

Abstract
Selection of a personalized dose for an individual patient can be informed by the patient’s preferences, translated as weights on each of the clinically relevant safety and efficacy drug attributes, based on results from a brief patient preference elicitation questionnaire. In this analysis, the weighted attributes were simulated to represent various endometriosis patient profiles. Exposure‐response simulations were performed for elagolix, a drug approved for management of moderate to severe pain associated with endometriosis, across a range of plasma exposures corresponding to a range of doses. The results were combined to calculate a personalized clinical utility index. An interactive user‐friendly online application was developed and envisioned as a physician’s desk tool to personalize the dose selection process based on individual patient preferences. This demonstration should serve as an example of how patient/physician conversation can be facilitated with quantitative tools for personalizing the dose.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Clinical utility index (CUI) calculations have been suggested and used to evaluate the risk‐benefit assessment of new drugs, in a quantitative manner, based on multiple safety and efficacy attributes.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Could patient preferences influence the CUI of drugs and enable personalized medicine?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Patient preferences from a discrete choice experiment study were quantitatively integrated in an exposure‐response modeling and simulation framework of elagolix phase III endometriosis data, to predict a CUI dose. The predicted CUI dose was personalized by introducing various weights on each safety and efficacy end point, derived from patient preferences data. This study is a demonstration of interdisciplinary model‐informed drug development in personalized medicine.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This work integrates the patient perspective into the quantitative process of dose selection and offers a novel approach to the assessment of benefit‐risk and personalized dosing at various stages of drug development.
Clinical utility index (CUI) calculations are a valuable tool for evaluating the benefit‐risk balance of a drug over a range of doses or concentrations. By utilizing multiple efficacy and safety attributes of a drug, CUI calculations can be used to assess new drugs or characterize a patient’s response to therapy in a quantitative manner. Currently, the primary uses of CUI calculations are to inform dose selection during drug development from early trial results (i.e., phase I or II) or for supporting the selection of a fixed dosing regimen. 1 , 2 , 3 , 4 To our knowledge, CUI has not been applied for calculation of individual patient utility to personalize dosing. In the era of personalized medicine, however, the best clinical utility of a drug for a patient should ideally be informed by the patient’s preferences for treating their disease or condition. For practical use of the CUI calculations in clinical practice, the proposed optimal treatment option may be provided by an interactive online application that can easily be used in a physician‐patient conversation.
For this concept, elagolix was used as an example of a drug with safety and efficacy attributes that can be integrated with patient preferences and quantitative modeling and simulation. Elagolix (Orilissa) was approved in 2018 by the US Food and Drug Administration for the management of moderate‐to‐severe pain associated with endometriosis. The approved dosages are 150 mg once‐daily (q.d.) for a maximum treatment duration of 24 months and 200 mg twice‐daily (b.i.d.) for a maximum treatment duration of 6 months, to provide flexibility for treating patients with endometriosis based on the severity of symptoms. 5 A clinical pharmacology overview of elagolix by Shebley et al. underscored the utility of plasma exposure‐response modeling in supporting the approved dosages of elagolix. 6 Population pharmacokinetics (PK) modeling by Winzenborg et al. 7 described factors that influence elagolix PK and dosing. The exposure‐efficacy modeling for the study primary pain end points dysmenorrhea (DYSM) responder rates and nonmenstrual pelvic pain (NMPP) responder rates by Winzenborg et al. 8 was able to describe the phase III results with reasonable accuracy. Further, the exposure‐safety modeling of bone mineral density (BMD) loss 9 and incidence of hot flashes 10 were able to describe the dose and time‐dependent changes in BMD and the rates of hot flash occurrence observed in phase III trials.
To integrate the available information from the exposure‐response modeling and demonstrate how patients’ preferences can inform a personalized clinical utility index (pCUI), exposure‐response simulations were developed for elagolix attributes of pain efficacy (DYSM, NMPP, and dyspareunia (DYSP)) and safety (BMD loss and incidence of hot flashes). To incorporate patients’ preferences in this study, the relative weighting of the safety and efficacy attributes of elagolix were derived from the results of a discrete choice experiment (DCE) study in endometriosis patients. The DCE quantified patient preferences for different attributes of treatments for endometriosis pain. 11 We then quantitatively integrated patient preferences from the DCE study into the exposure‐response modeling and simulation framework of elagolix to predict a CUI dose. Results were presented in a user‐friendly online application, which also allows users to specify individual patient preferences’ weights.
METHODS
DYSP model development
An exposure‐response model for clinical response of DYSP was developed using data from four phase III endometriosis clinical trials, as previously described. 8 The same Markov‐Chain discrete‐time model structure, as previously described for the DYSM and NMPP model, was applied. Covariates were evaluated for significance on transition probabilities or half‐maximal effective concentration/maximum effect (Emax) values. The model code and a dataset example are included in the Supplementary Material . Exposure‐response models for BMD changes and probability of experiencing hot flashed were as described from previous literature. 9 , 10
Utility simulation framework
A joint exposure‐response simulation framework for the three efficacy and two safety end points was implemented in Matlab R2015b (version 8.6.0). Dose administration was varied from 0 to 400 mg daily dose (in 10 mg dose increments) and simulations were performed for each dosing scenario for b.i.d. and q.d. dosing for 100 trials with 100 subjects each and for a treatment duration up to 24 months. The following assumptions and conventions were used to ensure relevant simulation results to the endometriosis patient population:
Patient demographics and baseline characteristics (baseline BMD, baseline BMD Z‐scores, race, and age) were simulated to resemble those of all subjects screened for enrollment in the phase III studies as a representative sample of the general endometriosis patient population. On the other hand, baseline body mass index, OATP1B1 genotype status, and baseline pain scores (DYSM, NMPP, and DYSP) were based on the observed distribution in the enrolled subjects, because these data were not available at screening.
Elagolix average concentrations as inputs to the safety and efficacy models were sampled based on the respective dosing scenario assuming full dosing compliance and the estimated elagolix clearance distribution as previously reported. 7
Simulation results were first summarized by replicate, month, and end point. The percentage responders for DYSM, NMPP, DYSP, percentage subjects not experiencing moderate or severe hot flashes, and the number of subjects having < 1% loss in BMD relative to baseline was calculated per replicate. Afterward, the mean end point result across replicates was calculated per daily dose group and per treatment duration.
The mean response rates per daily dose and per end point for a specific treatment duration were the entry point for the CUI calculation. The combined CUI U given specific weights w 1, …, w N per end point EP 1, …, EP N and per dose group d is calculated as:
| (1) |
R Shiny application for interactive utility predictions
The interactive application is based on the R Shiny technology from R Studio and programmed in R version 3.5.2. The above‐mentioned mean response rates per daily dose, treatment duration, and end point were stored as a lookup table for the interactive application to access. This allows the application to calculate the CUI in real time given the user‐specified weights. Information about the individual end point average response rates is given in addition to the CUI prediction. The R Shiny code is included in the Supplementary Material .
Discrete choice experiment survey
An online DCE survey was administered in 2017 to 250 women with self‐reported endometriosis diagnosis who experienced moderate‐to‐severe pain associated with endometriosis. The analysis outcome was estimated weights for each level of the treatment attributes (severity of DYSM, NMPP, DYSP, risk of experiencing moderate to severe hot flashes, mode of administration, and risk of adverse pregnancy outcomes or bone fracture) corresponding to the level of importance that patients put on each attribute level. Detailed methods are listed in ref. 11
The estimated weights from the survey were then used as input weights in Eq. 1 to inform the CUI from a patient’s perspective. As fracture risk was not directly modeled, the number of subjects experiencing > 1% BMD loss per year was used as surrogate for this end point.
RESULTS
Elagolix population PK and exposure‐response modeling
Previously described population PK and exposure‐response models for the primary elagolix‐efficacy and elagolix‐safety end points served as basis for the pCUI simulations.
An elagolix population PK model was developed based on data from four phase III clinical trials in patients with endometriosis and five phase I studies in healthy volunteers. The elagolix PK was best described by a two‐compartment model with OATP1B1 genotype status as covariate on apparent clearance as the only significant covariate. Details of the population PK analysis were previously described in ref. 7 and summarized in ref. 6
In brief, the exposure‐response relationships of elagolix for efficacy and safety were developed based on data from four phase III studies with placebo treatment up to 6 months and active drug treatment up to 12 months. Clinical efficacy responses for DYSM and NMPP were adequately described over time by discrete‐time first‐order Markov‐Chain models, which had states for nonresponse, response, and considered dropout for each state. 8 Baseline DYSM and NMPP scores were identified as significant covariates on respective placebo transition probabilities, with higher placebo response in subjects with higher baseline disease scores. Additionally, age was significant on the half‐maximal effective concentration parameter estimate in the NMPP model part leading to older age patients having better response rates for NMPP. Modeling results are summarized in ref. 6 and described in detail in ref. 8, including the rationale of selecting the Markov‐Chain model to describe the efficacy end points.
An additional exposure‐response model for a secondary clinical response of DYSP was developed using data from four phase III and five phase I studies, as described previously 8 for the primary efficacy end points DYSM and NMPP. Consistent with previous modeling results for DYSM and NMPP, baseline DYSP score was identified as significant covariate on placebo transition probability and age was identified as having a small but statistically significant influence on Emax parameters. The impact of age on DYSP maximum response in our model can be interpreted as patients with older age having a 5% better improvement of response (from the lowest age quartile from 18–27 years to the oldest age quartile from 36–49 years). The final model described the observed DYSP responder rates for the placebo group up to 6 months and the two active treatment groups (elagolix 150 mg q.d. and 200 mg b.i.d.) up to 12 months (Figure 1 ).
Figure 1.
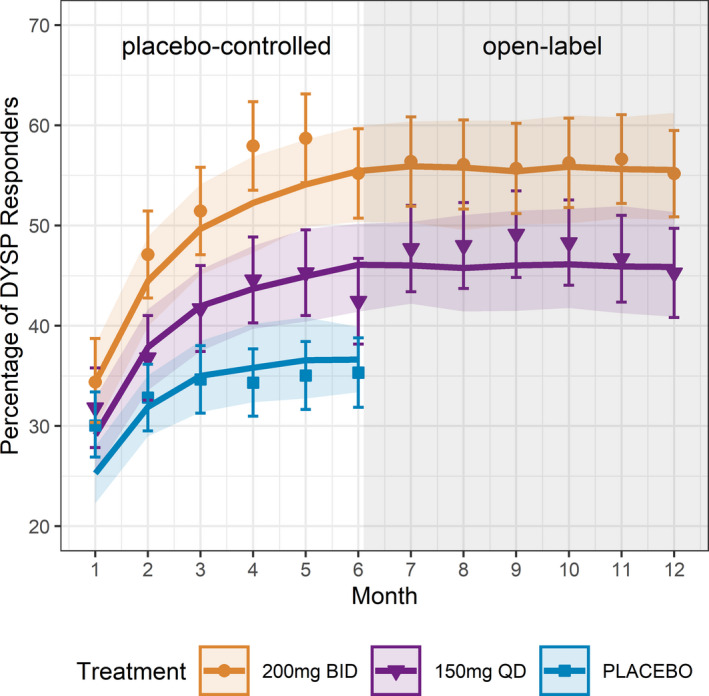
Graphical representation of the goodness of fit for the dyspareunia (DYSP) exposure‐response Markov‐Chain model.
For safety end points, the previously developed indirect response model described the effect of elagolix exposure on the hypoestrogenic adverse effect of loss in lumbar spine BMD over time using data from the endometriosis phase III clinical trials. 9 In addition, a logistic regression model was developed to describe the effect of elagolix exposure on the probability of experiencing moderate‐to‐severe hot flashes during treatment. 10 Both models were used without modification and combined with the exposure‐efficacy models to conduct simulations. Efficacy and safety simulations were combined for the development of the pCUI. A schematic illustration of the modeling concept is shown in Figure S1 and described each component of the overall pCUI approach.
Clinical utility index simulation
The exposure‐response models were used to simulate efficacy and safety responses for a range of elagolix exposures that correspond to doses up to 400 mg total daily dose. The benefit‐risk balance for a given dose of elagolix is inferred from the model‐predicted responses represented as a CUI prediction, with patient preference data implemented as relative weights 11 on each response end point to calculate a pCUI that corresponds to an individualized dose. Safety end point presentation is inverted for the pCUI calculation and the corresponding visualizations (i.e., probability of nonoccurrence rather than occurrence of the respective safety event is shown), in order to show increasing input for efficacy and decreasing input for safety end points on the utility for increasing dose amounts.
A user‐friendly online application was developed to allow specifying patient preferences as weights on each end point, and to conduct simulations of various scenarios to predict a pCUI dose. To enable easy access of the pCUI and for interactive use, an open source link was developed and provided for public access at the following link: https://abbviescience.shinyapps.io/Endometriosis_Utility_App/.
Figure 2a shows a screenshot of the pCUI app with options to implement weights for each safety and efficacy attribute based on individual patient preferences, as well as flexibility to select the duration of treatment. In this scenario, elagolix duration of treatment was fixed to 6 months according to the duration of the placebo‐controlled period (efficacy primary end point) of the phase III clinical trials. DYSM, NMPP, BMD loss, and incidence of hot flash end points were equally weighted to represent a clinical trial setting without considering patient preferences. DYSP weighting was set to zero because efficacy for this end point in the phase III trials was only observed with the higher elagolix dosage of 200 mg b.i.d. Under this scenario, the predicted pCUI dose was 150 mg q.d., an approved dosage for elagolix, where DYSP efficacy did not reach statistical significance. 5 Although statistical significance of the 150 mg q.d. for DYSP was not achieved at month 6, 12 the exposure‐response time‐course model for this end point suggests a possible benefit compared with placebo when the overall data are considered (Figure 1 ).
Figure 2.

pCUI app interface of a hypothetical scenario representing equal weighting of the primary efficacy end points (DYSM and NMPP) and safety end points (BMD loss and incidence of hot flash) (a) and CUI app interface of a scenario representing equal weighting of the primary efficacy end points (DYSM and NMPP), safety end points (BMD loss and incidence of hot flush), and higher weighting for a secondary efficacy end point DYSP, to account for patients with more severe symptoms (b). BMD, bone mineral density; CUI, clinical utility index; DYSM, dysmenorrhea; NMPP, nonmenstrual pelvic pain; pCUI, personalized clinical utility index.
Under another scenario where DYSP efficacy is desired for patients with more severe symptoms, DYSP weighting was increased to two. The predicted pCUI dose under this scenario is a total daily dose of 400 mg (or 200 mg b.i.d.; Figure 2b ), representing the approved elagolix 200 mg b.i.d. for treating patients with endometriosis associated DYSP. 5
CUI weighted simulations based on patients’ preferences
Results of a DCE survey conducted on 250 women with self‐reported endometriosis diagnosis who experienced moderate‐to‐severe pain associated with endometriosis, have shown that patients’ primary preference was for a medical treatment that does not induce risk of experiencing moderate‐to‐severe hot flashes, followed by preference for improvement in DYSM, NMPP, and DYSP severity levels, with relatively less concern for risk of adverse pregnancy outcomes, mode of administration, and finally risk of bone fracture later in life. 11
The reported ranges of preference weights from the DCE study were 3.66–3.58 for risk of moderate‐to‐severe hot flashes (depending on prior experience of moderate‐to‐severe hot flashes), 1.70 for improvement in DYSP, 1.49 for improvement in NMPP, 1.48 for improvement in DYSM, and 0.49 for increased risk of bone fracture later in life.
These observed preference weights for each treatment attribute were directly entered into the pCUI app, as shown in Figure 3 . Under this scenario of patient preferences for hypothetical medical treatment attributes, the predicted pCUI total daily dose of elagolix was 230 mg, representing a hypothetical dose that achieves balanced safety and efficacy influenced by the patient perspective. This hypothetical dose predicted by pCUI simulations is still within the range of approved elagolix dosages (150 mg q.d. and 200 mg), however, this is not an approved dose of elagolix for endometriosis‐associated pain.
Figure 3.
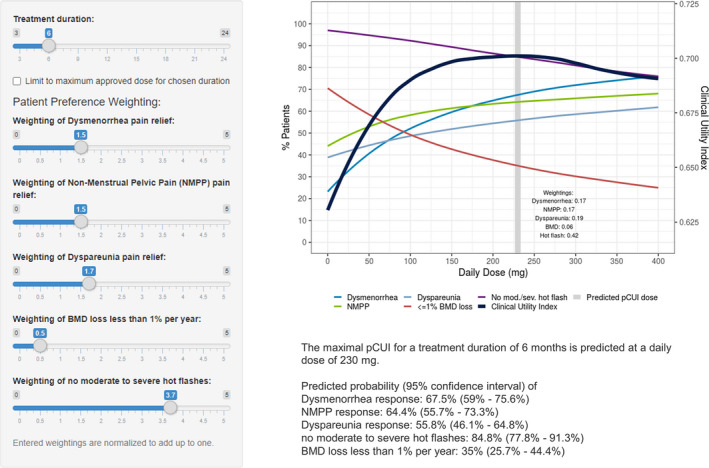
pCUI app interface for a hypothetical scenario representing weighted treatment attributes informed by results of the DCE study in patients with endometriosis. Equal weighting of primary efficacy DYSM and NMPP, relatively slightly higher weight for DYSP, relatively lower weight for not experiencing BMD loss, and highest weight for avoiding moderate‐to‐severe hot flashes. BMD, bone mineral density; CUI, clinical utility index; DCE, discrete choice experiment; DYSM, dysmenorrhea; DYSP, dyspareunia; NMPP, nonmenstrual pelvic pain; pCUI, personalized clinical utility index.
Personalized CUI simulations of hypothetical patient preferences
In addition to the clinical trial settings and the observed endometriosis patient population preferences from the DCE study, various hypothetical patient preference scenarios were simulated. Figure 4 describes multiple scenarios of virtual patients with personalized weightings. A longer treatment duration of 12 months was selected to represent a duration in between the approved 24 months for 150 mg q.d. and 6 months for 200 mg b.i.d. according to the elagolix US package insert. 5 For virtual patient 1, pain relief and nonoccurrence of moderate‐to‐severe hot flashes is more important than BMD loss, which results in predicted pCUI dose of 400 mg daily dose (e.g., 200 mg b.i.d.; Figure 4a ). Virtual patient 2 has higher preference and therefore higher weights for bone health and pain relief relative to occurrence of moderate‐to‐severe hot flashes. This hypothetical setting leads to a relatively flat pCUI profile, as can be seen from the small range of pCUI values in Figure 3b , where the 150 mg q.d. regimen has a slightly better utility than the 200 mg b.i.d. regimen. Given the flexibility and accessibility of the pCUI app, multiple other scenarios could be simulated to personalize dosing.
Figure 4.
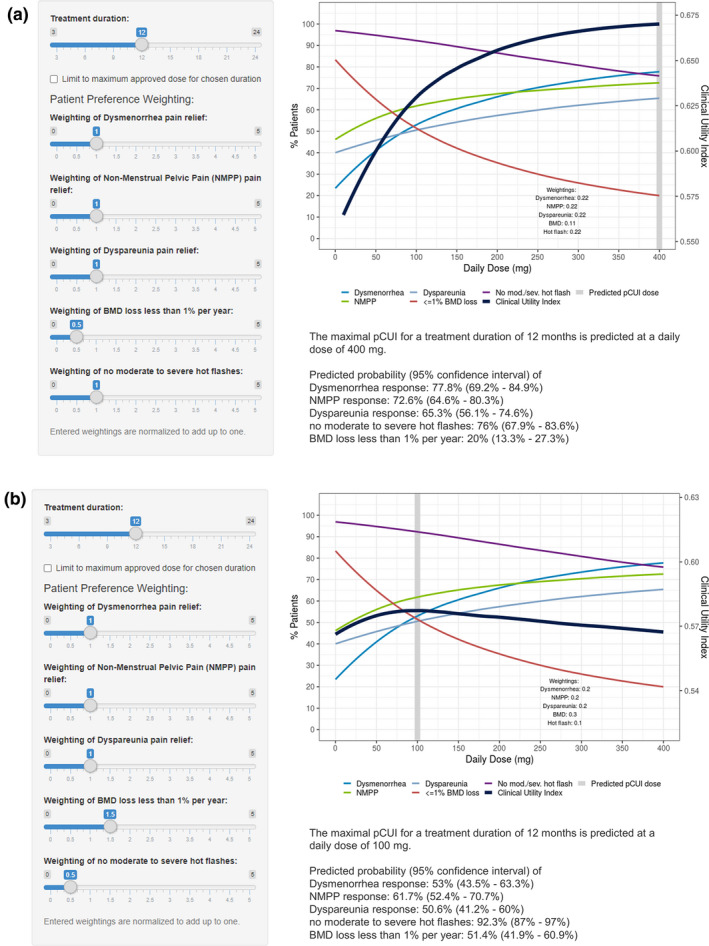
Pain relief and avoidance of hot flashes more important than BMD loss (a) and BMD loss and pain relief more important than avoidance hot flashes (b). BMD, bone mineral density; NMPP, nonmenstrual pelvic pain; pCUI, personalized clinical utility index.
DISCUSSION
Personalized medicine aims to individualize medical therapies based on the patient’s characteristics, with the potential to tailor therapy with the best response and highest safety margin to ensure better patient care. 13 Most personalized medicine approaches utilize genomics analysis to characterize patient’s response to therapy. In this work, a modeling and simulation approach for safety and efficacy attributes of elagolix, as an example, was combined with patient preferences for endometriosis medical treatment in an online user‐friendly app framework to personalize dosing. In the presented example with elagolix, the predicted CUI doses under the phase III clinical trials scenarios shown in Figure 2 support the approved dosages of elagolix 150 mg q.d. and 200 mg b.i.d. (represented as 400 mg total daily dose), and further support that the approved dosages are well‐suited to address the patients’ needs. For serving as a physician’s app, the application should be restricted to only provide dosing recommendation from the approved dosages and up to the approved dosing duration. Although elagolix was used in this work, it was selected to demonstrate the concept of applying patient preferences for a medical treatment by integrating clinical trials data, exposure‐response modeling and simulations, with patient preferences data, and not to specifically recommend the predicted pCUI doses for elagolix. This novel personalized medicine framework is envisioned as a quantitative tool to aid the physician’s selection of the dose for a given patient when a range of doses are available. The user‐friendly online interface was envisioned to facilitate the pCUI to be used as a physician’s desk tool during the physician‐patient conversation by selecting weights for each of the safety and efficacy attributes, to reflect the patient’s preference and physician’s art of practicing medicine simultaneously to personalize dosing. In addition, dose adjustments on later visits can be achieved based on the patient’s experience with the drug, which can be reflected with updated preferences to predict the new pCUI dose. This is consistent with the concept of “medication concordance” where a mutual agreement between the patient and healthcare provider is achieved on the basis of considering and respecting the preferences of the patient on whether, when, and how a medication is to be taken. 14 , 15
This application, however, is informative if a given drug has a diversity of clinically relevant end points, where dose and exposure response relationships are established. In the case of elagolix, dose and exposure‐dependent changes in hypoestrogenic effects (BMD loss and occurrence of hot flash) and endometriosis pain efficacy (DYSM and NMPP) have been reported and described using modeling approaches. 6 , 12 Therefore, improved patient satisfaction with the treatment may be possible if there is an opportunity to allow the physician and patient to individualize dosing using a quantitative tool, such as the presented pCUI. For a completed assessment using pCUI, the physician’s preference can also be incorporated in the weights for each safety and efficacy attribute to offer a balanced personalized dosing. In the case of elagolix, another discrete choice experiment study focusing on the physician’s preferences is currently ongoing and can be used in the future to include the physician’s perspective for a complete and balanced pCUI. This is not far from reality because existing approaches, such as the Clinical Decision Support System (CDSS), which has been in use since the early 1980s, and was envisioned as a way to improve healthcare delivery by enhancing medical decisions with targeted clinical knowledge, patient information, and other health information. 16 The CDSS includes software and other computerized and artificial intelligence tools that enable healthcare providers make informed decisions based on individual patient characteristics and clinical knowledge base to tailor care. Similar to the established CDSS tools, our proposed pCUI app allows for shared decision making between patient and provider and serves as an interactive tool to make patients more involved in their own care. 16
Another way to utilize this pCUI app is for internal decision making in early drug development (i.e., at the end of a phase II dose ranging study) to support the choice of dosing regimens and/or sample size to achieve a certain end point for a phase III study design, assuming DCE assessment of the patient preferences for the intended treatment is available. 17 , 18
This work is not without limitations. The pCUI application presented in this work is only a proof‐of‐concept and has not been used in a clinical setting, thus data are lacking regarding its applicability and use in the real‐world. In addition, the current pCUI application used BMD loss as one of the safety attributes, however, the DCE study question related to bone health was presented to patients as an increased risk of bone fracture later in life. 11 Although BMD loss is related to increased risk of bone fracture in postmenopausal women, the patient population for elagolix is premenopausal young women where changes in BMD are not expected to translate to a significantly increased risk of bone fractures. 19 , 20 To address the DCE results on patient preference for treatment that increased risk of bone fractures later in life, the modeled BMD loss due to treatment with elagolix was used as a conservative approach to increase the sensitivity of this safety end point on the predicted pCUI dose. A cutoff of BMD loss of at least 1% over 12 months was used to predict the percentage of patients and calculate the pCUI. This is considered an acceptable threshold based on the observed BMD changes with elagolix and the duration of therapy for each of the approved dosages. A potential extension to the current work would be to consider incorporating patient‐specific characteristics, such as demographics or baseline disease scores, for more specific pCUI predictions. In cases where dropout was observed to be significantly varying by drug exposure or treatment group, an additional factor to account for dropout could be considered in utility predictions. Additionally, dosing frequency was not considered in our pCUI analysis, because for most end points, dosing frequency did not have a significant effect. For the DYSM efficacy model, a drug‐dependent effect was observed that could not be described by average concentration alone, but it remained unclear whether this was related to dosing frequency as the daily doses were very different between the q.d. (150 mg daily dose) and the b.i.d. (400 mg daily dose) dosing regimen. Thus, this drug‐dependent effect for the DYSM model was averaged in the utility prediction.
Overall, the presented work provides a user‐friendly quantitative framework online application that combines exposure‐response modeling with patients’ preference data to inform specific weighting of each modeled drug attribute, and to personalize dosing based on the predicted clinical utility index.
Funding
This work was supported by AbbVie. AbbVie contributed to the study design, research, and interpretation of the data, and the writing, review, and approval of the manuscript.
Conflict of Interest
All authors are employees of AbbVie and may hold AbbVie stock or stock options.
Author Contributions
I.W., A.M.S., and M.S. wrote the manuscript. I.W., A.M.S., and M.S. designed the research. I.W., A.M.S., and M.S. performed the research. I.W., A.M.S., and M.S. analyzed the data.
Supporting information
Fig S1
Supplementary Material
Supplementary Material
Acknowledgments
Medical writing support was provided by Wesley Wayman, an AbbVie employee.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.
References
- 1. Ouellet, D. , Werth, J. , Parekh, N. , Feltner, D. , McCarthy, B. & Lalonde, R.L. The use of a clinical utility index to compare insomnia compounds: a quantitative basis for benefit‐risk assessment. Clin. Pharmacol. Ther. 85, 277–282 (2009). [DOI] [PubMed] [Google Scholar]
- 2. Ouellet, D. Benefit‐risk assessment: the use of clinical utility index. Expert Opin. Drug Saf. 9, 289–300 (2010). [DOI] [PubMed] [Google Scholar]
- 3. Holmes, E.A.F. et al Patient‐focused drug development methods for benefit‐risk assessments: a case study using a discrete choice experiment for antiepileptic drugs. Clin. Pharmacol. Ther. 105, 672–683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Poland, B. et al The clinical utility index as a practical multiattribute approach to drug development decisions. Clin. Pharmacol. Ther. 86, 105–108 (2009). [DOI] [PubMed] [Google Scholar]
- 5. AbbVie Inc . Orilissa™ (elagolix) [US package insert] (AbbVie Inc., North Chicago, IL, 2018). [Google Scholar]
- 6. Shebley, M. et al Clinical pharmacology of elagolix: an oral gonadotropin‐releasing hormone receptor antagonist for endometriosis. Clin. Pharmacokinet. 59, 297–309 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winzenborg, I. et al Population pharmacokinetics of elagolix in healthy women and women with endometriosis. Clin. Pharmacokinet. 57, 1295–1306 (2018). [DOI] [PubMed] [Google Scholar]
- 8. Winzenborg, I. , Polepally, A.R. , Nader, A. , Mostafa, N.M. , Noertersheuser, P. & Ng, J. Effect of elagolix exposure on clinical efficacy end points in phase III trials in women with endometriosis‐associated pain: an application of Markov model. CPT Pharmacometrics Syst. Pharmacol. 9, 466–475 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suleiman, A. et al Exposure‐safety analyses identify predictors of change in bone mineral density and support elagolix labeling for endometriosis‐associated pain. CPT Pharmacometrics Syst. Pharmacol. 10.1002/psp4.12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nader, A. et al Combined efficacy and safety modeling and simulation approach to inform drug development decisions for an oral gonadotropin‐releasing hormone (GnRH) receptor antagonist. J. Pharmacokinet. Pharmacodyn. 45(suppl. 1), S4 (2018). [Google Scholar]
- 11. Poulos, C. , Soliman, A.M. , Renz, C.L. , Posner, J. & Agarwal, S.K. Patient preferences for endometriosis pain treatments in the United States. Value Health 22, 728–738 (2019). [DOI] [PubMed] [Google Scholar]
- 12. Taylor, H.S. et al Treatment of endometriosis‐associated pain with elagolix, an oral GnRH antagonist. N. Engl. J. Med. 377, 28–40 (2017). [DOI] [PubMed] [Google Scholar]
- 13. Vogenberg, F.R. , Isaacson Barash, C. & Pursel, M. Personalized medicine: part 1: evolution and development into theranostics. P T 35, 560–576 (2010). [PMC free article] [PubMed] [Google Scholar]
- 14. Dickinson, D. , Wilkie, P. & Harris, M. Taking medicines: concordance is not compliance. BMJ 319, 787 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Settineri, S. , Frisone, F. , Merlo, E.M. , Geraci, D. & Martino, G. Compliance, adherence, concordance, empowerment, and self‐management: five words to manifest a relational maladjustment in diabetes. J. Multidiscip. Healthc. 12, 299–314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sutton, R.T. , Pincock, D. , Baumgart, D.C. , Sadowski, D.C. , Fedorak, R.N. & Kroeker, K.I. An overview of clinical decision support systems: benefits, risks, and strategies for success. NPJ Digit. Med. 3, 17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Overbeeke, E. et al Design, conduct, and use of patient preference studies in the medical product life cycle: a multi‐method study. Front. Pharmacol. 10, 1395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cook, N.S. , Cave, J. & Holtorf, A.P. Patient preference studies during early drug development: aligning stakeholders to ensure development plans meet patient needs. Front. Med. (Lausanne) 6, 82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Binkley, N. , Besuyen, R. , Fuerst, T. , Skillern, L. & Hans, D. Is drug‐induced bone loss acceptable in premenopausal women? A practical fracture risk modeling exercise. Osteoporos. Int. 28, 3501–3513 (2017). [DOI] [PubMed] [Google Scholar]
- 20. Chiuve, S.E. , Peloso, P. , Chand, D. , Patwardhan, M. , Snabes, M. & Kilpatrick, R. Risk factors for low bone mineral density in premenopausal women with endometriosis in the national health and nutrition examination survey (NHANES). Fertil. Steril. 110, e384–e385 (2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Supplementary Material
Supplementary Material
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.