

Abstract
Physiologically‐based pharmacokinetic (PBPK) modeling is increasingly used to predict drug disposition and drug–drug interactions (DDIs). However, accurately predicting the pharmacokinetics of transporter substrates and transporter‐mediated DDIs (tDDIs) is still challenging. Rosuvastatin is a commonly used substrate probe in DDI risk assessment for new molecular entities (NMEs) that are potential organic anion transporting polypeptide 1B or breast cancer resistance protein transporter inhibitors, and as such, several rosuvastatin PBPK models have been developed to try to predict the clinical DDI and support NME drug labeling. In this review, we examine five representative PBPK rosuvastatin models, discuss common challenges that the models have come across, and note remaining gaps. These shared learnings will help with the continuing efforts of rosuvastatin model validation, provide more information to understand transporter‐mediated drug disposition, and increase confidence in tDDI prediction.
Rosuvastatin, a β‐hydroxy β‐methylglutaryl‐CoA reductase inhibitor, is frequently prescribed to reduce low‐density lipoprotein cholesterol levels in patients with hyperlipidemia. 1 This hydrophilic statin is also commonly used as a probe in transporter‐mediated drug‐drug interaction (tDDI) studies due to its unique pharmacokinetic (PK) properties.
Rosuvastatin has a low oral bioavailability of 20%. 2 It is minimally metabolized by cytochrome P450 (CYP) 2C9 and CYP2C19 3 and uridine diphosphate glucuronosyltransferase (UGT) 1A1 and UGT1A3 4 , 5 and alternatively predominately undergoes biliary and renal excretion as an unchanged drug. 6 , 7 Rosuvastatin is extensively distributed to the liver, its site of action, 8 through active transport by the organic anion transporting polypeptide (OATP) 1B1, 1B3, and 2B1 9 and the sodium‐taurocholate cotransporting polypeptide (NTCP), 10 , 11 whereas its biliary excretion is believed to be mediated predominantly by the breast cancer resistance protein (BCRP) 12 (Figure 1 ). The clinical significance of these transporters has been noted in drug–drug interaction (DDI) studies where the area under the concentration‐time curve (AUC) of rosuvastatin increased in subjects treated with concomitant drugs including cyclosporine (and its inhibitory metabolite AM1), 13 rifampin, 14 and gemfibrozil (and its inhibitory metabolite gemfibrozil 1‐O‐ß glucuronide). 15
Figure 1.
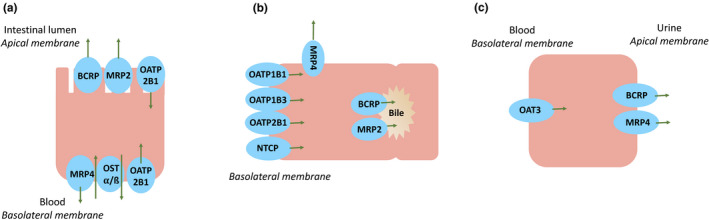
Proposed transporter involvement in rosuvastatin disposition in (a) enterocytes, (b) hepatocytes, and (c) kidney proximal tubule cells. BCRP, breast cancer resistance protein; MRP, multidrug resistance protein; NTCP, sodium‐taurocholate cotransporting polypeptide; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; OST α/ß, organic solute transporter alpha/beta.
To better describe the role of transporters in rosuvastatin’s disposition and ultimately predict clinical DDIs between new molecular entities (NMEs) (as transporter inhibitors) and rosuvastatin (as a transporter substrate), physiologically‐based PK (PBPK) models have been developed by different groups using mixed bottom‐up, top‐down, or middle‐out approaches. 16 Although some of these PBPK models were able to qualitatively assess rosuvastatin tDDI risk, it has been challenging to accurately predict the magnitude of AUC and maximum concentration (Cmax) changes. 17 Unlike the more established CYP‐based DDI prediction, the use of PBPK models for tDDI prediction is still under development. 18 Understanding and improving the in vitro to in vivo extrapolation (IVIVE) is the key. Although improvements of DDI prediction can be explored for both substrate and inhibitor aspects, this review focuses on the substrate (rosuvastatin).
As rosuvastatin is a commonly used probe substrate to determine the DDI potential for NMEs that are OATP1B or BCRP transporter inhibitors, questions on the performance of different rosuvastatin PBPK models have been raised from both industry and regulatory agencies. With the desire of understanding the differences among existing models and ultimately increase confidence in using a generically validated rosuvastatin model for DDI prediction to support regulatory decision, for the first time, we examined five previously reported rosuvastatin PBPK models to gain a better understanding of their differences and the impact on PK and tDDI prediction, noting remaining gaps. The objective of this review is to provide an in‐depth understanding of the current rosuvastatin PBPK models with regard to the input parameters, source of in vitro data support, and rationale of each model’s selection in describing the transporter‐mediated disposition based on our findings. The model gaps and common uncertainties as well as studies suggested for further model improvement are discussed. As a first step, a thorough mechanistic understanding of each existing PBPK model with shared learnings of common challenges and suggestions for future studies can provide guidance to the continued effort of model development and validation for rosuvastatin. 19
This review includes the following three sections: (i) Rosuvastatin model comparison, including our in‐depth findings and explanations of the selection of model input parameters associated with effective permeability, intestinal transport, distribution, hepatic metabolism and transport, and renal clearance in five models; (ii) rosuvastatin PK and DDI simulations to elucidate the impact of the key model parameters in the different rosuvastatin models described; and (iii) a discussion of common knowledge gaps and challenges in current rosuvastatin models with our mechanistic understanding and recommendations that can benefit future model development.
ROSUVASTATIN MODEL COMPARISON
A literature search was conducted through PubMed using search terms, including “rosuvastatin,” “physiologically based pharmacokinetic,” and “model,” and several unique human PBPK models developed for rosuvastatin were found as of July 2020. Five of these were unique rosuvastatin models developed using the Simcyp simulator (Simcyp, Sheffield, UK), whereas others were customized models built from scratch using different software and are listed in Table S1 . In addition, among published applications of rosuvastatin PBPK models for PK and DDI predictions, the majority used the PBPK models developed in the Simcyp simulator (Table S1 ). For the purpose of this review, the five models developed using the Simcyp simulator are focused on for several reasons: detailed information and model input parameters from the same model structure are available for a fair comparison; more application examples could be found since the simulator has frequently been used by pharmaceutical companies and adopted by regulatory agencies 20 ; there is reduced inconsistency in a large amount of information, such as the demographic, physiological, and genetic information of populations that the user is required to assign 21 ; and substrate/inhibitor information included within the simulator are updated with each version release to reflect the most recent literature information. For the final five models reviewed, 38 references were examined in addition for the original data sources.
The first of the models examined here, that of Jamei et al. 17 (referred to from here on as M‐I), reported the development of the rosuvastatin compound file built into the substrate library using the Simcyp simulator version 12, and the file remained consistent up to and including the rosuvastatin file included in version 18. The model of Emami Riedmaier et al. 22 (M‐II) included a permeability‐limited model for the kidney and investigated the sample size needed to detect an effect of OATP1B1 phenotype (using version 14). Wang et al. 23 (M‐III) developed a rosuvastatin model that included additional transporters to try to more accurately account for the mechanisms believed to govern rosuvastatin disposition to better capture tDDIs (using version 14). A recent model of Chan et al. 24 (M‐IV) focused on using a bottom‐up approach predominantly employing in vitro transporter and metabolism data (using version 17). Finally, the latest rosuvastatin model in the compound library of Simcyp version 19 (M‐V) will be discussed. Additional mechanistic data gathered and the strategies applied in the version 19 rosuvastatin compound file development will be presented by Simcyp scientists elsewhere. 25
These five rosuvastatin models were each developed with different primary purposes: M‐I and M‐V are commercially available and broadly used as fit for purpose to assess DDI risk in regulatory interactions, M‐II and M‐III are exploratory works to evaluate what is mechanistically possible in rosuvastatin’s disposition, and M‐IV is an exploratory exercise attempting to use as much in vitro data as possible for bottoM‐up modeling. Differences in the absorption, distribution, metabolism, transport, and elimination inputs of these models are described in the following section of comparison of rosuvastatin input parameters.
Comparison of Rosuvastatin input parameters
The physiochemical properties (molecular weight (MW), log of the octanol:water partition coefficient (logP), molecular species, pKa, etc.) of rosuvastatin were consistent across all models and can be found listed at the top of Table 1 . The differences in inputs between models can also be found in Table 1 , with more details described in the subsequent sections.
Table 1.
Summary of the physiologically‐based pharmacokinetic input parameters for M‐I to M‐V
| Rosuvastatin model parameters | |||||
|---|---|---|---|---|---|
|
MW fu Blood‐to‐plasma ratio Log of the octanol:water partition coefficient Compound type pKa Main plasma binding protein |
481.54 0.107 0.625 2.4 Monoprotic acid 4.27 Human serum albumin |
||||
| M‐I 17 | M‐II 22 | M‐III 23 | M‐IV 24 | M‐V 25 | |
| Absorption | |||||
| P app | Caco‐2 with inhibitors (passive) | Caco‐2 with inhibitors (passive) | Caco‐2 no inhibitors (passive) | Caco‐2 no inhibitors (passive + active) | MDCK II (passive) |
| Reference P app (10−6 cm/second) | Propranolol (measured) | Propranolol (measured) | Propranolol (from library file) | Propranolol (from library file) | Four reference compounds (measured) |
| P eff (10−4 cm/second) | 0.855 | 0.855 | 0.036 | 0.184 | 0.161 |
| Distribution | |||||
| V ss method |
Method 2 K p = 1 |
Method 2 K p = 1 |
Method 2 K p = 1 |
Method 2 + rat distribution data K p = 1 |
Method 2 K p = 4.85 |
| V ss (L/kg) | 0.117 | 0.117 | 0.117 | 0.70 | 0.385 |
| Metabolism | |||||
| HLM | HLM CLint | HLM CLint | HLM CLint |
CYP3A4 CLint UGT1A1 V max/K m UGT1A3 V max/K m |
HLM CLint |
| Intestinal transport | |||||
| Uptake | — | — | Apical uptake CLint,T a | — | OATP2B1 CLint,T b |
| Efflux | BCRP CLint,T b | BCRP CLint,T b | BCRP Jmax a/K m c | — | BCRP Jmax b/K m c |
| Hepatic transport | |||||
| Uptake |
OATP1B1 CLint,T d OATP1B3 CLint,T d NTCP CLint,T d |
OATP1B1 CLint,T d OATP1B3 CLint,T d NTCP CLint,T d |
OATP1B1 CLint,T d OATP1B3 CLint,T d NTCP CLint,T d |
OATP1B1 J max/K m (and REF) c OATP1B3 J max/K m (and REF) c NTCP CLint,T (and REF) c OATP2B1 J max/K m (and REF) c |
OATP1B1 CLint,T d OATP1B3 CLint,T d NTCP CLint,T d OATP2B1 CLint,T d |
| Efflux | BCRP CLint,T c | BCRP CLint,T c |
BCRP CLint,T c MRP4 CLint,T b |
Canalicular efflux CLint,T (and REF) c MRP4 J max/K m (and REF) c |
BCRP CLint,T c MRP4 CLint,T c |
| CLPD (mL/minute/106 cells) | SCHH (one study) | SCHH (one study) | SCHH (one study) | SCHH (one study) | SCHH (meta‐analysis) |
| Renal elimination | |||||
| CLR (L/hour) | In vivo value | — | — | — | In vivo value |
| Uptake transport | — | OAT3 CLint,T b | OAT3 CLint,T a | OAT3 J max/K m c | — |
| Efflux transport | — | BCRP CLint,T b | BCRP CLint,T a | MRP4 J max/K m c | — |
| CLPD (mL/minute/106 cells) | — | Included | Not included | Included | — |
BCRP, breast cancer resistance protein; Caco‐2, human colon cancer cell line; CLint, intrinsic clearance; CLPD, passive diffusion clearance; CLR, renal clearance; fu, fraction unbound; HLM, human liver microsomes; J max, maximum rate of transport; K m, Michaelis‐Menten constant; K p, tissue‐to‐plasma partition coefficient; MDCK II, Madin‐Darby canine kidney cell line II; MW, molecular weight; MRP, multidrug resistance protein; NTCP, sodiuM‐taurocholate cotransporting polypeptide; OATP, organic anion transporting polypeptide; P app, apparent permeability; P eff, effective permeability; REF, relative expression factor; SCHH, sandwich culture human hepatocytes; Vmax, maximum velocity of the metabolic reaction; V ss, steady‐state volume of distribution.
“Model fit.”
Sensitivity analysis.
In vitro data.
Global CLint,T fit using clinical data, fraction transported determined from in vitro data.
Absorption
The advanced dissolution, absorption, and metabolism model 26 was used in all five rosuvastatin models, and in vitro permeability data (P app) were taken from different sources to extrapolate to an effective permeability in human (P eff,man). M‐I, M‐II, M‐III, and M‐V used solution formulations, whereas M‐IV provided information about the dissolution of rosuvastatin to use either a solution or tablet formulation.
Effective permeability
M‐I (and M‐II) predicted a P eff,man value using data from a Caco‐2 study with chemical inhibitors to determine the passive uptake of rosuvastatin. 27 Propranolol permeability was measured in the same study and used as the reference compound. 27 Although M‐III also used data from a Caco‐2 study, 28 the permeability was from a different report and predicted a lower value P eff,man. It should be noted that M‐III assigned the permeability to represent passive permeability only; however, because inhibitors were not used in the in vitro study, both active and passive transports were present. In addition, the P app value used for the reference standard propranolol was the default value from the Simcyp simulator instead of one measured in the same Caco‐2 study to mitigate interlaboratory variability. 29 , 30
M‐IV used Caco‐2 data under noninhibited conditions and correctly assigned both passive and active transports to the data 27 ; similar to M‐III, the default P app value for propranolol was used instead of a measured value. In M‐V, instead of the previously used Caco‐2 data (which was published in 2011), more recent Madin‐Darby canine kidney II–low expression cell line data were input, 31 and four calibrator values were used from the same laboratory. 32
Intestinal transport
To account for intestinal transporter involvement, M‐I, M‐II, M‐III, and M‐V assigned transport values, whereas M‐IV used the Caco‐2 data mentioned previously to represent a lumped passive and active uptake and efflux. Although the four models included the efflux transporter BCRP, the rationale behind the final input selections varied.
M‐I (and also used in M‐II) performed a sensitivity analysis to recover the observed rosuvastatin time to reach maximum concentration (tmax) and maximum concentration (Cmax) values with other PK parameters (e.g., total clearance and steady‐state volume of distribution (V ss), defined using other data) fixed to obtain net transport clearance (CLint,T), which was assigned to BCRP (as there was no way to separate the contribution of BCRP vs. multidrug resistance protein (MRP) 2 based on the available data at the time).
M‐III focused on capturing rosuvastatin’s delayed absorption, and the authors explained that although the distribution of BCRP (higher in the duodenum and jejunum) could cause the delay, a clinical genotyping study of subjects with reduced BCRP activity did not shift the tmax as would have been expected. 33 The authors then postulated that basolateral transport by organic solute transporter alpha/beta (OST α/ß) 28 may explain the absorption delay if OST α/ß is highly expressed in the middle to terminal sections of the ileum (however, a recent meta‐analysis found the regional expression for OST α/ß to be more uniform 34 ). M‐III input a gut apical uptake CLint,T, which was “model fit.” Because OST α/ß is a basolateral membrane transporter, not apical, 35 this approach makes the assumption that the driving concentrations are the same. In addition, although apical uptake and basolateral efflux give the same net effect, the inclusion of an apical transporter would lead to an increase in the enterocyte concentration vs. the inclusion of basolateral efflux would lead to a decrease in the enterocyte concentration, and this enterocyte concentration affects the driving concentration of BCRP. To determine the intestinal contribution of BCRP, a Michaelis‐Menten constant (K m) value was taken from membrane vesicle studies, 12 and the maximum rate of transport (J max) was “model fit.” It should be noted that performing a sensitivity analysis and parameter estimation are modules available within the Simcyp Simulator, 36 and although M‐III reported to “model fit” data to an observed 10 mg oral dose, the details of the fitting were not described.
In M‐V, mechanistic modeling was done to determine the concentration at the binding site of BCRP using in vitro data from two studies and to obtain a K m input. The BCRP K m was then fixed while the BCRP J max and OATP2B1 (to account for an apical uptake transport mechanism) intrinsic clearance (CLint) were optimized simultaneously through sensitivity analysis fitting to in vivo data. 37 , 38 These inputs originally overpredicted part of the observed oral profiles, and it was hypothesized to be from too high an absorption in the colon, so an absorption scalar was added to the colon.
Distribution
To describe rosuvastatin’s distribution, every model used a full PBPK distribution model, and the V ss was predicted using the Rodgers and Rowland method (“Method 2” in the Simcyp Simulator). 39 This method calculates drug partitioning into a specific tissue by considering the partitioning into individual tissue components including neutral lipids and neutral phospholipids (and for compounds with at least one basic pKa ≥ 7, the electrostatic interactions with acidic phospholipids). This partitioning is then used to calculate the steady‐state tissue‐to‐plasma partition coefficient, K p, and calculate V ss. It is well known that active transport can lead to a larger observed volume of distribution, 40 and because the Rodgers and Rowland method 39 does not take into account active transport, it is expected that “Method 2” would not be representative of the V ss for acidic transporter substrates such as rosuvastatin. As seen with M‐I, M‐II, and M‐III, the estimated V ss from Method 2, reflective only of passive processes, was 0.117 L/kg, whereas the observed value is significantly higher (V ss observed = 1.73 L/kg). 2
Noting the discrepancy using “Method 2,” M‐IV tried a middle‐out approach for V ss prediction. Tissue concentrations of rosuvastatin were examined after a single oral dose of [14C]‐rosuvastatin in Sprague‐Dawley rats, 41 and these values were used to change the default K p values for individual tissues. This middle‐out approach led to a higher predicted V ss of 0.70 L/kg; however, the authors reported that altering these values did not improve the predicted concentration‐time profiles and did not significantly alter the PK parameters (AUC, Cmax, and tmax) of rosuvastatin compared with their model using the estimated K p values based on “Method 2.”
In M‐V, instead of changing individual tissue K p values, a global K p scalar of 4.85 was added to help capture the distribution phase of the intravenous (i.v.) infusion profile and this increased the predicted V ss to 0.385 L/kg.
Hepatic metabolism
Although rosuvastatin undergoes minor metabolism, the models used different strategies for the inputs. Because the kinetics were not determined in vitro at the time, M‐I, M‐II, and M‐III used the in vivo clearance to back‐calculate a global intrinsic hepatic clearance of which 10% was assigned to metabolism based on observed mass balance data. 6 M‐V also back‐calculated a value based on the in vivo mass balance data using the reverse translational tool and incorporated a liver unbound partition coefficient (K puu) to reflect the active transporter uptake at the peak portions of the observed profiles from positron emission tomography imaging data.
M‐IV included specific enzyme kinetics and assigned a CLint to CYP3A4 (although CYP3A4 did not have measured involvement in rosuvastatin acid in the cited study 42 ) and input maximum velocity of the metabolic reaction (Vmax) and K m values for UGT1A1/3 from supersomes of recombinant UGT. 5 The Vmax units reported by Schirris et al. 5 are in pmol/second/mg protein (vs. the pmol/minute/mg protein inputs of Simcyp), meaning the current input values may be 60‐fold too low and corrected values would lead to a lower AUC (1.3‐fold lower with an i.v. infusion simulation). Further clarification of the units used is needed from the original authors.
Hepatic transport
Different approaches were used to determine accurate inputs for rosuvastatin hepatic uptake (CLint,T) by each transporter in these models. For M‐I, the authors stated that in vitro experimental data could not recover the observed concentration‐time profiles. Therefore, a global intrinsic clearance for active hepatic uptake was estimated using the parameter estimation module and clinical i.v. data from healthy volunteers. 2 To then partition the global uptake CLint,T among different transporters, published in vitro data were used to assign a percentage contribution of each transporter. Using sandwich culture human hepatocytes (SCHH) and sodiuM‐containing vs. sodiuM‐free buffer, Ho et al. 10 estimated an NTCP uptake contribution of 35%. Using data from human embryonic kidney (HEK) 293‐OATP1B1 cells and hepatocytes, Kitamura et al. 11 determined a relative activity factor and found that the contribution of OATP1B1 to rosuvastatin uptake was 49% (range of 43–55% based on three different hepatocyte lots). M‐I attributed the remaining 16% of the global uptake clearance to OATP1B3 and reported that although OATP2B1 has also been implicated in rosuvastatin uptake, given its complexity to model without enough data, its hepatic uptake contribution was included in that of OATP1B3. The passive intrinsic clearance was measured in a study using sandwich culture human hepatocytes (SCHH) 43 and for hepatic canalicular efflux, the contribution of BCRP was also determined in SCHH. 44 These values were used in M‐II as well.
M‐III used the same fitted global uptake intrinsic clearance as mentioned previously, however with additional in vitro data then available, the percentages assigned to each transporter varied. In vitro data from three sources were referenced where uptake was measured in HEK293‐OATP1B1/3 cells and corrected for transporter activity or abundance. 11 , 45 , 46 The data from Kunze et al. 46 suggested that about 4–20% of rosuvastatin’s active uptake was not accounted for by OATP1B1/3, leading M‐III to assign 10% of uptake to NTCP. In terms of hepatic efflux transporters, a study found that rosuvastatin is a substrate of the basolateral MRP4 and that the basolateral efflux is about four times greater than the biliary efflux mediated by BCRP in vitro. 47 Although in vitro values were provided in the study, M‐III model fit the CLint,T value for MRP4 and used the relationship so the CLint,T of BCRP was increased in response to reflect the fourfold difference. It should be noted that the estimation of basolateral efflux from Pfeifer et al. 47 contains both active transport and passive diffusion, so the inputs for MRP4 and subsequently BCRP may be overestimated.
Instead of using the global CLint,T determined from fitting in vivo data and assigning percentages to each hepatic uptake transporter involved for input, M‐IV used in vitro transporter data and scaled the data up to predict in vivo values. The CLint from a suspension hepatocyte study with and without sodiuM‐containing buffer was input for NTCP, 48 while J max and K m values from HEK293 cells overexpressing transporters were input for OATP1B1, 1B3, and 2B1. 45 , 49 MRP4 was included for sinusoidal efflux as it was with M‐III, but instead of model fitting to obtain a value, here experimental J max and K m values from membrane vesicles were input. 47 It should be noted that the reported J max of OATP1B1 and MRP4 are in units of pmol/minute/mg protein (vs. the input of Simcyp is pmol/minute/106 cells) so a 1:1 assumption of mg protein:106 cells is being made. For efflux transporters involved in biliary secretion, although BCRP is thought to be involved, there are conflicting data about the role of P‐glycoprotein (P‐gp), 11 , 12 so M‐IV decided to assign an overall CLint for canalicular efflux using data from SCHH. 50
To account for transporter expression differences between the in vitro systems and in vivo, M‐IV incorporated two scaling factors (SF) into the Simcyp relative expression factor (REF) of each transporter: SF1 accounted for the difference in transporter expression/activity between expression systems (membrane vesicles or HEK293 cells) and isolated hepatocytes, and SF2 accounted for the difference in transporter expression between isolated hepatocytes and liver tissue. Using the in vitro data and correcting with REF, M‐IV had a global uptake CLint,T in line with the value derived from in vivo data by M‐I.
In M‐V, for the hepatic efflux transporters, the CLint of BCRP was increased 5.3‐fold compared with M‐I and the input was from SCHH data with activity corrections for absolute abundances of BCRP applied. 51 , 52 The contribution of MRP4 was included in this model using the relationship between rosuvastatin’s SCHH basolateral efflux and biliary clearance previously discussed 47 and correcting for SCHH‐to‐liver transporter expression differences. 25 Given these increases in biliary clearance and basolateral efflux, the global uptake CLint then needed to be increased (4.5‐fold) to fit the observed data. 2 The largest difference with hepatic uptake transporters in M‐V is seen with NTCP, where the contribution is decreased to 6% based on a recent hepatocyte solute carrier (SLC) phenotyping study. 53 For the other hepatic uptake transporters in this model, OATP2B1 was included with a 21% contribution based on the same phenotyping study 53 in addition to OATP1B1 and OATP1B3, where the fractions transported were assigned based on a meta‐analysis. The passive diffusion input was based on a meta‐analysis of five SCHH studies.
Renal clearance
To account for rosuvastatin’s renal clearance (CLR), M‐I and M‐V assumed perfusion‐limited distribution and used an input value based on a meta‐analysis of in vivo data, whereas M‐II explored the permeability‐limited kidney model (Mech KiM) 54 to account for transporter involvement.
To determine the passive diffusion, M‐II used Caco‐2 data previously mentioned 27 and scaled it using total nephron surface area, kidney weight, compound ionization, and proximal tubule cells/gram kidney. The passive diffusion from the blood‐to‐cell and cell‐to‐tubule and vice versa were assumed to be equal, and after accounting for this and glomerular filtration, the CLR was predicted as 1.13 L/hour. To account for the difference between the predicted and observed (CLR = 17 L/hour), a sensitivity analysis was performed to determine the active transport CLint,T, which was applied to the basolateral uptake transporter OAT3 (the rate‐limiting step 55 ), and the apical efflux transporter BCRP 12 , 56 (but input into the Simcyp simulator as MRP4 because there was not a BCRP input in the kidney). At the time, the contribution of BCRP vs. MRP4 could not be distinguished with in vitro data, so the efflux CLint,T was lumped into the one value. Because there is only relative transporter abundance scaling available in the kidney, exact efflux transporter assignment will not have an impact.
M‐III also used Mech KiM and included OAT3 and BCRP as kidney transporters. As a passive diffusion clearance was not assigned in this model, the authors assigned a lower value of CLint,T to both transporters based on fitting in vivo data from a 10 mg dose study.
M‐IV continued using the bottoM‐up approach with the Mech KiM model, and the J max and K m values for OAT3 were taken from a study using a mixed monolayer of human proximal and distal tubule/collecting duct cells. 55 The efflux was assigned to MRP4, and again the values from OAT3 were used, given that uptake is the rate‐limiting step. The passive diffusion from the blood‐to‐cell and cell‐to‐tubule were assumed to be equal and were estimated from the same study. 55
ROSUVASTATIN PK AND DDI SIMULATIONS
To elucidate the impact of the key model parameters in the different rosuvastatin models described previously on PK and DDI prediction, we conducted simulations using the Simcyp simulator version 18 release 2 in which the five different rosuvastatin models described previously were used for PK simulation, and transporter inhibitors in the compound library of version 18 release 2 were used when conducting DDI simulation. We recognize that different versions of simulator were used for each model during its development, and simulator changes may result in different simulation results. In addition, in the original published work, the transporter inhibition constant (Ki) was modified in some models for tDDI simulation to match the observed data. For the purpose of this review, which is not to compare the simulations in this work with that in the original publication of these models or to match the published clinical DDI data but, rather, to understand some common mechanistic uncertainties encountered by all five models and how different approaches perform in addressing these questions, using the same version of software provides a comparison that will allow us to explore what is mechanistically possible to better describe rosuvastatin’s disposition.
Pharmacokinetics
Rosuvastatin PK simulations were conducted using the five models and compared to clinical data for an i.v. infusion, single oral dose (10–80 mg), and multiple oral dosing. Simulations were run with rosuvastatin as a solution formulation. Details of the clinical studies examined and the simulation design can be found in Table S2 . Given the clinical potential of enterohepatic recirculation (EHR), 2 active EHR for the i.v. dose was allowed in every model with 100% available for reabsorption.
Drug–Drug Interactions
Simulations of tDDIs between rosuvastatin and the transporter inhibitors cyclosporine (with its inhibitory metabolite), rifampin (dosed orally and i.v.), and gemfibrozil (with its inhibitory metabolite) were conducted using the five rosuvastatin models with inhibitors from the Simcyp compound library (version 18 release 2). No optimization of inhibition Ki for each transporter inhibitor was performed even though it is known that the accuracy of DDI prediction is also driven by inhibition Ki. It is considered beyond the scope of this review. Details about the clinical data and simulation trial designs can be found in Table S3 and 10 trials were run for each simulation.
The differences between the model inputs and the PK and DDI simulation results are presented in Figure 2 and Tables 1 , 2 , 3 and discussed in the follow sections of simulated PK and DDI.
Figure 2.
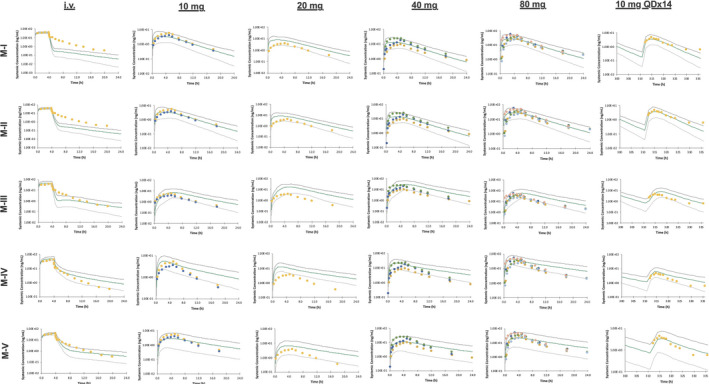
Mean rosuvastatin plasma concentration‐time profiles for an 8 mg intravenous infusion, single oral dosing (10–80 mg), and multiple oral dosing (10 mg once a day for 14 days). The simulated results are shown as a green line with the 5th and 95th percentiles shown as gray lines. The observed clinical data (detailed in Table S2 ) are plotted as points.
Table 2.
The observed and predicted geometric mean of the AUC and Cmax and median of the tmax for rosuvastatin
| Dose (mg) | Observed |
M‐I Predicted |
M‐II Predicted |
M‐III Predicted |
M‐IV Predicted |
M‐V Predicted |
|
|---|---|---|---|---|---|---|---|
| Oral | |||||||
| AUC (ng*hour/mL) | 10 | 31.6–45.9 | 30.9 (1.02–1.49) | 31.6 (1.00–1.45) | 62.7 (0.50–0.73) | 53.7 (0.59–0.85) | 28.5 (1.11–1.61) |
| 20 | 56.8 | 70.0 (0.81) | 71.4 (0.80) | 148.0 (0.38) | 123.6 (0.46) | 68.7 (0.83) | |
| 40 | 98.2–216 | 122.8 (0.80–1.76) | 126.1 (0.78–1.71) | 252.7 (0.39–0.85) | 212.1 (0.46–1.02) | 111.8 (0.89–1.93) | |
| 80 | 253–410 | 240.9 (1.05–1.70) | 246.6 (1.03–1.66) | 490.7 (0.52–0.84) | 416.0 (0.61–0.99) | 218.4 (1.16–1.88) | |
| 10 QDx14 | 40.1 | 31.6 (1.27) | 32.5 (1.23) | 67.9 (0.59) | 63.1 (0.64) | 34.0 (1.18) | |
| Cmax (ng/mL) | 10 | 3.75–5.80 | 3.27 (1.15–1.77) | 3.32 (1.13–1.75) | 6.74 (0.56–0.86) | 5.20 (0.72–1.12) | 2.34 (1.60–2.48) |
| 20 | 6.79 | 7.42 (0.92) | 7.53 (0.90) | 16.3 (0.42) | 12.1 (0.56) | 5.64 (1.20) | |
| 40 | 10.3–25.0 | 12.9 (0.80–1.94) | 13.2 (0.78–1.89) | 27.6 (0.37–0.91) | 20.5 (0.50–1.22) | 9.22 (1.12–2.71) | |
| 80 | 30.1–53.5 | 25.4 (1.19–2.11) | 25.9 (1.16–2.07) | 54.4 (0.55–0.98) | 40.5 (0.74−1.32) | 18.1 (1.66–2.96) | |
| 10 q.d. × 14 | 4.58 | 3.39 (1.35) | 3.47 (1.32) | 7.19 (0.64) | 5.89 (0.78) | 2.74 (1.67) | |
| tmax (hour) | 10 | 5.0 | 3.55 (1.41) | 3.63 (1.38) | 5.65 (0.88) | 2.50 (2.00) | 3.60 (1.39) |
| 20 | 5.0 | 3.58 (1.40) | 3.29 (1.52) | 5.55 (0.90) | 2.45 (2.04) | 3.58 (1.40) | |
| 40 | 5.0 | 3.58 (1.40) | 3.65 (1.37) | 5.65 (0.88) | 2.40 (2.08) | 3.55 (1.41) | |
| 80 | 3.0–5.0 | 3.48 (0.86–1.44) | 3.55 (0.85–1.41) | 5.60 (0.54–0.89) | 2.40 (1.25–2.08) | 3.50 (0.86–1.43) | |
| 10 q.d. × 14 | 3.0 | 3.30 (0.91) | 3.35 (0.90) | 5.55 (0.54) | 2.30 (1.30) | 3.35 (0.90) | |
| Intravenous | |||||||
| AUC (ng*hour/mL) | 8 | 164 | 143.3 (1.14) | 147.5 (1.11) | 186.5 (0.88) | 229.6 (0.71) | 157.3 (1.04) |
The fold difference (observed/predicted) is in parentheses. References for the observed data can be found in Table S2 . AUC, area under the concentration‐time curve; Cmax, maximum concentration; q.d., once a day; tmax, time to reach maximum concentration.
Table 3.
The observed and predicted CmaxR of rosuvastatin in the presence of absence of inhibitor, the AUCR, and the fold difference (obs/pred)
| Parameter | Observed |
M‐I Predicted |
M‐II Predicted |
M‐III Predicted |
M‐IV Predicted |
M‐V Predicted |
|
|---|---|---|---|---|---|---|---|
| Cyclosporine | CmaxR | 10.6 | 3.23 (3.08–3.38) | 3.19 (2.86–3.56) | 1.54 (1.47–1.63) | 1.57 (1.54–1.61) | 2.09 (2.01–2.18) |
| Obs/pred | 3.28 | 3.32 | 6.88 | 6.75 | 5.07 | ||
| AUCR | 7.1 | 1.48 (1.45–1.51) | 1.50 (1.44–1.57) | 1.50 (1.45–1.54) | 1.41 (1.38–1.43) | 1.55 (1.51–1.59) | |
| Obs/pred | 4.80 | 4.73 | 4.73 | 5.04 | 4.58 | ||
| Rifampin (oral) | CmaxR | 9.93 (7.25–13.6) | 5.50 (5.13–5.90) | 5.46 (5.09–5.86) | 3.75 (3.51–3.99) | 2.44 (2.34–2.54) | 5.41 (4.97–5.89) |
| Obs/pred | 1.81 | 1.82 | 2.65 | 4.07 | 1.84 | ||
| AUCR | 5.24 (3.66–7.49) | 2.27 (2.19–2.36) | 2.31 (2.22–2.41) | 2.79 (2.69–2.90) | 2.00 (1.95–2.06) | 3.21 (3.05–3.38) | |
| Obs/pred | 2.31 | 2.27 | 1.88 | 2.62 | 1.63 | ||
| Rifampin (intravenous) | CmaxR | 5.51 (4.38–6.93) | 2.38 (2.25–2.52) | 2.41 (2.28–2.56) | 2.94 (2.78–3.10) | 2.37 (2.29–2.46) | 4.20 (3.91–4.52) |
| Obs/pred | 2.32 | 2.29 | 1.87 | 2.32 | 1.31 | ||
| AUCR | 4.55 (2.95–7.02) | 1.78 (1.72–1.83) | 1.80 (1.75–1.86) | 2.26 (2.17–2.36) | 1.91 (1.86–1.97) | 2.74 (2.62–2.85) | |
| Obs/pred | 2.56 | 2.53 | 2.01 | 2.38 | 1.66 | ||
| Gemfibrozil | CmaxR | 2.21 (1.81–2.69) | 1.47 (1.44–1.50) | 1.52 (1.49–1.55) | 1.70 (1.66–1.74) | 1.45 (1.42–1.48) | 1.62 (1.58–1.66) |
| Obs/pred | 1.50 | 1.45 | 1.30 | 1.52 | 1.36 | ||
| AUCR | 1.88 (1.60–2.21) | 1.34 (1.32–1.36) | 1.37 (1.35–1.39) | 1.56 (1.53–1.59) | 1.33 (1.31–1.35) | 1.40 (1.37–1.42) | |
| Obs/pred | 1.40 | 1.37 | 1.21 | 1.41 | 1.34 |
Data are reported as geometric means (90% confidence intervals). References for the observed data can be found in Table S3. AUCR, area under the concentration‐time curve ratio; CmaxR, maximum concentration ratio; obs/pred, observed/predicted.
Simulated PK
For the i.v. simulations, although the predicted AUC value was within twofold of the observed value for all the models, there were large differences when trying to accurately capture the triphasic decline of the concentration‐time profile (which still needs improvement). When M‐II activated Mech KiM, with the inclusion of the rate‐limiting OAT3 in the renal clearance, the elimination phase of rosuvastatin’s concentration‐time profile appeared more gradually than when Mech KiM was not activated in M‐I. With M‐III, the inclusion of MRP4 in this model helped capture more of the distribution phase, and a comparison of the predictions with and without MRP4 can be seen in Figure 3a,b . Although the triphasic decline is not fully captured, the simulated profiles of M‐IV and M‐V captured many of the observed data points. To explore what led to the improved profile of M‐V, the K p scalar of 4.85 was decreased to 1 (Figure 4 ), showing that the K p scalar helped smooth the profile and capture the distribution phase.
Figure 3.
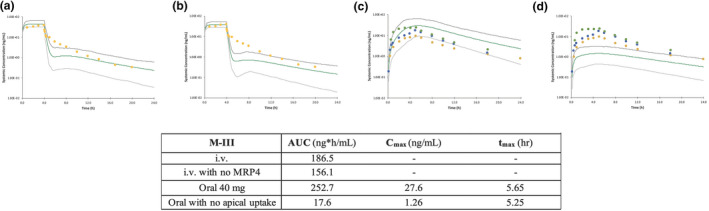
The effect of including the hepatic sinusoidal efflux transporter MRP4 with the intravenous dose of rosuvastatin (a) vs. removing MRP4 involvement in M‐III (b) and the effect of including intestinal apical uptake with the 40 mg oral dose of rosuvastatin (c) vs. removing the apical uptake (d) in M‐III. AUC, area under the concentration‐time curve; Cmax, maximum concentration; hr, hour; i.v., intravenous; MRP, multidrug resistance protein; tmax, time to reach maximum concentration.
Figure 4.
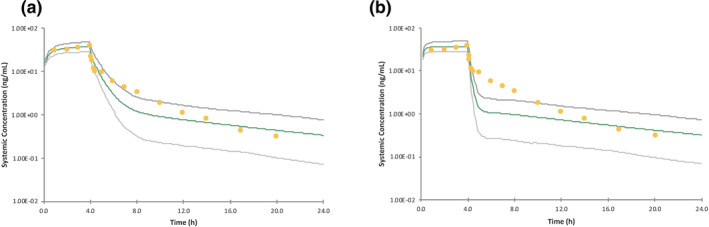
The effect of including a tissue‐to‐plasma partition (K p) scalar for the predictions of an intravenous dose of rosuvastatin (a) vs. removing the K p scalar (b) in M‐V.
For the oral doses, the predicted PK parameters (AUC, Cmax, and tmax) generally fell within twofold of the observed clinical data for all models. A consistent issue was that although the simulated concentration‐time profiles matched the observed data reasonably well, often the absorption delay was not fully captured. M‐III helped improve the prediction of the absorption delay by including a gut apical uptake CLint,T; however, the mechanistic reasoning behind the improvement needs further consideration. The effect of including this gut apical uptake can be seen in Figure 3c,d —when the apical uptake component is removed, the AUC and Cmax are dramatically decreased, and the tmax is shifted slightly earlier. Another issue that can be seen with the results of M‐IV and M‐V is that the elimination phase was slower than the observations in some cases, which could be due to EHR not being captured effectively.
Simulated DDI
All rosuvastatin models examined underpredicted the cyclosporine DDI (Table 3 ) using the cyclosporine inhibitor file (with its inhibitory metabolite) in Simcyp version 18 release 2. However, it was noted that the in vivo study examined was not a dedicated crossover DDI study but, rather, it was a comparison of healthy volunteers taking rosuvastatin and patients taking rosuvastatin and cyclosporine after heart transplants. 13 In addition, the cyclosporine inhibition Ki values could be a cause of underprediction, and in an original model of cyclosporine, needing to empirically lower in vitro values was described. 17 Although all models significantly underpredicted the DDI, the extent of Cmax underprediction was less for M‐I and M‐II, where inhibition of intestinal BCRP can occur, compared with the predicted interaction using M‐IV, which does not have intestinal transporters activated.
For rifampin, all models were able to predict a mild to moderate DDI; however, a trend of underprediction, in some cases more than twofold, was observed. As expected with intestinal BCRP inhibition, the ratios were accurately higher with oral compared with i.v. rifampin dosing for all except M‐IV, where the similarity between the oral and i.v. interaction could be because this model does not specifically include intestinal transporters.
For gemfibrozil (with its inhibitory metabolite), the least potent DDI in terms of magnitude of change from baseline, there is minimal difference in the predicted magnitude of DDI among the five models, and the predicted interaction ratios fell within twofold of the observed for all models.
DISCUSSION
Further qualification of the rosuvastatin model for transporter DDI prediction of NMEs is desired from both the pharmaceutical industry and regulatory agencies. Examination of five previously reported PBPK models allows us to understand how rosuvastatin disposition is currently being described, what the model gaps and common uncertainties are, and what studies are required for prediction improvement. These five models were originally assessed to qualify their specific purpose of use. M‐I and M‐V are the first and the most recent model compound files in the Simcyp simulator, respectively, and have been commonly used for DDI risk assessment in regulatory interactions. M‐II was exploratory and activated the Mech KiM model to include kidney transporter involvement. M‐III was also exploratory and included more transporters in the intestine and liver to try to better describe absorption and more accurately capture tDDIs. M‐IV was more of an academic exercise attempting bottoM‐up modeling with in vitro metabolism and transporter data. Although several mechanistic aspects of rosuvastatin’s disposition have been explored with these model developments, disconnections still exist (Table 4 ), and further optimization with better mechanistic understanding will increase our confidence and further qualify the model for broader application in addressing regulatory questions.
Table 4.
Aspects for improvement of rosuvastatin models
| Area | Issue | Potential ways to improve and outstanding questions |
|---|---|---|
| Absorption | Absorption delay is missed | Include OST α/ß; what is the driving concentration? |
| Include OATP2B1; where is it localized? | ||
| Include additional efflux transporters such as MRP2; how can this contribution be separated out from BCRP? | ||
| Include information for a tablet/capsule formulation | ||
| Consider food effects, were all observed subjects truly fasted? How does feeding impact enterohepatic recirculation? | ||
| Distribution | Method 2 prediction of V ss does not account for active transport | Use tissue data from preclinical animals to alter K p values; middle‐out approach |
| Apply a K p scalar | ||
| Run simulation, independently obtain a value for V ss with NCA | ||
| Develop a method that incorporates the role of transporters into the prediction | ||
| Hepatic uptake | Inputting in vitro transporter data alone does not capture the concentration‐time profile | Apply REF scalars to account for expression differences between systems; scaling factors should be from same laboratory |
| Explore other mechanistic reasons for in vitro/in vivo disconnects | ||
| Hepatic basolateral efflux | MRP4 may be included, but low expression in the liver | Need additional information to know if other transporters could be involved |
| tDDIs | Often still qualitatively but do not quantitively predict DDIs | Gather more information about preincubation effect with in vitro systems and additional reasons for current discrepancies |
| Additional information about transporter contributions may improve predictions |
One of the areas that has been focused on in the different models is trying to more accurately capture the rate of absorption in the single oral dose concentration‐time profiles. Although the extent of absorption is accurately predicted (the simulated fraction absorbed ranged from 0.4–0.7 in the five models vs. the observed absorption was 0.52), the simulated tmax occurred too early in all of the models with the exception of M‐III. M‐III included an apical uptake clearance (to account for OST α/ß transport); however, the reason for this improvement could be because of M‐III’s data input selection. Revisiting the absorption inputs of this model, the passive permeability was taken from uninhibited Caco‐2 data (already accounting for uptake and efflux). If the apical uptake is removed as shown in Figure 3c,d , efflux is left and double counted as there is a specific BCRP efflux transporter input and efflux present in the passive permeability input, causing the lower AUC and Cmax without the uptake transporter to offset the double counting. As future model development is done, sensitivity analyses on additional parameters can be conducted to understand their effect in driving the model’s prediction of rosuvastatin disposition.
In M‐V, OATP2B1 uptake is included in the file; however, the simulated tmax remains too early. The details of specific intestinal transporter involvement in rosuvastatin transport are still not very clear. It has been noted that OATP2B1 is likely involved in the intestinal transport of rosuvastatin, and one study found that uptake decreased when buffer pH was raised from 5.5 to 7.4, demonstrating its relevance as an intestinal transporter 57 ; however, there is debate about whether OATP2B1 an apical or basolateral transporter in the intestine. 58 , 59 Other transporters that have been considered to explain absorption delay are BCRP and MRP2. Although the regional distribution differences of BCRP in the intestine could explain the delay, a genotyping study did not support this. 33 There has been in vitro evidence of additional efflux transporter involvement with MRP2 11 ; however, there is not currently a way to separate this potential contribution from that of BCRP and verify this from a clinical perspective. More definitive studies on rosuvastatin intestinal uptake/efflux are one aspect needed to help capture the observed tmax. It should be noted that the regional intestinal BCRP distribution was updated in Simcyp version 19 (and BCRP phenotypes were added), which may also change model performance. In addition, food effects should be considered in case subjects were not truly fasted with the observed data and should also be considered for their impact on modeling EHR. Formulation aspects should also be further explored since oral rosuvastatin is often given as a tablet or capsule (not solution).
Another disconnection present for all five models is the prediction of rosuvastatin’s volume of distribution, as a prediction method that accounts for transporter effects does not currently exist. The role of active transport is not incorporated, which likely explains the discrepancy between the predicted values and the observed V ss for rosuvastatin. Some empirical approaches were tried, e.g., adding a global K p scalar or incorporating some K p values from animal data, to alter the prediction. While the middle‐out approach of using rat values is interesting to consider, more should be explored for potential future use including the impact of species differences between rat and human (including plasma protein binding and the blood:plasma ratio) and time‐point selection of these tissue concentrations. A way around this is to independently calculate the V ss with noncompartmental analysis (NCA) of the simulated plasma concentrations, which M‐III did and reported a value of 2.0 L/kg, much closer to the observed value of 1.73. Until another prediction method is developed, caution should be taken in relying on Method 2 V ss predictions of transporter substrates.
In terms of hepatic transporter modeling, all models used an approach of fitting in vivo data and then assigning transporter percentages based on in vitro studies, except M‐IV, which assigned transporter values scaled directly from in vitro data. It is currently difficult to separate out transporter involvement as many inhibitors are not transporter specific. 53 Instead, cells overexpressing a transporter can be used and subsequently corrected for the expression and/or activity levels in hepatocytes and liver tissue. As reported by the authors of M‐IV, without applying a scaling factor (such as REF) to the in vitro data, the simulated exposure of i.v. infusion would be 10.3‐fold over the observed data. Although the bottoM‐up approach is encouraging for new compounds, we are still facing significant challenges. 18 There must be caution when directly using in vitro data due to interlaboratory variability, and ideally all transporter expression levels would need to be measured under the same conditions since transporter quantification is known to vary with digestion procedures and surrogate peptide sequence selection. 60 , 61 If such variability occurred and caused a twofold increase in the REF values applied, the predictions shift as shown in Figure S1 .
Switching focus to other transporters, in terms of hepatic basolateral efflux, while MRP4 is currently assigned (in M‐III, M‐IV, and M‐V), there is low MRP4 expression in the healthy liver, 62 and an additional transporter may be involved. However, given that there is no additional information at this time except that MRP3 does not appear to transport rosuvastatin, 47 the inputs are reasonable. For the renal transporters, M‐II and M‐III fit in vivo data for transporter inputs to help capture the renal clearance, while with the bottoM‐up approach of M‐IV (no REF was included), the renal clearance was underpredicted by fivefold, emphasizing the challenges with current renal in vitro systems. 63
When examining tDDI prediction accuracy, the largest error was for the cyclosporine interaction; however, healthy volunteers were compared with patients who had heart transplants. Vildhede et al. 64 showed that variability in hepatic transporter levels could cause variation in DDI impacts, so the in vivo interaction could be due to pathophysiological differences between the healthy volunteers and patients as well as the DDI. In general, for all DDIs examined here, there was underprediction, which has previously been seen when in vitro Ki values are used. 18 Simcyp version 18 inhibitor files were used for this review to allow for a consistent comparison across models; however, M‐III evaluated their own inhibitor files with generally lower, optimized Ki values and reported more accurate predictions. Simcyp version 19 has an updated rifampin file where instead of using the previous in vitro values optimized with clinical data as done for the version 18 file, updated measured values (with the inhibitor preincubated for OATP1B1/3 for instance 65 ) were applied when possible. DDI predictions were the most accurate for gemfibrozil, where in vitro inhibition data were directly input into the compound file without optimization, suggesting that these values may have less uncertainty.
The tDDI underpredictions could be improved with more understanding of rosuvastatin’s transporter involvement by using specific transporter inhibitors or conducting additional i.v. inhibitor studies to understand the contribution of intestinal vs. hepatic transporters. Adding MRP2 to rosuvastatin models and including potential MRP2, OATP2B1, and/or NTCP inhibition may help improve the current tDDI underpredictions. This has also been suggested for a pravastatin PBPK model, which has a similar disposition pathway as rosuvastatin. 66 The pravastatin model was similarly unable to capture the DDI with cyclosporine when using the most potent reported in vitro Ki value but was able to more accurately capture DDIs with rifampin and gemfibrozil. 66 Learnings from PBPK simulations of tDDI with other drugs with similar disposition pathways can be leveraged for future rosuvastatin model development. As more information about the contribution of specific transporters, the effect of inhibitor preincubation, 67 and the impact of inhibitory metabolites becomes available, the tDDI predictions should be revisited.
These challenges are not unique to the rosuvastatin models developed using the Simcyp simulator, and additional references can be found in Table S1 . Jones et al. 50 found that SCHH data alone could not accurately capture rosuvastatin’s i.v. concentration‐time profile, and an empirical scaling factor was needed. In addition, although Bosgra et al. 45 were able to capture the i.v. and oral profiles reasonably well using HEK293 cells overexpressing hepatic uptake transporters along with REF values from liquid chromatography tandem mass spectrometry transporter quantitation, the triphasic decline of the i.v. profile and the absorption delay of the oral profile were missed. Further assessment of other customized models built from scratch using different software (Table S1 ) could provide more information that may benefit future rosuvastatin model development.
This current work provides an overview of five rosuvastatin models with hopes that the shared learnings will support further model improvement with better mechanistic understanding of transporter‐mediated drug disposition and ultimately increase our confidence in tDDI prediction for NMEs. Additional information about rosuvastatin’s absorption in terms of transporter involvement and formulation and food effects, additional in vitro and in vivo transporter studies to more accurately capture DDI predictions, and development of a volume of distribution prediction method that incorporates the role of transporters are aspects that could be considered with future model development. The five existing rosuvastatin PBPK models were developed for different purposes, and although there is still more that can be explored to enhance qualification, the models described here have made significant progress toward capturing the disposition of this transporter substrate, and bottoM‐up modeling may be attainable in the future. A highly qualified probe substrate model will enable mechanistic understanding of transporter‐mediated drug disposition and increase the confidence in regulatory decision making based on PBPK model predictions of tDDI for NMEs.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
Supporting information
Figure S1
Table S1
Table S2
Table S3
Acknowledgments
The authors would like to thank Matthew Harwood and Sibylle Neuhoff from Certara and Buyun Chen for their review and discussions as well as Eugene Chen and Kenta Yoshida for their review.
Contributor Information
Christine M. Bowman, Email: bowman.christine@gene.com.
Yuan Chen, Email: chen.yuan@gene.com.
References
- 1. Olsson, A.G. , McTaggart, F. & Raza, A. Rosuvastatin: a highly effective new HMG‐CoA reductase inhibitor. Cardiovasc. Drug Rev. 20, 303–328 (2002). [DOI] [PubMed] [Google Scholar]
- 2. Martin, P.D. , Warwick, M.J. , Dane, A.L. , Brindley, C. & Short, T. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin. Ther. 25, 2553–2563 (2003). [DOI] [PubMed] [Google Scholar]
- 3. McTaggart, F. et al Preclinical and clinical pharmacology of rosuvastatin, a new 2‐hydroxy‐3‐methylglutaryl coenzyme a reductase inhibitor. Am. J. Cardiol. 87, 28B–32B (2001). [DOI] [PubMed] [Google Scholar]
- 4. Prueksaritanont, T. , Tang, C. , Qiu, Y. , Mu, L. , Subramanian, R. & Lin, J.H. Effects of fibrates on metabolism of statins in human hepatocytes. Drug Metab. Dispos. 30, 1280–1287 (2002). [DOI] [PubMed] [Google Scholar]
- 5. Schirris, T.J. , Ritschel, T. , Bilos, A. , Smeitnik, J.A. & Russel, F.G. Stain lactonization by uridine 5’‐diphospho‐glucuronosyltransferases (UGTs). Mol. Pharm. 12, 4048–4055 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Martin, P.D. et al Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin. Ther. 25, 2822–2835 (2003). [DOI] [PubMed] [Google Scholar]
- 7. Bergman, E. et al Biliary secretion of rosuvastatin and bile acids in humans during the absorption phase. Eur. J. Pharm. Sci. 29, 205–214 (2006). [DOI] [PubMed] [Google Scholar]
- 8. Nezasa, K.‐I. et al Liver‐specific distribution of rosuvastatin in rats: comparison with pravastatin and simvastatin. Drug Metab. Dispos. 30, 1158–1163 (2002). [DOI] [PubMed] [Google Scholar]
- 9. Kalliokoski, A. & Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 158, 693–705 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ho, R.H. et al Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology 130, 1793–1806 (2006). [DOI] [PubMed] [Google Scholar]
- 11. Kitamura, S. , Maeda, K. , Wang, Y. & Sugiyama, Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 36, 2014–2023 (2008). [DOI] [PubMed] [Google Scholar]
- 12. Huang, L. , Wang, Y. & Grimm, S. ATP‐dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab. Dispos. 34, 738–742 (2006). [DOI] [PubMed] [Google Scholar]
- 13. Simonson, S.G. et al Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin. Pharmacol. Ther. 76, 167–177 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Prueksaritanont, T. et al Pitavastatin is a more sensitive and selective organic anion‐transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schneck, D.W. et al The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin. Pharmacol. Ther. 75, 455–463 (2004). [DOI] [PubMed] [Google Scholar]
- 16. Tsamandouras, N. , Rostami‐Hodjegan, A. & Aarons, L. Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 79, 48–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jamei, M. et al A mechanistic framework for in vitro‐in vivo extrapolation of liver membrane transporters: prediction of drug‐drug interaction between rosuvastatin and cyclosporine. Clin. Pharmacokinet. 53, 73–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taskar, K. et al PBPK models for evaluating membrane transporter mediated DDIs: current capabilities, case studies, future opportunities and recommendations. Clin. Pharmacol. Ther. 107, 1081–1115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen, Y. et al Physiologically‐based pharmacokinetic model‐informed drug development for fenebrutinib: understanding complex drug‐drug interactions. CPT Pharmacometrics Syst. Pharmacol. 9, 332–341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grimstein, M. et al Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J. Pharm. Sci. 108, 21–25 (2019). [DOI] [PubMed] [Google Scholar]
- 21. Chen, Y. , Jin, J.Y. , Mukadam, S. , Malhi, V. & Kenny, J.R. Application of IVIVE and PBPK modeling in prospective prediction of clinical pharmacokinetics: strategy and approach during the drug discovery phase with four case studies. Biopharm. Drug Dispos. 33, 85–98 (2012). [DOI] [PubMed] [Google Scholar]
- 22. Emami Riedmaier, A. , Burt, H. , Abduljalil, K. & Neuhoff, S. More power to OATP1B1: an evaluation of sample size in pharmacogenetic studies using a rosuvastatin PBPK model for intestinal, hepatic, and renal transporter‐mediated clearance. J. Clin. Pharmacol. 56, S132–S142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang, Q. , Zheng, M. & Leil, T. Investigating transporter‐mediated drug‐drug interactions using a physiologically based pharmacokinetic model of rosuvastatin. CPT Pharmacometrics Syst. Pharmacol. 6, 228–238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan, J.C.Y. , Tan, S.P.F. , Upton, Z. & Chan, E.C.Y. BottoM‐up physiologically‐based biokinetic modelling as an alternative to animal testing. Altex 36, 597–612 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Harwood et al, Harwood preparation. Simcyp compound file. Version 19 (2020).
- 26. Jamei, M. et al Population‐based mechanistic prediction of oral drug absorption. AAPS J. 11, 225–237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li, J. et al Use of transporter knockdown Caco‐2 cells to investigate the in vitro efflux of statin drugs. Drug Metab. Dispos. 39, 1196–1202 (2011). [DOI] [PubMed] [Google Scholar]
- 28. Li, J. , Wang, Y. , Zhang, W. , Huang, Y. , Hein, K. & Hidalgo, I.J. The role of a basolateral transporter in rosuvastatin transport and its interplay with apical breast cancer resistance protein in polarized cell monolayer systems. Drug Metab. Dispos. 40, 2102–2108 (2012). [DOI] [PubMed] [Google Scholar]
- 29. Volpe, D.A. Variability in Caco‐2 and MDCK cell‐based intestinal permeability assays. J. Pharm. Sci. 97, 712–725 (2007). [DOI] [PubMed] [Google Scholar]
- 30. Larregieu, C.A. & Benet, L.Z. Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco‐2 as a surrogate for human intestinal permeability measurements. AAPS J. 15, 483–497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riccardi, K.A. et al A novel unified approach to predict human hepatic clearance for both enzyme‐ and transporter‐mediated mechanisms using suspended human hepatocytes. Drug Metab. Dispos. 47, 484–492 (2019). [DOI] [PubMed] [Google Scholar]
- 32. Di, L. et al Development of a new permeability assay using low‐efflux MDCKII cells. J. Pharm. Sci. 100, 4974–4985 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Keskitalo, J.E. , Zolk, O. , Fromm, M.F. , Kurkinen, K.J. , Neuvonen, P.J. & Niemi, M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 86, 197–203 (2009). [DOI] [PubMed] [Google Scholar]
- 34. Harwood, M.D. , Zhang, M. , Pathak, S.M. & Neuhoff, S. The regional‐specific relative and absolute expression of gut transporters in adult caucasians: a meta‐analysis. Drug Metab. Dispos. 47, 854–864 (2019). [DOI] [PubMed] [Google Scholar]
- 35. Ballatori, N. Biology of a novel organic solute and steroid transporter, OSTalpha‐OSTbeta. Exp. Biol. Med. 230, 689–698 (2005). [DOI] [PubMed] [Google Scholar]
- 36. Jamei, M. et al The Simcyp population based simulator: architecture, implementation, and quality assurance. Silico Pharmacol. 1, 9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cooper, K.J. , Martin, P.D. , Dane, A.L. , Warwick, M.J. , Raza, A. & Schneck, D.W. The effect of erythromycin on the pharmacokinetics of rosuvastatin. Eur. J. Clin. Pharmacol. 59, 51–56 (2003). [DOI] [PubMed] [Google Scholar]
- 38. Cooper, K.J. , Martin, P.D. , Dane, A.L. , Warwick, M.J. , Raza, A. & Schneck, D.W. Lack of effect of ketoconazole on the pharmacokinetics of rosuvastatin in healthy subjects. Br. J. Clin. Pharmacol. 55, 94–99 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodgers, T. & Rowland, M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm. Res. 24, 918–933 (2007). [DOI] [PubMed] [Google Scholar]
- 40. Grover, A. & Benet, L.Z. Effects of drug transporters on volume of distribution. AAPS J. 11, 250–261 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nezasa, K. , Takao, A. , Kimura, K. , Takaichi, M. , Inazawa, K. & Koike, M. Pharmacokinetics and disposition of rosuvastatin, a new 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor, in rat. Xenobiotica 32, 715–727 (2002). [DOI] [PubMed] [Google Scholar]
- 42. Fujino, H. , Saito, T. , Tsunenari, Y. , Kojima, J. & Sakaeda, T. Metabolic properties of the acid and lactone forms of HMG‐CoA reductase inhibitors. Xenobiotica 34, 961–971 (2004). [DOI] [PubMed] [Google Scholar]
- 43. Kotani, N. et al Culture period‐dependent changes in the uptake of transporter substrates in sandwich‐cultured rat and human hepatocytes. Drug Metab. Dispos. 39, 1503–1510 (2011). [DOI] [PubMed] [Google Scholar]
- 44. Abe, K. , Bridges, A.S. & Brouwer, K.L. Use of sandwich‐cultured human hepatocytes to predict biliary clearance of angiotensin II receptor blockers and HMG‐CoA reductase inhibitors. Drug Metab. Dispos. 37, 447–452 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bosgra, S. et al Predicting carrier‐mediated hepatic disposition of rosuvastatin in man by scaling from individual transfected cell‐lines in vitro using absolute transporter protein quantification and PBPK modeling. Eur. J. Pharm. Sci. 65, 156–166 (2014). [DOI] [PubMed] [Google Scholar]
- 46. Kunze, A. , Huwyler, J. , Camenisch, G. & Poller, B. Prediction of organic anion‐transporting polypeptide 1B1‐ and 1B3‐mediated hepatic uptake of statins based on transporter protein expression and activity data. Drug Metab. Dispos. 42, 1514–1521 (2014). [DOI] [PubMed] [Google Scholar]
- 47. Pfeifer, N. , Yang, K. & Brouwer, K.L.R. Hepatic basolateral efflux contributes significantly to rosuvastatin disposition I: characterization of basolateral versus biliary clearance using a novel protocol in sandwich‐cultured hepatocytes. J. Pharmacol. Exp. Ther. 347, 727–736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bi, Y.A. et al Quantitative assessment of the contribution of sodiuM‐dependent taurocholate co‐transporting polypeptide (NTCP) to the hepatic uptake of rosuvastatin, pitavastatin, and fluvastatin. Biopharm. Drug Dispos. 34, 452–461 (2013). [DOI] [PubMed] [Google Scholar]
- 49. Izumi, S. et al Relative activity factor (RAF)‐based scaling of uptake clearance mediated by organic anion transporting polypeptide (OATP) 1B1 and OATP1B3 in human hepatocytes. Mol. Pharmaceutics. 15, 2277–2288 (2018). [DOI] [PubMed] [Google Scholar]
- 50. Jones, H.M. et al Mechanistic pharmacokinetic modeling for the prediction of transporter‐mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab. Dispos. 40, 1007–1017 (2012). [DOI] [PubMed] [Google Scholar]
- 51. Li, N. , Bi, Y.A. , Duignan, D.B. & Lai, Y. Quantitative expression profile of hepatobiliary transporters in sandwich cultured rat and human hepatocytes. Mol. Pharm. 6, 1180–1189 (2009). [DOI] [PubMed] [Google Scholar]
- 52. Burt, H.J. , Emami Riedmaier, A. , Harwood, M.D. , Kim Crewe, H. , Gill, K.L. & Neuhoff, S. Abundance of hepatic transporters in caucasians: a meta‐analysis. Drug Metab. Dispos. 44, 1550–1561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bi, Y.A. et al Quantitative contribution of six major transporters to the hepatic uptake of drugs: “SLC‐Phenotyping: using primary human hepatocytes. J. Pharmacol. Exp. Ther. 370, 72–83 (2019). [DOI] [PubMed] [Google Scholar]
- 54. Neuhoff, S. et al Accounting for transporters in renal clearance: toward a mechanistic kidney model (Mech KiM) In Transporters in Drug Development Vol. 7 (eds. Sugiyama Y. & Steffansen B.) AAPS Advances in the Pharmaceutical Sciences Series (Springer, New York, NY, 2013). [Google Scholar]
- 55. Verhulst, A. , Sayer, R. , De Broe, M.E. , D‐Haese, P.C. & Brown, C.D.A. Human proximal tubular epithelium actively secretes but does not retain rosuvastatin. Mol. Pharmacol. 74, 1084–1091 (2008). [DOI] [PubMed] [Google Scholar]
- 56. Windass, A.S. , Lowes, S. , Wang, Y. & Brown, C.D. The contribution of organic anion transporters OAT1 and OAT3 to the renal uptake of rosuvastatin. J. Pharmacol. Exp. Ther. 322, 1221–1227 (2007). [DOI] [PubMed] [Google Scholar]
- 57. Varma, M.V. et al pH‐sensitive interaction of HMG‐CoA reductase inhibitors (statins) with organic anion transporting polypeptide 2B1. Mol. Pharm. 8, 1303–1313 (2011). [DOI] [PubMed] [Google Scholar]
- 58. Kobayashi, D. , Nozawa, T. , Imai, K. , Nezu, J. , Tsuji, A. & Tamai, I. Involvement of human organic anion transporting polypeptide OATP‐B (SLC21A9) in pH‐dependent transport across intestinal apical membrane. J. Pharmacol. Exp. Ther. 306, 703–708 (2003). [DOI] [PubMed] [Google Scholar]
- 59. Keiser, M. et al The organic anion‐transporting peptide 2B1 is localized in the basolateral membrane of the human jejunum and Caco‐2 monolayers. J. Pharm. Sci. 106, 2657–2663 (2017). [DOI] [PubMed] [Google Scholar]
- 60. Wegler, C. et al Variability in mass spectrometry‐based quantification of clinically relevant drug transporters and drug metabolizing enzymes. Mol. Pharm. 5, 3142–3151 (2017). [DOI] [PubMed] [Google Scholar]
- 61. Chen, B. et al Strategies of drug transporter quantitation by LC‐MS: importance of peptide selection and digestion efficiency. AAPS J. 19, 1469–1478 (2017). [DOI] [PubMed] [Google Scholar]
- 62. Borst, P. , de Wolf, C. & van de Wetering, K. Multidrug resistance‐associated proteins 3, 4 and 5. Pflugers Arch. 453, 661–673 (2007). [DOI] [PubMed] [Google Scholar]
- 63. Bajaj, P. , Chowdhury, S.K. , Yucha, R. , Kelly, E.J. & Guangqing, X. Emerging kidney models to investigate metabolism, transport, and toxicity of drugs and xenobiotics. Drug Metab. Dispos. 46, 1692–1702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vildhede, A. et al Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug‐drug interactions. Drug Metab. Dispos. 42, 1210–1218 (2014). [DOI] [PubMed] [Google Scholar]
- 65. Pahwa, S. et al Pretreatment with rifampicin and tyrosine kinase inhibitor dasatinib potentiates the inhibitory effects toward OATP1B1‐ and OATP1B3‐mediated transport. J. Pharm. Sci. 106, 2123–2135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Varma, M.V. , Lai, Y. , Feng, B. , Litchfield, J. , Goosen, T.C. & Bergman, A. Physiologically based modeling of pravastatin transporter‐ mediated hepatobiliary disposition and drug‐ drug interactions. Pharm. Res. 29, 2860–2873 (2012). [DOI] [PubMed] [Google Scholar]
- 67. Tatrai, P. et al A systematic in vitro investigation of the inhibitor preincubation effect on multiple classes of clinically relevant transporters. Drug Metab. Dispos. 47, 768–778 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Table S2
Table S3