

Abstract
Aims
Thioredoxin 1 (Trx1) is an evolutionarily conserved oxidoreductase that cleaves disulphide bonds in oxidized substrate proteins such as mechanistic target of rapamycin (mTOR) and maintains nuclear-encoded mitochondrial gene expression. The cardioprotective effect of Trx1 has been demonstrated via cardiac-specific overexpression of Trx1 and dominant negative Trx1. However, the pathophysiological role of endogenous Trx1 has not been defined with a loss-of-function model. To address this, we have generated cardiac-specific Trx1 knockout (Trx1cKO) mice.
Methods and results
Trx1cKO mice were viable but died with a median survival age of 25.5 days. They developed heart failure, evidenced by contractile dysfunction, hypertrophy, and increased fibrosis and apoptotic cell death. Multiple markers consistently indicated increased oxidative stress and RNA-sequencing revealed downregulation of genes involved in energy production in Trx1cKO mice. Mitochondrial morphological abnormality was evident in these mice. Although heterozygous Trx1cKO mice did not show any significant baseline phenotype, pressure-overload-induced cardiac dysfunction, and downregulation of metabolic genes were exacerbated in these mice. mTOR was more oxidized and phosphorylation of mTOR substrates such as S6K and 4EBP1 was impaired in Trx1cKO mice. In cultured cardiomyocytes, Trx1 knockdown inhibited mitochondrial respiration and metabolic gene promoter activity, suggesting that Trx1 maintains mitochondrial function in a cell autonomous manner. Importantly, mTOR-C1483F, an oxidation-resistant mutation, prevented Trx1 knockdown-induced mTOR oxidation and inhibition and attenuated suppression of metabolic gene promoter activity.
Conclusion
Endogenous Trx1 is essential for maintaining cardiac function and metabolism, partly through mTOR regulation via Cys1483.
Keywords: Heart, Redox, Metabolism, Thioredoxin-1(Trx1), Mechanistic target of rapamycin (mTOR)
Graphical Abstract
Graphical Abstract.
Time for primary review: 18 days
1. Introduction
Trx1 is an evolutionarily conserved protein disulphide reducing enzyme. Utilizing two cysteines positioned at 32 and 35 in the catalytic centre, Trx1 cleaves disulphide bonds of oxidized proteins while concurrently forming a disulphide bond in Trx1. The oxidized Trx1 is reduced by Trx reductase 1 (Txnrd1). Trx1 itself does not effectively scavenge H2O2 but indirectly scavenges it through peroxiredoxins (Prdxs), Trx1-dependent peroxidases.1 Peroxiredoxin reduces H2O2 to H2O in association with sulfenic acid (S–OH) formation at a catalytic cysteine residue. The sulfenic acid, in turn, forms a disulphide bond with other Prdx molecules, resulting in oligomerization. Trx1 reduces the disulphide bond in Prdx. Under excessive oxidative stress conditions, the thiol group of Prdx forms either sulfinic (S–OOH) or sulphonic (S–OOOH) acid, which do not form a disulphide. Sulfinic acid is reduced by sulfiredoxin 1 (Srxn1) rather than Trx1, but Trx1 is a potential electron donor for Srxn1.2 Trx1 also mediates both transnitrosylation and denitrosylation. S-nitrosylation refers to covalent attachment of nitric oxide (NO) to a cysteine thiol group (S–NO); removal of NO is denitrosylation. In addition, the NO group may be transferred to another molecule, termed transnitrosylation. Trx1 plays a central role in denitrosylation through its catalytic centre3 and mediates transnitrosylation with alternative cysteine residues at positions 69 and 73.4 Thus, Trx1 plays a crucial role in cellular redox homeostasis.
Global Trx1 knockout in mice resulted in embryonic lethality at 3.5 days of gestation (E3.5), presumably corresponding to the 32- to 64-cell stage of blastocyst.5 Thus, Trx1 plays an essential role in early embryogenesis. However, partly due to the early embryonic lethality, no pathophysiological roles of Trx1 have been demonstrated with a loss-of-function model in vivo. Although liver-specific Trx1 knockout mice have recently been generated, these mice do not show any abnormalities in liver development or function.6 Thus, besides during early embryogenesis, there is thus far no evidence that endogenous Trx1 has an indispensable biological role in any context in vivo. Although cardioprotective effects of Trx1 have been repeatedly reported in studies utilizing cardiac-specific overexpression of intact and dominant negative Trx1 (Trx1CC32/35SS),7 the significance of endogenous Trx1 in the heart has not been investigated with a loss-of-function model.
Trx1 regulates cellular signal transduction through specific substrates or binding proteins, including apoptosis signal regulating kinase 1 (ASK1), phosphatase and tensin homologue (PTEN), class II histone deacetylases (HDACs), caspase, AMP-activated protein kinase (AMPKα), and mechanistic target of rapamycin (mTOR).7 Trx1 negatively regulates ASK1 through direct binding; under oxidative stress conditions, Trx1 is oxidized, releasing it from ASK1 and leading to ASK1 activation. On the other hand, Trx1 maintains the function of PTEN, Class II HDACs, AMPKα, and mTOR through reduction of specific cysteine residues. Class II HDACs play an important role in preventing cardiac hypertrophy, while AMPKα and mTOR confer stress resistance.8–10
mTOR is an evolutionarily conserved serine/threonine kinase and catalytic subunit in protein complexes including mTORC1 and mTORC2, which regulates cell growth, survival, and mRNA translation. In addition, mTOR plays a crucial role in mitochondrial energy production through transcriptional and translational regulation of nuclear-encoded mitochondrial genes.11,12 Muscle-specific deletion of mTOR results in fatal cardiomyopathy with early death.13 Using unbiased screening with cardiac-specific Trx1C35S substrate trapping mutant mice, we previously showed that mTOR is a major Trx1 substrate.10 In cultured cardiomyocytes, mTOR is inhibited through intermolecular disulphide formation at Cys1483 in the presence of H2O2, whereas Trx1 prevents mTOR inhibition.9 Cells expressing mTOR-C1483F, an oxidation-resistant mutant, are protected against H2O2-induced intermolecular disulphide bond formation in mTOR and consequent inhibition of mTOR and downstream signalling mechanisms, including mitochondrial gene expression. Thus, mTOR is inhibited by oxidation at Cys1483, whereas Trx1 protects mTOR against oxidation and inactivation. However, whether downregulation of endogenous Trx1 alone, in the absence of exogenous oxidants such as H2O2, induces oxidation at Cys1483 remains unknown. In addition, the specific importance of the Trx1-mTOR pathway, one of multiple targets of Trx1, in the regulation of cardiac function and metabolism at baseline and during stress has never been defined in vivo.
In this study, we have generated cardiac-specific Trx1 knockout mice and demonstrated that endogenous Trx1 in the heart is indispensable for cardiac function, animal survival, and mitochondrial function. In vivo and in vitro experiments suggest that Trx1-mediated mTOR regulation via Cys1483 plays a crucial role in maintaining mitochondrial function, thus representing the major signalling pathway mediating endogenous Trx1 function in cardiomyocytes. This study is the first to prove an indispensable biological role of Trx1 and demonstrate the underlying molecular mechanisms at a time point later than the blastocyst stage in a mammalian system.
2. Methods
2.1 Animal experiments
The Trx1flox/flox mouse was generated on the C57BL/6 background by Cyagen Bioscience (Santa Clara, CA, USA) with a standard gene knockout strategy. Global Trx1+/− mice were described previously.5 Due to early death of Trx1cKO mice, echocardiographic measurements and collection of heart samples were conducted at 2–5 weeks of age. For pressure overload (PO) experiments, we used 3- to 9-month-old mice. To collect heart samples, mice were euthanized with pentobarbital (120 mg/kg body weight, i.p.). All procedures involving animals were performed in accordance with NIH guidelines and protocols approved by Rutgers Biomedical and Health Sciences.
2.2 Transverse aortic constriction
Mice were anaesthetized with pentobarbital (60–70 mg/kg, i.p.) and mechanically ventilated. The left side of the chest was opened at the second intercostal space. Aortic constriction was performed by ligation of the transverse thoracic aorta between the innominate artery and left common carotid artery with a 28-gauge needle using a 7-0 braided polyester suture under an operating microscope. After surgery, mice were allowed to recover in a thermocare unit. Echocardiographic measurements and collection of heart samples were conducted after 2 weeks.
2.3 Echocardiography
Mice were anaesthetized with 2.5% avertin (12 μL/g body weight, i.p.), since isoflurane occasionally lowers blood pressure and induces irregular heartbeat. Echocardiography was performed using ultrasonography (Acuson Sequoia C256, Siemens Medical Solutions USA Inc., Malvern, PA, USA).
2.4 Histological and electron microscopic analyses
Heart specimens were fixed with 10% formalin and sectioned at 6 μm thickness. Fibrosis was evaluated by picric acid sirius red (PASR) staining. The positively stained fibrotic area was measured with the ImageJ program (National Institute of Health, Bethesda, MD, USA) and normalized by total area. Cell size was evaluated by wheat germ agglutinin (WGA) staining using the ImageJ program. The number of TUNEL-positive nuclei was normalized by total area. Mitochondrial size and aspect ratio were evaluated by electron microscopy using the ImageJ program.
2.5 Western blot analysis
Heart homogenates were prepared using a lysis buffer [50 mM Tris–HCl (pH 7.6), 1% Triton X-100, 10 mM EDTA, 150 mM NaCl, 50 mM NaF, 100 mM N-ethylmaleimide (NEM), and protease inhibitor cocktail (Sigma, St. Louis, MO, USA)]. SDS sample buffer with or without 2-mercaptoethanol was used for SDS-PAGE under reducing or non-reducing conditions, respectively. The signal intensity of western blot signals was quantified using the ImageJ program. The signal intensity of a specific protein with modification such as phosphorylation or sulphonic acid formation was normalized with the total protein without modification. The signal intensity of other proteins was normalized by a loading control (GAPDH, Tubulin, or alpha actin). NS indicates non-specific bands serving as loading controls.
2.6 Primary cultures of neonatal rat ventricular myocytes
Primary cultures of ventricular cardiomyocytes were prepared from 1-day-old Crl: (WI) BR-Wistar rats (Harlan, Indianapolis, IN, USA). A discontinuous Percoll gradient was employed to isolate a cardiomyocyte-enriched fraction.
2.7 Plasmid vector transfection
Plasmid vectors were transfected into primary cultured rat neonatal cardiomyocytes and H9c2 cells, a rat cardiac myoblast cell line, plated on 3.5 cm dishes using LipofectAmine 2000 (Invitrogen, Carlsbad, CA, USA). A total of 2.5 μg of mammalian expression vectors (e.g. 0.5 μg of pRK5-HA-GST-p85S6K and 2 μg of pRK5-Myc-mTOR or pRK5-Myc-mTOR-C1483F) and 5 μL LipofectAmine 2000 were used per dish.
2.8 Adenovirus vectors
Adenovirus harbouring shTrx1 and its control were generated using the AdMax system (Microbix, Mississauga, ON, USA) as described previously.8 All cell culture experiments with shTrx1 were conducted 4–7 days after adenovirus transduction.
2.9 Luciferase assay
Luciferase assays were performed in primary cultured rat cardiomyocytes. Reporter plasmids (0.3 μg/well) and mammalian expression vectors (0.7 μg/well), including pDC316-Flag-mTOR and pDC316-Flag-mTOR-C1483F, were transfected into cells plated on 12-well plates. The total amount of plasmids was kept at 1 μg/well using pDC316 control vector. After 24 h of transfection, cells were transduced with shTrx1 adenovirus (20 MOI). Luciferase assays were performed 4–5 days after transduction with a luciferase assay system (Promega, Madison, WI, USA).
2.10 Oxygen consumption
Oxygen consumption rate was measured with a Seahorse XFe96 Analyzer (Agilent, Santa Clara, CA, USA) 5 days after shTrx1 (20 MOI) transduction.
2.11 RNA-sequencing
Total RNA for RNA-seq was prepared from 4-week-old Trx1cKO and control mice. Biological pathways associated with significant genes were analysed by Gene Ontology (GO) analysis (http://www.geneontology.org). Mapping between genes and GO entries was obtained from the NCBI Gene database (http://www.ncbi.nlm.nih.gov/gene).
2.12 Accession numbers
RNA-seq data are available on the Gene Expression Omnibus website (https://www.ncbi.nlm.nih.gov/geo) using accession number GSE129286.
2.13 Data analyses and presentation
Normality was tested with the Shapiro–Wilk normality test. Statistical comparisons of data exhibiting a normal distribution were conducted using the Student’s t test for pairwise testing and one-way ANOVA followed by post hoc hypothesis testing between interventions using a Tukey test for multiple comparisons. If a group failed normality testing, pairwise testing was performed with a non-parametric Mann–Whitney U test. Kaplan–Meier curves were analysed using the log rank test. P < 0.05 was defined as statistically significant and is indicated by filled asterisks. *P < 0.05, **P < 0.01, ***P < 0.001. NS = not significant. All error bars represent SEM. In all western blot images shown, individual lanes correspond to individual mice or independent samples from cell culture experiments. Molecular weights associated with the bands are shown.
3. Results
3.1 Cardiac-specific Trx1 deletion induces early death
The strategy used for conditional knockout of mouse Trx1 is shown in Figure 1A. Ideally, an exon floxed to generate a conditional knockout should, when deleted, induce a frameshift, thereby generating an artificial stop codon. mRNA with an artificial stop codon is often degraded by non-sense mediated mRNA decay. However, deletion of none of the exons of Trx1 induces a frameshift because all use the same frame. Targeting Exon 1 is also a possible strategy, but it occasionally affects or diminishes promoter activity because a loxP sequence is inserted in the promoter region. Therefore, we floxed Exons 2 and 3, deleting more than half of Trx1, including the catalytic centre (Cys 32 and 35) (Figure 1B). To achieve cardiac-specific deletion of Trx1, we crossed Trx1flox/flox mice with Tg-Myh6-Cre. To elucidate how the deletion of Exons 2 and 3 affects Trx1 mRNA structure and expression, RNA-sequencing was performed. Exon level visualization showed reduced expression of mRNA corresponding to Exons 2 and 3 in the Trx1cKO mice, whereas expression of other exons was intact (Figure 1C). These results suggest that the Trx1cKO mice express a truncated form of Trx1 mRNA, which may produce a truncated Trx1 protein (Trx1Δ9-64) that, in theory, lacks the catalytic centre but retains two cysteine resides (Cys 69 and 73) (Figure 1D). As shown in Figure 1E, cardiac-specific Trx1 homozygous knockout (Trx1cKO) mice on the C57BL/6 genetic background were viable. In contrast, embryonic lethality was reported for global Trx1 knockout mice on the 129 genetic background.5 Embryonic lethality could be specific to mice with the 129 genetic background but not C57BL/6. To address this, we used a global Trx1 knockout mouse line that had been backcrossed to the C57BL/6 genetic background more than 12 times. However, even after backcrossing, we could not obtain any Trx1 homozygous knockout mice, suggesting that global Trx1 homozygous knockout causes a development defect leading to embryonic lethality in both the 129 and C57BL/6 genetic backgrounds, but cardiac-specific Trx1 knockout does not. Reduced Trx1 protein was observed by western blot analysis using an anti-Trx1 antibody with an antigen that includes the floxed region (Figure 1F). As shown in Figure 1G, Trx1cKO mice started dying at 13 days of age. The median survival age was 25.5 days. None of the mice survived longer than 58 days. This high mortality rate was not observed in the heterozygous knockout mice. These results indicate that cardiac-specific Trx1 homozygous deletion causes early death.
Figure 1.

Cardiac-specific Trx1 deletion causes early death. (A) Schematic representation of the strategy for knockout of Trx1. (B) Exon 2 contains the catalytic centre (Cys 32 and 35), indicated by yellow triangles. Other cysteine residues are indicated by grey triangles. (C) Trx1 mRNA lacking Exons 2 and 3 are expressed in Trx1cKO mice. (D) Schematic representation of the truncated Trx1 mRNA expressed in the cKO mice. (E) Mendelian ratios of Trx1cKO and global Trx1KO mice. Genotype of mating pairs and number of animals for the indicated mouse genotypes are shown. Genomic DNA for genotyping of pups was extracted at 1–3 weeks of age. (F) Full length Trx1 protein is down-regulated in the cKO mice. (G) High mortality of the Trx1cKO mice. Statistical analysis was performed with the Kaplan–Meier log rank test.
3.2 Cardiac-specific Trx1 deletion induces heart failure
The hearts of Trx1cKO mice were enlarged even at 6 days of age (Figure 2A). To evaluate cardiac function, echocardiography was performed at 2–5 weeks of age. Trx1cKO mice showed cardiac systolic dysfunction compared to wild-type mice, as evidenced by a reduced left ventricular (LV) ejection fraction (Figure 2B and C). Trx1cKO mice showed an increased LV end diastolic diameter and decreased ventricular wall thicknesses compared to wild-type mice (Supplementary material online, Table S1). The lung weight/tibia length and heart weight/tibia length ratios, indicators of lung congestion, a sign of heart failure, and cardiac hypertrophy, respectively, were enhanced in Trx1cKO mice compared to in wild-type mice (Figure 2D and E). Organ weight measurements are summarized in Supplementary material online, Table S2. Histological analyses with WGA staining showed increased cardiomyocyte size, indicating cardiac hypertrophy (Figure 2F). PASR staining showed increased cardiac fibrosis in Trx1cKO mice compared to in wild-type mice (Figure 2G). There were significantly more TUNEL-positive cardiomyocytes in Trx1cKO mice than in wild-type mice (Figure 2H). In addition, the level of cleaved caspase 3, a marker of apoptosis, was significantly elevated in Trx1cKO mice (Figure 2I). These results suggest that cardiac-specific homozygous deletion of Trx1 results in heart failure with increased cardiac hypertrophy and apoptosis. In contrast, the cardiac phenotype of cardiac-specific heterozygous Trx1 KO mice was not significantly different from that of wild-type mice.
Figure 2.

Cardiac-specific Trx1 deletion induces failing heart phenotypes. (A) H&E stained images of samples from Trx1cKO mice at 6 days of age. (B) Representative M-mode echocardiography images taken from Trx1cKO mice at 4 weeks of age. WT, wall thickness’ LVEDD, LV end diastolic dimension; LVESD, LV end systolic dimension; DSEP, diastolic septal; DP, diastolic posterior; SP, systolic posterior; SSEP, systolic septal. (C) Contractile dysfunction in Trx1cKO mice. Ejection fraction, an index of cardiac contractile function, was determined by echocardiographic analysis. (D) Lung congestion, characterized by lung weight/tibia length ration, is induced in Trx1cKO mice. (E) Cardiac hypertrophy, characterized by heart weight/tibia length ratio, is induced in Trx1cKO mice. (F) WGA (wheat germ agglutinin) staining was performed to assess cardiomyocyte size in Trx1cKO mice. (G) Cardiac fibrosis in Trx1cKO mice. Representative PASR staining. Relative cardiac fibrosis was measured using the ImageJ program. (H) Increased apoptotic cell death in Trx1cKO mice. TUNEL staining was performed in Trx1cKO mice. (I) Increased cleaved caspase 3 in Trx1cKO mice. The numbers of mice examined in each experimental group were: 5–7 (C), 7–13 (D–E), and 5–6 (F), 5 (G), and 5–6 (H). Statistical significance was determined with the Student’s t test (C, D, F, G, H and I) and Mann–Whitney U test (E). *P<0.05, **P<0.01, and ***P<0.001.
3.3 Cardiac-specific Trx1 deletion induces oxidative stress
To investigate whether loss of Trx1 affects expression of other antioxidants, we conducted RNA-sequencing analyses (Figure 3A). The apparent absence of a reduction in the expression of Trx1 (Txn1) in the Trx1cKO mice is due to detection of the undeleted portions of the Trx1 mRNA (Figure 1C). Txnrd1, a major Trx1 reductase, was significantly up-regulated in Trx1cKO mice, suggesting the presence of a compensatory mechanism. In addition, catalase (Cat), glutathione peroxidase 1 (Gpx1), and Srxn1 were also up-regulated in Trx1cKO mice. On the other hand, Prdx2 and SOD2 were down-regulated. These results suggest that the loss of Trx1 differentially regulates expression of reducing enzymes. To investigate whether oxidative stress is induced in Trx1cKO mice, we examined the redox status of Prdx1, a Trx1 substrate. It has been suggested that Prdx1 forms oligomers upon oxidation due to disulphide bond formation and that Trx1 reduces Prdx1 oligomers to monomers. Contrary to expectation, however, non-reducing SDS-PAGE showed that Prdx1 formed dimers in cardiomyocytes under basal conditions and that H2O2 treatment increased monomers and decreased dimers (Figure 3B). These results indicate that the redox cycle of Prdx1 in cardiomyocytes is distinct from the conventional model. Consistently, Prdx1 exhibited an increased monomer/dimer ratio in Trx1cKO mice (Figure 3C). The increased Prdx1 monomers in Trx1cKO mice may be due to sulfinic or sulphonic acid formation, since these oxidative modifications prevent disulphide bond formation. Indeed, we observed increased sulphonic acid formation on Prdx1 in Trx1cKO mice (Figure 3D). Biotin-iodoacetamide labeling indicated that there was less protein with free thiols in Trx1cKO mice than in wild-type mice (Figure 3E). Moreover, the amount of protein oxidative adducts, such as 4HNE (4-Hydroxynonenal) and DiTyrosine, was significantly elevated in Trx1cKO mice compared to in wild-type mice (Figure 3F and G). These results suggest that loss of Trx1 induces oxidative stress in the heart.
Figure 3.

Trx1 is required for redox regulation in the heart. (A) Expression levels of reducing enzymes in Trx1cKO mice. Expression levels of indicated reducing enzymes were examined with RNA-seq. The mean value from wild-type mice was expressed as 1 (N = 3). (B) Increased monomer form of Prdx1 in response to H2O2 in primary cultured cardiomyocytes. Cardiomyocytes were incubated with 100 μM H2O2 for 30 min. Cell lysates were resolved by SDS-PAGE under non-reducing conditions and subjected to western blot analysis. (C) Increased monomer/dimer ratio of Prdx1 in Trx1cKO mice. Heart lysates from Trx1cKO mice were subjected to SDS-PAGE under non-reducing conditions and subsequent western blot analysis. Monomer and dimer signal intensities were quantified (Right). (D) Increased sulphonic acid formation in Prdx1 in Trx1cKO mice. (E) Decrease in reduced protein thiols in Trx1cKO mice. Reduced protein thiols were labelled with biotin-IAM (iodoacetamide). The labelled proteins were visualized with HRP-streptavidin. (F–G) Increased oxidative protein adducts such as 4HNE (4-hydroxynonenal) (F) and DiTyrosine (G). N5–6 (C and D) and 6 (E–G). Statistical significance was determined with the Student’s t test (A–G). *P<0.05, **P<0.01, and ***P<0.001.
3.4 Cardiac-specific Trx1 deletion impairs metabolic gene expression
To gain insight into the reduced cardiac function and elevated oxidative stress observed in Trx1cKO mice, we performed a bioinformatics investigation of the RNA-sequencing results. Gene ontology analysis revealed that downregulation of genes involved in energy production, referred to as oxidation-reduction process, ATP metabolic process and hydrogen transport in this analysis, was the most significant change in Trx1cKO mice (Supplementary material online, Table S3A). Transcription factors/co-factors whose targets were significantly down-regulated in Trx1cKO mice included PPARα, PPARγ, PGC-1α, and ERRα (Supplementary material online, Table S3B). These transcriptional regulators play a crucial role in metabolic gene expression in the heart. On the other hand, transcription factors whose targets were significantly up-regulated in Trx1cKO mice included HTT, p53, MTPN, TWIST1, and NR3C2 (Supplementary material online, Table S3C). The extent of downregulation of metabolic genes in Trx1cKO mice is shown in Figure 4A. The metabolic genes involved in fatty acid metabolism, the mitochondrial citric acid cycle, and the electron transport chain were widely down-regulated in Trx1cKO mice. We confirmed the downregulation of mitochondrial proteins, such as Atp5a, Uqcrc, Sdhb, and Ndfub8, with immunoblot analyses (Figure 4B). In contrast, the levels of Drp1 and Mfn2, regulators of mitochondrial fission and fusion, respectively, did not change significantly. To investigate whether Trx1 deletion induces mitochondrial abnormality, we examined the mitochondrial morphology by electron microscopy (Figure 4C). Mitochondria were significantly smaller in Trx1cKO mice, whereas the aspect ratio (long axis/short axis) was significantly enlarged. The number of mitochondria did not differ significantly between control and Trx1cKO mice. Thus, downregulation of metabolic genes is associated with mitochondrial morphological abnormalities in Trx1cKO mice.
Figure 4.

Downregulation of genes involved in fatty acid metabolism and mitochondrial ATP production in Trx1cKO mice. (A) Expression levels of the indicated genes were examined with RNA-seq. The mean value from wild-type mice was expressed as 1 (N = 3). (B) Downregulation of indicated mitochondrial proteins, as shown by western blot analyses. (C) Electron microscopic analyses of Trx1cKO mice. Representative electron microscopic images are shown. Mitochondrial size, aspect ratio, and number were determined from the electron microscopic images (N = 5). Statistical significance was determined with the Student’s t test (A and C). *P<0.05, **P<0.01, and ***P<0.001.
3.5 Endogenous Trx1 protects against stress
Since homozygous Trx1cKO mice develop heart failure with early death, we used cardiac-specific Trx1 heterozygous knockout mice to examine the role of endogenous Trx1 in regulating cardiomyocyte responses to stresses. The hetero Trx1cKO mice were subjected to PO by transverse aortic constriction (TAC), a model of heart failure. The results of echocardiographic and organ weight measurements in hetero Trx1cKO mice under PO conditions are summarized in Supplementary material online, Tables S4 and S5. PO-induced cardiac dysfunction, indicated by reduced ejection fraction and increased lung congestion, was exacerbated in hetero Trx1cKO mice (Figure 5A and B). Histological analyses indicated that the PO-induced increase in cardiomyocyte size, an index of cardiomyocyte hypertrophy, was enhanced in hetero Trx1cKO mice (Figure 5C). In contrast, PO-induced increases in cardiac fibrosis and apoptotic cell death were not significantly changed in Trx1cKO mice compared to in wild-type mice (Figure 5D and E). Immunoblot analyses of cardiac homogenates indicated that the ratio of Prdx1-SO3H/total Prdx1 was significantly increased in Trx1cKO mice under PO conditions (Figure 5F). Mitochondrial proteins, including Atp5a, Uqcrc2, Sdhb and Ndufb8, were decreased in hetero Trx1cKO mice at baseline and in response to PO (Figure 5G). Thus, modest mitochondrial dysfunction may exist in hetero Trx1cKO mice. Autophagy may affect survival and death of cardiomyocytes.14 The level of LC3-II tended to decrease at baseline, whereas it was increased in response to PO in hetero Trx1cKO mice compared to in control mice. The level of p62, a protein degraded by autophagy, was greater in hetero Trx1cKO mice than in control mice at baseline and in response to PO (Figure 5H). These results suggest that autophagy is negatively regulated in hetero Trx1cKO mice at baseline and in response to PO. Taken together, these results suggest that endogenous Trx1 confers stress resistance to the heart and maintains mitochondrial protein expression and autophagy under stress conditions.
Figure 5.

Cardiac-specific Trx1 heterozygous knockout mice are susceptible to pressure overload (PO)-induced heart failure. Pressure overload was applied to mice at 3–9 months of age (Wild: 5.5 ± 2.0, Wild TAC: 6.1 ± 0.9, Trx1cKO: 5.6 ± 1.4, Trx1cKO TAC: 5.9 ± 1.2) (A) Ejection fraction, (B) lung congestion, (C) cardiomyocyte cell size, (D) cardiac fibrosis, and (E) apoptotic cell death were examined in hetero Trx1cKO mice under PO conditions. (F) Increased Prdx1 with sulphonic acid formation in hetero Trx1cKO mice under PO conditions. (G) Downregulation of mitochondrial proteins in hetero Trx1cKO mice under PO conditions. (H) The level of LC3-II and p62 was evaluated with immunoblot analyses. The numbers of mice examined in each experimental group were: 5–8 (A), 5–6 (B), 5–6 (C), 5 (D), and 5–6(E–G). Statistical significance was determined with the Student’s t test (B, D, E, and F) and one-way ANOVA followed by Tukey test (A, C, G, and H). *P<0.05, **P<0.01, and ***P<0.001.
3.6 Dysregulation of mTOR signalling in Trx1cKO mice
Trx1 reduces proteins with disulphide bonds through thiol disulphide exchange reactions.15 Among Trx1 substrates, we focused on mTOR because muscle- and cardiac-specific mTOR knockout mice show a cardiac phenotype similar to that of Trx1cKO mice, including heart failure and early death.13,16 As we reported previously,9 upon H2O2 treatment, mTOR exhibited band shifts to higher molecular weights on SDS-PAGE gels under non-reducing conditions (Figure 6A). The band shifts were abolished under standard SDS-PAGE conditions using a sample loading buffer containing 2-mercaptoethanol (2ME), a reducing agent that cleaves disulphide bonds. These results indicate that mTOR forms intermolecular disulphide bonds upon oxidation. The band shift was also induced by Trx1 knockdown with an adenovirus vector carrying Trx1 short hairpin RNA (shTrx1) in cardiomyocytes. Unlike the mTOR band shift caused by either H2O2 or shTrx1, the band shift in Trx1cKO mice was not obvious after a short exposure (SE) of the gel. However, after a long exposure (LE), the band shift of mTOR to higher molecular weights (indicated by white arrow heads) was observed in Trx1cKO under baseline conditions but not in wild-type mice. These results suggest that endogenous Trx1 reduces and cleaves disulphide bonds in mTOR and that mTOR is more oxidized in Trx1cKO mice. To investigate whether mTOR signalling is inhibited in Trx1cKO mice, phosphorylation levels of mTOR and its substrates, including S6K and 4EBP1, were evaluated. Whereas phosphorylation of mTOR at Ser 2481 was not significantly affected, that of S6K and 4EBP1 was inhibited in Trx1cKO mice (Figure 6B), suggesting that loss of Trx1 inhibits mTOR signalling in the heart in vivo. Since Trx1cKO mice developed heart failure and mitochondrial dysfunction is a hallmark of heart failure, we investigated whether loss of Trx1 induces mitochondrial dysfunction in cardiomyocytes in a cell autonomous manner. To this end, we knocked down Trx1 in cardiomyocytes with shTrx1 and performed reporter gene assays, using luciferase reporter genes driven by the promoters of metabolic genes. Since mTOR enhances protein translation through a 5′ untranslated region (UTR)-dependent mechanism,17 we included both the promoter region and the majority of the 5′ UTR in all reporter constructs used in this study (Figure 6C). We chose promoters of metabolic genes whose mRNA levels are down-regulated in Trx1cKO mice, including Ndufa1, Ndufs1, Acot2, and Fbp2 (Figure 4 and data not shown). The IL-6 promoter served as a negative control because IL-6 was not down-regulated in Trx1cKO mice (data not shown). Both shTrx1 and rapamycin inhibited the promoter activity of Ndufa1, Ndufs1, Acot2, and Fbp2, but did not significantly inhibit that of IL-6 (Figure 6D). These results suggest that both loss of Trx1 and rapamycin suppress mitochondrial gene expression in cardiomyocytes in a cell autonomous manner. We investigated mitochondrial oxygen consumption to evaluate to what extent downregulation of the metabolic genes affects mitochondrial function. As shown in Figure 6E, shTrx1 inhibited the basal, ATP-production-coupled, and maximum oxygen consumption rates, indicating impaired mitochondrial function. These results suggest that downregulation of endogenous Trx1 suppresses mTOR signalling and mitochondrial function in cardiomyocytes. Although mTOR was inhibited in homozygous Trx1cKO mice, LC3II, or p62 levels did not differ significantly between homozygous Trx1cKO and control mice at baseline (Figure 6F), suggesting that autophagy is not altered significantly in homozygous Trx1cKO mice at baseline.
Figure 6.

mTOR signalling is inhibited in Trx1cKO mice. (A) Enhanced oxidation of mTOR in Trx1cKO mice. Oxidation of mTOR was examined by SDS-PAGE under non-reducing conditions and subsequent western blot analysis. (Top) Cardiomyocytes were treated with 100 μM H2O2 for 30 min and shTrx1 for 7 days. The H2O2-induced mTOR band shift was abolished with 2ME. (Bottom) The band shift of mTOR in Trx1cKO mice. Ratio of oxidized (higher molecular weight) and reduced form of mTOR band intensity is indicated. (B) Inhibition of mTOR signalling in Trx1cKO mice. The phosphorylation status of mTOR substrates such as S6K and 4EBP1 was examined. (C) Schematic representation of reporter gene constructs. TSS, transcription start site; UTR, untranslated region. (D) Reporter gene assays were performed with luciferase reporter genes driven by the endogenous promoter sequences of the indicated genes. (E) Trx1 knockdown inhibits mitochondrial respiration. Oxygen consumption was measured in cardiomyocytes treated with shTrx1. Basal, ATP-production-coupled and maximum oxygen consumption rates were measured. (F) Autophagy is not significantly altered in homozygous Trx1cKO mice. The level of LC3-II and p62 was evaluated with immunoblot analyses. N=5 (A), 6–7 (B), 6–8 (D) 8 (E, upper), 24 (E, lower), and 7 (F). Statistical significance was determined with Mann–Whitney U test (D, Ndufa1 and F, p62/Tubulin). Besides this, statistical significance was determined with the Student’s t test. *P<0.05, **P<0.01, and ***P<0.001.
3.7 Oxidative inhibition of mTOR via Cys1483 contributes to impaired mitochondrial gene expression resulting from loss of Trx1
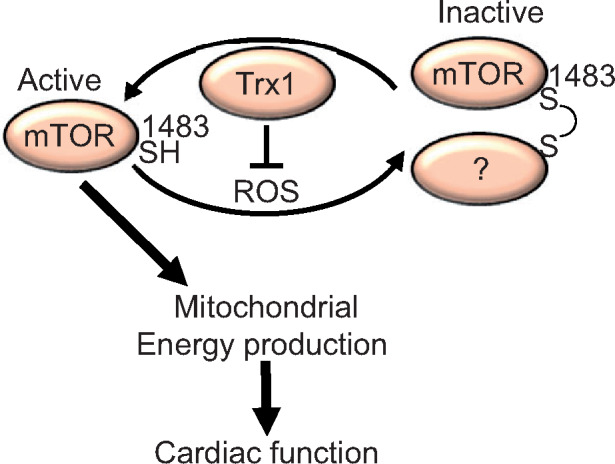
Since H2O2-induced oxidation at Cys1483 inhibits mTOR,9 we hypothesized that the loss of Trx1 promotes oxidation at Cys1483, leading to inhibition of mTOR. To test this, Myc-tagged mTOR-C1483F, an oxidation-resistant mutant, was expressed in the presence or absence of shTrx1. Since an adenovirus vector harbouring mTOR does not work in primary cultured cardiomyocytes and H9c2 cells due to technical difficulties, we employed lipofection to introduce mTOR and its mutant. Although Trx1 knockdown induced a band shift of wild-type mTOR, it did not shift the C1483F mTOR mutant, suggesting that mTOR is oxidized at Cys1483 when Trx1 is down-regulated (Figure 7A). To investigate the role of Cys1483 oxidation in mTOR inhibition in the absence of Trx1, HA-, and GST-tagged S6K was co-transfected with the mTOR-C1483F mutant. Although HA-GST-S6K phosphorylation by exogenous Myc-mTOR was inhibited in the presence of shTrx1, phosphorylation by mTOR-C1483F was not affected by shTrx1 in H9c2 cells or primary cultured cardiomyocytes (Figure 7B and C). These results suggest that Cys1483 is responsible for mTOR oxidation and inhibition in the absence of Trx1. To test whether oxidation at Cys1483 is responsible for metabolic gene suppression in the presence of Trx1 downregulation, reporter gene assays were performed. Downregulation of Trx1 inhibited Ndufa1 and Ndufs1 promoter activity even when wild-type mTOR was overexpressed, but not in the presence of mTOR-C1483F (Figure 7D). These results suggest that Trx1 maintains metabolic gene expression partly through maintenance of mTOR function by preventing oxidation of mTOR at Cys1483 (Figure 7E).
Figure 7.

Oxidation at Cys1483 of mTOR mediates inhibition of metabolic gene promoter activity in the absence of Trx1. (A) Cys1483 forms an intermolecular disulphide in response to Trx1 knockdown. Myc-mTOR-C1483F was expressed in H9c2 cells via plasmid vector transfection, and then Trx1 was knocked down with shTrx1 adenovirus. Ratio of oxidized/reduced form of mTOR is indicated (N = 5). (B–C) Trx1 knockdown-induced mTOR inhibition is normalized by mTOR-C1483F. Myc-mTOR-C1483F and GST-HA-S6K were expressed in H9c2 cells (B) and cardiomyocytes (C) via plasmid vector transfection, and then Trx1 was knocked down with shTrx1 adenovirus. Phosphorylation of GST-HA-S6K was examined. (B, Right) Densitometric analysis of phosphorylated S6K in H9c2 cells (N = 8–9). (D) Trx1 knockdown-induced inhibition of Ndfua1 and Ndufs1 reporter gene activity is normalized by mTOR-C1483F (N = 6). (E) Schematic representation of Trx1-mediated metabolic gene expression. Trx1 prevents oxidation at Cys1483 in mTOR, thereby maintaining mTOR-induced metabolic gene expression. Statistical significance was determined with one-way ANOVA followed by Tukey test (A, B, and D). *P<0.05 and ***P<0.001.
4. Discussion
Our study demonstrates, for the first time, that Trx1 is required to maintain pump function and redox homeostasis in the adult heart at baseline and in response to stress. Using unbiased gene expression analyses, we further demonstrate that endogenous Trx1 is an essential mediator of metabolic gene expression. In addition, the current study clearly demonstrates that mitochondrial dysfunction is the major functional consequence of cardiomyocyte-specific downregulation of endogenous Trx1 in vivo. Furthermore, we show that mTOR oxidation at Cys1483 causally mediates mitochondrial dysfunction induced by Trx1 downregulation in cardiomyocytes.
4.1 Trx1 is indispensable for redox homeostasis in the heart
RNA-sequencing demonstrated that loss of Trx1 affects expression of other antioxidants (Figure 3A). A trend of overall upregulation of glutathione peroxidases (Gpxs) and glutathione reductase (GSR) in Trx1cKO mice suggests that upregulation of the glutathione-dependent reducing system may be compensatory for loss of Trx1. In Escherichia coli, genetic deletion of both Trx and the glutathione system is lethal, whereas E.coli with either Trx1 or the glutathione system is viable.18 Whether and how glutathione is regulated in Trx1cKO mice is an important subject for future investigation. Although several anti-oxidative systems, including catalase and glutathione, were up-regulated, oxidative stress was consistently found to be increased in Trx1cKO mice (Figure 3), suggesting that Trx1 is indispensable for redox homeostasis in the heart.
4.2 Trx1 is indispensable for metabolic gene expression in the heart
Unbiased gene expression profiling revealed that Trx1 is indispensable for metabolic gene expression in the heart. Since metabolic gene downregulation is commonly observed during heart failure, it could arguably be a secondary effect due to heart failure in Trx1cKO mice. However, Trx1 knockdown in cardiomyocytes also down-regulated mitochondrial function in a cell autonomous manner (Figure 6C), and we have shown previously that cardiac-specific Trx1 overexpression up-regulates metabolic genes without significantly affecting cardiac function under basal conditions.19 Thus, Trx1 affects metabolic genes independently of cardiac function. Then what is the underlying mechanism? We propose that Trx1 maintains metabolic gene expression through mTOR. Muscle-specific mTOR knockout with MCK-Cre in mice produces a cardiac phenotype similar to that of Trx1cKO mice, including early death with a 26.4-day-median life span, cardiac dilation, apoptotic cell death, and downregulation of several metabolic genes and regulators, including Cpt1, PPARα, and PGC-1α.13 Cardiac-specific mTOR deletion (mTORcKO) at 8 weeks of age using an inducible knockout strategy also caused cardiac contractile dysfunction, cardiac hypertrophy, mitochondrial morphological abnormality, apoptotic cell death, early death, and impaired mTOR signaling.16 These results are consistent with the notion that the cardiac phenotypes and early death observed in Trx1cKO mice may be attributed, in part, to impaired mTOR function. Importantly, we have shown previously that H2O2 inhibits mTOR via intermolecular disulphide bond formation at Cys1483, thereby downregulating metabolic genes in cultured cardiomyocytes.9 Trx1 protects mTOR against exogenous oxidants such as H2O2. We here show that loss of endogenous Trx1 alone is sufficient to induce mTOR Cys1483 oxidation and mitochondrial dysfunction. Trx1 downregulation-induced inhibition of Ndufa1 and Nsufs1 transcription and mTOR signalling was rescued by overexpression of oxidation-resistant mTOR (mTOR-C1483F), but not wild-type mTOR, in cardiomyocytes in vitro. These results suggest that downregulation of mitochondrial genes in Trx1cKO mice in vivo may also be mediated primarily through oxidation-induced suppression of mTOR.
4.3 Phenotypic variations between cardiac- and liver-specific Trx1 knockout mice
In contrast to cardiac-specific Trx1 knockout mice, liver-specific Trx1 knockout mice do not exhibit redox imbalance or abnormalities in liver function.6 The heart is metabolically more active than the liver and harbours a large volume of mitochondria to support continuous pump function. Mitochondrial damage in cardiomyocytes in the absence of Trx1 would thus dramatically facilitate a vicious cycle of mitochondrial dysfunction and oxidative stress and readily cause cardiac dysfunction. We propose that reliance on Trx1 at baseline differs substantially among organs and that Trx1 may be more important in organs with a high abundance of mitochondria.
4.4 Trx1 substrates may mediate cardiac phenotypes in Trx1cKO mice
Trx1 is an essential co-factor for its binding partners and substrates, including ASK1, PTEN, RelA, p53, class II HDACs, AMPK, and mTOR.7 Abnormalities observed in Trx1cKO mice may be due, in part, to loss of Trx1 interaction with other binding partners besides mTOR as well. Although protein synthesis is negatively regulated in the presence of mTOR inhibition, cardiac hypertrophy was observed in homozygous Trx1cKO mice. Since inducible mTOR cKO mice also exhibit cardiac hypertrophy,16 hypertrophy may develop through mTOR-independent mechanisms to compensate for LV dysfunction. In addition, class II HDACs play a crucial role in inhibition of cardiac hypertrophy20 and, thus, may be involved in the cardiac hypertrophy observed in Trx1cKO mice. Since Class II HDAC function is also regulated through phosphorylation, ubiquitination, and sumoylation, the extent to which Trx1 regulates Class II HDACs in the heart in vivo remains to be elucidated. Moreover, several lines of evidence suggest that oxidation of sarcomere proteins decreases contractile force and calcium sensitivity.21 Thus, loss of Trx1 may promote contractile dysfunction through oxidation of sarcomere proteins. Trx1cKO mice would be a useful tool to elucidate the role of endogenous Trx1 and its interaction partners.
4.5 Limitation of the Trx1 conditional knockout strategy
In this study, we deleted Exons 2 and 3 of Trx1 to generate conditional Trx1 KO mice. However, a truncated form of Trx1 mRNA lacking Exons 2 and 3 is expressed in these mice (Figure 1C). Unfortunately, because the commercially available anti-Trx1 antibody (which recognizes the amino acid stretch encoded by Exons 4 and 5) we used could not detect endogenous Trx1 even from wild-type mice (data not shown), we could not determine whether the truncated Trx1 protein is expressed in our Trx1cKO mice. Our results clearly indicate, however, that the cardiac-specific complete loss of oxidoreductase activity achieved by deletion of Exons 2 and 3 is sufficient to stimulate oxidative stress and suppress mTOR, thereby causing cardiac dysfunction. Although we wished to test whether increased oxidative stress precedes heart failure in homozygous Trx1cKO mice, LV dysfunction was already observed at Day 6 after birth, the earliest time point at which we could evaluate LV function. Thus, whether LV and mitochondrial dysfunction is secondary to elevated oxidative stress or vice versa remains to be elucidated.
In summary, cardiac-specific deletion of Trx1 results in cardiac dysfunction and early death, suggesting that endogenous Trx1 is essential for maintaining cardiac function and animal survival. We here showed that downregulation of Trx1 in the heart induces prominent downregulation of metabolic genes. mTOR was oxidized and inhibited in Trx1cKO mice, which may contribute to mitochondrial abnormality and failing cardiac phenotypes. Trx1cKO mice are useful to evaluate the functional significance of endogenous Trx1 and its targets during cardiac stress.
Authors’ contributions
S.-I.O. performed the majority of experiments, including design of the conditional knockout strategy, maintenance of animal colonies with breeding and genotyping, generation and preparation of adeno and plasmid vectors, and experiments with cultured cells such as exogenous gene expression and reporter gene assays. S.-I.O. and A.C. measured organ weights. A.C., F.T., and M.Z. analysed histochemical and electron microscopic images. J.N. evaluated autophagy. J.Y.P. and B.T. analysed RNA-sequencing results. S.I. and W.M. conducted echocardiographic measurements. P.Z. performed TAC surgery. M.T. took electron microscopic images. T.K. measured oxygen consumption in cardiomyocytes. S.-I.O., A.C., G.R., J.B., F.T., Y.E., and C.-Y.H. performed western blot analyses. J.Y. oversaw generation of systemic Trx1 knockout mice. Y.H. backcrossed the systemic Trx1 knockout mice to C57BL/6 genetic background. J.S. conceived and oversaw the study. S.-I.O. and J.S. wrote the manuscript.
Supplementary Material
Acknowledgement
The authors thank Daniela Zablocki for critical reading of the manuscript.
Conflict of interest: none declared.
Funding
This work was supported in part by American Heart Association (AHA) Scientist Developmental Grant (12SDG11890014 to S.-I.O.), Grant in Aid (17GRNT33440031 to S.-I.O.), Transformational Project Award (19TPA34850170 to S.-I.O.), New Jersey Health Foundation research grants (PC56-16, PC80-17 to S.-I.O.), the Fondation Leducq Transatlantic Network of Excellence (15CBD04 to J.S.), and US Public Health Service Grants (HL67724, HL91469, HL112330, HL138720, AG23039 to J.S.).
Translational perspective
Although cell protective effects of Trx1 have been demonstrated previously, the in vivo function and the direct target of endogenous Trx1 remain to be elucidated. Using cardiac-specific Trx1 KO mice, this study demonstrates that endogenous Trx1 plays an essential role in maintaining cardiac function and redox homeostasis and confers stress resistance to the heart. The salutary effect of Trx1 in the heart is primarily mediated through reduction of mTOR in vivo.
References
- 1. Rhee SG. Overview on peroxiredoxin. Mol Cells 2016;39:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jeong W, Bae SH, Toledano MB, Rhee SG.. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic Biol Med 2012;53:447–456. [DOI] [PubMed] [Google Scholar]
- 3. Benhar M, Forrester MT, Hess DT, Stamler JS.. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 2008;320:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu C, Liu T, Chen W, Oka S, Fu C, Jain MR, Parrott AM, Baykal AT, Sadoshima J, Li H.. Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol Cell Proteomics 2010;9:2262–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM.. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol 1996;178:179–185. [DOI] [PubMed] [Google Scholar]
- 6. Prigge JR, Coppo L, Martin SS, Ogata F, Miller CG, Bruschwein MD, Orlicky DJ, Shearn CT, Kundert JA, Lytchier J, Herr AE, Mattsson A, Taylor MP, Gustafsson TN, Arner ESJ, Holmgren A, Schmidt EE.. Hepatocyte hyperproliferation upon liver-specific co-disruption of thioredoxin-1, thioredoxin reductase-1, and glutathione reductase. Cell Rep 2017;19:2771–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nagarajan N, Oka S, Sadoshima J.. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic Biol Med 2017;109:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J.. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 2008;133:978–993. [DOI] [PubMed] [Google Scholar]
- 9. Oka SI, Hirata T, Suzuki W, Naito D, Chen Y, Chin A, Yaginuma H, Saito T, Nagarajan N, Zhai P, Bhat S, Schesing K, Shao D, Hirabayashi Y, Yodoi J, Sciarretta S, Sadoshima J.. Thioredoxin-1 maintains mechanistic target of rapamycin (mTOR) function during oxidative stress in cardiomyocytes. J Biol Chem 2017;292:18988–19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shao D, Oka S, Liu T, Zhai P, Ago T, Sciarretta S, Li H, Sadoshima J.. A redox-dependent mechanism for regulation of AMPK activation by thioredoxin1 during energy starvation. Cell Metabol 2014;19:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P.. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007;450:736–740. [DOI] [PubMed] [Google Scholar]
- 12. Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St-Pierre J, Topisirovic I, Sonenberg N.. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab 2013;18:698–711. [DOI] [PubMed] [Google Scholar]
- 13. Zhang P, Shan T, Liang X, Deng C, Kuang S.. Mammalian target of rapamycin is essential for cardiomyocyte survival and heart development in mice. Biochem Biophys Res Commun 2014;452:53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J.. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 2016;133:1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holmgren A. Thioredoxin. Annu Rev Biochem 1985;54:237–271. [DOI] [PubMed] [Google Scholar]
- 16. Zhang D, Contu R, Latronico MVG, Zhang J, Zhang JL, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan K-L, Brown JH, Chen J, Sonenberg N, Condorelli G.. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest 2010;120:2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nandagopal N, Roux PP.. Regulation of global and specific mRNA translation by the mTOR signaling pathway. Translation (Austin) 2015;3:e983402.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Toledano MB, Kumar C, Le Moan N, Spector D, Tacnet F.. The system biology of thiol redox system in Escherichia coli and yeast: differential functions in oxidative stress, iron metabolism and DNA synthesis. FEBS Lett 2007;581:3598–3607. [DOI] [PubMed] [Google Scholar]
- 19. Ago T, Yeh I, Yamamoto M, Schinke-Braun M, Brown JA, Tian B, Sadoshima J.. Thioredoxin1 upregulates mitochondrial proteins related to oxidative phosphorylation and TCA cycle in the heart. Antioxid Redox Signal 2006;8:1635–1650. [DOI] [PubMed] [Google Scholar]
- 20. Oka S, Ago T, Kitazono T, Zablocki D, Sadoshima J.. The role of redox modulation of class II histone deacetylases in mediating pathological cardiac hypertrophy. J Mol Med 2009;87:785–791. [DOI] [PubMed] [Google Scholar]
- 21. Steinberg SF. Oxidative stress and sarcomeric proteins. Circ Res 2013;112:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.