103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Gao Z., Yin J., Quon M. J. and Ye J. (2008). S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 283, 35375-35382. 10.1074/jbc.M806480200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Wang J. L., Lu J., Song Y., Kwak K. S., Jiao Q., Rosenfeld R., Chen Q., Boone T., Simonet W. S. et al. (2010). Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142, 531-543. 10.1016/j.cell.2010.07.011 [DOI] [PubMed] [Google Scholar]
- Zimmers T. A., Davies M. V., Koniaris L. G., Haynes P., Esquela A. F., Tomkinson K. N., McPherron A. C., Wolfman N. M. and Lee S.-J. (2002). Induction of cachexia in mice by systemically administered myostatin. Science 296, 1486-1488. 10.1126/science.1069525 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.