

Abstract
Mutation and recombination are the primary sources of genetic variation. To better understand the evolution of genetic variation, it is crucial to comprehensively investigate the processes involving mutation accumulation and recombination. In this study, we performed mutation accumulation experiments on four heterozygous diploid yeast species in the Saccharomycodaceae family to determine spontaneous mutation rates, mutation spectra, and losses of heterozygosity (LOH). We observed substantial variation in mutation rates and mutation spectra. We also observed high LOH rates (1.65–11.07×10−6 events per heterozygous site per cell division). Biases in spontaneous mutation and LOH together with selection ultimately shape the variable genome-wide nucleotide landscape in yeast species.
Keywords: base substitutions, mutation rate, mutational spectrum, GC content, diploid genome, LOH
Introduction
Mutation processes are largely dictated by specific genetic mechanisms involved in DNA replication and repair (Garcia-Diaz and Kunkel 2006; Lujan et al. 2015), which themselves are subject to mutation and selection. Spontaneous mutations can be directly measured via mutation accumulation (MA) experiments. Recent studies have documented variable mutation rates among different organisms for different types of mutation, for example, substitutions and insertions/deletions (indels) (for reviews, see Lynch et al. 2016; Katju and Bergthorsson 2019), diverse mutation spectra (Ossowski et al. 2010; Denver et al. 2012; Weller et al. 2014; Keightley et al. 2015; Long et al. 2016), and variable context-dependent mutation patterns (Zhu et al. 2014; Sung et al. 2015). Theory predicts that natural selection plays a role in lowering the mutation rate to the point that the selective advantage of further reduction in the error rate is overcome by the power of genetic drift (Lynch 2011; Lynch et al. 2016).
Recombination promotes genetic variation, facilitates selection in some cases, and is widely appreciated as a powerful evolutionary force in sexually reproducing organisms (Smith 1974; Barton and Charlesworth 1998; Lynch 2007). Even in the absence of sexual reproduction, mitotic recombination can affect genome stability (Hiraoka et al. 2000; Lee et al. 2009) and facilitate selection (Omilian et al. 2006). In mitotic recombination, reciprocal interhomolog crossovers and nonreciprocal gene conversions (or simply gene conversions) lead to loss of heterozygosity (LOH) (Symington et al. 2014). Unfortunately, previous MA studies focused primarily on haploid and/or homozygous diploid organisms, which exclude the possibility of detecting LOH processes. Some recent studies have made attempts to investigate LOH processes in naturally heterozygous Daphina pulex (Omilian et al. 2006; Flynn et al. 2017) and experimentally constructed yeast (Dutta et al. 2017; James et al. 2019), but there remains a need for quantifying the role of LOH relative to spontaneous mutation processes.
Yeast genomes vary substantially among species in GC-content, genome size, ploidy, and life styles (Butler et al. 2004; Knop 2006; Dujon 2010). The strikingly variable yeast genomes become uniquely informative for identifying and quantifying processes of mutation and recombination. The yeast family Saccharomycodaceae has two main genera, Saccharomycodes and Hanseniaspora, with nuclear genomes ranging from 22.6% to 36.8% in GC-content (Riley et al. 2016; Sternes et al. 2016; Shen et al. 2018; Tavares et al. 2018). Species in this yeast family have different ascus propensities (ascus-persistent vs. ascus-releasing), and intratetrad mating (mating among spores from the same tetrad) occurs primarily in ascus-persistent species (Knop 2006). Within the Hanseniaspora genus, there are two main lineages (faster-evolving lineage FEL vs. slower-evolving lineage SEL). FEL species have lost more cell-cycle and DNA repair genes than SEL species (Steenwyk et al. 2019). In this study, we leverage heterozygous diploid genomes in the Saccharomycodaceae family (fig. 1) to address how spontaneous mutation and LOH shape genomic variation, and infer the effect of differential gene loss on mutation and LOH. These species exhibit different mutation spectra, rapid LOH rates, and different spectra of nucleotide changes in LOH. Finally, mtDNA mutations in Hanseniaspora yeasts are biased toward A/T, which is in contrast to G/C-biased mtDNA mutations in the budding yeast Saccharomyces cerevisiae.
Fig. 1.

Phylogenetic relationship of seven yeast species that have available spontaneous mutation460 rate estimated. G + C content of each nuclear genome is shown. Hanseniaspora uvarum and H. valbyensis belong to the fast-evolving lineage (FEL) in Hanseniaspora. The phylogenetic tree is constructed based on 1,020 single-copy orthologous genes using PhyML (Guindon et al. 2010). The tree is rooted using Rhizophagus irregularis as outgroup. The outgroup is removed from the presentation for clarity.
Results
Mutation Rates of Base Substitution
Our MA experiments generated 371 base-substitution mutations (BSMs) in Hanseniaspora uvarum, 208 in H. valbyensis, 520 in H. osmophila, and 87 in Saccharomycode ludwigii (details of all BSMs are shown in supplementary tables S1, S8, S15, and S22, Supplementary Material online). These result in a genome-wide BSM rate of 1.60 ± 0.09×10−10 (mutations per site per cell division) in H. uvarum, 0.96 ± 0.08×10−10 in H. valbyensis, 1.66 ± 0.11×10−10 in H. osmophila, and 0.73 ± 0.09×10−10 in Saccharomycode ludwigii (fig. 2). The BSM rate per genome is 2.25×10−3 (mutations per genome per cell division) in H. uvarum, 1.10×10−3 in H. valbyensis, 2.91×10−3 in H. osmophila, and 0.73×10−3 in Saccharomycode ludwigii. These BSM rates are close to the Drake’s prediction (0.0033 mutations per genome per generation) (Drake 1991). Within Hanseniaspora, H. uvarum and H. valbyensis belong to the FEL lineage, whereas H. osmophila is a SEL species. BSM rates show no clear distinction between the FEL and SEL lineages. One SEL species H. osmophila has a higher BSM rate per genome than FEL species H. uvarum or H. valbyensis. This is consistent with the notion of a burst of accelerated sequence evolution in stem Hanseniaspora lineages followed by a reduction in the pace of sequence evolution (Steenwyk et al. 2019). This is similar to the phenomenon that mutator strains evolved a decreased mutation rate (McDonald et al. 2012). Among the seven yeast species with available MA data (table 1), Saccharomycode ludwigii has the lowest BSM rate, and haploid budding yeast Saccharomyces cerevisiae has the highest BSM rate (Sharp et al. 2018).
Fig. 2.
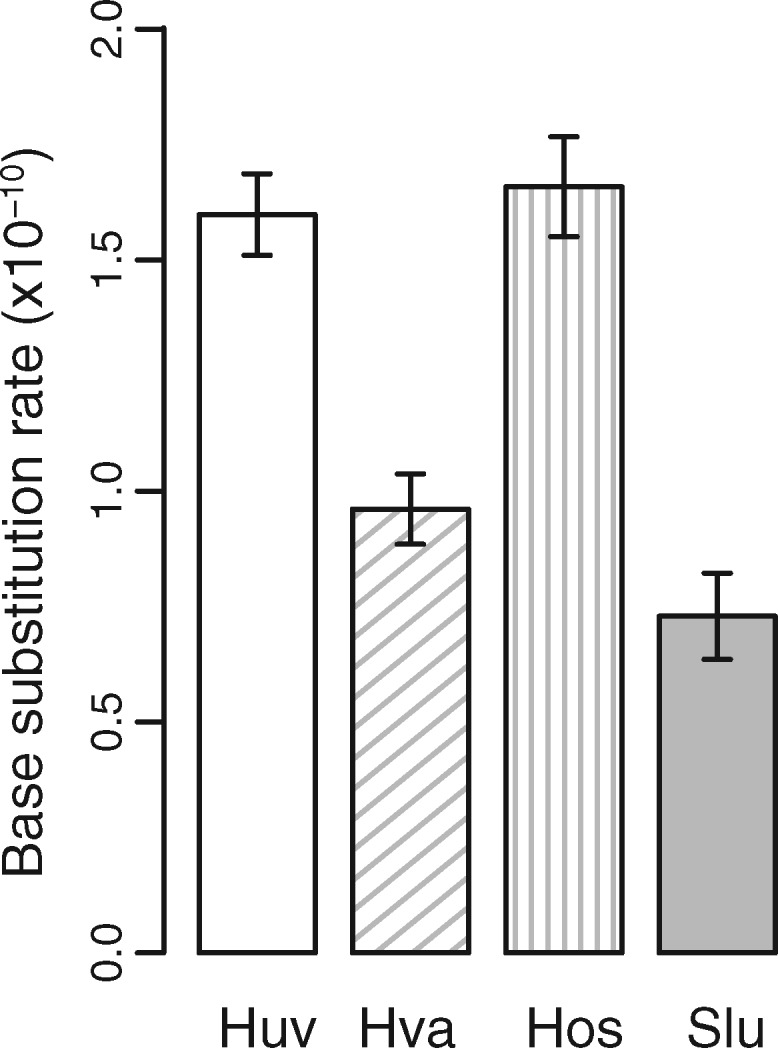
Nuclear-genome base-substitution mutation rates (per cell division) of four yeast species in Saccharomycodaceae on YM medium at 25 °C. Error bars are SEs calculated from all MA lines. Species names are shown in abbreviations: Huv (Hanseniaspora uvarum), Hva (H. valbyensis), Hos (H. osmophila), and Slu (Saccharomycode ludwigii).
Table 1.
Comparison of Measured Genome-Wide Mutation Rates (per nucleotide site per cell division) among Yeast Species.
| Organisms | Ploidy | Media | Nuclear (×10−10) |
mtDNA (×10−10) |
References | ||
|---|---|---|---|---|---|---|---|
| BSMsa | Indels | BSMs | Indels | ||||
| Hanseniaspora uvarum | Diploid | YM | 1.60 | 0.42 | 13.10 | — | This study |
| H. valbyensis | Diploid | YM | 0.97 | 0.59 | 5.94 | — | This study |
| H. osmophila | Diploid | YM | 1.67 | 0.39 | 3.65 | 19.9 | This study |
| Saccharomycode ludwigii | Diploid | YM | 0.75 | 0.21 | — | — | This study |
| Saccharomyces cerevisiae | Diploid | YPD | 1.67 | 0.05 | — | — | Zhu et al. (2014) |
| Saccharomyces cerevisiae | Diploid | YPDb | 1.95 | 0.19 | 58.3 | 121.5 | Liu and Zhang (2019) |
| Saccharomyces cerevisiae | Diploid | YPD | 2.89 | 0.20 | 4.47 | 4.47 | Sharp et al. (2018) |
| Saccharomyces cerevisiae | Haploid | YPD | 4.04 | 0.16 | 48.2 | 57.1 | Sharp et al. (2018) |
| Saccharomyces cerevisiae | Haploid | YPD | 3.30 | 0.20 | 122.3 | 74.8 | Lynch et al. (2008) |
| Schizosaccharomyces pombe | Haploid | YES | 2.00 | 0.60 | — | — | Farlow et al. (2015) |
| Schizosaccharomyces pombe | Haploid | YPD | 1.70 | 1.74 | — | — | Behringer and Hall (2015) |
| Rhodotorula toruloides | Haploid | YM | 1.90 | 0.22 | — | — | Long et al. (2016) |
Base-substitution mutations.
Mutation rates were measured in seven media, the rates measured on YPD are shown.
Test for Selection in MA Experiments of Diploid Organisms
The BSM rates in protein-coding regions are also variable: 1.33 ± 0.08×10−10 mutations per site per cell division in H. uvarum, 0.78 ± 0.07×10−10 in H. valbyensis, 1.40 ± 0.11×10−10 in H. osmophila, 0.59 ± 0.06×10−10 in Saccharomycode ludwigii (details of all BSMs are shown in supplementary tables S6, S13, S20, and S27, Supplementary Material online). The BSM rates in protein-coding sequences are consistently lower than their corresponding genome-wide BSM rates. The nonsynonymous/synonymous mutation ratios were calculated and compared against the expected ratios based on the empirical base composition and mutation spectrum (supplementary tables S4, S11, S18, and S25, Supplementary Material online). There is no evidence of selection in protein-coding sequences (Fisher’s exact test, P values 0.45, 0.76, 0.48, and 0.62, in H. uvarum, H. valbyensis, H. osmophila, or Saccharomycode ludwigii, respectively). Thus, the difference in BSM rate among genomic regions is likely due to nucleotide composition in coding versus noncoding sequences.
Different Mutation Spectra and Mutation Biases
The ratios of transition to transversion mutations (Ts/Tv) are 0.92 in H. uvarum, 1.26 in H. valbyensis, 1.16 in H. osmophila, and 1.72 in Saccharomycode ludwigii, all of which are greater than the expected 0.5 (when there is no transition–transversion bias) (details are shown in table 2 and supplementary tables S2, S9, S16, and S23, Supplementary Material online). These findings are in good agreement with previous MA studies on yeast mutations biased toward transition over transversion (Farlow et al. 2015; Long et al. 2016).
Table 2.
Spontaneous Base-Substitution Mutations in Nuclear DNAs.
| Average No. of Analyzed-Sites Per MA Line | Transitions |
Transversions |
Mutations | Ratea (×10−10) | |||||
|---|---|---|---|---|---|---|---|---|---|
| G:C→A:T | A:T→G:C | G:C→T:A | A:T→C:G | G:C→C:G | A:T→T:A | ||||
| Hanseniaspora uvarum | 7,025,336 | 129 | 49 | 48 | 49 | 35 | 49 | 371 | 1.60 |
| H. valbyensis | 5,636,213 | 72 | 44 | 26 | 30 | 14 | 22 | 208 | 0.97 |
| H. osmophila | 8,692,565 | 142 | 137 | 77 | 71 | 36 | 57 | 520 | 1.67 |
| Saccharomycode ludwigii | 4,894,429 | 30 | 25 | 9 | 12 | 5 | 6 | 87 | 0.75 |
Substitutionper nucleotide site per cell division
The seven yeast species show different mutation spectra. For instance, H. uvarum has the highest rate of G:C→A:T transition, and Rhodotorula toruloides has the highest rate of A:T→G:C transition (fig. 3). The relative mutation rates of all six base mutation types were calculated to better compare mutation bias among yeast species (fig. 4). The relative mutation rate of G:C→A:T transition is high in all four Saccharomycodaceae species. The relative mutation rate of G:C→T:A transversion is high in Schizosaccharomyces pombe. As oxidative DNA damage leads to G:C→T:A mutations (Cheng et al. 1992), the overrepresentation of G:C→T:A mutations suggests elevated oxidative damage and/or impaired repair of oxidative DNA damage in Schizosaccharomyces pombe.
Fig. 3.

Mutation spectra of nuclear genomes among the seven yeast species. The abbreviations of the Saccharomycodaceae species are per figure 2. The full names and data references of three additional yeast species are Sce (Saccharomyces cerevisiae; Zhu et al. 2014), Spo (Schizosaccharomyces pombe; Farlow et al. 2015), and Rto (Rhodotorula toruloides; Long et al. 2016).
Fig. 4.

Relative substitution rates of six possible nucleotide changes among the seven yeast species. Species abbreviations are per figure 3.
Variable Equilibrium G + C Contents
Based on the spontaneous mutation rates, the expected equilibrium G + C contents under mutation pressure alone, that is, neutrality, are 20.7 ± 7.1% in H. uvarum, 23.6 ± 10.6% in H. valbyensis, 35.6 ± 5.9% in H. osmophila, and 32.5 ± 11.0% in Saccharomycode ludwigii. They are significantly <50%, suggesting strong mutation bias toward A/T. Furthermore, the expected equilibrium %(G + C) values are lower than observed genome-wide %(G + C) (fig. 5).
Fig. 5.

Plot of expected equilibrium G + C contents based on spontaneous mutation bias in seven yeast species against their observed genome-wide %(G + C). The diagonal line denotes agreement with expected equilibrium %(G + C). Species abbreviations are per figure 3.
Context-Dependent Mutations
To better understand the differences in mutation spectra, we categorized the mutation rates of focal nucleotides in the context of their immediate flanking nucleotides (e.g., xYz sites are for a Y focal nucleotide flanked by x and z). Like in Saccharomyces cerevisiae (Zhu et al. 2014) and Schizosaccharomyces pombe (Behringer and Hall 2015), C/G focal nucleotides have higher mutation rates than A/T nucleotides in Saccharomycodaceae (fig. 6). Among C/G focal nucleotides, gCg sites consistently have the highest mutation rate (two-tailed Z-test, P value <0.01 in each species in figure 6, and supplementary tables S3, S10, S17, and S24, Supplementary Material online).
Fig. 6.

Neighbor-dependent single nucleotide mutation rates in the nuclear genomes of (A) Hanseniaspora uvarum, (B) H. valbyensis, (C) H. osmophila, and (D) Saccharomycode ludwigii. The rates reflect the impact of flanking nucleotides on mutation at A and C bases. For instance, xYz represents both xYz and its reverse complement triplets. For each rate, the 95% Poisson confidence interval is shown.
High mutation rates of C nucleotides at CG dinucleotide sites (or called CpG sites) have been previously reported in Saccharomyces cerevisiae (Zhu et al. 2014), Schizosaccharomyces pombe (Behringer and Hall 2015; Farlow et al. 2015) despite the lack of clear evidence for cytosine DNA methylation in these two species. The existence of cytosine methylation in Saccharomycodaceae is unknown. We first performed TBlastN searches for homologs of human Dnmt1 and Dnmt3a, two key methyltransferase enzymes in DNA methylation (Gowher and Jeltsch 2001), and found no matches in H. uvarum, H. valbyensis, H. osmophila, or Saccharomycode ludwigii using a relaxed criterion (E-value <10−3). We then examined mutation rates at C focal nucleotides at CpG sites. In Escherichia coli, methylated cytosines have order-of-magnitude higher rates of C→T mutations than unmethylated cytosines (Cooper and Youssoufian 1988). With active cytosine methylation, one would expect C nucleotides at CpG sites to exhibit higher rates of C→T mutations than C nucleotides not followed by a G nucleotide (i.e., followed by A, C, or T). In H. uvarum, there are 27 mutations at CpG sites with 15 mutations being C→T and 12 mutations being C→A or C→G. When mutations at all G/C sites are compared, there are 212 mutations at G/C sites with 129 mutations being G:C→A:T and 83 mutations being G:C→T:A or G:C→C:G. There is no evidence of elevated C→T mutations at CpG sites (Fisher’s exact test, P value = 0.42). Similarly, there is no evidence of elevated C→T mutations at CpG sites when compared with C nucleotides followed by non-G nucleotides in H. valbyensis, H. osmophila, and Saccharomycode ludwigii (Fisher’s exact test, P values = 0.65, 0.82, 0.91, respectively).
Small Indel Mutations
The rates of indel mutations are low compared with BSM rates (table 3). Indel mutation rates are 4.17 ± 0.49×10−11 indels per site per generation in H. uvarum, 5.93 ± 0.59×10−11 in H. valbyensis, 3.88 ± 0.44×10−11 in H. osmophila, and 2.08 ± 0.38×10−11 in Saccharomycode ludwigii. They are 26.1%, 61.6%, 23.2%, and 27.7% of their corresponding BSM rates. Furthermore, we compared insertions versus deletions by events and by nucleotides in Saccharomycodaceae (table 3). There is no significant bias toward either insertion or deletion events in any species. For numbers of inserted nucleotides and deleted nucleotides, significant differences are evident in H. uvarum, H. valbyensis, and H. osmophila, with more nucleotides in deletions than in insertions (χ2 test, P value <0.05 in each of the three species).
Table 3.
Comparison of Indel Mutations among Different Nuclear Genomes.
|
Hanseniaspora uvarum
|
H. valbyensis
|
H. osmophila
|
Saccharomycode ludwigii
|
|||||
|---|---|---|---|---|---|---|---|---|
| Deletions | Insertions | Deletions | Insertions | Deletions | Insertions | Deletions | Insertions | |
| No. of events | 59 | 37 | 72 | 52 | 70 | 47 | 13 | 11 |
| No. of nucleotides | 207 | 83 | 255 | 194 | 293 | 212 | 50 | 45 |
LOH at Heterozygous Sites
Hanseniaspora valbyensis and H. osmophila have abundant LOH sites, and show similar relative LOH rates among the four types of heterozygous patterns, C/T, C/A, C/G, A/T (table 4 and supplementary fig. S2, Supplementary Material online). Bias is evident in LOH directionality. In H. osmophila, there are significantly more A:T→G:C than G:C→A:T at C/T sites (χ2 test, P value <0.01), and more T:A→G:C than G:C→T:A at C/A sites (χ2 test, P value <0.01), suggesting a gene-conversion bias toward G/C.
Table 4.
Loss of Heterozygosity (LOH) at Heterozygous Sites.
| Transitions |
Transversions |
Subtotal | |||||
|---|---|---|---|---|---|---|---|
| G:C→A:T | A:T→G:C | G:C→T:A | A:T→C:G | G:C→C:G | A:T→T:A | ||
| Hanseniaspora uvarum | 5 | 3 | 0 | 4 | 0 | 2 | 14 |
| H. valbyensis | 6,815 | 6,681 | 1,969 | 1,917 | 756 | 3,935 | 22,251 |
| H. osmophila | 2,356 | 2,615 | 464 | 586 | 410 | 810 | 7,241 |
| Saccharomycode ludwigii | 8 | 11 | 2 | 0 | 0 | 3 | 24 |
The LOH rate ranges from 1.65×10−6 events per heterozygous site per cell division in Saccharomycode ludwigii to 11.07×10−6 events per heterozygous site per cell division in H. valbyensis (table 5). The median length of LOH tracts ranges from 1 bp in Saccharomycode ludwigii to 1,216 bp in H. osmophila (fig. 7). The finding of most LOH events involved short tracts is consistent with previous reports in Saccharomyces cerevisiae (Dutta et al. 2017) and Candida albicans (Ene et al. 2018). The short LOH tracts (<1 kb) are likely to have arisen through local gene conversions (Palmer et al. 2003; Laureau et al. 2016). The long LOH tracts may have arisen through mitotic crossovers, whose median tract lengths are 8–12 kb in Saccharomyces cerevisiae (St Charles and Petes 2013). It is noteworthy that LOH detection and tract estimation are limited by the number and density of heterozygous single nucleotide polymorphisms. The inferred LOH tract lengths are the minimal LOH lengths.
Table 5.
Details of LOH Events.
| LOH Events | LOH Tract Length (nt) |
LOH Ratesa (×10−6) | ||||
|---|---|---|---|---|---|---|
| Mean | 1st Quartile | Median | 3rd Quartile | |||
| Hanseniaspora uvarum | 14 | 1 | 1 | 1 | 1 | 4.09 |
| H. valbyensis | 1,911 | 429 | 1 | 46 | 3,131 | 11.07 |
| H. osmophila | 202 | 14,422 | 1 | 1,216 | 15,948 | 6.93 |
| Saccharomycode ludwigii | 21 | 260 | 1 | 1 | 1 | 1.65 |
LOH event per site per cell division.
Fig. 7.

Frequency distribution of conversion tract lengths in three Saccharomycodaceae species. Hanseniaspora uvarum is not shown as the lengths of all conversion tracts are 1 bp (table 5). Species abbreviations are per figure 3.
LOH can also be caused by deletion of chromosomal regions, which ideally can be detected by on an average half the depth of normal coverage. Due to the nature of the Saccharomycodaceae genomes and/or sequencing methods, there is a high variation (i.e., over 2-fold) of coverage depth along sequences in this study (see supplementary fig. S1, Supplementary Material online). Unfortunately, our data could not reliably and systematically infer chromosomal deletion and aneuploidy. As LOH can be caused by deletion of chromosome regions of various length, the contribution of possible chromosomal deletion to LOH was assessed by comparing read depths among homozygous BSMs (see below).
LOH at Spontaneous Mutation Sites
Spontaneously arisen mutations can subsequently undergo LOH events and become homozygous. There are collectively 28 homozygous BSMs (table 6). No bias was detected among LOH changes, given the low number. The LOH rates at substitution sites are 10.47×10−6 event per substitution site per cell division in H. uvarum, 13.38×10−6 in H. valbyensis, 11.46×10−6 in H. osmophila and 6.18×10−6 in Saccharomycode ludwigii. The LOH rates are four to five orders of magnitude higher than the BSM rates.
Table 6.
Loss of Heterozygosity (LOH) among Spontaneous Base-Substitution Mutations.
| Transitions |
Transversions |
Subtotal | (%)a | Rateb(×10−6) | |||||
|---|---|---|---|---|---|---|---|---|---|
| G:C→A:T | A:T→G:C | G:C→T:A | A:T→C:G | G:C→C:G | A:T→T:A | ||||
| Hanseniaspora uvarum | 6 | 1 | 0 | 1 | 0 | 0 | 8 | 2.15 | 10.47 |
| H. valbyensis | 0 | 2 | 0 | 0 | 2 | 2 | 6 | 2.88 | 13.38 |
| H. osmophila | 2 | 6 | 1 | 0 | 2 | 2 | 13 | 2.50 | 11.46 |
| Saccharomycode ludwigii | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1.15 | 6.18 |
Out of total substitution sites.
LOH per substitution site per cell division.
Among the 28 LOH substitution mutation sites, four (14.3%) mutation sites have low read depths (0.56–0.61 times) relative to the average read depth of all heterozygous substitution mutation sites from the respective MA line (fig. 8). Some of these low-coverage LOH sites could have resulted from chromosomal deletion. The remaining 24 (85.7%) LOH substitution sites have comparable or higher read depths than the average read depth of all heterozygous substitution sites from the respective MA line. This supports that a vast majority of LOH events associated with substitution sites result from mitotic gene conversion.
Fig. 8.

Comparison read depths between each LOH substitution site and the average of heterozygous substitution sites from the same MA line. The diagonal line is when the read depth of an LOH substitution site is equal to the average read depth, whereas the dashed line is when the read depth of an LOH substitution site is half of the average read depth, presumably due to chromosomal deletion.
Mutation Validation
Mutation calls were validated by additional Illumina sequencing with longer sequencing read lengths (pair-end 250 nt) using separately prepared PCR-free sequencing libraries for two MA lines from each species. Two MA lines from Saccharomycode ludwigii were sequenced by PCR-free sequencing, but one MA line was excluded due to low sequencing quality in the mutation analysis data. In total, 54 mutation calls in seven MA lines were validated by PCR-free sequencing, with 48 mutation calls covered by ≥4 PCR-free sequencing reads and six covered by two or three reads. All 48 mutation calls covered by ≥4 PCR-free sequencing reads are supported (supplementary table S29, Supplementary Material online).
Spontaneous Mutations in mtDNAs
In yeasts, the mtDNA mutation rate is only available in the budding yeast Saccharomyces cerevisiae (Lynch et al. 2008). No mtDNA mutations were found in Schizosaccharomyces pombe (Farlow et al. 2015) or R. toruloides (Long et al. 2016). This study observed three mtDNA BSMs in H. uvarum, three mtDNA BSMs in H. valbyensis, yielding a BSM rate of 13.1 ± 7.4×10−10 mutations per site per cell division in H. uvarum and 5.94 ± 3.60×10−10 mutations per site per cell division in H. valbyensis. The rates of mtDNA BSMs are higher than nuclear BSM rates. In H. osmophila, there are four mtDNA BSMs and five mtDNA indels, resulting in a genome-wide BSM rate of 9.57 ± 6.96×10−10 mutations per site per cell division and an indel rate of 12.49 ± 6.45×10−10 mutations per site per cell division. Three BSMs and one indel are from one MA line, whose mtDNA polymerase MIP1 gene carries a single nonsynonymous mutation causing Pro-to-Ser change (supplementary table S20, Supplementary Material online). This mtDNA mutator strain has three times the number of BSMs in mtDNA than all the remaining MA lines. No mtDNA mutation data were generated in Saccharomycode ludwigii due to poor sequencing coverage. There are collectively five G/C→A/T mtDNA mutations and two A/T→G/C mtDNA mutations in Hanseniaspora, whereas there are 13 G/C→A/T mtDNA mutations and 34 A/T → G/C mtDNA mutations in Saccharomyces cerevisiae (table 7). The mtDNA mutation pattern in Hanseniaspora is significantly different from that in Saccharomyces cerevisiae (Fisher’s exact test, P value <0.05), which is biased toward G/C (Lynch et al. 2008).
Table 7.
Raw Counts of Base-Substitution Mutations in mtDNAs.
| Transitions |
Transversions |
Reference | |||||
|---|---|---|---|---|---|---|---|
| G:C→A:T | A:T→G:C | G:C→T:A | A:T→C:G | G:C→C:G | A:T→T:A | ||
| Hanseniaspora uvarum | 2 | 1 | 0 | 0 | 0 | 0 | This study |
| H. valbyensis | 2 | 1 | 0 | 0 | 0 | 0 | This study |
| H. osmophila | 1 | 0 | 0 | 0 | 1 | 2 | This study |
| Saccharomyces cerevisiae | 0 | 10 | 0 | 4 | 0 | 7 | Lynch et al. (2008) |
| Saccharomyces cerevisiae a | 12 | 9 | 0 | 9 | 14 | 14 | Sharp et al. (2018) |
| Saccharomyces cerevisiae b | 1 | 1 | 0 | 0 | 2 | 2 | Sharp et al. (2018) |
Haploid.
Diploid.
Discussion
Variation of Nuclear Mutations within and between Different Species in Yeast
Among seven yeast species, nuclear BSM rates varied 5.4-fold ranging from 0.75×10−10 in Saccharomycode ludwigii to 4.04×10−10 in Saccharomyces cerevisiae (table 1). Within Saccharomyces cerevisiae, haploid strains have higher BSM rates than diploid strains (Sharp et al. 2018). BSM rates are similar among yeasts when cultured in rich media and at optimal temperatures (Long et al. 2016), whereas BSM rates of the same strain can vary significantly among media with different carbon and nitrogen sources, and especially for those containing chemical stressors (Liu and Zhang 2019). Even though all media in yeast MA studies use dextrose as carbon source (table 1), the influence of different media on mutation rates cannot be ruled out. High mutation rate variation in sexually reproduced Saccharomyces cerevisiae lines has been suggested to result from different mating patterns (Nishant et al. 2010) and possible mutagenic effects of meiosis (Magni and Von Borstel 1962). Mitotic recombination can also be mutagenic (Rattray et al. 2001), but the influence of LOH on genome-wide mutation rate is likely complex. In this study, H. valbyensis has the highest LOH rate, yet its mutation rate is lower than H. uvarum or H. osmophila.
Indel mutation rates among yeasts are more variable than BSM rates. Indel mutation rates range from 0.05×10−10 in Saccharomyces cerevisiae (Zhu et al. 2014) to 1.74×10−10 in Schizosaccharomyces pombe (Behringer and Hall 2015) (∼34-fold). Indel mutations are highly dependent on sequence-context and GC-content (Montgomery et al. 2013; Williams and Wernegreen 2013; Jiang et al. 2015; Xiao et al. 2017), which are indeed variable among yeast species. Indel-mutation rates are known to vary significantly among different genetic backgrounds and environmental conditions (Behringer and Hall 2016).
Higher Mutation Rates at G/C Sites than at A/T Sites
All yeast species, but R. toruloides, show a mutation bias toward A/T with higher mutation rates at C/G nucleotides than at A/T nucleotides. In transition mutations, the different rates between G:C→A:T and A:T→G:C mutations can often be dictated by frequencies of nucleotide deamination. In E. coli, cytosine deamination (leading to G:C→A:T transition mutations) is significantly more frequent than adenine deamination (leading to A:T→G:C transition mutations) (Lindahl and Nyberg 1974). However, it is important to note that mutations in Saccharomycodaceae show no evidence of elevated C→T mutations of focal C nucleotides at CpG sites compared with C nucleotides followed by non-G nucleotides. Thus, cytosine methylation is unlikely to play an important role in causing mutations at C/G nucleotides. In transversion mutations, the higher rates of G:C→T:A than A:T→C:G may be due to oxidative DNA damage, such as 8-oxo-dG (leading to G:C→T:A transversion mutations) (Cheng et al. 1992).
Variable Equilibrium G + C Contents
Equilibrium %(G + C) values predicted from mutation spectra vary among yeast species and reflect difference in their mutation spectra. Expected equilibrium %(G + C) values are lower than the observed G + C contents in all yeast species, suggesting mechanism(s) consistently operating to increase G + C content. There may be two reasons for this behavior: 1) selection being nearly universal in favor of G + C content (Long et al. 2018) and 2) gene conversion being mostly biased toward G/C during DNA mismatch repair (Pessia et al. 2012; Lassalle et al. 2015). LOH events in H. osmophila show a bias toward G/C, whereas LOH sites in H. valbyensis do not show such a bias toward G/C. Furthermore, G + C content, per se, is a parameter influencing mutation rates and the rates of mitotic and meiotic recombination (Kiktev et al. 2018). Since G + C content change alters nucleotide landscape, the rates and spectra of mutation and recombination can be further influenced in a context-dependent manner.
Rapid Genome-Wide LOH Rates
This study, to our knowledge, is the first on genome-wide LOH rates in naturally heterozygous yeast. The LOH rates are four orders of magnitude higher than the BSM rates. LOH events have been previously measured ranging from 3.4×10−4 to 6.0×10−3 per heterozygous site per cell division in constructed heterozygous Saccharomyces cerevisiae S288c/YJM789 strains (Dutta et al. 2017). The LOH rates in S288c/YJM789 are at least one order of magnitude higher than the measured LOH rates in this study, likely due to dynamic chromosome change and extensive LOH in hybrid genomes (Li et al. 2012). As LOH can convert a mutated allele back to the ancestral type, rapid LOH can lead to underestimation of mutation rates. Among detected spontaneous mutations in this study, 1.15–2.88% are homozygous due to LOH (table 6). If mutations converting back to the reference allele take place at the same rate as to the alternate allele, mutation rates would have been underestimated by 1.15–2.88%. Such small underestimation by LOH alone is insufficient to explain the significant difference of mutation rates between diploid and haploid genomes (Sharp et al. 2018).
LOH Variation
Differential loss of recombination-associated genes might lead to LOH variation. The presence or absence of 23 recombination proteins (Krogh and Symington 2004) was determined for the four Saccharomycodaceae species (supplementary table S30, Supplementary Material online). More gene losses are evident in FEL species, which are lacking RAD55 and MMS4. There are also differences between the two FEL species, with YKU70 missing in H. uvarum, and RMI1 missing in H. valbyensis. RMI1 is part of the Sgs1-Top3-Rmi1 antirecombination complex, which functions to remove double Holiday junctions (Krogh and Symington 2004). The lack of antirecombination RMI1 could have contributed to the high LOH rates in H. valbyensis.
Rates and Spectra of Yeast mtDNA Mutations
Hanseniaspora mtDNA mutations collectively have more G/C→A/T mutations than A/T→G/C mutations (table 7). This is consistent with the near-universal mutation bias toward A/T in bacteria (Hershberg and Petrov 2010) and in mtDNAs of nonyeast organisms such as Drosophila melanogaster (Haag-Liautard et al. 2008), Caenorhabditis briggsae (Howe et al. 2010), and C. elegans (Konrad et al. 2017), but in contrast to budding yeast Saccharomyces cerevisiae, which has mtDNA mutations biased toward G/C (Lynch et al. 2008). As the Saccharomyces cerevisiae FY10 strain (isogenic to S288c) used in the Lynch et al. (2008) study contains a single nonsynonymous mutation in the mtDNA polymerase (MIP1) linked to reduced fidelity of mtDNA replication (Baruffini et al. 2007), it would be of great interest to determine the exact contribution of the MIP1 gene to the G/C biased mtDNA mutation spectrum in Saccharomyces cerevisiae. All of these observations make yeast mtDNAs an excellent model system to investigate genetic mechanisms driving the evolution of nucleotide composition.
b Substitutionper nucleotide site per cell division
Conclusion
In this study, we quantified mutations and LOH events in naturally heterozygous yeast genomes. Mutation rates are variable among yeast species, between haploid and diploid strains. LOH rates are four to five orders of magnitude higher than base-substitution rates. The difference in LOH rates between naturally heterozygous strains and experimentally constructed strains raises the need to identify mechanisms and processes that dictate LOH rates. LOH events influence the accumulation of mutations. Even though LOH has only a very minor impact on mutation-rate estimates during MA courses (∼4,000 cell divisions) in this study, the cumulative impact of LOH on mutations would be significant. Further studies are needed to quantify the role of LOH in causing mutations.
Materials and Methods
Yeast Strains and MA Experiments
Four diploid yeast strains, H. uvarum Y-1612, H. valbyensis Y-1626, H. osmophila Y-1381, and Saccharomycode ludwigii Y-12793, were kindly provided by the USDA-ARS National Center for Agricultural Utilization Research (IL). For each strain, 60 MA lines were established from a single cell ancestor and underwent single-cell bottleneck every 48 h for 200 passages. MA lines were cultured on Universal Medium for yeast and mold (YM) without pH adjustment (1% dextrose, 0.3% malt extract, 0.5% peptone, 0.3% yeast extract, 2% agar) at 25 °C. During passages, only well-isolated colonies were picked to minimize contamination. In every 50 passages, 10 MA lines were selected to estimate generation times by counting colony-forming units. The estimated generation times were 2.35 ± 0.03, 2.26 ± 0.05, 2.21 ± 0.03, and 2.60 ± 0.04 h per cell division for H. uvarum, H. valbyensis, H. osmophila, and Saccharomycode ludwigii, respectively. These resulted in 4,119, 4,312, 4,364, and 3,718 cell divisions, respectively, after 200 passages.
Genome Sequencing and Reference Genome Construction
Genomic DNAs of the MA lines and their progenitor from each species were phenol–chloroform extracted from a 2-day culture of a single colony inoculation following Sambrook et al. (1989). Sequencing libraries were constructed using the Nextera DNA Library Preparation Kit and sequenced on an Illumina HiSeq2500 platform (paired-end 150 bp, Hubbard Center for Genome Studies, University of New Hampshire) as done in Long et al. (2016). To improve the reference genomes and validate mutation calls in the MA data, additional library preparation using TruSeq DNA PCR-Free Library Prep kit and sequencing (paired-end 250 bp) were performed on the progenitor and two selected MA lines for each species. Genomes were assembled using SPAdes v3.7.1 (Bankevich et al. 2012) with k-mers of 55, 75, 89, 97, and 127. Each assembly was validated by visualizing mapping of raw reads versus assembled genome using BWA-MEM v0.7.12 (Li and Durbin 2009) and Integrative Genomics Viewer (IGV v2.3.60) (Robinson et al. 2011). The assembled haploid genome sizes are 8.7 Mb for H. uvarum, 9.5 Mb for H. valbyensis, 10.9 Mb for H. osmophila, and 11.8 Mb for Saccharomycode ludwigii (supplementary fig. S1, Supplementary Material online). The nuclear genomes were annotated using the Yeast Genome Annotation Pipeline (YGAP) (Proux-Wera et al. 2012). Annotated protein genes were used to determine synonymous and nonsynonymous mutations in each MA line.
Mutation Analyses and Validation
Raw sequencing reads were trimmed for adaptor sequences and low quality reads using Trimmomatic v0.36 (Bolger et al. 2014), mapped to reference genomes using BWA-MEM v0.7.12 (Li and Durbin 2009), and indexed with Samtools v1.2 (Li et al. 2009). To reduce mapping errors, mapping quality was set to be >59 using “samtools view -bp 59.” Duplicate reads were removed using PicardTools-1.119 (http://broadinstitute.github.io/picard/). Analyzed nucleotide-sites in each strain were required to have read depths at least 15×, between 5th and 95th percentiles of all nucleotide coverages and at least half of the median coverage in the corresponding genome (see supplementary fig. S1, Supplementary Material online). MA lines were required to share ≥50% of analyzed nucleotide-sites with the progenitor. The progenitor genomes of H. valbyensis and Saccharomycode ludwigii exhibit high variation in read depth, sites with read depth <50% of the average coverage were excluded. The final numbers of MA lines in mutation analysis are 40 for H. uvarum, 44 for H. valbyensis, 41 for H. osmophila, and 32 for Saccharomycode ludwigii. The average read depths of analyzed nucleotides are 97× in H. uvarum, 69× in H. valbyensis, 68× in H. osmophila, and 57× in Saccharomycode ludwigii. The three H. mitogenomes was covered in nearly full length and by average read depths >700×. The Saccharomycode ludwigii mitogenome was poorly covered in length (only 10% of the total nucleotides were covered by >10× read depths) and excluded from mtDNA mutation analysis.
Mutation calls were made for BSMs and insertions/deletions (indels) using GATK UnifiedGenotyper (McKenna et al. 2010) with recommended parameters (DePristo et al. 2011; Van der Auwera et al. 2013). Mutation calls were made only on nucleotide positions of ≥100 Phred-scaled quality (QUAL) score, ≥20 genome quality (GQ) score, and ≥5 quality per depth (QD) score. To reduce detection errors, mutation calls were required to be supported by ≥3 forward reads, ≥3 reverse reads, and ≥25% of covered reads. Heterozygous sites were excluded from mutation analyses, but analyzed specifically for rates of LOH (see below). Following previous studies (Zhu et al. 2014; Long et al. 2016), mutations were called only when present in one MA line due to their low frequencies. Multinucleotide mutations are evident, two double-nucleotide polymorphisms (DNPs) in H. uvarum, five DNPs and one triple-nucleotide polymorphism in H. osmophila, and one DNP in Saccharomycode ludwigii, whereas there are no multinucleotide mutations in H. valbyensis. To further reduce sequencing and/or mapping errors, we took advantage of the diploid nature, where reads of alternative allele are expected to be half of the total mapped reads. For indel mutation calling, low-quality calls have been associated with mixed insertion and deletion in the same position due to sequencing and mapping errors (Fang et al. 2014). For each initial indel call made by GATK, we counted the number of reads supporting insertion and reads supporting deletion. Since each bona fide indel mutation in a diploid organism against its own reference genome is expected to be either an insertion or a deletion (but not both), we excluded indel mutation calls with reads supporting both insertion and deletion and sites where reference- and alternative alleles together were supported by <80% of covered reads.
Mutation rate (per generation per site) is calculated by , where m is the number of mutations, N is the number of nucleotides analyzed, G is the number of generations, and 2 is for diploid genomes. Standard error (SE) is , where s is the SD of mutation rates across MA lines and n is the number of analyzed MA lines. Similarly, the rates of neighbor-dependent nucleotide mutations were calculated using the number of triplets. The equilibrium G + C contents (±SE) were calculated as the ratio, where μ and ν represent the A/T to G/C and G/C to A/T substitution rates, respectively (Lynch and Walsh 1998; Lynch 2007). Mutations in mitogenomes were observed and the rates were analyzed using , where fm is the fraction of each mutated allele among the sequencing reads, N is the number of nucleotides analyzed, G is the number of generations. Mutations in mitogenomes were always specifically stated in this study. For simplicity, mutations in nuclear genomes might not always be specified.
Quantification of the LOH
Heterozygous sites were identified in the progenitor strains by fulfilling the criteria for mutation calls and alternative-allele frequency ≥35%. The numbers of identified heterozygous sites are 44 in H. uvarum, 20442 in H. valbyensis, 3488 in H. osmophila, and 239 in Saccharomycode ludwigii. In principle, LOH generates homozygous alleles, where all reads can be either identical with (no BSM called) or different from (100% BSM) the reference sequences. In practice, mutation calling relies on statistical tests to distinguish real alternative alleles from mapping/sequencing errors, thus no mutation calls cannot be reliably interpreted as being identical with the reference. Because of this, we only detected LOH events toward BSM alleles (i.e., half of the total LOH events). Since the rates of Illumina sequencing errors range from ∼0.1–1% (Meacham et al. 2011; Jünemann et al. 2013) and could be high in AT-rich genomes (Chen et al. 2013), we applied a cutoff of BSM-containing reads >95% in MA lines to detect LOH sites. Each LOH event was inferred by consecutive LOH sites. Gene conversion tract was defined by the maximum distance of LOH sites in each LOH event. The LOH rate (per site per generation) is calculated by , where l is the number of heterozygous sites becoming homozygous, N is the number of heterozygous sites analyzed, G is the number of generations, and 2 is to count for unconsidered LOH changes toward the reference genomes.
Selection Detection in MA Lines
MA experiments through single-cell bottlenecks have been shown to be effective in minimizing selection against deleterious mutations in haploid yeast strains (Farlow et al. 2015; Long et al. 2016). To test whether mutations in this study were under selection, we conducted Fisher’s exact tests to compare the expected numbers of nonsynonymous and synonymous mutations in the absence of selection against the observed ones in MA experiments. The expected numbers of nonsynonymous and synonymous mutations in each genome can be derived from its base composition, mutation rate, and mutation spectrum following Yang (2014).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We would like to thank the associate editor and three anonymous reviewers for very helpful comments on a previous version of this article. We are grateful for the technical support from IEMB-1 computation cluster at OUC. This work was supported by grants from Pilot National Laboratory for Marine Science and Technology (Qingdao), the Marine S & T Fund of Shandong Province (No. 2018SDKJ0406-5) and Distinguished Scholars Support Program of Laboratory for Marine Biology and Biotechnology (YJ2019NO04), the Taishan Scholars Program for Early Career Experts of Shandong Province (tsqn201812024) to H.L., the Multidisciplinary University Research Initiative Award from the US Army Research Office (W911NF-09-1-0444) to M.L., P. Foster, H. Tang, and S. Finkel, and National Institutes of Health award (R35-GM122566) to M.L., and University Research Grant (from Wayne State University) to W.H.
Author Contributions
M.L. and W.H. designed the project; D.T.N., B.W., H.L., N.Z., C.P., S.S., K.M., and W.K.T. performed research; D.T.N. and W.H. analyzed data; D.T.N. and W.H. wrote the article, with contributions from B.W., H.L., and M.L.
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH, Charlesworth B.. 1998. Why sex and recombination? Science 281(5385):1986–1990. [PubMed] [Google Scholar]
- Baruffini E, Lodi T, Dallabona C, Foury F.. 2007. A single nucleotide polymorphism in the DNA polymerase gamma gene of Saccharomyces cerevisiae laboratory strains is responsible for increased mitochondrial DNA mutability. Genetics 177(2):1227–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer MG, Hall DW.. 2015. Genome-wide estimates of mutation rates and spectrum in Schizosaccharomyces pombe indicate CpG sites are highly mutagenic despite the absence of DNA methylation. G3 (Bethesda) 6:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer MG, Hall DW.. 2016. The repeatability of genome-wide mutation rate and spectrum estimates. Curr Genet. 62(3):507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G, Kenny C, Fagan A, Kurischko C, Gaillardin C, Wolfe KH.. 2004. Evolution of the MAT locus and its Ho endonuclease in yeast species. Proc Natl Acad Sci U S A. 101(6):1632–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Liu T, Yu CH, Chiang TY, Hwang CC.. 2013. Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS One 8(4):e62856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA.. 1992. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G->T and A->C substitutions. J Biol Chem. 267(1):166–172. [PubMed] [Google Scholar]
- Cooper DN, Youssoufian H.. 1988. The CpG dinucleotide and human genetic disease. Hum Genet. 78(2):151–155. [DOI] [PubMed] [Google Scholar]
- Denver DR, Wilhelm LJ, Howe DK, Gafner K, Dolan PC, Baer CF.. 2012. Variation in base-substitution mutation in experimental and natural lineages of Caenorhabditis nematodes. Genome Biol Evol. 4(4):513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43(5):491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW. 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci U S A. 88(16):7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. 2010. Yeast evolutionary genomics. Nat Rev Genet. 11(7):512–524. [DOI] [PubMed] [Google Scholar]
- Dutta A, Lin G, Pankajam AV, Chakraborty P, Bhat N, Steinmetz LM, Nishant KT.. 2017. Genome dynamics of hybrid Saccharomyces cerevisiae during vegetative and meiotic divisions. G3 (Bethesda) 7:3669–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Farrer RA, Hirakawa MP, Agwamba K, Cuomo CA, Bennett RJ.. 2018. Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc Natl Acad Sci U S A. 115(37):E8688–E8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Wu Y, Narzisi G, ORawe JA, Barrón LTJ, Rosenbaum J, Ronemus M, Iossifov I, Schatz MC, Lyon GJ.. 2014. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 6(10):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow A, Long H, Arnoux S, Sung W, Doak TG, Nordborg M, Lynch M.. 2015. The spontaneous mutation rate in the fission yeast Schizosaccharomyces pombe. Genetics 201(2):737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Chain FJ, Schoen DJ, Cristescu ME.. 2017. Spontaneous mutation accumulation in Daphnia pulex in selection-free vs. competitive environments. Mol Biol Evol. 34(1):160–173. [DOI] [PubMed] [Google Scholar]
- Garcia-Diaz M, Kunkel TA.. 2006. Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem Sci. 31(4):206–214. [DOI] [PubMed] [Google Scholar]
- Gowher H, Jeltsch A.. 2001. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol. 309(5):1201–1208. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321. [DOI] [PubMed] [Google Scholar]
- Haag-Liautard C, Coffey N, Houle D, Lynch M, Charlesworth B, Keightley PD.. 2008. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 6(8):e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2010. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 6(9):e1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka M, Watanabe K, Umezu K, Maki H.. 2000. Spontaneous loss of heterozygosity in diploid Saccharomyces cerevisiae cells. Genetics 156(4):1531–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe DK, Baer CF, Denver DR.. 2010. High rate of large deletions in Caenorhabditis briggsae mitochondrial genome mutation processes. Genome Biol Evol. 2:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TY, Michelotti LA, Glasco AD, Clemons RA, Powers RA, James ES, Simmons DR, Bai F, Ge S.. 2019. Adaptation by loss of heterozygosity in Saccharomyces cerevisiae clones under divergent selection. Genetics 213(2):665–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Turinsky AL, Brudno M.. 2015. The missing indels: an estimate of indel variation in a human genome and analysis of factors that impede detection. Nucleic Acids Res. 43(15):7217–7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jünemann S, Sedlazeck FJ, Prior K, Albersmeier A, John U, Kalinowski J, Mellmann A, Goesmann A, von Haeseler A, Stoye J, et al. 2013. Updating benchtop sequencing performance comparison. Nat Biotechnol. 31(4):294–296. [DOI] [PubMed] [Google Scholar]
- Katju V, Bergthorsson U.. 2019. Old trade, new tricks: insights into the spontaneous mutation process from the partnering of classical mutation accumulation experiments with high-throughput genomic approaches. Genome Biol Evol. 11(1):136–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Pinharanda A, Ness RW, Simpson F, Dasmahapatra KK, Mallet J, Davey JW, Jiggins CD.. 2015. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol Biol Evol. 32(1):239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiktev DA, Sheng Z, Lobachev KS, Petes TD.. 2018. GC content elevates mutation and recombination rates in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 115(30):E7109–E7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M. 2006. Evolution of the hemiascomycete yeasts: on life styles and the importance of inbreeding. Bioessays 28(7):696–708. [DOI] [PubMed] [Google Scholar]
- Konrad A, Thompson O, Waterston RH, Moerman DG, Keightley PD, Bergthorsson U, Katju V.. 2017. Mitochondrial mutation rate, spectrum and heteroplasmy in Caenorhabditis elegans spontaneous mutation accumulation lines of differing population size. Mol Biol Evol. 34(6):1319–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh BO, Symington LS.. 2004. Recombination proteins in yeast. Annu Rev Genet. 38(1):233–271. [DOI] [PubMed] [Google Scholar]
- Lassalle F, Perian S, Bataillon T, Nesme X, Duret L, Daubin V.. 2015. GC-Content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLoS Genet. 11(2):e1004941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laureau R, Loeillet S, Salinas F, Bergstrom A, Legoix-Ne P, Liti G, Nicolas A.. 2016. Extensive recombination of a yeast diploid hybrid through meiotic reversion. PLoS Genet. 12(2):e1005781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PS, Greenwell PW, Dominska M, Gawel M, Hamilton M, Petes TD.. 2009. A fine-structure map of spontaneous mitotic crossovers in the yeast Saccharomyces cerevisiae. PLoS Genet. 5(3):e1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Averette AF, Desnos-Ollivier M, Ni M, Dromer F, Heitman J.. 2012. Genetic diversity and genomic plasticity of Cryptococcus neoformans AD hybrid strains. G3 (Bethesda) 2:83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Nyberg B.. 1974. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 13(16):3405–3410. [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang J.. 2019. Yeast spontaneous mutation rate and spectrum vary with environment. Curr Biol. 29(10):1584–1591.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Behringer MG, Williams E, Te R, Lynch M.. 2016. Similar mutation rates but highly diverse mutation spectra in Ascomycete and Basidiomycete yeasts. Genome Biol Evol. 8(12):3815–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Sung W, Kucukyildirim S, Williams E, Miller SF, Guo W, Patterson C, Gregory C, Strauss C, Stone C, et al. 2018. Evolutionary determinants of genome-wide nucleotide composition. Nat Ecol Evol. 2(2):237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Clark AB, Kunkel TA.. 2015. Differences in genome-wide repeat sequence instability conferred by proofreading and mismatch repair defects. Nucleic Acids Res. 43(8):4067–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2007. The origins of genome architecture. Sunderland (MA): Sinauer Associates.
- Lynch M. 2011. The lower bound to the evolution of mutation rates. Genome Biol Evol. 3:1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Ackerman MS, Gout JF, Long H, Sung W, Thomas WK, Foster PL.. 2016. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet. 17(11):704–714. [DOI] [PubMed] [Google Scholar]
- Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 105(27):9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Walsh B.. 1998. Genetics and analysis of quantitative traits. Sunderland: Sinauer.
- Magni GE, Von Borstel RC.. 1962. Different rates of spontaneous mutation during mitosis and meiosis in yeast. Genetics 47(8):1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MJ, Hsieh YY, Yu YH, Chang SL, Leu JY.. 2012. The evolution of low mutation rates in experimental mutator populations of Saccharomyces cerevisiae. Curr Biol. 22(13):1235–1240. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham F, Boffelli D, Dhahbi J, Martin DI, Singer M, Pachter L.. 2011. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics 12(1):451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Goode DL, Kvikstad E, Albers CA, Zhang ZD, Mu XJ, Ananda G, Howie B, Karczewski KJ, Smith KS, The 1000 Genomes Project Consortium, et al. 2013. The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res. 23(5):749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant KT, Wei W, Mancera E, Argueso JL, Schlattl A, Delhomme N, Ma X, Bustamante CD, Korbel JO, Gu Z, et al. 2010. The baker’s yeast diploid genome is remarkably stable in vegetative growth and meiosis. PLoS Genet. 6(9):e1001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omilian AR, Cristescu ME, Dudycha JL, Lynch M.. 2006. Ameiotic recombination in asexual lineages of Daphnia. Proc Natl Acad Sci U S A. 103(49):18638–18643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledo JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M.. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327(5961):92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S, Schildkraut E, Lazarin R, Nguyen J, Nickoloff JA.. 2003. Gene conversion tracts in Saccharomyces cerevisiae can be extremely short and highly directional. Nucleic Acids Res. 31(4):1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia E, Popa A, Mousset S, Rezvoy C, Duret L, Marais GA.. 2012. Evidence for widespread GC-biased gene conversion in eukaryotes. Genome Biol Evol. 4(7):675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proux-Wera E, Armisen D, Byrne KP, Wolfe KH.. 2012. A pipeline for automated annotation of yeast genome sequences by a conserved-synteny approach. BMC Bioinformatics 13(1):237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, McGill CB, Shafer BK, Strathern JN.. 2001. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics 158:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley R, Haridas S, Wolfe KH, Lopes MR, Hittinger CT, Goker M, Salamov AA, Wisecaver JH, Long TM, Calvey CH, et al. 2016. Comparative genomics of biotechnologically important yeasts. Proc Natl Acad Sci U S A. 113(35):9882–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP.. 2011. Integrative genomics viewer. Nat Biotechnol. 29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T.. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Sharp NP, Sandell L, James CG, Otto SP.. 2018. The genome-wide rate and spectrum of spontaneous mutations differ between haploid and diploid yeast. Proc Natl Acad Sci U S A. 115(22):E5046–E5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XX, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MAB, Wisecaver JH, Wang M, Doering DT, et al. 2018. Tempo and mode of genome evolution in the budding yeast subphylum. Cell 175(6):1533–1545.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM. 1974. Recombination and the rate of evolution. Genetics 78(1):299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Charles J, Petes TD.. 2013. High-resolution mapping of spontaneous mitotic recombination hotspots on the 1.1 Mb arm of yeast chromosome IV. PLoS Genet. 9(4):e1003434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenwyk JL, Opulente DA, Kominek J, Shen X-X, Zhou X, Labella AL, Bradley NP, Eichman BF, Čadež N, Libkind D, et al. 2019. Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. PLoS Biol. 17(5):e3000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternes PR, Lee D, Kutyna DR, Borneman AR.. 2016. Genome sequences of three species of Hanseniaspora isolated from spontaneous wine fermentations. Genome Announc. 4:e01287–e01316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Gout JF, Miller SF, Williams E, Foster PL, Lynch M.. 2015. Asymmetric context-dependent mutation patterns revealed through mutation-accumulation experiments. Mol Biol Evol. 32(7):1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M.. 2014. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198(3):795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares MJ, Guldener U, Esteves M, Mendes-Faia A, Mendes-Ferreira A, Mira NP.. 2018. Genome sequence of the wine yeast Saccharomycodes ludwigii UTAD17. Microbiol Resour Announc. 7(18):e01195–e01218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 43:11.10.11–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller AM, Rodelsperger C, Eberhardt G, Molnar RI, Sommer RJ.. 2014. Opposing forces of A/T-biased mutations and G/C-biased gene conversions shape the genome of the nematode Pristionchus pacificus. Genetics 196(4):1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LE, Wernegreen JJ.. 2013. Sequence context of indel mutations and their effect on protein evolution in a bacterial endosymbiont. Genome Biol Evol. 5(3):599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Nguyen DT, Wu B, Hao W.. 2017. Genetic drift and indel mutation in the evolution of yeast mitochondrial genome size. Genome Biol Evol. 9(11):3088–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2014. Molecular evolution: a statistical approach. New York: Oxford University Press. [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA.. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 111(22):E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.