

Abstract
ADP-ribosylation is a post-translational modification generated by members of the superfamily of ADP-ribosyltransferases, known as the Parp enzymes. Depending on the superfamily member, Parp enzymes can conjugate a single ADP-ribose to a protein substrate, or extend the product and generate poly-ADP-ribose. Parp superfamily members confer regulation to a variety of biological processes that include cell signaling, DNA repair, transcription, and stress responses. Here, we describe biochemical methods for detection of ADP-ribose conjugated to the androgen receptor (AR) using the archaeal macrodomain, AF1521, from Archaeoglobus fulgidus. The utility of AF1521 is based on its highly selective recognition of ADP-ribose conjugated to protein. AF1521 immobilized on beads can be used to enrich for ADP-ribosylated proteins, which in our application results in recovery of ADP-ribosylated AR from prostate cancer cell extracts. We engineered tandem AF1521 macrodomains and found this improves the recovery of ADP-ribosylated AR under native conditions, and it enabled development of an assay for detection of ADP-ribosylation on blots. Thus, AF1521 can be used to query ADP-ribosylation of protein under both native and denaturing conditions. Our assays should prove useful for understanding how ADP-ribosylation regulates AR function.
Keywords: ADP-ribosylation, Androgen receptor, Post-translational modification, Parps, Prostate cancer, Macrodomain, AF1521
Introduction
The androgen receptor (AR) is a member of the nuclear receptor family and plays important physiological roles in male sexual differentiation and maturation. As a nuclear receptor, AR acts as a ligand-regulated transcription factor and has the shared domain structure of an N-terminal transactivation domain, a central DNA-binding domain, and a C-terminal ligand-binding domain [1]. Binding of the androgen ligand to AR induces a conformational change in the receptor that allows dimerization and nuclear import [2, 3]. Activated AR binds to androgen response elements to regulate expression of its target genes. As part of the regulation of AR signaling, AR undergoes a range of post-translational modifications including phosphorylation, acetylation, SUMOylation, ubiquitination, and methylation [4]. These modifications impact AR-dependent transcription output through various mechanisms, such as regulation of AR nuclear export, protein stability, and association with transcription co-activators.
AR is a key driver of prostate cancer (PCa), the second leading cause of cancer deaths among US men [5]. The reliance of PCa on the AR signaling pathway is targeted by androgen deprivation therapy where drugs compete with androgen binding to AR or reduce androgen synthesis, in both cases reducing AR signaling output [6]. While there is an initial therapeutic response to androgen deprivation therapy, resistance develops, and finding new ways to treat resistant forms of PCa is an important area of research. One promising approach is the use of Parp inhibitors in PCa. These drugs target Parps (also referred to as ADP-ribosyltransferases) which is a class of enzymes that use NAD+ as a donor molecule for generating ADP-ribose and catalyze mono- or poly-ADP-ribosylation of protein substrates [7]. The human genome encodes seventeen Parps, with Parp1 being the most well-studied member, especially in the context of DNA damage repair [8]. Parp inhibitors are efficacious in breast and ovarian cancer patients because of synthetic lethal interactions between loss-of-function mutations in the DNA repair gene BRCA1/2 and Parp1 inhibition [9]. This logic has been applied to PCa where a clinical trial showed that the use of the Parp inhibitor olaparib improves survival in patients harboring mutations in DNA damage repair genes [10]. While the focus has been on Parp inhibitor therapeutic effect from a DNA damage perspective, there is also pre-clinical data suggesting that Parp1 inhibition in PCa reduces AR-dependent transcription and impairs PCa growth [11]. This is in line with a growing appreciation that Parp-mediated ADP-ribosylation plays diverse roles beyond DNA damage repair, including transcription regulation, cellular stress response, cell signaling, and chromatin structure modulation [12].
Herein, we describe methods that we developed to assess whether AR is modified by ADP-ribosylation because of a recent precedent of nuclear hormone receptor ADP-ribosylation. Bindesbøll et al. found that the nuclear hormone receptors liver X receptor (LXR) α and β are ADP-ribosylated where this post-translational modification was shown to enhance LXR-dependent transcription activity [13]. Thus, we hypothesized that ADP-ribosylation of AR could represent yet another post-translational modification that regulates AR function.
We have taken advantage of the ADP-ribose binding property of the macrodomain protein AF1521 for our studies on AR ADP-ribosylation. The crystal structure for AF1521, a protein isolated from the bacteria Archaeoglobus fulgidus, displays a macrodomain which is a compact globular protein structure consisting of a mixed α/β fold [14, 15]. A binding cleft on the surface of AF1521 that tightly accommodates an ADP-ribose molecule is apparent on the solved structure with the ligand ADP-ribose bound [15]. The binding cleft contains a deep hydrophobic pocket where the adenine moiety of ADP-ribose fits in. Meanwhile, AF1521 makes specific hydrogen bond contacts with the phosphates and distal ribose ring of ADP-ribose. Macrodomains are found in viruses as well as organisms ranging from bacteria to humans, and residues that interact with ADP-ribose in the binding pocket are highly conserved, suggesting that this domain functions generally as a ADP-ribose recognition module [16]. Binding studies showed that AF1521 binds with a high affinity to ADP-ribose (KD = 0.13 μM), compared with ADP (KD = 5.63 μM) or NAD+ (KD > 100 μM) [15]. Because of these biochemical properties, AF1521 has been widely used in the Parp field for selective enrichment of ADP-ribosylated substrates in multiple proteomic studies [17–20]. Unlike with the study of other post-translational modifications where specific antibodies against the modifications exist, developing ADP-ribose-specific antibodies has been challenging because of the labile nature of the ADP-ribose-amino acid linkage for ADP-ribosylated antigens injected into antibody-producing animals [21]. Thus, AF1521 continues to be a useful tool for investigating protein ADP-ribosylation.
To examine the ADP-ribosylation status of AR, we utilized in a pulldown assay the highly selective ADP-ribose binding capacity of AF1521 (Fig. 1A). We expressed and purified GST-AF1521 from Escherichia coli and prepared glutathione-agarose beads coated with GST-AF1521 for a pulldown assay. Extract from androgen-treated cells was incubated with the prepared beads, and after bead washes, bound fraction was analyzed by SDS-PAGE and immunoblotting (Fig. 1B). We found that AR from androgen-treated cells is ADP-ribosylated as detected by AF1521 pulldown (Fig. 1C). The site of modification and the identity of the specific Parp mediating AR ADP-ribosylation are currently under investigation. The enrichment of ADP-ribosylated AR is not observed when extract from androgen-treated cells is incubated with mutant AF1521 in key residues critical for ADP-ribose recognition (Fig. 2).
Figure 1.
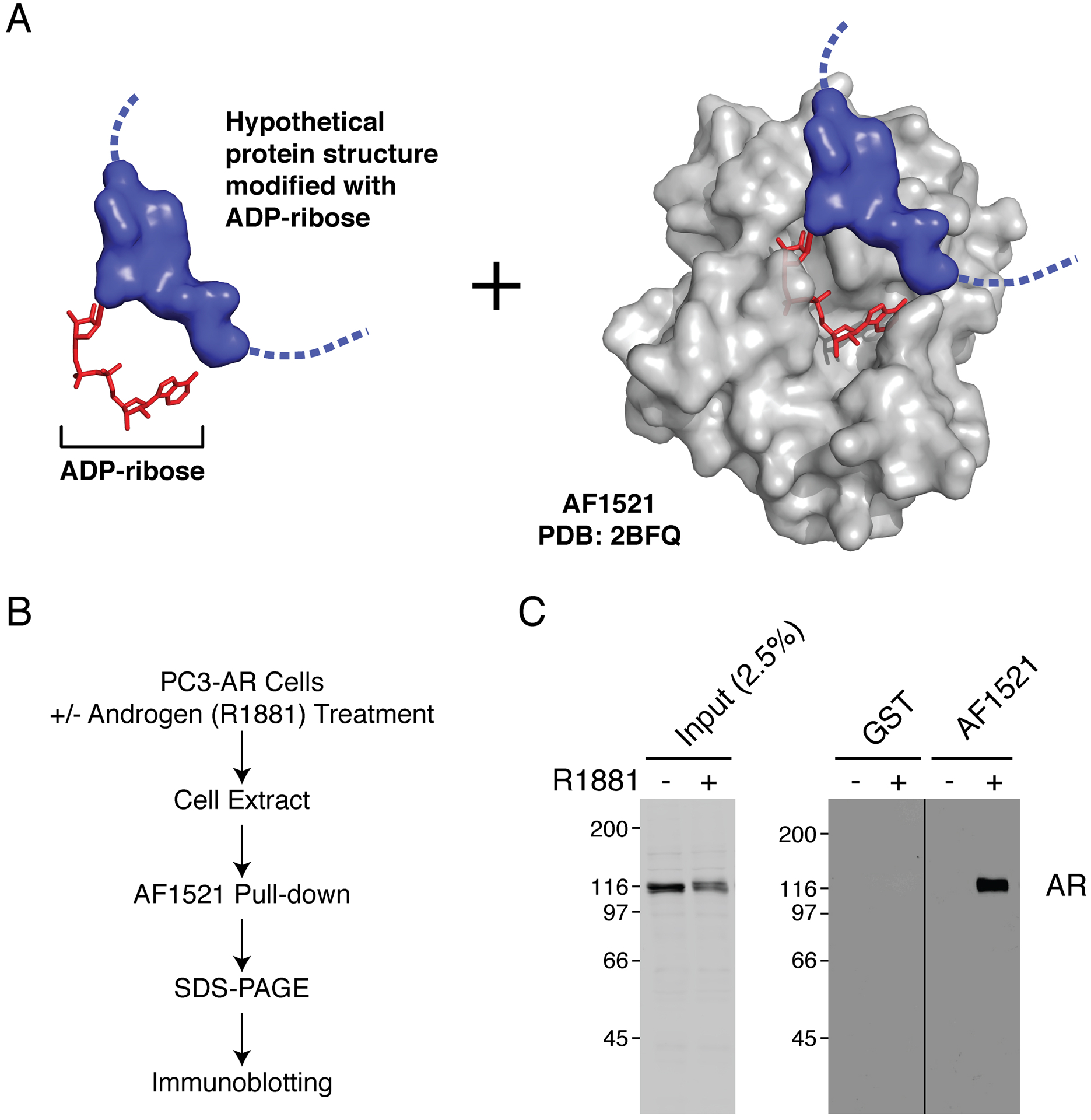
Immobilized AF1521 can selectively bind AR from extract of androgen-treated PC3-AR cells. A) Schematic of ADP-ribosylated hypothetical protein (blue) bound by AF1521 (gray, PDB: 2BFQ). B) Flowchart for AF1521 pulldown protocol. C) Cell extracts from androgen (R1881)- or vehicle-treated PC3-AR cells were analyzed by AF1521 pulldown.
Figure 2.
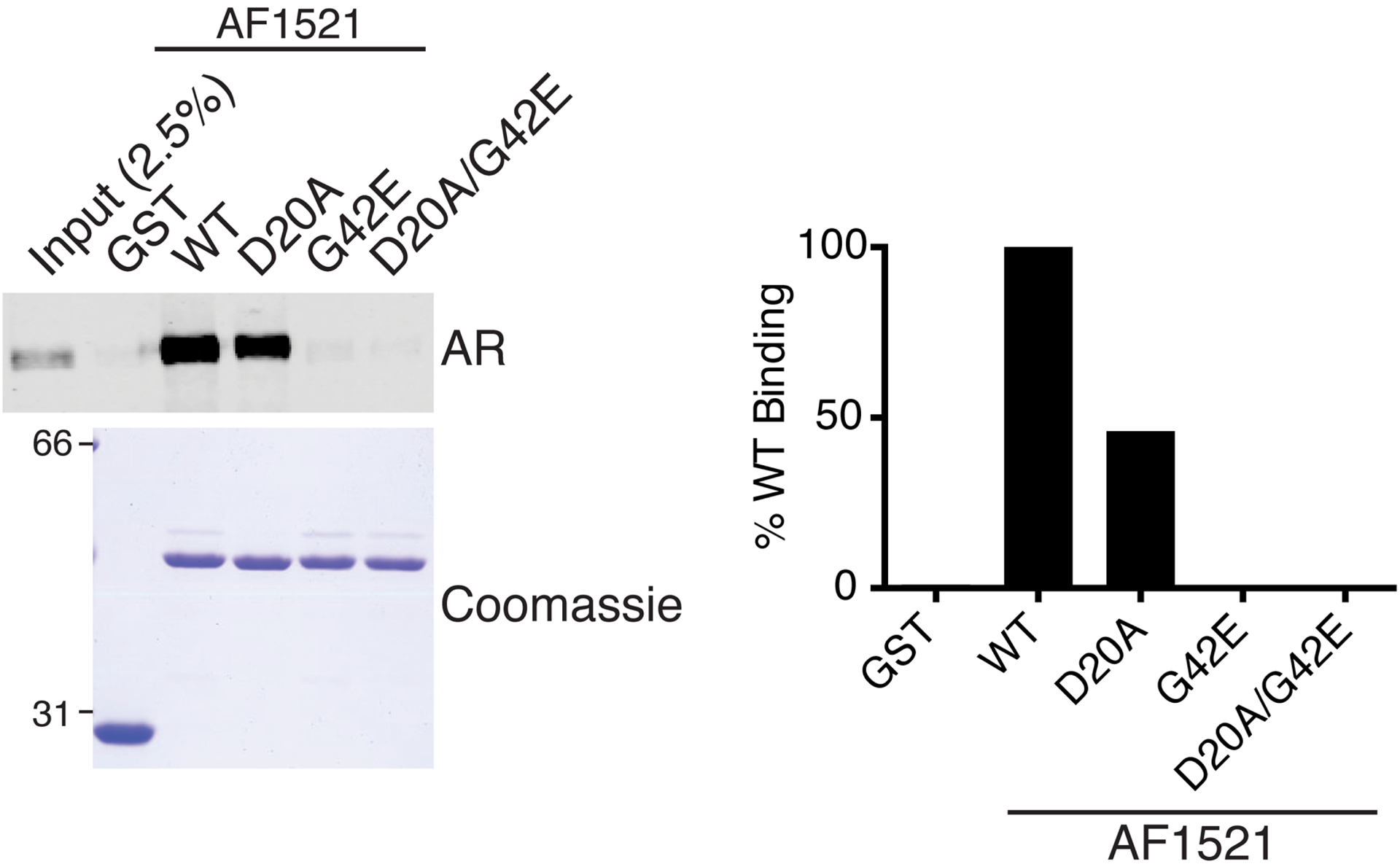
Loss-of function mutations for ADP-ribose binding abrogate AF1521 binding to AR. Extract from androgen (R1881)-treated PC3-AR cells were analyzed by pulldowns using glutathione-agarose beads coated with GST-AF1521 wildtype (WT) or indicated mutants. GST-coated glutathione-agarose beads served as negative control. Band intensities from AR blot were quantified as percent of AR bound by AF1521 WT.
We further explored the nature of the enrichment of AR by AF1521 pulldown using snake venom phosphodiesterase. This enzyme is a pyrophosphatase that cleaves between the two phosphates in ADP-ribose, generating AMP and ribose-5-phosphate [22]. Treatment of ADP-ribosylated protein substrate with snake venom phosphodiesterase would leave behind a phosphoribosylated protein that is no longer recognized by AF1521 (Fig. 3A). As expected, snake venom phosphodiesterase-treated samples showed no AR binding to AF1521 (Fig. 3B).
Figure 3.
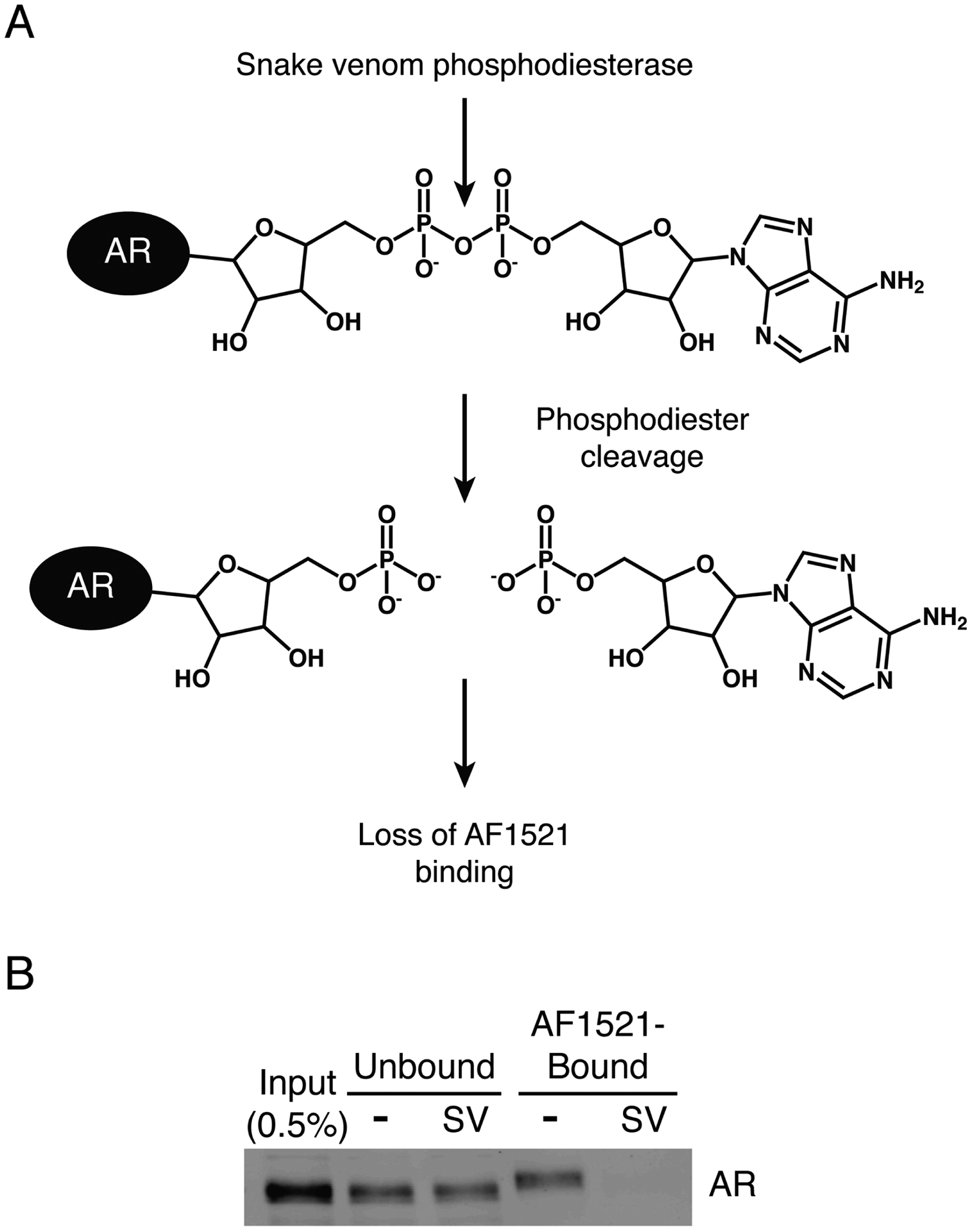
Phosphodiester cleavage of ADP-ribosylated AR by snake venom phosphodiesterase eliminates AR binding to AF1521. A) Schematic of snake venom phosphodiesterase cleavage of phosphodiester bond in ADP-ribosylated AR. B) Extract from androgen (R1881)-treated PC3-AR cells was treated with snake venom phosphodiesterase (SV) and analyzed by AF1521 pulldown.
We improved the AF1521 pulldown assay by generating a recombinant protein with two AF1521 fused in tandem (GST-AF1521tandem) to increase avidity for ADP-ribosylated target proteins. Using glutathione-agarose beads coated with GST-AF1521tandem, we observed increased recovery of AR from extract of androgen-treated cells (Fig. 4A). There are two scenarios for the interaction between ADP-ribosylated AR and GST-AF1521tandem: 1) the two AF1521 macrodomains bind individually to two ADP-ribosylated AR, or 2) the two AF1521 macrodomains bind to two separate ADP-ribosylation sites on a single AR (Fig. 4B).
Figure 4.
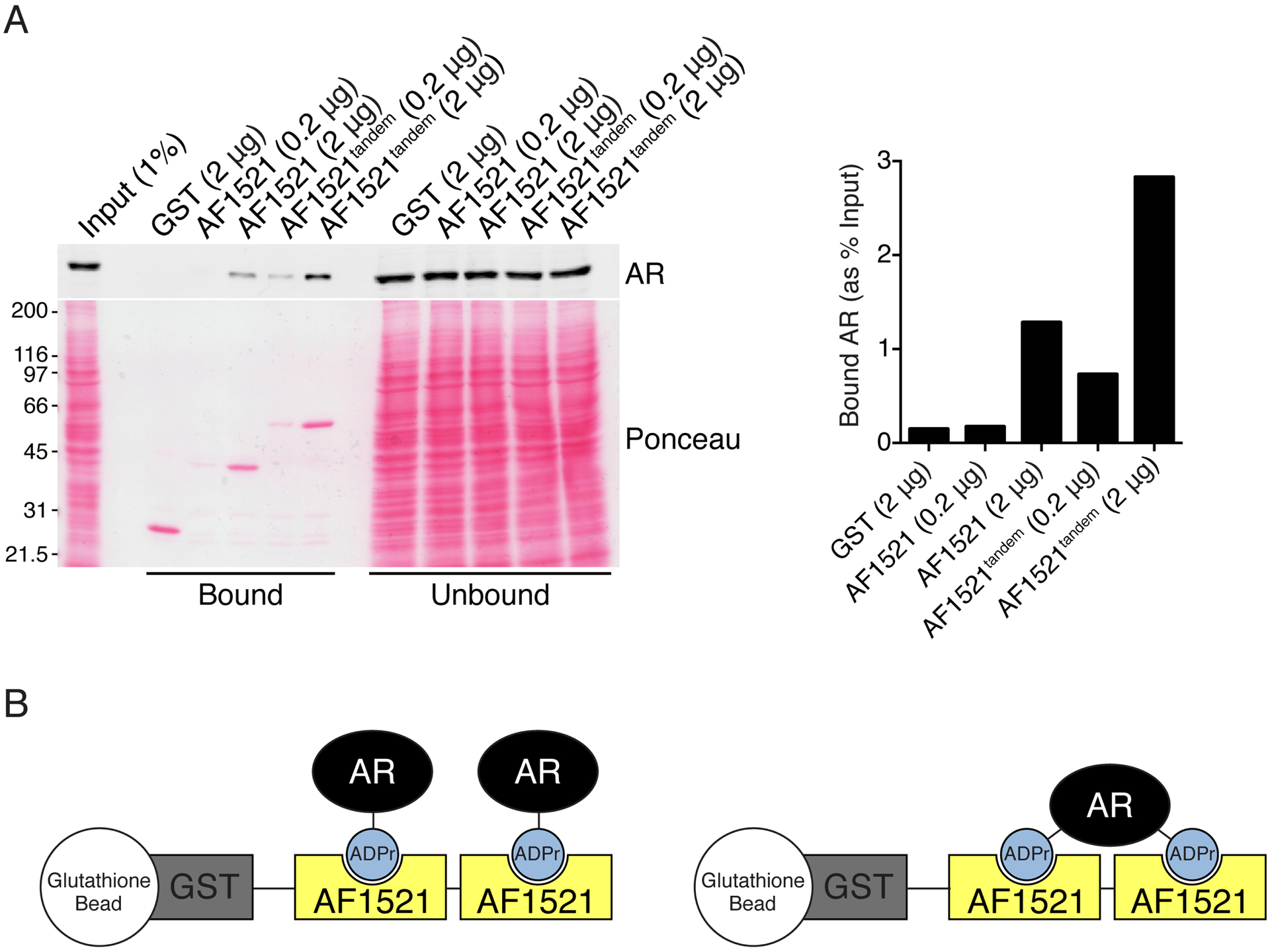
Expression of tandem AF1521 macrodomains improves recovery of ADP-ribosylated AR from extract of androgen-treated cells. A) Extract from androgen (R1881)-treated PC3-AR cells were analyzed by AF1521 pulldown using the indicated amounts of AF1521 or AF1521tandem. From AR blot, band intensities for bound AR were quantified as percent input. B) Schematic of two possible conformations of AF1521tandem binding to ADP-ribosylated (ADPr) AR.
Finally, we fluorescently labeled GST-AF1521tandem and developed it as a blotting reagent for detection of ADP-ribosylated AR on membrane. Using this reagent, we detected a band that runs near the molecular weight of AR in extract from androgen-treated cells (Fig. 5A). Detection of this band was lost when the fluorescently-labeled AF1521tandem probe was pre-incubated with ADP-ribose, while NAD+ pre-incubation had no effect (Fig. 5A). These observations are consistent with the idea that the fluorescently-labeled AF1521tandem probe is specifically recognizing ADP-ribosylated protein on membrane. We confirmed that the detected band is ADP-ribosylated AR through immunoprecipitation of AR from androgen-treated cells. AF1521 blotting showed that this band originally detected in whole-cell extract is reduced in the unbound, AR-depleted fraction while it is enriched in the bound AR fraction (Fig. 5B).
Figure 5.
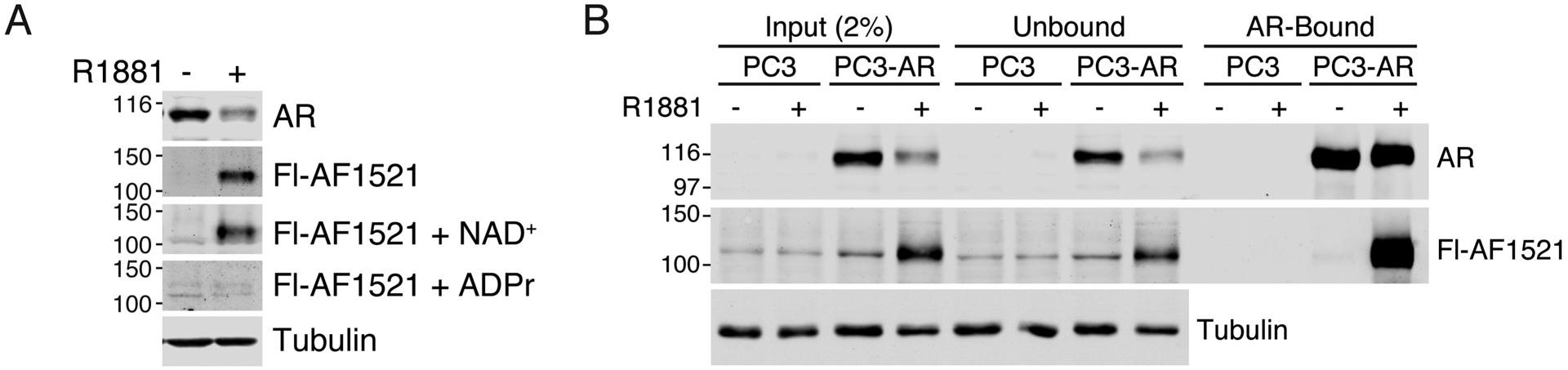
Fluorescently-labeled AF1521tandem can detect ADP-ribosylated AR by blot overlay. A) Extracts from androgen (R1881)- or vehicle-treated PC3-AR cells were analyzed by blotting using anti-AR, anti-tubulin, and fluorescently-labeled AF1521tandem probe (Fl-AF1521). As indicated, Fl-AF1521 was pre-incubated with 10 μM NAD+ or ADP-ribose (ADPr) before application on membrane. B) AR immunoprecipitation was conducted on extracts from androgen (R1881)- or vehicle-treated PC3 and PC3-AR cells and analyzed as in A).
To summarize, we have developed both a pulldown and blotting method for assessing the ADP-ribosylation of AR. Using these approaches, we found that in response to androgen, AR is ADP-ribosylated in PCa cells. We used controls such as AF1521 mutants, snake venom phosphodiesterase, and pre-incubation of fluorescently-labeled AF1521tandem probe with ADP-ribose or NAD+ to confirm that the AR enrichment by AF1521 pulldown or AF1521 blot signal we detect is a consequence of specific recognition of ADP-ribosylation by the macrodomain protein AF1521. Both the AF1521 pulldown and blotting are useful techniques for investigating the role of ADP-ribosylation of AR in PCa biology.
Materials
2.1. Cell Culture
PC3 (human prostate cancer) cells.
PC3-AR cells (see Subheading 3.3).
Cell culture medium: Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 5% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin.
100 mm tissue culture dish (Corning Incorporated, catalog #353003).
6-well tissue culture plate (Corning Incorporated, catalog #3516).
Synthetic androgen R1881 (methyltrienolone, metribolone) (PerkinElmer, catalog #NLP005005MG): To make 18 mM stock solution, dilute 5 mg R1881 with 977 μL 100% ethanol. From the 18 mM stock solution, dilute 1:1,800 in 100% ethanol to make up 10 μM R1881 working solution. For androgen treatment of cells, dilute the working solution 1:5,000 in cell culture medium for a final concentration of 2 nM R1881.
1X Phosphate-buffered saline (PBS): 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4. To prepare 1 L of 10X PBS, combine 800 mL of deionized water with 80 g NaCl, 2 g KCl, 14.2 g Na2HPO4, and 2.4 g KH2PO4. Adjust pH to 7.4 and bring to a final volume of 1 L. Dilute 1:10 from 10X stock to make 1X PBS.
2.2. Escherichia coli Expression and Purification for GST-AF1521 Proteins
BL21 E. coli competent cells.
Plasmid DNA for expression of GST-AF1521 and GST-AF1521tandem.
LB broth, Lennox medium: To make 1 L, dissolve 20 g LB powder (Becton, Dickinson and Company, catalog #240230) in 1 L of deionized water and autoclave.
100% Ethanol.
1X PBS.
Elution buffer: 50 mM Tris-HCl pH 8, 10 mM glutathione, 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 2 mM DTT.
Dialysis buffer: 50 mM Tris-HCl pH 7.5, 50 mM NaCl, 14.3 mM β-mercaptoethanol.
Coomasie stain.
1 M isopropyl-β-D-thiogalactoside (IPTG): To make 1 M stock solution, dissolve 2.38 g IPTG in 8 mL deionized water and bring to a final volume of 10 mL. Sterilize with 0.22 μm syringe filter. Aliquot and store at −20°C.
Glutathione-agarose resin/beads (Sigma-Aldrich, catalog #G4510).
LB + ampicillin (100 μg/mL) agar plate.
2 L flask.
Spectrophotometer.
French press.
Sorvall RC-5B Plus centrifuge.
Sorvall SS-34 rotor.
Sorvall SLA-3000 rotor.
Chromatography column.
2.3. SDS-PAGE and Immunoblotting
1X PBST: 1X PBS with 0.15% Tween 20 (v/v).
Antibody/probe dilution buffer: 1X PBST with 1% BSA (w/v).
Standard SDS-PAGE running buffer.
Standard transfer buffer.
Blocking buffer: 1X PBST with 5% nonfat dry milk (w/v).
10 mM NAD+: Dissolve 33.2 mg NAD+ (Sigma-Aldrich, catalog #N1511) in 4.8 mL molecular biology grade water. Adjust pH to ~8 and bring to a final volume of 5 mL. Aliquot and store at −80°C.
50 mM ADP-ribose: Dissolve 100 mg of ADP-ribose (Sigma-Aldrich, catalog #A0752) in 3.576 mL 50 mM HEPES pH 7.5, 50 mM MgCl2. Aliquot and store at −80°C.
- Primary antibodies:
- Anti-AR rabbit polyclonal antibody: Custom antibody was generated using AR residues 656 to 669 (TQKLTVSHIEGYEC) as immunogen and affinity purified.
- Anti-tubulin mouse monoclonal antibody (Sigma-Aldrich, catalog #T9028).
- Secondary antibodies:
- Alexa Fluor® 680-labeled donkey anti-rabbit IgG antibody (Life Technologies, catalog #A10043).
- IRDye® 800CW goat anti-mouse IgG antibody (LI-COR Biosciences, catalog #926–32210).
Tris-glycine 1.5 mm, 15-well, 8% gel.
Gel electrophoresis system for SDS-PAGE and wet/tank transfer.
Nitrocellulose membrane (0.2 μm) (GE Healthcare, catalog #10600001).
Odyssey® CLx imaging system (LI-COR Biosciences).
IRDye® 800CW Protein Labeling Kit – High MW (LI-COR Biosciences, catalog #928–38040).
2.4. Cell Lysis and Sample Preparation
Lysis buffer: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% Triton X-100 (v/v), 5 mM EDTA, 5 μg/mL aprotinin, 5 μg/mL leupeptin, 5 μg/mL pepstatin, 1 mM PMSF, 1 mM DTT.
1X SDS-PAGE sample buffer: 62.5 mM Tris-HCl pH 6.8, 2% SDS (w/v), 0.05% bromophenol blue (w/v), 10% glycerol (v/v), 50 mM DTT.
Cell scraper.
Eppendorf 5417R refrigerated microcentrifuge.
Eppendorf FA-45-30-11 rotor.
Sonicator.
2.5. Snake Venom Phosphodiesterase Purification
Snake venom phosphodiesterase (Worthington Biochemical Corporation, catalog #LS003926).
Wash buffer: 10 mM Tris-HCl pH 7.5, 50 mM NaCl, 10% glycerol (v/v).
Elution buffer: 10 mM Tris-HCl pH 7.5, 50 mM NaCl, 150 mM K2HPO4, 10% glycerol (v/v).
Blue sepharose (GE Healthcare, catalog #17094801).
2.6. Snake Venom Phosphodiesterase Treatment
3-aminobenzamide (Acros Organics, catalog #339080050).
2.7. AF1521 Pulldown
Wash buffer: 25 mM Tris-HCl pH 7.5, 50 mM NaCl, 0.1% NP-40 (v/v), 0.1 mM EDTA, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 1 mM DTT.
Low binding plastic 1.7 mL microcentrifuge tube (Corning Incorporated, catalog #C3207).
2.8. AR Immunoprecipitation
Anti-FLAG M2 magnetic beads (Sigma-Aldrich, catalog #M8823).
Magnetic separation tube rack.
Methods
3.1. GST-AF1521 Cloning
AF1521 cDNA clone (AfCD00370825) was obtained from the DNASU Plasmid Repository (Arizona State University, Tempe, AZ) and used as template for cloning into bacterial expression vector. Full-length AF1521 CDS was PCR amplified and cloned into the pGEX-4T-2 vector via BamHI and EcoRI sites (pGEX-4T-2/GST-AF1521). Site-directed mutagenesis was conducted on pGEX-4T-2/GST-AF1521 to introduce D20A and G42E mutations alone or in combination, which are critical residues for binding to ADP-ribose [16]. To generate a construct where the GST tag was fused to two AF1521 proteins in tandem, full-length AF1521 CDS was PCR amplified and cloned into pGEX-4T-2/GST-AF1521 via AccI and NotI sites (pGEX-4T-2/GST-AF1521tandem). The parental empty pGEX-4T-2 vector was used for expression of GST control protein.
3.2. E. coli Expression and Purification for GST-AF1521 Proteins
Transform BL21 competent cells using expression plasmid DNA, plate onto LB + ampicillin agar plate, and incubate at 37°C overnight.
Pick a single colony from the transformation plate and inoculate 5 mL LB medium supplemented with 100 μg/mL ampicillin.
Incubate the culture overnight at 37°C with shaking (220 rpm).
Add 5 mL of the overnight culture into a 2 L flask containing 500 mL of LB medium supplemented with 100 μg/mL ampicillin.
Incubate at 37°C with shaking (220 rpm). Monitor bacterial growth by measuring the OD600 nm using a spectrophotometer.
Once the OD600 nm reaches between 0.6 and 0.8 (see Note 1), cool down the culture by incubating at 4°C for 10 min.
Add 100% ethanol to the culture to a final concentration of 2% (v/v).
Induce recombinant protein expression with 1 mM IPTG (i.e. dilute stock IPTG 1:1,000).
Incubate the culture at 18°C overnight with shaking (220 rpm).
Harvest bacterial cell pellet by centrifuging 5,000 rpm (~4,225 × g) at 4°C for 20 min in a Sorvall RC-5B Plus centrifuge with a SLA-3000 rotor. Cell pellets can be stored at −80°C until ready for lysis and protein purification.
Resuspend cell pellet in 10 mL 1X PBS with 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 1 mM PMSF, and 2 mM DTT.
Lyse cells by passing them through a French press three to four times.
To clarify lysate, centrifuge 18,000 rpm (~38,724 × g) at 4°C for 30 min in a Sorvall RC-5B Plus centrifuge with a SS-34 rotor. Meanwhile, prepare chromatography column by loading with 1.5 mL of glutathione-agarose resin and washing with 15 mL 1X PBS.
Save an aliquot of the clarified lysate as input sample for analysis by SDS-PAGE. Apply the remaining clarified lysate to the prepared chromatography column.
Add Triton X-100 to a final concentration of 1% (v/v), cap the column, and rotate at 4°C for 1.5 h.
Remove end-cap and allow unbound fraction to drain until meniscus reaches the top of the resin. Save an aliquot of the unbound fraction for analysis by SDS-PAGE.
Wash the resin with 50 mL 1X PBS with 1% Triton X-100 (v/v).
Wash the resin with 50 mL 1X PBS with 500 mM NaCl.
Wash the resin with 50 mL 1X PBS.
Apply 1 mL elution buffer to washed resin, incubate 10 min, and collect elution fraction. Repeat as necessary (see Note 2).
Measure the OD280 nm of the collected elution fractions using a spectrophotometer.
Pool together peak elution fractions and dialyze in 1 L dialysis buffer at 4°C overnight.
Replace with fresh 1 L dialysis buffer and dialyze at 4°C for 2 h. Repeat this step with fresh dialysis buffer.
Check concentration of dialyzed protein by Bradford or BCA assay (see Note 3).
Check purity of dialyzed protein by running on SDS-PAGE, followed by Coomassie stain (see Note 4). Processing samples saved during lysis and purification procedures (i.e. input and unbound fractions) can also be run and analyzed on same gel.
Aliquot purified proteins, flash freeze in liquid nitrogen, and store at −80°C.
3.3. Cell Culture
PC3 is a PCa cell line that does not express AR. We generated a stable cell line that expresses AR (PC3-AR) through lentiviral transduction of PC3 cells. Starting with the pWPI lentiviral vector (a gift from Dr. Didier Trono, Addgene plasmid #12254), the EGFP marker was replaced with a hygromycin resistance selectable marker. Subsequently, FLAG epitope-tagged AR was subcloned, and the resulting vector was used for lentivirus production. All cells were grown at 37°C, 5% CO2.
3.4. Cell Lysis
Carry out all centrifugation steps at 4°C. All centrifugation speeds noted below are for an Eppendorf 5417R refrigerated microcentrifuge with a FA-45-30-11 rotor.
Grow PC3-AR cells in a 100 mm dish to 70–80% confluency (~7 × 106 cells per dish).
Treat cells for 24 h with androgen by replacing with fresh cell culture medium containing 2 nM R1881. For vehicle control, use fresh cell culture medium with ethanol.
Wash cells with ice-cold 1X PBS twice.
Add 1 mL of ice-cold 1X PBS to dish and scrape off cells.
Transfer cell suspension to microcentrifuge tube. Pellet cells by centrifuging 3,000 rpm (~956 × g) for 5 min.
Carefully aspirate off buffer without disturbing cell pellet. Cell pellet can be flash frozen in liquid nitrogen and stored at −80°C until ready for lysis.
Lyse cell pellet with 400 μL of lysis buffer on ice for 20 min.
Centrifuge samples at 14,000 rpm (~20,817 × g) for 15 min.
Keep clarified lysate for AF1521 pulldown or AR immunoprecipitation. Save an aliquot as input sample for SDS-PAGE analysis.
3.5. Preparation of AF1521-Coated Glutathione-Agarose Beads
For each pulldown sample, prepare 10 μL of GST- or GST-AF1521-coated glutathione-agarose beads (see Note 5).
Aliquot appropriate amount of glutathione-agarose beads into a low binding plastic 1.7 mL microcentrifuge tube and wash three times with 1X PBS with 0.1% Triton X-100 (v/v) (see Note 6). Use at least 500 μL buffer per wash.
Incubate 1 μg recombinant protein per 1 μL of glutathione-agarose beads with rotation at 4°C overnight in 1X PBS with 0.1% Triton X-100 (v/v) (see Note 7).
Wash beads three times with 1X PBS. Use at least 500 μL buffer per wash. Prepared beads are stable for at least one month when stored in 1X PBS with 5 mM sodium azide at 4°C.
3.6. AF1521 Pulldown
Set up pulldowns in low binding plastic 1.7 mL microcentrifuge tubes. Add 195 μL of clarified lysate (prepared from Subheading 3.4) to 10 μL of GST-coated or 10 μL of GST-AF1521-coated glutathione-agarose beads.
Incubate with rotation at 4°C for 4 h.
Centrifuge briefly at 4°C to collect beads.
Save an aliquot of the unbound fraction for SDS-PAGE analysis. Carefully aspirate off remaining unbound fraction, while avoiding loss of beads.
Wash beads by adding 500 μL of wash buffer and inverting tube several times to disrupt bead pellet.
Centrifuge briefly at 4°C to collect beads.
Carefully aspirate off wash buffer, while avoiding loss of beads.
Repeat steps 5 to 7, four additional times (i.e. five total washes).
After final wash, aspirate off as much buffer as possible and resuspend beads in 15 μL 1X SDS-PAGE sample buffer.
Boil samples at 95°C for 5 min.
Load 15 μL of pulldown sample per lane on a Tris-glycine 1.5 mm, 15-well, 8% gel, and run at 120 V until dye front reaches the bottom of the gel.
Transfer the proteins onto nitrocellulose membrane using a wet/tank blotting apparatus.
After transfer, incubate membrane with blocking buffer for at least 1 h.
Wash membrane three times 5 min each with 1X PBST.
Incubate membrane with primary antibodies diluted in antibody/probe dilution buffer: anti-AR (1 μg/mL) and anti-tubulin (1:10,000).
Wash membrane three times 5 min each with 1X PBST.
Incubate membrane with secondary antibodies diluted 1:20,000 in antibody/probe dilution buffer. Keep covered to avoid exposing the secondary antibodies to light.
Wash membrane three times 5 min each with 1X PBST, rinse membrane briefly with deionized water, and image on an Odyssey® CLx imaging system.
3.7. Snake Venom Phosphodiesterase Purification
Resuspend a vial of snake venom phosphodiesterase in 0.5 mL wash buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 10% glycerol (v/v)). Save an aliquot as input sample for SDS-PAGE analysis.
Add 0.5 mL blue sepharose to a microcentrifuge tube and wash several times with wash buffer.
Add resuspended snake venom phosphodiesterase to washed sepharose and incubate with rotation at 4°C for 1 h.
Centrifuge briefly to collect sepharose beads.
Save an aliquot of the unbound fraction for gel analysis and discard the remainder.
Wash sepharose beads five times with 750 μL of wash buffer.
Resuspend sepharose beads with wash buffer and transfer to chromatography column. Let beads settle and allow any excess buffer to drain by gravity flow until meniscus reaches the top of the resin.
Add elution buffer and elute in 0.5 mL fractions.
Measure the OD280 nm of the collected elution fractions using a spectrophotometer.
Pool together peak elution fractions and dialyze in 0.5 L wash buffer at 4°C overnight.
Replace with fresh 0.5 L wash buffer and dialyze at 4°C for 2 h.
Check concentration of dialyzed protein by Bradford or BCA assay.
Check purity of dialyzed protein by running on SDS-PAGE, followed by Coomassie stain. Processing samples saved during purification procedures (i.e. input and unbound fractions) can also be run and analyzed on same gel.
Aliquot purified proteins, flash freeze in liquid nitrogen, and store at −80°C.
3.8. Snake Venom Phosphodiesterase Treatment
Prepare clarified lysate as described in Subheading 3.4 with the exception of using 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% Triton X-100 (v/v), 5 μg/mL aprotinin, 5 μg/mL leupeptin, 5 μg/mL pepstatin, 1 mM PMSF, 1 mM DTT, 20 mM 3-aminobenzamide, 15 mM MgCl2 for cell lysis.
Add 50 μg of purified snake venom phosphodiesterase (from Subheading 3.7) to 195 μL of clarified lysate. For control, add equivalent volume of snake venom phosphodiesterase storage buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 10% glycerol (v/v)) to 195 μL of clarified lysate.
Incubate at room temperature for 2 h.
Transfer samples to low binding plastic 1.7 mL microcentrifuge tubes containing 10 μL of GST-AF1521-coated glutathione-agarose beads each.
Continue with AF1521 pulldown procedures as described in Subheading 3.6, starting with step 2.
3.9. AF1521 Direct Blotting
Fluorescently label 1 mg of purified GST-AF1521tandem using IRDye® 800CW Protein Labeling Kit according to manufacturer’s instructions. Store as single-use aliquots at −80°C (see Note 8).
Grow PC3-AR cells in a 6-well plate to 70–80% confluency (~1 × 106 cells per well).
Treat cells for 24 h with 2 nM R1881 or ethanol vehicle control.
Harvest cells using 300 μL 1X SDS-PAGE sample buffer per well.
Boil samples at 95°C for 5 min.
Cool, spin briefly, and sonicate samples to shear DNA and reduce sample viscosity (see Note 9).
Load 15 μL of lysate per lane on a Tris-glycine 1.5 mm, 15-well, 8% gel and run at 120 V until dye front reaches the bottom of the gel.
Transfer the proteins onto nitrocellulose membrane using a wet/tank blotting apparatus.
After transfer, wash nitrocellulose membrane three times 5 min each with 1X PBST (see Note 10).
Incubate membrane with blocking buffer at room temperature for at least 1 h.
Wash membrane three times 5 min each with 1X PBST.
Incubate membrane in fluorescently-labeled GST-AF1521tandem diluted to 1 μg/mL in antibody/probe dilution buffer at 4°C overnight (see Note 11). Keep covered to avoid exposing fluorescently-labeled probe to light. As controls, fluorescently-labeled GST-AF1521tandem dilution can be pre-incubated with 10 μM NAD+ (dilute NAD+ stock 1:1,000) or ADP-ribose (dilute ADP-ribose stock 1:5,000) at 4°C for 1 h before applying to membrane.
Wash membrane four times 5 min each with 1X PBST, rinse membrane briefly with deionized water, and image on an Odyssey® CLx imaging system.
3.10. AR Immunoprecipitation
For each immunoprecipitation sample, use 2 μL of packed anti-FLAG M2 magnetic beads. Add beads to low binding plastic 1.7 mL microcentrifuge tube and wash three times with 500 μL 1X PBS with 0.1% Triton X-100 (v/v).
Add 360 μL of clarified lysate (prepared from Subheading 3.4) to the washed beads and incubate with rotation at 4°C for 4 h.
Place tubes on magnetic separation tube rack to immobilize beads. Save an aliquot of the unbound fraction for analysis by SDS-PAGE.
Wash beads five times with 500 μL wash buffer (same composition as the wash buffer used for AF1521 pulldown).
After final wash, resuspend beads in 15 μL 1X SDS-PAGE sample buffer and boil at 95°C for 5 min.
Use 7.5 μL of sample for analysis by SDS-PAGE and immunoblotting as described in Subheading 3.6, starting with step 11. Use the remaining 7.5 μL of sample for AF1521 blotting as described in Subheading 3.9, starting with step 7.
Notes
Culture takes typically 3 to 4 h to reach the appropriate OD600 nm.
Most of the recombinant protein elutes in the first four fractions.
Approximate yield of purified recombinant GST-AF1521 from a 500 mL culture is 6 mg.
Expected molecular weights for recombinant proteins are as follows: GST-AF1521 = 49 kDa and GST-AF1521tandem = 70.5 kDa.
Because some of the beads stick to the pipette tip and are lost during dispensing/aliquoting, we account for this by preparing extra AF1521-beads.
To makes it easier to draw up and dispense glutathione-agarose bead slurry, cut off the pipette tip end with scissors to make the opening larger.
After adding recombinant protein, add enough buffer to form a ~30% bead slurry or the total volume (including beads) is at least 300 μL.
We store 30 μL 0.5 mg/mL aliquots that can be diluted into 15 mL of antibody/probe dilution buffer for a final 1 μg/mL concentration. Fluorescently-labeled GST-AF1521tandem is not stable at −20°C storage.
We sonicate samples one to two pulses on a Branson Sonifier 250 with output 3 and duty cycle 40% settings.
If membrane is stained with Ponceau S after transfer, wash membrane well (e.g., five times 5 min each with 1X PBST) before proceeding with blocking. This is to minimize residual acid from the staining solution being carried over into the subsequent steps involving the fluorescently-labeled GST-AF1521tandem probe.
Diluted fluorescently-labeled GST-AF1521tandem can be saved and reused; add 5 mM sodium azide and store at 4°C away from light. The probe is stable for at least 4 months under these conditions.
References
- 1.Gao W, Bohl CE, Dalton JT (2005) Chemistry and structural biology of androgen receptor. Chem Rev 105:3352–3370. doi: 10.1021/cr020456u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett NC, Gardiner RA, Hooper JD, et al. (2010) Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol 42:813–827. doi: 10.1016/j.biocel.2009.11.013 [DOI] [PubMed] [Google Scholar]
- 3.Heinlein CA, Chang C (2002) Androgen receptor (AR) coregulators: an overview. Endocr Rev 23:175–200 [DOI] [PubMed] [Google Scholar]
- 4.Gioeli D, Paschal BM (2012) Post-translational modification of the androgen receptor. Mol Cell Endocrinol 352:70–78. doi: 10.1016/j.mce.2011.07.004 [DOI] [PubMed] [Google Scholar]
- 5.Siegel Rebecca L, Miller Kimberly D, Jemal Ahmedin (2018) Cancer statistics, 2018. CA: Cancer J Clin 68:7–30. doi: 10.3322/caac.21442 [DOI] [PubMed] [Google Scholar]
- 6.Balk SP, Knudsen KE (2008) AR, the cell cycle, and prostate cancer. Nucl Recept Signal 6:e001. doi: 10.1621/nrs.06001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schreiber V, Dantzer F, Ame J-C, de Murcia G (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7:517–528. doi: 10.1038/nrm1963 [DOI] [PubMed] [Google Scholar]
- 8.Gibson BA, Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13:411–424. doi: 10.1038/nrm3376 [DOI] [PubMed] [Google Scholar]
- 9.Rouleau M, Patel A, Hendzel MJ, et al. (2010) PARP inhibition: PARP1 and beyond. Nat Rev Cancer 10:293–301. doi: 10.1038/nrc2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mateo J, Carreira S, Sandhu S, et al. (2015) DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373:1697–1708. doi: 10.1056/NEJMoa1506859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schiewer MJ, Goodwin JF, Han S, et al. (2012) Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov 2:1134–1149. doi: 10.1158/2159-8290.CD-12-0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupte R, Liu Z, Kraus WL (2017) PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31:101–126. doi: 10.1101/gad.291518.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bindesbøll C, Tan S, Bott D, et al. (2016) TCDD-inducible poly-ADP-ribose polymerase (TIPARP/PARP7) mono-ADP-ribosylates and co-activates liver X receptors. Biochem J 473:899–910. doi: 10.1042/BJ20151077 [DOI] [PubMed] [Google Scholar]
- 14.Allen MD, Buckle AM, Cordell SC, et al. (2003) The crystal structure of AF1521 a protein from Archaeoglobus fulgidus with homology to the non-histone domain of macroH2A. J Mol Biol 330:503–511. doi: 10.1016/S0022-2836(03)00473-X [DOI] [PubMed] [Google Scholar]
- 15.Karras GI, Kustatscher G, Buhecha HR, et al. (2005) The macro domain is an ADP‐ribose binding module. EMBO J 24:1911–1920. doi: 10.1038/sj.emboj.7600664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Till S, Ladurner AG (2009) Sensing NAD metabolites through macro domains. Front Biosci 14:3246–3258 [DOI] [PubMed] [Google Scholar]
- 17.Dani N, Stilla A, Marchegiani A, et al. (2009) Combining affinity purification by ADP-ribose-binding macro domains with mass spectrometry to define the mammalian ADP-ribosyl proteome. Proc Natl Acad Sci USA 106:4243–4248. doi: 10.1073/pnas.0900066106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gagné J-P, Pic É, Isabelle M, et al. (2012) Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res 40:7788–7805. doi: 10.1093/nar/gks486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jungmichel S, Rosenthal F, Altmeyer M, et al. (2013) Proteome-wide identification of poly(ADP-ribosyl)ation targets in different genotoxic stress responses. Mol Cell 52:272–285. doi: 10.1016/j.molcel.2013.08.026 [DOI] [PubMed] [Google Scholar]
- 20.Martello R, Leutert M, Jungmichel S, et al. (2016) Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat Commun 7:12917. doi: 10.1038/ncomms12917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bock FJ, Chang P (2016) New directions in poly(ADP-ribose) polymerase biology. FEBS J 283:4017–4031. doi: 10.1111/febs.13737 [DOI] [PubMed] [Google Scholar]
- 22.Matsubara H, Hasegawa S, Fujimura S, et al. (1970) Studies on poly (adenosine diphosphate ribose) V. Mechanism of hydrolysis of poly(adenosine diphosphate ribose) by snake venom phosphodiesterase. J Biol Chem 245:3606–3611 [PubMed] [Google Scholar]