

Abstract
In the current study, a structure-based virtual screening paradigm was used to screen a small molecular database against the Non-structural protein 15 (Nsp15) endoribonuclease of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). The SARS-CoV-2 is the causative agent of the recent outbreak of coronavirus disease 2019 (COVID-19) which left the entire world locked down inside the home. A multi-step molecular docking study was performed against antiviral specific compounds (~8722) collected from the Asinex antiviral database. The less or non-interacting molecules were wiped out sequentially in the molecular docking. Further, MM-GBSA based binding free energy was estimated for 26 compounds which shows a high affinity towards the Nsp15. The drug-likeness and pharmacokinetic parameters of all 26 compounds were explored, and five molecules were found to have an acceptable pharmacokinetic profile. Overall, the Glide-XP docking score and Prime-MM-GBSA binding free energy of the selected molecules were explained strong interaction potentiality towards the Nsp15 endoribonuclease. The dynamic behavior of each molecule with Nsp15 was assessed using conventional molecular dynamics (MD) simulation. The MD simulation information was strongly favors the Nsp15 and each identified ligand stability in dynamic condition. Finally, from the MD simulation trajectories, the binding free energy was estimated using the MM-PBSA method. Hence, the proposed final five molecules might be considered as potential Nsp15 modulators for SARS-CoV-2 inhibition.
Keywords: Nsp15 endoribonuclease, SARS-CoV-2, COVID-19, Virtual screening, Molecular dynamics
Graphical abstract
1. Introduction
With a clinical feature as “pneumonia of unknown etiology”, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection created an emergency situation since its outbreak was first testified in Wuhan City of Hubei Province in central China at end of December 2019 [1]. The disease caused by the SARS-CoV-2 was termed as novel coronavirus disease 2019 (COVID-19). Considering the severity of the disease and rapid infection scenario, the World Health Organization (WHO) announced SARS-CoV-2 infection/COVID-19 as pandemic on March 11, 2020 [2]. As of now, due to the lack of any therapeutic option to combat the SARS-CoV-2 infection or curative measures for COVID-19, it has been spread to almost every country in the world except very few nations are left to report infections and deaths [3]. As of January 11, 2021, the total number of confirmed COVID-19 cases was 88 828 387 across the globe among which 1 926 625 confirmed deaths were reported in ‘weekly operational updates on COVID-19’ by WHO (https://www.who.int/publications/m/item/). Hence, COVID-19 exigence triggers scientific communities to find out potential drug-like molecules to cure or prevent such pandemic disease. Although tremendous and continuous efforts are being incremented to get an effective way out of this pandemic situation, however no proper medication strategy or drug is available so far to confront COVID-19.
The proteome structure of SARS-CoV-2 consists of four structural proteins, two polyproteins and possibly nine accessory proteins [4]. The four structural proteins are Spike protein (S), Nucleocapsid protein (N), Membrane protein (M) and Envelope protein (E) which majorly responsible for viral assembly to virion structure constructions and maintaining structural integrity [5]. Interestingly, these protein sequences are highly similar to the sequences of the corresponding protein of SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) [6]. The sequence identity and similarity between SARS-CoV-2 and SARS-CoV were found to be 88 and 95%, respectively [7]. Such high-level sequence similarity can reveal a common pathogenesis mechanism, thus might help in developing potential drug therapeutics by targeting these proteins. On the other hand, two large polyproteins namely pp1a and pp1ab are further processed by viral proteases viz. 3C-like protease (3CLpro) and papain-like protease (PLP) [8]. In particular, the two polyproteins further resulted in the formation of 16 non-structural proteins (Nsps) [9]. Ultimately, the cleavage generated ~16 viral Nsps assembled into a large Replication-Transcription Complex (RTC) or replicase complex assembled in the double-membrane vesicles and demonstrates multiple enzymatic activities. Mostly, the functions of Nsps are associated with RNA replication and processing of subgenomic RNAs, however, the roles of some Nsps are still poorly understood or remain unknown.
Among such several Nsps, Nsp15 is one of the enigmatic enzymes that precisely belong to the EndoU family and it is a nidoviral RNA uridylate-specific endoribonuclease (NendoU) consisting of C-terminal catalytic domain [10]. The EndoU family enzymes are found in all the living kingdoms and participate in a number of biological functions linked to RNA processing. Whereas, the NendoU protein is found to be preserved with arteriviruses, toroviruses and coronaviruses, but it is lacking in mesoniviruses and roniviruses – which usually are the nonvertebrate-infecting representatives of nidoviruses order. Initially, it was assumed that Nsp15 straightforwardly join in viral replication, but further, it was revealed that Nsp15-deficient coronaviruses were also viable and capable of replication that creates confusion about its biological function [11]. Another recent study also proposed that NendoU activity of Nsp15 is highly accountable for protein interfering with the innate immune response [7]. It was also presumed that Nsp15 degrades viral RNA in order to enshroud it from the host defenses and act independently as exhibiting endonuclease activity [7]. In addition to the endoribonuclease activity poses by Nsp15, an animal model experiment revealed that it has shown immunomodulating properties during early viral infection [11]. Moreover, Nsp15 plays an important role in suppressing the type I IFN (Type I interferons) associated with innate immune response by infecting macrophages [12]. Further, being a unique nidoviral genetic marker, modulating the biological role of SARS-CoV-2 Nsp15 by small molecule can be expected to inhibit its close homolog of SARS-CoV or MERS-CoV Nsp15 as well. Nevertheless, Nsp15 is someway very essential in coronavirus biology, and hence strategy to inhibit Nsp15 of SARS-CoV-2 might lead towards a strong therapeutic option against COVID-19 [13].
Looking at the amino acid sequences and structural or conformational features of Nsp15 of SARS-CoV-2, it was indicated that six key residues (His235, His250, Lys290, Thr341, Tyr343 and Ser294) forming the catalytic site of Nsp15 are universally conserved among SARS-CoV, SARS-CoV-2 and MERS-CoV. The important chain architecture of this catalytic region is the side-chain conformations that are highly conserved except Lys290 of active site residues among all three proteins belonged to SARS-CoV, SARS-CoV-2 and MERS-CoV [7]. In particular, two basic amino acid residues, His235 and His250 are contributed to form the helical layer of the domain and each amino acid behaves as general acid and base, respectively. On the other hand, pair residues Lys290/Ser294 and Thr341/Tyr343 representing to form edges of the β-sheets [7]. Two residues, Ser294 and Tyr343 together are believed to govern Uridyl specificity [7]. Most importantly, based on the mutual arrangement to the active site residues of ribonuclease, three basic amino acids viz. His235, His250, and Lys290 of Nsp15 protein have been represented to establish as the catalytic triad [7,14]. Notably, a recent study suggested that amino acid residues, His235, His250, Lys290, and Thr341 have some putative role on the formation of intermolecular interactions which follow some triggering effect on interactions stabilization of the EndoU domain [7].
Like other viral Nsp15, the SARS-CoV-2 Nsp15 monomer is composed of three distinct domains such as N-terminal, middle and C-terminal domain [7]. More precisely, the N-terminal domain is consist of β-sheets (strands β1, β2, and β3) which enwrapped around two α-helices (α1 and α2). The consecutive middle domain is constituted by 3 β-hairpins (β5–β6, β7–β8, and β12–β13), a mixed β-sheet (β4, β9, β10, β11, β14, and β15), and 3 short helices [15]. The C-terminal catalytic NendoU domain comprises of two β-sheets (β16–β17–β18 and β19–β20–β21) [15]. Overall, the active or catalytic site of Nsp15 is situated in a shallow groove between the two β-sheets. Secondary structural elements of SARS-CoV-2 Nsp15 monomer have been presented in Figure S1 (Supplementary data). In the hexamer conformation, the Nsp15 of SARS-CoV-2 and SARS-CoV appears to differ structurally in the position of middle domain only which seems to be transposed out for creating a concave surface like structure that might be required for other protein interactions [7]. In general, the hexamer conformation of Nsp15 is majorly stabilized by the interaction formations at N-terminal oligomerization domains. Although, each subunit domain also contributed to the formation of an oligomer interface region for Nsp15 [7,16]. More specifically, hexamer conformation is potentially very sensitive as monomers have interacted extensively with all five other subunits of the hexamer [7,16,17]. Hence disrupting the monomeric or oligomeric assembly of Nsp15 by small molecules can be an eminent and distinct approach for modulating or inhibiting the Nsp15 endoribonuclease activity. The overall molecular surface model of the Nsp15 hexamer conformation (top and side view) has been displayed in Figure S2 (Supplementary data). Current global crusades to conquer COVID-19 pandemic, several computational approaches including drug repurposing or therapeutic switching or computational screening of large databases, and de novo designing of new molecules are being used and implemented using the world's most advanced high computing resources [[18], [19], [20], [21], [22], [23]]. Moreover, the drug development protocol possesses various key challenges and a high degree of uncertainty, and the success of computational drug discovery approaches will highly depend on the behavior of selective biomolecular targets and fetched out drug-like molecules. Therefore, a better understanding of the bottleneck challenges is very important before constructing a hypothesis to find out some potentially active drug-like molecules against COVID-19 in a limited time span with a lower risk of toxicity and higher efficacy. The prime objective of this pharmacoinformatics-based study is to identify a few potential small chemical entities from the Asinex antiviral database which can capable to firmly interact with active site residues of SARS-CoV-2 Nsp15 protein and hence disruption or modulation or inhibition of the endoribonuclease activity can be achieved. Therefore, in the present study, although a conventional structure-based drug design strategy has been followed, however, a rigorous and extensive analyses have been performed for Nsp15 of SARS-CoV-2 against compounds of Asinex antiviral database (http://www.asinex.com/antiviral/) through several advanced computational techniques such as hierarchical molecular docking, binding free energy estimation using Molecular Mechanics Generalized-Born Surface Area (MM-GBSA) approach, in silico pharmacokinetic assessment and all atomistic long-range molecular dynamics (MD) simulation. The study has been revealed the five potential drug-like compounds (N1–N5) that can modulate or inhibit the Nsp15 of SARS-CoV-2 upon interacting at the active site region with high binding affinity precision. However, their preclinical optimization may be necessary which can be evaluated further through various in-vivo and/or in-vitro experimental assays before considering as efficient therapeutic measures for the COVID -19 by targeting Nsp15.
2. Materials and methods
Pharmacoinformatics is an effective and widely accepted approach to the drug discovery scientific community due to its fast and trustworthy attitude towards finding and designing potential small molecules for a specific target. Structure-based virtual screening (SBVS) is an approach of pharmacoinformatics that has become a fast and cost-effective essential tool in drug discovery research [24]. Herein, the Asinex antiviral database was screened through Nsp15 endoribonuclease protein as a selective target. A number of advanced computational approaches included multi-step molecular docking, binding free energy calculation using MM-GBSA approach, in-silico pharmacokinetic assessment and MD simulation were used to select the best molecules.
2.1. Ligands and protein preparation
A total of 8722 small molecules were downloaded from the Asinex antiviral database in a structured data format (.sdf). Available molecules in the Asinex database belong to the natural product-like chemical entities with mainly polar functional groups and appropriate for exploration of hit-to-lead as well as fragment-based and receptor-based drug design, etc. Before using those molecules for any molecular modelling purposes, it is extremely essential to prepare the molecules such as to convert into the three-dimensional (3D) format, add the hydrogens and charge, and remove any bad valencies. For this purpose, the “LigPrep” [25] module of Schrödinger Suite was considered to prepare the molecules by following the default parameters and allowing them to generate a maximum number of 32 stereoisomers. The “Epik” tool [26] was applied to generate the protonation states of the molecules at physiological pH of 7.4.
The 3D crystal structure of the Nsp15 endoribonuclease was collected from the RCSB-Protein Data Bank (PDB) [27] (PDB ID: 6W01) [7]. Two important parameters such as resolution (1.90 Å) and R-value (0.185) of the protein were checked. The amino acid sequence length of the protein was 370 with no mutation. The “Protein Preparation Wizard” [28,29] tool of Schrödinger suite was used to prepare the protein. The missing atoms, side and backbone chains were repaired. The hydrogen atoms were added and water molecules deleted. Different parameters related to the connectivity of the molecules were corrected by the appropriate assignment of bond orders, formal charges and capping the protein terminals. To repair the missing and invalid amino acid residues the loops were refined. With the help of the PROPKA function of ‘Protein Preparation Wizard’, the protonation state of the protein was determined at close to the physiological pH. Finally, the protein molecule was minimized through the molecular mechanics force field, OPLS3 [30] and remove the steric clashes present in the protein structure. Followed by the protein preparation, the ‘Receptor Grid Generation’ panel of Glide (Grid-Based Ligand Docking with Energetics) [31] module of Schrödinger's suite interface was used to generate the grid. The binding or active site was selected as per the report of the published article on the crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2 (PDB ID: 6W01) [7]. As per the article information, the binding site of the Nsp15 present around the co-crystal citric acid and also confined the residues His235, His250, Lys290, Thr341, Tyr343 and Ser294. Hence, in the current study, the coordinate of one of the atoms of co-crystal citric acid was considered as the center of the grid and dimension selected in such a way so that all the above-mentioned amino acids should be confined within the grid box. Hence, the coordinates of the grid were selected as −64.929, 72.335, 29.026 Å along X-, Y- and Z-axes, respectively. Grid box dimension was considered as 19 × 19 × 19 Å along X-, Y- and Z-axes, respectively.
2.2. Virtual screening using multi-step molecular docking
The “Virtual Screening Workflow” (VSW) module available in the Schrödinger suite was adopted to screen the prepared molecular dataset obtained from the Asinex antiviral database. In particular, the VSW tool is projected to execute an entire sequence of user-defined jobs for screening large library databases of chemical compounds against one or more receptors or protein targets. The VSW utility module is integrated with the multi-step sequential molecular docking programs that included Glide-HTVS (high throughput virtual screening), Glide-SP (standard precision), and Glide-XP (extra precision). Each docking program follows the Emodel scoring function which has much weightage to pick the “best” pose of a ligand. Emodel [31] scoring function is one of the important molecular docking scoring functions and it is used by the Glide algorithm of the Schrödinger suite. The GlideScore [32] is a crucial function to differentiate between active over inactive small molecules. Glide score and protein-ligand coulomb-vdW energy are the main components of the Emodel scoring function. Moreover, GlideScore belongs to the empirical scoring function that amplifies the separation of small molecules with strong binding affinity from those with a little to no binding ability. In each step of docking, a systematic search was carried out to achieve the best orientation of the molecule. The VSW execution was performed in the CHPC server (https://www.chpc.ac.za/index.php/resources/lengau-cluster) available in Cape Town, South Africa. Particularly, the ligand set was given as .sdf file format under the “Input” tab of the VSW panel. The QikProp module was selected to filter out the molecules violating Lipinski's rule of five (LoF) [33] and other drug-like characteristics. The LoF describes a molecule being lead-like if it possesses molecular weight and hydrophobicity (logP) less than or equals to 500 kDa and 5, respectively. In addition to the above, the number of hydrogen bond donors and acceptors should be less than or equals to 5 and 10, respectively. The generated grid was inserted via the “Receptor” tab of the VSW panel. In the “Docking” tab of the VSW panel, the option was selected to retain 30% best-docked molecules after HTVS. Compounds remained after HTVS was considered as input of SP and similar to the previous step, the best 10% docked molecules were considered for the next step. In XP docking, also best 10% molecules were considered. Further, the binding free energy of the remaining molecules was calculated using the Prime-MM-GBSA approach. Based on high Glide-XP score and binding free energy the top-ranked molecules were selected for further assessment.
2.3. In-silico pharmacokinetics, drug-likeliness and toxicity analysis studies
Pharmacokinetic assessment of selected molecules was performed through the freely available online SwissADME web tool [34]. Several parameters including physiochemical, lipophilicity, water-solubility, pharmacokinetic, drug-like properties, LoF [33] and Veber's rule [35] were calculated. The SwissADME webserver becomes a favorite pharmacokinetic analysis tool for the scientific community. Molecules obtained afterward the VSW approach were checked for LoF and Veber's rule. The drug-likeness of any molecule can be explained by the LoF. Veber's rule is crucial to assess the flexibility as well as the surface area of any potential compound. According to Veber's rule, being a promising molecule, the total polar surface area (TPSA) and the number of rotatable bonds should be below or equals to 140 Å2 and 10, respectively. The human intestinal absorption (HIA) and blood-brain barrier (BBB) are crucial pharmacokinetic parameters and play a vital role in helping the appropriate selection of good candidate drug-like compounds [36]. The orally administered drugs are mainly absorbed by the intestine. The HIA parameter elucidates that for a given molecule how much percentage will be absorbed through the human intestine [36]. On the other side, the BBB parameter signifies the competence of the compound to penetrate in the brain cells.
Further, toxicity properties were evaluated employing ‘pkCSM’ – a freely accessible web server-based extensively used in silico toxicity prediction tool [37] and it is available at http://biosig.unimelb.edu.au/pkcsm/. In particular, ‘pkCSM’ relies on graph-based signatures i.e. mathematical illustration of any given compound to develop predictive models of different pharmacokinetics and toxicity properties. Upon submission of SMILES format as an input of respective chemical entities, the pkCSM was successfully generated several toxicity parameters includes AMES toxicity, maximum tolerated dose (human), hERG-I/hERG-II inhibitor, oral rat acute toxicity, oral rat chronic toxicity (LOAEL), hepatotoxicity, skin sensitization, T. Pyriformis toxicity, and Minnow toxicity. All these attributes are very important and well-known toxicity associated properties that needed to be considered during drug development processes for evaluating toxicity related safety properties or filtering out unsatisfactory properties for any chemical entities.
2.4. Molecular dynamic simulation
The behavior of the protein-ligand complex in the dynamic state can be explored through MD simulation study. Proposed modulators docked with Nsp15 endoribonuclease were considered for classical MD simulation study for 100 ns simulation time. The Gromacs-2018-2 was used to execute the simulations and it is installed at the Lengau CHPC server (https://www.chpc.ac.za/index.php/resources/lengau-cluster). In the simulation, the time step, constant temperature and constant pressure were considered as 2 fs, 300 K and 1 atm, respectively. The CHARMM36 all-atoms force field [38] was used to generate protein topology. The SwissParam, an online tool [39] was adopted to develop the ligand topology. The simulation box was considered as a cubic box with a diameter of 1 Å from the center of the system. Each protein-ligand system in the cubic box was solvated through the TIP3P water model [40]. A required quantity of Na+ and Cl− ions were adjusted to neutralize the system prior to the energy minimization and production. The close-contacts and overlapped atoms were removed using the steepest descent algorithm. Before starting the production phase, each of the systems was equilibrated with NVT (constant number of particles, volume and temperature) as well as NPT (constant number of particles, pressure and temperature) to ensure the equal distribution of solvent and ions around the protein-ligand complex. Several parameters included RMSD (root mean square deviation), RMSF (root mean square fluctuation) and RoG (radius of gyration) were estimated from the MD simulation trajectories to explore the conformational changes and steadiness of the system.
2.5. Binding free energy calculation through MM-PBSA method
The g_mmpbsa tool [41] is a Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) based binding free energy (ΔG bind) estimation. The above tool was considered to estimate the ΔG bind of the final proposed molecules. This method is widely used and already proved its significant application to biomolecular complex studies. The MM-PBSA approach taken into consideration the conformational fluctuation and entropic contributions [41]. The g_mmpbsa tool is a compiled and standalone tool that is extremely user friendly and doesn't need any dependency. Herein, ΔG bind of all final molecules was calculated. The ΔG bind can be obtained through the following equation.
| (1) |
Here, G complex signifies the total free energy of the complex between protein and ligand. Individual, G protein and G ligand are the free energy of the protein and ligand, respectively in the solvent. Moreover, the free energy of individual complex, protein and ligand can be obtained as below.
| (2) |
E MM represents the average molecular mechanics (MM) potential energy in a vacuum. T and S are the temperature and entropy, respectively. The free energy of solvation is represented by G solvation.
EMM can be defined as follows.
| (3) |
E bonded is the summation of bonded interactions included bond-length, bond-angle and dihedral angle. E nonbonded defines the nonbonding interactions included electrostatic and van der Waals interactions.
The G solvation is described by the energy needed to move a solute from a vacuum to the solvent. It can be expressed as follows.
| (4) |
The G polar and G nonpolar are denoted by the electrostatic and non-electrostatic contribution to the solvation free energy, respectively.
3. Results and discussion
3.1. Virtual screening
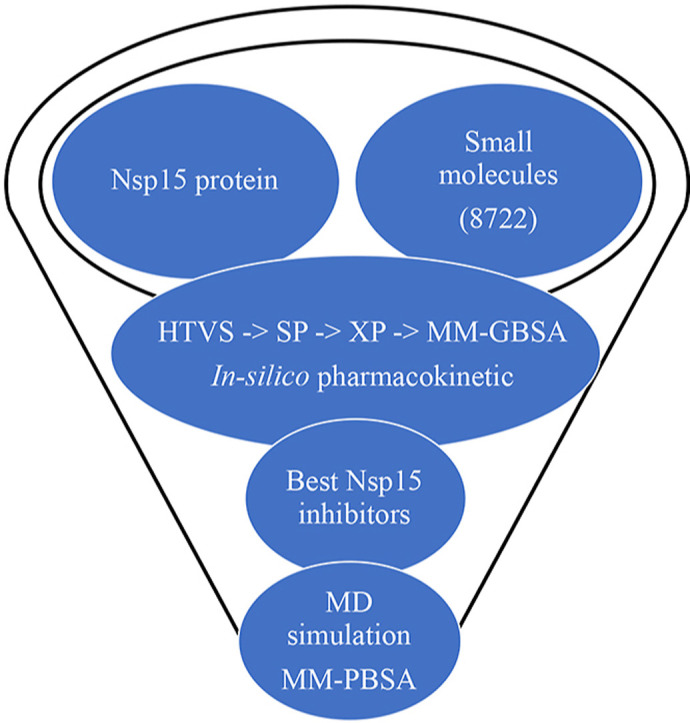
A structure-based virtual screening paradigm is an excellent and efficient computational approach to search for large chemical databases for a specific target. This approach needs 3D-coordinates of macromolecules obtained either from an experimental approach or homology modelling followed by docking the small molecules to reduce the chemical space based on active and inactive. To explore the potential Nsp15 endoribonuclease modulators, the multi-step molecular docking approaches were carried out to screen the Asinex antiviral database consisting of more than eight thousand compounds. A step-wise workflow of the work is given in Fig. 1 .
Fig. 1.

Schematic representation of screening steps of Nsp15 endoribonuclease modulators. More than eight thousand molecules considerd for VSW followed by binding free enrgy, pharmacokinetics assessment and MD simulation.
Three levels of sequential molecular docking study included HTVS, SP and XP, and finally, the estimation of binding free energy using the Prime-MM-GBSA method were performed. Arbitrarily, after docking of the entire set through HTVS, the best 30% molecules were considered for the next level of docking. Compounds remained after HTVS were given as input for the SP docking. To reduce the chemical space, the best 10% of docked molecules were considered for the next level of docking such as XP. Finally, on the successful completion of XP docking, about 26 molecules were retained (approximately 10%). The binding free energy of the above compounds was estimated using the Prime-MM-GBSA method. Molecules having binding free energy higher than −35 kcal/mol were removed. A total of 20 molecules were retained by following the previous criteria. A number of pharmacokinetic and drug-likeness parameters of above 20 molecules were retrieved from the SwissADME webserver. It was found that 5 molecules possessed a low GI value and removed. Further, molecules having moderate and poor soluble profiles were also deleted for further assessment. After the above filtering step, the five best molecules remained and were considered to be the best Nsp15 endoribonuclease modulators. Final Nsp15 endoribonuclease modulators in the 2D format are given in Fig. 2 . From Fig. 2, it can be observed that all molecules consisting of a diverse kind of functional groups, and phenyl and heterocyclic rings. All the molecules possess multiple -oxo, hydroxyl and amine groups those might be important to establish a number of crucial binding interactions in the form of hydrogen bonds with Nsp15 endoribonuclease. The presence of methyl groups in N1, N2 and N3 increased the chance of non-hydrogen bond interactions such as hydrophobic contacts. Moreover, the cyclic rings in all proposed molecules can be critical components to form hydrophobic contacts with Nsp15 endoribonuclease. In short, proposed molecules were found to consist of interesting functional groups or pharmacophores definitely help to the potential inhibition/modulation of Nsp15 endoribonuclease.
Fig. 2.

Two-dimensional representation of final Nsp15 endoribonuclease modulators. Each of the molecules represents with diverse kind of functional groups.
3.2. Binding interaction analysis
The XP GScore of N1, N2, N3, N4 and N5 was found to be −11.539, −10.639, −10.507, −10.347 and −10.257 kcal/mol, respectively. The binding free energy calculated using MM-GBSA approach of the Prime module was found to be −49.370, −37.170, −43.140, −41.340 and −51.020 kcal/mol for N1, N2, N3, N4 and N5, respectively. The molecular binding interactions were explored through an online server, PLIP (protein-ligand interaction profiler) [42]. From the molecular docking study, the best-docked pose of each molecule was initially selected by considering the lowest XP-dock score among all the generated poses and afterward investigating the number of binding interactions between each molecule and involvement of ligand-binding catalytic amino acid residues of the Nsp15. In particular, XP-GlideScore, H-bond energy, ligand efficiency, and the total number of intermolecular interactions (non-covalent) between all the generated poses of each molecule and active site residues of Nsp15 protein were considered for best poses selection. The binding interactions profile between the proposed molecules and ligand-binding amino acid residues of Nsp15 endoribonuclease are given in Fig. 3 .
Fig. 3.

Binding interactions of proposed modulators with Nsp15 endoribonuclease. A number of important ligand-binding amino acid residues are labeled.
From the binding interaction analysis, it can be observed that the hydroxyl group attached with phenyl ring in N1 was successfully formed two and one hydrogen bond (H-bond) interactions with Asn278 and Leu346, respectively. An oxo group in between the linear chain and another at the terminal position were found to be critical to establishing H-bond with Gln245 and Thr341, respectively. In addition to the above, Gln245 was also found to connect the terminal hydroxyl group via H-bond interaction. A number of hydrophobic contacts were also found between N1 and, Val292 and Tyr343 amino acids present in the active site of the Nsp15 endoribonuclease. Interestingly, a few amino acids included such as His235, Gln245 and Lys290 (active site residues) were found to establish the salt bridges with N1. The salt bridge interaction is formed between two groups of opposite charge and plays an important role to form the stable protein-ligand complex. In the case of N2, the amine group present in the long-chain was found to form H-bond interaction with His235 and Thr341. Asp240 and Gln245 were seen to interact with N2 through H-bond interaction. A number of salt bridges were established with His235 and Gly248. A pi-cation interaction was also found to form between N2 and His235.
The putative Nsp15 endoribonuclease modulator, N3 was successfully formed H-bond and hydrophobic interaction with Thr341 and Lys345, respectively. Beyond the above, His235, Gly248 and His250 were also found important to form salt bridges with N3. The binding interaction profile of N4 was revealed that Gly248, Ser294 and Thr341 were important to interact with N4 through H-bond interactions. One hydrophobic contact between N4 and Lys345 was observed. The salt bridges were also seen with His235 and Lys290. A pi-stacking was observed between the pyridine ring of N4 and Thr343. Quite interesting binding interactions were observed between N5 and ligand-binding amino acid residues of Nsp15 endoribonuclease. Individually, Asp278 and Leu346 were found to form two H-bonds with N5. Another two crucial amino acid residues, Thr341 and Gly248 were seen to interact with N5 via an independent single H-bond. The piperidine and pyrimidine rings of N5 were critically formed hydrophobic contacts with Thr343 and Lys345 respectively. Beyond the above, a number of crucial salt bridges were also observed. Therefore, the above crucial observation about diverse kind binding interactions between the proposed molecules and ligand-binding amino residues of Nsp15 endoribonuclease undoubtedly explained the stability of protein-ligand complexes. Although, the present computational studies have been carried out using monomer of Nsp15 protein, however, an extensive study is required to understand the conformational variability of the endoU domain of Nsp15 hexamer in ligand bound-state, because active site residues of each Nsp15 protomer dwell very close to the interface with a neighboring endoU domain and moreover the conformational stability for the individual Nsp15 protomers are also assisted by the Nsp15 oligomerization or hexamer conformation. Nevertheless, the exact mechanism of hexamer conformation constitution and/or molecular requirement for Nsp15 oligomerization is still unknown, however, it has been postulated that each individual Nsp15 protomer possibly interacts through the mechanism of allosteric communication [43]. Therefore, any ligand bound state of Nsp15 hexamer may display heterogeneity in the conformation of the endoU domain and also independent movement in residues of individual domains of Nsp15.
A recent Nsp16 crystal structure with co-crystal uridine-5′-monophosphate was submitted with PDB ID: 6WLC [44]. The binding interactions between uridine-5′-monophosphate and amino residues of the Nsp15 were explored. His250, Ser294 and Leu346 were found to form H-bonds with uridine-5′-monophosphate. Moreover, hydrophobic interactions were also seen between uridine-5′-monophosphate, and Gly248 and Tyr343. A similar kind of binding profile was also found in the proposed Nsp15 molecules. Compound N1 was found to form one of each hydrogen and hydrophobic bond interactions with Leu346 and Tyr343 respectively. Similar to uridine-5′-monophosphate, N2 has established a hydrophobic contact with Gly248. His250 was seen to form an H-bond with N3. Moreover, N3 was formed one of each H-bond and hydrophobic bond with Gly248. Compound N4 was also found to form hydrogen and hydrophobic bonds with Ser294 and Glu248 respectively. Similar to uridine-5′-monophosphate, His250, Gly248, Tyr343 and Leu346 were formed binding interactions with N5. Hence, the above observations clearly suggested that proposed molecules were shown a similar binding interactions profile as of uridine-5′-monophosphate.
In order to explore the binding mode of the proposed molecules inside the receptor cavity of Nsp15, a three-dimensional surface view of each complex was deduced and it is given in Fig. 4 . All five reported Nsp15 molecules were perfectly occupied inside the receptor cavity.
Fig. 4.

Binding mode of proposed modulators in Nsp15 endoribonuclease. All molecules were found fitted inside the receptor cavity of Nsp15.
3.3. Pharmacokinetics and drug-likeness assessment
Several pharmacokinetic and drug-likeness parameters of the final Nsp15 endoribonuclease molecules were calculated and these are given in Table 1 . Not a single molecule was found to violate the LoF which indicates that the molecular weight of each molecule less than or equals to 500 g/mol, the number of hydrogen bond acceptors and donors are not more than 5 and 10, respectively, and logP value less than or equals to 5. It is reported that a molecule having TPSA less than 140 Å2 might be potential in nature. The TPSA of N1, N2, N3, N4 and N5 was found to be 99.13, 119.73, 99.13, 82.44 and 128.03 Å2, respectively that clearly indicated the potentiality of the molecules. All the molecules were found soluble or very soluble in nature and highly absorbable in the gastrointestinal tract. The number of rotatable bonds of compounds N1, N2, N3, N4 and N5 was found to be 4, 8, 5, 4 and 5, respectively. The above observation suggested that N4 is more flexible compared to others and the remaining four molecules possess an almost similar pattern of rigidity. The synthetic accessibility (SA) parameter explains how ease or challenging to synthesize the molecule and the value varies from 0 to 10. Higher the SA value more difficult to synthesis. The SA of all five molecules was found to be less than 5 that clearly indicated the feasibility of synthesis. Hence, from the above discussion, it is clear that the final proposed molecules are potential enough for Nsp15 endoribonuclease inhibition.
Table 1.
Physiochemical and drug-likeness parameters of selected Nsp15 endoribonuclease modulators.
| Parameters | N1 | N2 | N3 | N4 | N5 |
|---|---|---|---|---|---|
| Formula | C20H24NO6 | C20H26N4O4 | C22H28NO6 | C12H13N2O4 | C15H18N5O3 |
| aMW(g/mol) | 374.41 | 386.44 | 402.46 | 249.24 | 316.34 |
| bNHA | 27 | 28 | 29 | 18 | 23 |
| cNAHA | 6 | 10 | 6 | 6 | 9 |
| dNRB | 4 | 8 | 5 | 4 | 5 |
| eMR | 99.23 | 107.72 | 108.84 | 65.91 | 86.44 |
| fTPSA(Å2) | 99.13 | 119.73 | 99.13 | 82.44 | 128.03 |
| gLogS | −2.81 | −2.24 | −3.25 | −1.15 | −1.74 |
| hSC | Soluble | Soluble | Soluble | Very Soluble | Very Soluble |
| iGI | High | High | High | High | High |
| jvROF | 0 | 0 | 0 | 0 | 0 |
| kBS | 0.56 | 0.55 | 0.56 | 0.56 | 0.56 |
| lSA | 4.29 | 3.19 | 4.42 | 2.08 | 3.12 |
| LogP | 2.31 | 2.2 | 2.87 | 1.57 | 0.99 |
Molecular weight.
No of heavy atoms.
No of aromatic heavy atoms.
No of rotatable bonds.
Molar refractivity.
Topological polar surface area.
Solubility.
Solubility class.
Gastrointestinal absorption.
Violation of Lipinski's rule of five.
Bioavailability score.
Synthetic accessibility.
Further, the in-silico predicted toxicity properties of all five compounds (N1–N5) were assessed and results are given in Table 2 . It was found that all compounds showed no indication of AMES toxicity, hence can be considered as non-mutagenic compounds. Evaluated maximum tolerated toxic dose of compounds was found to be −0.63, −0.19, −0.86, 0.65 and 0.30 log(mg/kg/day) for N1, N2, N3, N4 and N5, respectively. It was suggested that in human maximum recommended tolerated toxic dose for any given compound considered to be low when value appears as less than or equal to 0.477 log(mg/kg/day) otherwise high when value found to be greater than 0.477 log(mg/kg/day). In terms of evaluating the cardiotoxicity nature of all compounds, the hERG-I/hERG-II inhibition profile was checked based on the inhibition of potassium channels encoded by hERG (human ether-a-go-go gene) and found as negative or no indication of ventricular arrhythmia upon administration of those compounds. Moreover, a safety concern for drug-induced liver injury was measured by means of evaluating hepatotoxic indication for all compounds, which bring out as negative and suggesting no disruption of normal function of the liver upon administration. Another important safety concern is known as skin sensitivity or sensitization which was also found as negative indicating no potential skin irritation or allergic effect on the treatment of these compounds. The oral acute toxicity (LD50) was found to be 2.33, 1.47, 2.21, 1.50 and 2.07 mol/kg for N1, N2, N3, N4 and N5, respectively. All these values were found within the accepted or recommended range i.e. LD50 < 2.5 mol/kg. On the other hand, oral chronic toxicity was also found within the recommended value (i.e. log 4.4.mg/kg_bw/day) for all compounds (Table 2). Hence, overall hypothetical ADMET properties justify that all the screened antiviral compounds showed potential lead-like characteristics and can be used for further evaluation for SARS-CoV-2 Nsp15 modulation and biological activity.
Table 2.
Predicted toxicity properties of selected Nsp15 endoribonuclease modulators – inhibitors.
| Toxicity properties | N1 | N2 | N3 | N4 | N5 |
|---|---|---|---|---|---|
| AMES toxicity | No | No | No | No | No |
| Max. tolerated dose (human) | −0.63 | −0.19 | −0.86 | 0.65 | 0.30 |
| hERG I/hERG II inhibitor | No/No | No/No | No/No | No/No | No/No |
| Oral Rat Acute Toxicity (LD50) | 2.33 | 1.47 | 2.21 | 1.50 | 2.07 |
| Oral Rat Chronic Toxicity (LOAEL) | 1.04 | 0.62 | 0.89 | 0.96 | 1.02 |
| Hepatotoxicity | No | No | No | No | No |
| Skin Sensitization | No | No | No | No | No |
| T.Pyriformis toxicity | 0.41 | 0.27 | 0.44 | 0.09 | 0.27 |
| Minnow toxicity | 2.53 | 2.00 | 2.62 | 3.06 | 3.10 |
3.4. Ligand efficiency assessment
The ligand efficiency and potentiality of the Nsp15 endoribonuclease modulators were explored through a number of parameters such as Ligand Efficiency (LE), Ligand Efficiency Scale (LE_Scale), Fit Quality (FQ) and LE dependent Lipophilicity (LELP). The value of each parameter of the five final proposed molecules are given in Table 3 .
Table 3.
Bioactivity and efficiency parameters of proposed Nsp15 endoribonuclease modulators.
| Molecule | 1BE | 2LE | 3LE_Scale | 4FQ | 5LELP |
|---|---|---|---|---|---|
| N1 | −11.539 | 0.425 | 0.368 | 1.154 | 5.435 |
| N2 | −10.639 | 0.378 | 0.356 | 1.061 | 5.820 |
| N3 | −10.507 | 0.362 | 0.346 | 1.046 | 7.928 |
| N4 | −10.347 | 0.572 | 0.482 | 1.186 | 2.744 |
| N5 | −10.361 | 0.443 | 0.415 | 1.067 | 2.234 |
Binding energy.
Ligand efficiency.
Ligand efficiency scale.
Fit quality.
Ligand-efficiency-dependent-lipophilicity.
The LE parameter proposed by Hopkins et al. [45] and it was estimated by the following expression (Equation (5)). It is the negative ratio of the docking binding energy in kcal/mol to the number of heavy atoms. All molecules were found to have LE value of less than 0.6 which indicates the potentiality and drug-likeness of the compounds.
| (5) |
Another parameter known as LE_Scale is defined by Reynolds et al. [46] and can be deduced using equation (6). It is a size-dependent parameter and a low value indicates the druglike characteristics of the molecule. The LE_Scale of N1, N2, N3, N4 and N5 was found to be 0.368, 0.356, 0.346, 0.482 and 0.415 respectively.
| (6) |
The FQ is one of the important parameters to assess the binding ability of the small molecules in the receptor cavity. This is the ratio between LE and LE_Scale (Equation (7)) and being a potent molecule value should be near 1. From Table 3, the FQ of N1, N2, N3, N4 and N5 can be seen as 1.154, 1.061, 1.046, 1.186 and 1.1067 respectively. The above value of FQ undoubtedly favors in drug-likeness nature of the molecules.
| (7) |
Another parameter, LELP defined by Keseru and Makara and can be calculated by deducing the ratio between logP and LE of the molecule (Equation (4)). The LELP value of N1, N2, N3, N4 and N5 was found to be 5.435, 5.820, 7.928, 2.744 and 2.234 respectively. The above data was clearly indicated the drug-likeness characteristics of the molecules.
3.5. Molecular dynamics simulation
MD simulation is widely used as an excellent and powerful computational simulation to investigate the behavioral and dynamical characteristics of the protein-ligand complex. An all-atoms MD simulation of each docked complex of proposed modulators and Nsp15 was explored for a 100ns time span. Several parameters such as RMSD, RMSF and RoG were obtained from the MD simulation trajectory. Each of the above parameters explains the stability of the protein-ligand complex in the dynamics states. The entire trajectory was further used to calculate the binding free energy through the MM-PBSA approach. Average, maximum and minimum RMSD, RMSF and RoG of each complex were calculated and these are given in Table 4 .
Table 4.
Average, maximum and minimum RMSD, RMSF and RoG values of Nsp15 endoribonuclease bound with N1, N2, N3, N4 and N5.
| RMSD(nm) | |||||
|---|---|---|---|---|---|
| Complex | N1 | N2 | N3 | N4 | N5 |
| aMin | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 |
| bMax | 0.368 | 0.525 | 0.423 | 0.591 | 0.420 |
| Average | 0.192 | 0.252 | 0.209 | 0.239 | 0.218 |
| RMSF (nm) | |||||
| aMin | 0.052 | 0.066 | 0.077 | 0.062 | 0.071 |
| bMax | 0.463 | 0.431 | 0.559 | 0.631 | 0.593 |
| Average | 0.122 | 0.147 | 0.171 | 0.136 | 0.158 |
| RoG (nm) | |||||
| aMin | 2.327 | 2.327 | 2.320 | 2.327 | 2.328 |
| bMax | 2.465 | 2.507 | 2.514 | 2.470 | 2.506 |
| Average | 2.384 | 2.402 | 2.404 | 2.381 | 2.396 |
Minimum.
Maximum.
3.5.1. Root-mean-square deviation
The protein backbone RMSD obtained from the simulation trajectory of protein-ligand complex explains the stability of the complex in the dynamic environment. The higher the protein-backbone RMSD value indicates unfolding and contrariwise the lower value favors the compactness. The low fluctuation of the backbone RMSD also substantiates the equilibration of the protein-ligand complex. The backbone RMSD of each frame was extracted and it is given in Fig. 5 . Except for the Nsp15 backbone bound with N4, all were found to be consistent throughout the simulation. In the case of the Nsp15-N4 complex, the backbone was deviated in between 12 and 30 ns and afterward remained consistent. The average RMSD of Nsp15 backbone was found to be 0.192, 0.252, 0.209, 0.239 and 0.218 nm when bound with N1, N2, N3, N4 and N5 respectively. The difference between the maximum and minimum RMSD can give an idea about the backbone deviation. From Table 4, it can be seen the difference between the maximum and minimum RMSD as of 0.367, 0.524, 0.422, 0.590 and 0.419 nm for Nsp15 backbone bound with N1, N2, N3, N4 and N5 respectively. Such low RMSD value of each complex undoubtedly explained the stability and consistency of the complex in the dynamic environment.
Fig. 5.

RMSD of Nsp15 endoribonuclease backbone bound with N1, N2, N3, N4 and N5. All Nsp15 backbone except bound with N4 found to be equilibrated from the beginning of the simulation. Nasp15 backbone bound with N4 was seen equilibrated after about 30ns.
3.5.2. Root-mean-square fluctuation
Each amino acid residue belongs to the protein-ligand complex is responsible for the stability of the dynamic system. The fluctuation of any particular amino acid concerning the reference or native structure can be measured through the RMSF parameter calculated from the MD simulation trajectories. In any trajectory, a large RMSF value indicates the instability, otherwise, the residue remained stable. The RMSF of each amino residue of all five complexes was calculated from the MD simulation trajectory and these are given in Fig. 6 . The average, maximum and minimum RMSF of each complex are given in Table 4. Amino acid residues of all Nsp15 endoribonuclease bound with N1, N2, N3, N4 and N5 were seen to fluctuate in an almost similar manner throughout the simulation. With a few exemptions, not much fluctuation was observed which clearly favors the stability of the amino residues in dynamic states. Such observation might be due to conformation changes of amino acids to accommodate the ligand in the receptor cavity. The difference between maximum and average RMSF values was found to be 0.341, 0.284, 0.388, 0.495 and 0.435 nm for amino residues of Nsp15 bound with N1, N2, N3, N4 and N5 respectively. Such a low value of RMSF undoubtedly indicated the stability of each complex in the MD simulation.
Fig. 6.

RMSF of individual amino acid residues of Nsp15 endoribonuclease. Amino acids of Nsp15 bound with all small molecules were fluctuated almost similar manner.
3.5.3. Radius of gyration
The RoG is one of the crucial parameters obtained from the MD simulation trajectory to check the firmness of the protein-ligand system. Less deviation and steady variation of the RoG explains the steady folding of protein during the execution of MD simulation. The complex systems of Nsp15 endoribonuclease bound with N1, N3 and N5 were found very low deviation with respect to the initial structure (see Fig. 7 ). The Nsp15 endoribonuclease protein complex with N2 was found to deviate compare to other systems but most importantly no abnormal or unusual deviation was observed (see Fig. 7). Another protein-ligand complex of Nsp15 and N4 was observed to deviate up to about 30ns and afterward, the system achieved stability that suggests the compactness of the system (see Fig. 7). The difference between the highest and lowest RoG value can give an idea about the deviation of each system. The above value was found to be 0.138, 0.180, 0.194, 0.143 and 0.178 nm for the Nsp15 endoribonuclease bound with N1, N2, N3, N4 and N5 respectively. Hence, the low RoG value of each system explained the compactness of the protein-ligand complexes.
Fig. 7.

Radius of gyration for Nsp15 endoribonuclease. The range of the radius of gyration was found 2.30–2.50 nm. All trajectories showed the compactness of each complex throughout the simulation.
3.5.4. Binding free energy estimation using the MM-PBSA method
The potentiality and binding affinity of the molecules can be evaluated through the estimation of binding free energy (ΔGbind). Accurate and trustworthy methods are extremely essential to predict precise ΔGbind. The MM-PBSA is one of the trusted and widely used approaches that combine the molecular mechanics and continuum solvent models to calculate the ΔGbind of small molecules. The protein-ligand snapshots retrieved from the MD simulation trajectory of Nsp15 endoribonuclease bound with N1, N2, N3, N4 and N5 were used to calculate ΔGbind and these are given in Fig. 8 . The average, maximum and minimum ΔGbind of each complex were calculated and these are given in Table 5 . From Fig. 8, it can be observed that N2 was found to show a higher binding affinity compared to the remaining 4. For all molecules, it is interesting to note that not a single frame was found to have positive ΔGbind which undoubtedly substantiated the strong affinity towards the receptor.
Fig. 8.

Binding free energies of Nsp15 endoribonuclease modulators. All molecules showed high negative binding free energy that signify the strong affinity of each molecules towards Nsp15.
Table 5.
Minimum, maximum and average values for binding free energy.
| Compounds | N1 | N2 | N3 | N4 | N5 |
|---|---|---|---|---|---|
| Minimum (kJ/mol) | −633.190 | −1145.630 | −803.690 | −788.870 | −891.860 |
| Maximum (kJ/mol) | −124.450 | −39.471 | −15.998 | −28.991 | −43.135 |
| Average (kJ/mol) | −345.654 | −721.906 | −349.705 | −388.002 | −513.641 |
The average ΔGbind of N1, N2, N3, N4 and N5 was found to be −345.654, −721.906, −349.705, −388.002 and −513.641 kJ/mol respectively. Therefore, it is clear without any doubt that all proposed modulators possess strong binding affinity.
3.6. Comparative study
A few recent studies on the screening of Nsp15 modulators for inhibition of SARS-CoV-2 were published. Krishnan et al. [47] were explored the enamine for screening against the Nsp15. They have reported promising eight molecules for the inhibition of SARS-CoV-2. The authors reported a number of crucial binding interactions between proposed molecules from the Enamine database and Nsp15. It is quite interesting that all the ligand-binding amino acids except Trp333 reported in the study were also found important in the current study. Particularly, Lys290, Val292, His235, Ser294, Leu246, Gly248, Ser294, Tyr343, His235 and Thr341 were found to be common ligand-binding amino residues to form critical binding interactions. In another study, Barag et al. [48] have explored the computational re-purposing study on RdPd and Nsp15. The binding energy in the molecular docking was reported in the study in the range of −9 to −11 kcal/mol. Proposed molecules in the current study were showed binding energy in the range of −10 to −12 kcal/mol. Moreover, the binding interaction profile of the study was also found almost similar to the current study. The authors have performed a 30ns MD simulation study to check the stability of the protein-ligand systems. The RMSD and RMSF profile was quite similar to our observations. Moreover, the authors calculated the binding energy of the molecules through MM-PBSA approach from the MD simulation trajectory. They have reported the range of binding free energy of about −375 to −200 kJ/mol. In our observation, the average binding free energy range was found to be −722 to −345 kJ/mol. Batool et al. [49] were used the Nsp15 as a target to screen more than a hundred thousand molecules. They have developed a common-feature pharmacophore model from the known standard drug molecules followed by screening the above dataset. Through a number of imposed criteria, the authors finally were reported promising 10 molecules. In the molecular docking study, a number of catalytic amino acid residues were reported as important for binding interactions. Interestingly most of the amino residues reported in the study were found common in the binding interaction profile in our study. The MD simulation observations were quite similar to the current study in regards to stability, convergence and energetics. Khan et al. [50] was performed an in-silico drug repurposing approach to find promising therapeutics targeting the Nsp15. From starting with more than a hundred known drugs, the authors reported three promising drug molecules effective against Nsp15. The molecular docking and MD simulation profiles of the reported molecules were corroborated the outcomes in the current study. Hence, reported data in the current study undoubtedly were given the conclusive similarity with the published data.
3.7. Future directions
Although the computational screening of molecular datasets for promising molecules is extensively and effectively used in drug discovery research there is extremely essential to check the potential efficiency of the proposed Nsp15 modulators using several experimental validation methods. The thermal shift assay is one of the crucial approaches that can be considered to assess the binding affinity of the molecules. Moreover, other reaction kinetic studies can be performed to explore the binding and unbinding mechanism of the selected molecules in real states. The molecule may need further modification and optimization based on the experimental outcomes to improve the therapeutic efficacy of the molecules.
4. Conclusion
The virtual screening based on a multi-steps molecular docking approach was used to screen potential Nsp15 endoribonuclease molecules for the inhibition of SARS-CoV-2. About nine thousand small chemical compounds were obtained from the Asinex database and docked all molecules sequentially through HTVS, SP and XP in the Nsp15 endoribonuclease. The Prime-MMGBSA based binding free energy of the remaining molecules obtained in the above step was calculated. Molecules having low binding free energy were discarded. Finally, the in-silico pharmacokinetic analysis was carried out and five molecules were found to be crucial for Nsp15 endoribonuclease inhibition. The binding interaction analysis was revealed a number of important binding interactions in the form of H-bond, hydrophobic and salt bridges. Further, the MD simulation study was performed to explore the stability of protein-ligand complexes in the dynamic environment. A number of parameters were calculated from the MD simulation trajectory indicated stability. During the entire time span of simulation small molecules were retained inside the receptor cavity. The binding affinity of the molecules was explored by the calculation of binding free energy through the MM-PBSA approach. The range of binding free energy was found to be −345 to −722 kJ/mol that substantiated the strong affinity of the molecules towards Nsp15 endoribonuclease. Hence, the proposed molecules might be important chemical components for successful inhibition of Nsp15 endoribonuclease subjected to experimental validation.
CRediT authorship contribution statement
Rutuja Umesh Savale: Data curation, Formal analysis, Investigation, Methodology, Writing - original draft, Writing - review & editing. Shovonlal Bhowmick: Data curation, Formal analysis, Investigation, Methodology, Writing - original draft, Writing - review & editing. Sameh Mohamed Osman: Methodology, Writing - original draft, Writing - review & editing. Fatmah Ali Alasmary: Methodology, Writing - original draft, Writing - review & editing. Tahani Mazyad Almutairi: Methodology, Writing - original draft, Writing - review & editing. Dalal Saied Abdullah: Methodology, Writing - original draft, Writing - review & editing. Pritee Chunarkar Patil: Methodology, Writing - original draft, Writing - review & editing. Md Ataul Islam: Design, Formal analysis, Investigation, Methodology, Writing - original draft, Writing - review & editing.
Declaration of competing interest
Authors declare that there is no competing interest.
Acknowledgment
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group No (RG-1441-431).
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.abb.2021.108771.
Computational resource
The CHPC (www.chpc.ac.za), Cape Town, South Africa is thankfully acknowledged for computational resources and tools.
Availability of data and material
Not availability.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.MacKenzie J.S., Smith D.W. COVID-19: a novel zoonotic disease caused by a coronavirus from China: what we know and what we don't. Microbiol. Aust. 2020 doi: 10.1071/MA20013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai C.C., Shih T.P., Ko W.C., Tang H.J., Hsueh P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int. J. Antimicrob. Agents. 2020;55:105924. doi: 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han E., Tan M.M.J., Turk E., Sridhar D., Leung G.M., Shibuya K., Asgari N., Oh J., García-Basteiro A.L., Hanefeld J., Cook A.R., Hsu L.Y., Teo Y.Y., Heymann D., Clark H., McKee M., Legido-Quigley H. Lessons learnt from easing COVID-19 restrictions: an analysis of countries and regions in Asia Pacific and Europe. Lancet. 2020;396:1525–1534. doi: 10.1016/S0140-6736(20)32007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naqvi A.A.T., Fatima K., Mohammad T., Fatima U., Singh I.K., Singh A., Atif S.M., Hariprasad G., Hasan G.M., Hassan M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2020;1966:165878. doi: 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mukherjee S., Bhattacharyya D., Bhunia A. Host-membrane interacting interface of the SARS coronavirus envelope protein: immense functional potential of C-terminal domain. Biophys. Chem. 2020;266:106452. doi: 10.1016/j.bpc.2020.106452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Wit E., Van Doremalen N., Falzarano D., Munster V.J., SARS. MERS Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y., Jedrzejczak R., Maltseva N.I., Wilamowski M., Endres M., Godzik A., Michalska K., Joachimiak A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020;29:1596–1605. doi: 10.1002/pro.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanck S., Stinn A., Tsiklauri L., Zirkel F., Junglen S., Ziebuhr J. Characterization of an alphamesonivirus 3C-like protease defines a special group of nidovirus main proteases. J. Virol. 2014;88:13747–13758. doi: 10.1128/jvi.02040-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham R.L., Sparks J.S., Eckerle L.D., Sims A.C., Denison M.R. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008;133:88–100. doi: 10.1016/j.virusres.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Posthuma C.C., Nedialkova D.D., Zevenhoven-Dobbe J.C., Blokhuis J.H., Gorbalenya A.E., Snijder E.J. Site-directed mutagenesis of the nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J. Virol. 2006;80:1653–1661. doi: 10.1128/jvi.80.4.1653-1661.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng X., Hackbart M., Mettelman R.C., O'Brien A., Mielech A.M., Yi G., Kao C.C., Baker S.C. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E4251–E4260. doi: 10.1073/pnas.1618310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senanayake S.L. Overcoming nonstructural protein 15-nidoviral uridylate-specific endoribonuclease (nsp15/NendoU) activity of SARS-CoV-2. Futur. Drug Discov. 2020;2:FDD42. doi: 10.4155/fdd-2020-0012. [DOI] [Google Scholar]
- 13.Kumar S., Kashyap P., Chowdhury S., Kumar S., Panwar A., Kumar A. Identification of phytochemicals as potential therapeutic agents that binds to Nsp15 protein target of coronavirus (SARS-CoV-2) that are capable of inhibiting virus replication. Phytomedicine. 2020 doi: 10.1016/j.phymed.2020.153317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spadone C. Neurophysiology of cannabis. Encephale. 1991;17:17–22. [PubMed] [Google Scholar]
- 15.Polekhina G., Board P.G., Gali R.R., Rossjohn J., Parker M.W. Molecular basis of glutathione synthetase deficiency and a rare gene permutation event. EMBO J. 1999;18:3204–3213. doi: 10.1093/emboj/18.12.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhardwaj K., Palaninathan S., Alcantara J.M.O., Yi L.L., Guarino L., Sacchettini J.C., Kao C.C. Structural and functional analyses of the severe acute respiratory syndrome coronavirus endoribonuclease Nsp15. J. Biol. Chem. 2008;283:3655–3664. doi: 10.1074/jbc.M708375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joseph J.S., Saikatendu K.S., Subramanian V., Neuman B.W., Buchmeier M.J., Stevens R.C., Kuhn P. Crystal structure of a monomeric form of severe acute respiratory syndrome coronavirus endonuclease nsp15 suggests a role for hexamerization as an allosteric switch. J. Virol. 2007;81:6700–6708. doi: 10.1128/jvi.02817-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chikhale R.V., Gupta V.K., Eldesoky G.E., Wabaidur S.M., Patil S.A., Islam M.A. Identification of potential anti-TMPRSS2 natural products through homology modelling, virtual screening and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1798813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chatterjee S., Maity A., Chowdhury S., Islam M.A., Muttinini R.K., Sen D. In silico analysis and identification of promising hits against 2019 novel coronavirus 3C-like main protease enzyme. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1787228. [DOI] [PubMed] [Google Scholar]
- 20.Cao L., Goreshnik I., Coventry B., Case J.B., Miller L., Kozodoy L., Chen R.E., Carter L., Walls A.C., Park Y.-J., Strauch E.-M., Stewart L., Diamond M.S., Veesler D., Baker D. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370:426–431. doi: 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y., Wang F., Tang J., Nussinov R., Cheng F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health. 2020;2:e667–e676. doi: 10.1016/s2589-7500(20)30192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ke Y.Y., Peng T.T., Yeh T.K., Huang W.Z., Chang S.E., Wu S.H., Hung H.C., Hsu T.A., Lee S.J., Song J.S., Lin W.H., Chiang T.J., Lin J.H., Sytwu H.K., Chen C.T. Artificial intelligence approach fighting COVID-19 with repurposing drugs. Biomed. J. 2020;43:355–362. doi: 10.1016/j.bj.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kandeel M., Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020;251:117627. doi: 10.1016/j.lfs.2020.117627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lionta E., Spyrou G., Vassilatis D., Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr. Top. Med. Chem. 2014;14:1923–1938. doi: 10.2174/1568026614666140929124445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schrödinger . 2018. LigPrep | Schrödinger, Schrödinger Release 2018-1. [Google Scholar]
- 26.Shelley J.C., Cholleti A., Frye L.L., Greenwood J.R., Timlin M.R., Uchimaya M., Epik A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 27.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrödinger . 2018. Protein Preparation Wizard | Schrödinger, Schrödinger Release 2018-1. [Google Scholar]
- 29.Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 30.Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J.Y., Wang L., Lupyan D., Dahlgren M.K., Knight J.L., Kaus J.W., Cerutti D.S., Krilov G., Jorgensen W.L., Abel R., Friesner R.A. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theor. Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 31.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., Shaw D.E., Francis P., Shenkin P.S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 32.Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., Banks J.L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 33.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012;23:3–25. doi: 10.1016/j.addr.2012.09.019. [DOI] [PubMed] [Google Scholar]
- 34.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veber D.F., Johnson S.R., Cheng H.Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 36.de Vrieze M., Janssens P., Szucs R., van der Eycken J., Lynen F. In vitro prediction of human intestinal absorption and blood–brain barrier partitioning: development of a lipid analog for micellar liquid chromatography. Anal. Bioanal. Chem. 2015;407:7453–7466. doi: 10.1007/s00216-015-8911-z. [DOI] [PubMed] [Google Scholar]
- 37.Pires D.E.V., Blundell T.L., Ascher D.B., pkCSM Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., De Groot B.L., Grubmüller H., MacKerell A.D. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2016;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zoete V., Cuendet M.A., Grosdidier A., Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011;32:2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 40.Harrach M.F., Drossel B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014;140:174501. doi: 10.1063/1.4872239. [DOI] [PubMed] [Google Scholar]
- 41.Kumari R., Kumar R., Lynn A. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 42.Salentin S., Schreiber S., Haupt V.J., Adasme M.F., Schroeder M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pillon M.C., Frazier M.N., Dillard L.B., Williams J.G., Kocaman S., Krahn J.M., Perera L., Hayne C.K., Gordon J., Stewart Z.D., Sobhany M., Deterding L.J., Hsu A.L., Dandey V.P., Borgnia M.J., Stanley R.E. Cryo-EM structures of the SARS-CoV-2 endoribonuclease Nsp15. BioRxiv Prepr. Serv. Biol. 2020 doi: 10.1101/2020.08.11.244863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim Y., Wower J., Maltseva N., Chang C., Jedrzejczak R., Wilamowski M., Kang S., Nicolaescu V., Randall G., Michalska K., Joachimiak A. Tipiracil binds to uridine site and inhibits Nsp15 endoribonuclease NendoU from SARS-CoV-2. BioRxiv. 2020:2020. doi: 10.1101/2020.06.26.173872. 06.26.173872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hopkins A.L., Groom C.R., Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov. Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 46.Reynolds C.H., Bembenek S.D., Tounge B.A. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007;17:4258–4261. doi: 10.1016/j.bmcl.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 47.Krishnan D.A., Sangeetha G., Vajravijayan S., Nandhagopal N., Gunasekaran K. Structure-based drug designing towards the identification of potential anti-viral for COVID-19 by targeting endoribonuclease NSP15. Informatics Med. Unlocked. 2020;20:100392. doi: 10.1016/j.imu.2020.100392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barage S., Karthic A., Bavi R., Desai N., Kumar R., Kumar V., Lee K.W. Identification and characterization of novel RdRp and Nsp15 inhibitors for SARS-COV2 using computational approach. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1841026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batool A., Bibi N., Amin F., Kamal M.A. Drug designing against NSP15 of SARS-COV2 via high throughput computational screening and structural dynamics approach. Eur. J. Pharmacol. 2020;892:173779. doi: 10.1016/j.ejphar.2020.173779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khan R.J., Jha R.K., Singh E., Jain M., Amera G.M., Singh R.P., Muthukumaran J., Singh A.K. Identification of promising antiviral drug candidates against non-structural protein 15 (NSP15) from SARS-CoV-2: an in silico assisted drug-repurposing study. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1814870. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.