


Abstract
Defects in the posttranscriptional modifications of mitochondrial tRNAs have been linked to human diseases, but their pathophysiology remains elusive. In this report, we investigated the molecular mechanism underlying a deafness-associated tRNAIle 4295A>G mutation affecting a highly conserved adenosine at position 37, 3′ adjacent to the tRNA’s anticodon. Primer extension and methylation activity assays revealed that the m.4295A>G mutation introduced a tRNA methyltransferase 5 (TRMT5)-catalyzed m1G37 modification of tRNAIle. Molecular dynamics simulations suggested that the m.4295A>G mutation affected tRNAIle structure and function, supported by increased melting temperature, conformational changes and instability of mutated tRNA. An in vitro processing experiment revealed that the m.4295A>G mutation reduced the 5′ end processing efficiency of tRNAIle precursors, catalyzed by RNase P. We demonstrated that cybrid cell lines carrying the m.4295A>G mutation exhibited significant alterations in aminoacylation and steady-state levels of tRNAIle. The aberrant tRNA metabolism resulted in the impairment of mitochondrial translation, respiratory deficiency, decreasing membrane potentials and ATP production, increasing production of reactive oxygen species and promoting autophagy. These demonstrated the pleiotropic effects of m.4295A>G mutation on tRNAIle and mitochondrial functions. Our findings highlighted the essential role of deficient posttranscriptional modifications in the structure and function of tRNA and their pathogenic consequence of deafness.
INTRODUCTION
Posttranscriptional modifications of transfer RNA (tRNA) affect all aspects of tRNA structure and function, including the proper processing, stability, folding and decoding properties of tRNAs, fidelity and efficiency of translation (1–3). Defects in the posttranscriptional modifications of mitochondrial tRNAs have been linked to an array of human diseases, including neurological and muscular disorders, cardiovascular diseases, diabetes and deafness (3–9). In human mitochondria, 18 types of nucleotide modifications are present in the 137 positions of 22 tRNA species, encoded by mitochondrial DNA (mtDNA) (10–12). The nucleotide modifications of tRNAs were catalyzed by tRNA modifying enzymes, such as TRMU, GTPBP3 and MTO1, encoded by nuclear genome (13–17). Of core modifications, the pseudouridine (Ψ) at position 55 is required for the proper folding and stability of tRNAs (18). The loss of pseudouridinylation at position 55 at the TΨC loop of tRNAGlu perturbed the tRNA biogenesis and translation and caused maternally inherited deafness and diabetes (8). The anticodon loop modifications of tRNAs, including nucleotides at positions 34 and 37, impact the stabilization of anticodon structure, fidelity and efficiency of translation (19–23). The deficient modifications at U34 of tRNALys, tRNAGlu and tRNAGln, caused by deficiencies of tRNA modifying enzymes GTPBP3, MTO1 and TRMU, were associated with several human diseases including deafness (24–27). However, pathophysiology underlying the deficient tRNA modifications remains poorly understood.
The nucleotides at position 37 (A or G) of mammalian mitochondrial tRNAs contain a diverse range of complex modifications including N6-threonylcarbamoyladenosine (t6A), N6-isopentenyladenosine (i6A), 2-methylthio-N6-isopentenyladenosine (ms2i6A), and 1-methylguanosine (m1G) (10,28). However, nucleotide modification at this position is absent in five mitochondrial tRNAs such as tRNAMet and tRNAAsp (10,28). In the tRNALeu(CUN), tRNAPro and tRNAGln, the m1G37 modification is synthesized by YRDC and OSGEPL1 (10,14). In contrast, the biosynthesis of t6A37 present in the tRNAIle, tRNALys, tRNASer(AGY), tRNAAsn and tRNAThr was catalyzed by tRNA dimethylallyltransferase (TRIT1) (10,14,28,29). Deficiencies in the YRDC/OSGEPL1 or TRMT5 perturbed the t6A37 or m1G37 modifications leading to several clinical presentations (29, 30). Furthermore, mitochondrial tRNA mutations at position 37 including the tRNAAsp 7551A>G, tRNAMet 4435A>G and tRNAIle 4295A>G mutations were associated with hypertension, diabetes, visual loss, and deafness (31–37). In particular, the tRNAAsp 7551A>G and tRNAMet 4435A>G mutations introduced the new modified nucleotide m1G37 and altered the structure and function of corresponding tRNA (31,32). The tRNAIle 4295A>G mutation was identified in several French and Chinese pedigrees with maternal transmission of deafness (35,36). It was hypothesized that the substitution of A with G at position 37 of the tRNAIle may replace the t6A37 synthesized by YRDC/OSGEPL1 with the m1G37 modification catalyzed by TRMT5, thereby altering the structure and function of tRNAIle. Especially, the m.4295A>G mutation impacts the processing of tRNA precursors, aminoacylation capacity and stability of tRNAIle and mitochondrial translation. It was also anticipated that impairment of mitochondrial translation caused by the m.4295A>G mutation altered the respiration, production of ATP and reactive oxygen species (ROS). To further investigate the molecular mechanism of m.4295A>G mutation, cybrid cell lines were constructed by transferring mitochondria from lymphoblastoid cell lines derived from an affected matrilineal relative in a Chinese family bearing the m.4295A>G mutation and from a control individual lacking the mtDNA mutation, into human mtDNA-less (ρ°) cells (38–39). In the present study, we first examined if the m.4295A>G mutation introduced the m1G37 modification of tRNAIle by using primer extension and in vitro methylation assays. These resultant cybrid cell lines were then assessed for the effects of the m.4295A>G mutation on the aminoacylation capacity and stability of this tRNA, mitochondrial translation, respiration, and the production of ATP and oxidative reactive species (ROS), mitochondrial membrane potential and autophagy.
MATERIALS AND METHODS
Subjects and audiological examinations
A total of 2651 genetically unrelated Han Chinese subjects with hearing impairment and 574 normal hearing Han Chinese control subjects for this study were described elsewhere (40). One hearing-impaired Chinese Han pedigree for this study was ascertained at the Otology Clinic of the Children's Hospital, Zhejiang University School of Medicine. This study was in compliance with the Declaration of Helsinki. Informed consent, blood samples and clinical evaluations were obtained from all participants and families, under protocols approved by Ethic Committees of Zhejiang University School of Medicine. Audiological and neurological examinations of hearing impairment were performed as detailed previously (41,42). All available members of the pedigree and control subjects were evaluated at length to identify both of personal and family medical history of hearing loss, the history of the use of aminoglycosides and other clinical abnormalities.
Analysis of mitochondrial DNA
Genomic DNA was isolated from whole blood of participants using QIAamp DNA Blood Mini Kit (Qiagen, No. 51104). The subject's DNA fragments spanning the tRNAIle genes were PCR amplified by use of oligodeoxynucleotides corresponding to mtDNA at 3796–4693 (12). Fragment was purified and then analyzed by direct sequencing. These sequence results were compared with the updated consensus Cambridge sequence (GenBank accession number: NC_012920) (12). The entire mtDNAs of proband WZD104 III-8 and one Chinese control subject (C59) were PCR amplified in 24 overlapping fragments using sets of the light (L) and heavy (H) strand oligonucleotide primers, as described previously (43). These sequence results were compared with the updated consensus Cambridge sequence, as described above. To quantify the m.4295A>G mutation, PCR segment (251 bp) spanning the tRNAIle gene was amplified and subsequently digested with restriction enzyme NlaIII, as the m.4295A>G mutation and mismatched oligodeoxynucleotides primer together created the site for this enzyme (44). Equal amounts of various digested samples were then analyzed by electrophoresis through 10% polyacrylamide/8 M urea gel. The proportions of digested and undigested PCR products were determined by Image-Quant program after ethidium bromide staining to determine if the m.4295A>G mutation is in homoplasmy in these subjects.
Cell lines and culture conditions
Immortalized lymphoblastoid cell lines were generated from one hearing-impaired matrilineal relative (III-8) of the Chinese family carrying the m.4295A>G mutation and one genetically unrelated Chinese control individual (C59) belonging to the same mtDNA haplogroup D4j but lacking the mutation (45). These cell lines were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS). The bromodeoxyuridine (BrdU) resistant 143B.TK− cell line was grown in Dulbecco's Modified Eagle Medium (DMEM) (Life Technologies) (containing 4.5 mg of glucose and 0.11 mg pyruvate/ml), supplemented with 100 μg of BrdU/ml and 5% FBS. The mtDNA-less ρo206 cell line, derived from 143B.TK− was grown under the same conditions as the parental line, except for the addition of 50 μg of uridine/ml. Transformation by cytoplasts of mtDNA-less ρo206 cells using one affected subject (III-8) carrying the m.4295A>G mutation and one control individual (C59) was performed as described elsewhere (38–39). All cybrid cell lines constructed with enucleated lymphoblastoid cell lines were maintained in the same medium as the 143B.TK− cell line. Three mutant cybrids (III-8.46, III-8.47 and III-8.48) carrying the m.4295A>G mutation and three control cybrids (C59.21, C59.23 and C59.32) lacking the mutation with similar mtDNA copy numbers and the same karyotype were used for the biochemical characterization described below.
Molecular dynamics (MD) simulation
The anticodon stem and loop (ASL) of tRNAIle containing 17 nucleotide bases (Figure 1A) were constructed as the model of simulations. The initial coordinates for wild type tRNAIle molecule were obtained from the crystal structure of human mitochondrial tRNAPhe–PheRS complex (Protein Data Bank entry 3TUP). The coordinates of the backbone were maintained, and nucleotide bases were substituted according to tRNAIle sequence by using Chimera software (46). Atomic coordinates of the m1G (N1-methylguanosine) were obtained from X-ray structures (PDB ID: 2TRA). Based on the anticodon stem and loop structure of wild type tRNAIle, A37 was substituted to m1G37 in Chimera to establish the initial coordinates of the mutant. For the wild type tRNA molecule, MD simulation were performed with Amber14 utilizing ff14SB force field parameters (47). For the mutant molecule, Antechamber was used to generate complete unusual force field files of the modified nucleotide. Finally, 100 ns production simulations were carried out with a time step of 2 fs for both systems, as detailed previously (48).
Figure 1.

MD simulations on the anticodon stem-loop of wild-type and mutated tRNAIle. (A) Cloverleaf structure of human mitochondrial tRNAIle. An arrow indicated the location of the m.4295A>m1G mutation. Nucleotides at the dashed box in the anticodon stem–loop of tRNAIle were used for MD simulation. (B) The tertiary structures of the anticodon stem-loop for the wild-type (gray) and mutated (dodger blue) tRNAIle. (C) Time evolution of the root mean square deviation (RMSD) values of all backbone atoms on the anticodon stem–loop for the wild-type (WT) (black curve) and mutant (MT) (red curve) of tRNAIle. (D) RMSF curves calculated from the backbone atoms for the wild-type (black lines) and mutated (red lines) anticodon stem-loop of tRNAIle.
Primer extension assays of in vivo m1G37 modification
A primer extension experiment to analyze the m1G37 modification of tRNAIle was carried out as detailed elsewhere (31,32). Total mitochondrial RNAs were obtained using TOTALLY RNATM kit (Ambion) from mutant and control cybrids (∼2.0 × 108 cells) (49). A DNA primer (5′-TGGTAAGATATATAGGATTTAGCCTATAAT-3′) complementary to the 3′ end of the tRNAIle was the 5′ end labeled with non-radioactive digoxigenin (DIG). Primescript II 1st Strand cDNA Synthesis Kit (TAKARA) was used for reverse transcription with DIG-labeled oligodeoxynucleotide probe specific for the tRNAIle by using total mitochondrial RNA as templates. Extension reactions were performed as detailed previously (31). The samples were applied onto 15% polyacrylamide gel electrophoresis (PAGE) with 8 M urea in Tris–borate–EDTA (TBE) buffer and electroblotted onto a positively charged nylon membrane (Roche).
In vitro methylation activity assays
The fragments spanning tRNAIle (corresponding to mtDNA at positions 4263–4331) were PCR-amplified with genomic DNAs from the proband (III-8) carrying the m.4295A>G mutation and control subject (C59) lacking the mutation and cloned into the EcoRI/BamHI sites of pUC19. The unmodified tRNAs were generated by in vitro transcription using T7 RNA polymerase, as detailed elsewhere (50). The purification of archaeal Trm5 (aTrm5) from Methanocaldococcus jannaschii (Mj-Trm5) was carried out as detailed previously (32,51,52). The methylation reaction contained 200 μM 3H-SAM, 50 mM Tris–Cl, pH 7.0, 100 mM KCl, 10 mM MgCl2, 100 μg/ml bovine serum albumin (BSA), 5 mM DTT and 5 μM wild type or mutant unmodified tRNAIle. The reaction was undertaken by the addition of 2 μM Mj-Trm5 at 37°C. At time intervals ranging between 2 and 8 min, aliquots of 5 μl were removed, absorbed on paper discs and precipitated in trichloroacetic acid. The relative modification efficiency was calculated from the initial phase of the reaction.
Mitochondrial RNase P assay
The wild type and mutant precursors of tRNAIle corresponding to mtDNA at positions 4235 (5′) to 4350 (3′) were cloned into the pCRII-TOPO vector carrying SP6 and T7 promoters (Clonetech). After HindIII digestion, the RNA substrates (116 nt) were transcribed with T7 RNA polymerase, in the presence of 10 μM ATP, CTP, GTP and UTP, pH 7.5 and 10 units RNase inhibitor at 20°C. Transcripts were purified by denaturing polyacrylamide gel electrophoresis (PAGE) (8 M urea, 8% polyacrylamide/bisacrylamide [19:1]) and were dissolved in 1 mM EDTA. Mitochondrial RNase P was reconstituted from purified recombinant proteins MRPP1, MRPP2 and MRPP3 as described previously (53–55). Processing assays were carried out in parallel for wild type and mutant substrates in 40 μl reaction mixtures containing 30 mM TrisCl, 30 mM NaCl, 4.5 mM MgCl2, 200 μg/ml BSA, 600 ng RNA substrates and 25 nM RNase P, at 30°C. After 5, 10, 15, 20 and 25 min, 5 μl aliquots were withdrawn and stopped by addition of 5 μl loading buffer (85% formamide, 10 mM EDTA). Reaction products were resolved via denaturing PAGE, then electroblotted onto a nylon membrane (Roche) and hybridized with DIG-labeled oligodeoxynucleotide probes for tRNAIle precursor. DIG-labeled probes were generated by using DIG-oligonucleotide Tailing kit (Roche). The hybridization and quantification of density in each band were performed as detailed previously (53).
UV melting assays
UV melting assays were carried out as described elsewhere (32). The wild type and mutant tRNAIle transcripts were generated as described above. The tRNAIle transcripts were diluted in the buffer including 50 mM sodium phosphate (pH 7.0), 50 mM NaCl, 5 mM MgCl2 and 0.1 mM EDTA. Absorbance against temperature melting curves were measured at 260 nm with a heating rate of 1°C /min from 25 to 95°C through Agilent Cary 100 UV Spectrophotometer.
Mitochondrial tRNA analysis
Total mitochondrial RNAs were obtained from mitochondria isolated from cybrid cell lines (∼2.0 × 108 cells), as described previously (49). The tRNA Northern blot analysis was performed as detailed elsewhere (32). DIG-labeled oligodeoxynucleotide probes specific for tRNAIle, tRNAMet, tRNAAsp, tRNAGlu, tRNALys and 12S rRNA were as detailed elsewhere (55). The hybridization and quantification of density in each band were carried out as detailed previously (53–55).
For the aminoacylation assays, total mitochondrial RNAs were isolated under acid conditions, and 4 μg of total mitochondrial RNAs were electrophoresed at 4°C through an acid (pH 5.0) 10% polyacrylamide–8 M urea gel to separate the charged and uncharged tRNA as detailed elsewhere (32,56). To further distinguish nonaminoacylated tRNA from aminoacylated tRNA, samples of tRNAs were deacylated by being heated for 10 min at 60°C at pH 8.3 and then run in parallel (32,56). The gels were then electroblotted onto a positively charged nylon membrane (Roche) for the hybridization analysis with oligodeoxynucleotide probes as described above. Quantification of density in each band was performed as detailed previously (32).
For the tRNA mobility shift assay, two μg of total mitochondrial RNAs were electrophoresed through a 10% polyacrylamide native gel at 4°C with 50 mM Tris-glycine buffer. After electrophoresis, the gels were treated according to the Northern blot analysis as described above (32,48).
Mitochondrial translation assays
Pulse-labeling of the mutant and control cell lines for 30 min with [35S] methionine–[35S] cysteine in methionine-free DMEM in the presence of emetine, electrophoretic analysis of the translation products, and quantification of radioactivity in the whole-electrophoretic patterns or in individual well-resolved bands were carried out as detailed previously (57).
Western blot analysis
Western blotting analysis was performed as detailed previously (32,55). Twenty micrograms of total cell proteins obtained from various cell lines were denatured and loaded on sodium dodecyl sulfate (SDS) polyacrylamide gels. The gels were electroblotted onto a polyvinylidene difluoride (PVDF) membrane for hybridization. The antibodies used for this investigation were from Abcam [TOM20 (ab56783), ND1 (ab74257), ND5 (ab92624) and CO2 (ab110258), ND6 (ab81212), Total OXPHOS Human WB Antibody Cocktail (ab110411), NDUFS3 (ab14711), p62 (ab56416)], Cell signaling [SOD1 (4266T), SOD2 (13141T) and Catalase (12980T)], Novus [ND4 (NBP2–47365), LC3 (NB100-2220)] and Proteintech [CYTB (55090-1-AP), ATP8 (26723-1-AP), CO10 (10611-2-AP), β-actin (20536-1-AP), MRPS18B (16139-1-AP), MRPL44 (16394-1-AP)]. Peroxidase AffiniPure goat anti-mouse IgG and goat anti-rabbit IgG (Jackson) were used as a secondary antibody and protein signals were detected using the ECL system (CWBIO). Quantification of density in each band was performed as detailed elsewhere (55).
Blue native electrophoresis analysis
Blue native polyacrylamide gel electrophoresis (BN-PAGE) was performed by isolating mitochondrial proteins from mutant and control cell lines, as detailed previously (58,59). Samples containing 30 μg of total cellular proteins were separated on 3–12% Bis–Tris Native PAGE gel. The total OXPHOS Human WB Antibody Cocktail was applied as primary antibodies for this experiment. Peroxidase AffiniPure goat anti-mouse IgG and goat anti-rabbit IgG (Jackson) were used as secondary antibodies and protein signals were detected using the ECL system (CWBIO).
Assays of activities of respiratory complexes
The enzymatic activities of complex I, II, III and IV were assayed as detailed elsewhere (39,60). In brief, citrate synthase activity was analyzed by the reduction of 5,5'-dithiobis-2-nitrobenzoic acid at 412 nm in the assay buffer containing 0.1 mM 5,5'-dithiobis-2-nitrobenzoic acid, 50 μM acetyl coenzyme A, and 250 μM oxaloacetate. Complex I activity was determined with 10 μg/ml antimycin A and 2 mM KCN by following the decrease in the absorbance due to the NADH oxidation at 340 nm in assay buffer. The activity of complex II was analyzed by tracking the secondary reduction of DCPIP by decylubiquinone at 600 nm in the assay buffer. Complex III activity was determined in the presence of 2 μg/ml antimycin A and 2 mM KCN by measuring the reduction of cytochrome c at 550 nm with reduced decylubiquinone in the assay buffer. Complex IV activity was measured by monitoring the oxidation of reduced cytochrome c as a decrease of absorbance at 550 nm in the assay buffer. All assays were performed by using Synergy H1 (Biotek, Winooski, VT). Complex I–IV activities were normalized by citrate synthase activity.
Measurements of oxygen consumption
The rates of oxygen consumption (OCR) in various cybrid cell lines were measured with a Seahorse Bioscience XF-96 extracellular flux analyzer (Seahorse Bioscience), as detailed previously (61,62). Cybrid cells were seeded at a density of 2 × 104 cells per well on Seahorse XF96 polystyrene tissue culture plates (Seahorse Bioscience). Inhibitors were used at the following concentrations: oligomycin (1.5 μM), carbonyl cyanide 4-trifluoromethoxy-phenylhydrazone (FCCP) (0.8 μM), antimycin A (1.5 μM) and rotenone (3 μM).
ATP measurements
The CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega) was used for the measurement of cellular and mitochondrial ATP levels, according to the modified manufacturer's instructions (62). Briefly, the assay buffer and substrate were equilibrated at room temperature, transferred to and gently mixed with the substrate to obtain a homogeneous solution. After a 30 min equilibration of the cell plate at room temperature, 100 μl of the assay reagents were added into each well with 2 × 104 cells and the content was mixed for 2 min on an orbital shaker to induce cell lysis. After 10 min incubation in room temperature, the luminescence was read on a microplate reader (Syneregy H1, Bio-Tek).
Assessment of mitochondrial membrane potential
Mitochondrial membrane potential was assessed with JC-10 Assay Kit-Microplate (Abcam) following general manufacturer's recommendations with some modifications, as detailed elsewhere (32,63). In brief, ∼2 × 106 cells of each cybrid cell line were harvested, resuspended in 200 μl 1 × JC-10 Assay buffer and then incubated at 37°C for 30 min. Alternatively, harvested cells were preincubated with 10 μM of FCCP for 30 min at 37°C prior to staining with JC-10 dye. After washing with PBS twice, cells were resuspended in 200 μl PBS. The fluorescent intensities for both J-aggregates and monomeric forms of JC-10 were measured at Ex/Em = 490/530 and 490/590 nm with DB-LSR II flow cytometer system (Beckton Dickson, Inc.).
Measurement of ROS production
The levels of mitochondrial reactive oxygen species (ROS) generation were determined using MitoSOX assay as detailed previously (53,64). Briefly, approximate 2 × 106 cells of each cell line were harvested, resuspended in PBS supplemented with 100 μM of 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) and then incubated at 37°C for 20 min. After washing with PBS twice, cells were resuspended in PBS in the presence of 2 mM freshly prepared H2O2 and 2% FBS and then incubated at room temperature for another 45 min. Cells were further washed with PBS and resuspended with 1 ml of PBS with 0.5% paraformaldehyde. Samples with or without H2O2 stimulation were analyzed by BD-LSR II flow cytometer system (Beckton Dickson, Inc.), with an excitation at 488 nm and emission at 529 nm. Ten thousand events were analyzed in each sample.
Autophagy assessment
The status of autophagy in cybrids were measured via CYTO-ID® Autophagy Detection Kit (Enzo), according to the modified manufacturer's instructions (53). In brief, 5 × 105 cells were trypsinized, washed with 1 × Assay buffer and resuspended with CYTO-ID® Green stain solution. After incubated at 37°C for 30 min in the dark, cells were washed and resuspended in 1 × Assay Buffer. Samples were analyzed by BD-LSR II flow cytometer system (Beckton Dickson, Inc.), with an excitation at 488 nm and emission at 529 nm. Ten thousand events were analyzed in each sample.
Statistical analysis
Statistical analysis was performed using the unpaired, two-tailed Student's t-test contained in the Microsoft-Excel program (version 2017). Differences were considered significant at a P < 0.05.
RESULTS
Clinical presentations and derived cell lines of a hearing-impaired Chinese pedigree carrying the m.4295A>G mutation
One Han Chinese hearing-impaired proband carrying the m.4295A>G mutation was identified among 2651 Chinese hearing-impaired probands but absent in 574 Chinese normal hearing controls (35,40). As shown in Supplemental Figure S1, the Chinese pedigree exhibited the maternal inheritance of hearing loss. As shown in Supplemental Figure S2 and Supplemental Table S1, 9 of 14 matrilineal relatives exhibited the variable degree of hearing impairment (one with mild hearing loss, four with moderate hearing loss, three with severe hearing loss and one with profound hearing loss), whereas none of other members in this family had hearing loss. The age-at-onset of hearing loss ranged from 23 to 50 years, with the average of 35 years old. There was no evidence that any of other members of this family had any other causes to account for hearing loss. These matrilineal relatives showed no other clinical presentations. Further analysis showed that the m.4295A>G mutation was present in homoplasmy in all matrilineal relatives but not in other members of this family (Supplemental Figure S1B, C).
Immortalized lymphoblastoid cell lines were derived from one affected matrilineal relative carrying the m.4295A>G mutation (III-8; male, 23 years) and from one genetically unrelated control subject lacking the m.4295A>G mutation belonging to the same mtDNA haplogroup D4 (C59, male, 20 years) (Supplemental Table S2). The lymphoblastoid cells were enucleated, and subsequently fused to a large excess of mtDNA-less human ρo206 cells, derived from the 143B.TK- cell line (38–39). These cybrid clones were isolated by growing the fusion mixtures in the selective DMEM medium, containing BrdU and lacking uridine and subsequently analyzed for the presence and levels of the m.4295A>G mutation (44). The results confirmed the absence of the mtDNA mutation in the control clones and its presence in homoplasmy in all cybrids derived from the mutant cell line. Three cybrids derived from each donor cell line with similar mtDNA copy number and same karyotype were used for the biochemical characterization described below.
MD simulation analysis
As shown in Figure 1A, the m.4295A>G mutation affected the highly conserved adenine (A37) of the anticodon loop in tRNAIle, and led to the substitution of t6A37 with m1G37 modification. To assess the impact of m.4295A>G mutation on the tertiary structure of tRNAIle, MD simulations were carried out using the anticodon stem-loop (ASL) (17nt) of wild type and mutant tRNAIle by 100-ns all-atom. Figure 1B shows the tertiary structures of ASL region in the wild type and mutant tRNAIle after simulation. As illustrated in Figure 1C, the RMSD (root mean square deviation) values in the mutant ALS region were much less than those in wild type counterpart, indicating that mutant ASL structure of tRNAIle was less flexible than that of wild type counterpart. Furthermore, RMSF (root mean square fluctuation) was calculated from the two trajectories to analyze how this mutation affected the stability of ASL structure in the tRNAIle. As shown in Figure 1D, there were no significant differences of RMSF values between tRNAIle with m1G37 and A37. However, the RMSF values of G34 and U36 in mutant tRNAIle were much lower than those of wild type tRNAIle, indicating that the induced m1G37 affected the stability of anticodon structure. These results implied that the m.4295A>G mutation affected the structure and function of tRNAIle.
The m.4295A>G mutation substituted the t6A37 modification to m1G37 of tRNAIle
To investigate if m.4295A>G mutation caused the substitution of t6A37 with m1G37 modification, we subjected mitochondrial RNAs from mutant and control cybrids to the reverse transcription with DIG-labeled oligonucleotide probe specific for tRNAIle (Figure 2A). This resulted in a stop band one residue 3' to the methylation on 15% polyacrylamide gel. As shown in Figure 2B, the m1G37 modification was present in tRNAIle derived from mutant cell line but absent in the tRNAIle derived from control cell line.
Figure 2.
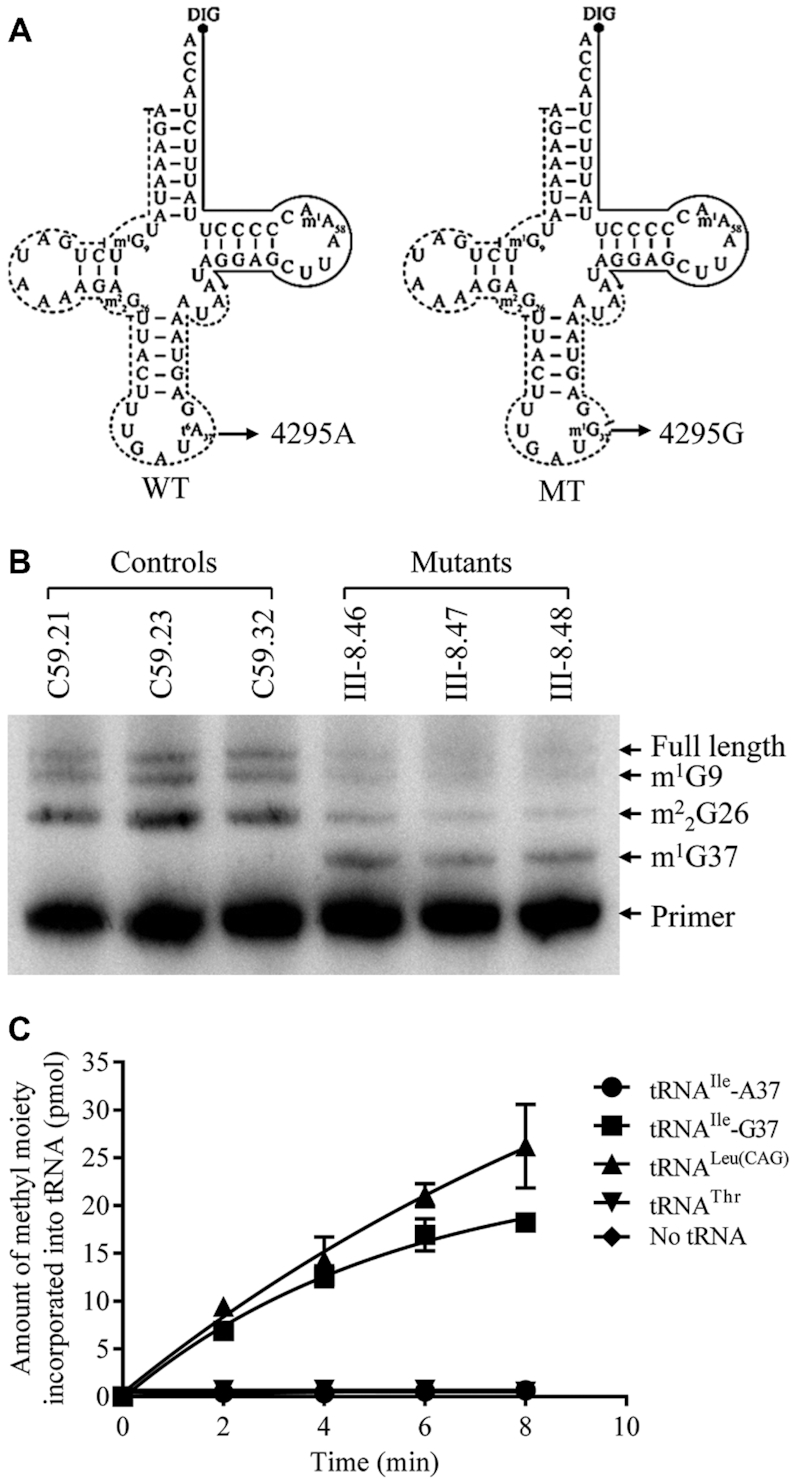
The m.4295A>G mutation introduced the m1G37 modification of tRNAIle. (A) Schematic of methylation shown in the cloverleaf structures of the human mitochondrial tRNAIle. An arrow denotes the location of the m.4295A>G mutation. Solid lines represent the DIG-labeled oligonucleotide probe specific for mt-tRNAIle. Broken lines represent the potential stops of primer extension and m1G or m2G. (B) Primer extension demonstrated the creation of m1G37 in the tRNAIle carrying the m.4295A>G mutation. The primer extension termination products are showed as m1G9, m22G26 and m1G37. (C) Methylation activity assays. The unmodified human mitochondrial wild type (A37) and mutant (G37) tRNAIle, cytosolic tRNALeu(CAG) and tRNAThr were generated from in vitro transcription. The unmodified tRNA transcripts were incubated with M. jannaschii (Mj-Trm5) in the presence of S-adenosyl-l-methionine. Samples were withdrawn and stopped after 2, 4, 6 or 8 min, respectively. The relative modification efficiency was calculated from the initial phase of the reaction. The calculations were based on three independent determinations.
To further examine if the m.4295A>G mutation introduced the m1G37 modification of tRNAIlein vitro, we prepared with wild type and mutant tRNAs by in vitro transcription and evaluated the methylation activity catalyzed by the recombinant M. jannaschii Trm5 (Mj-Trm5) (32,51,52). As shown in Figure 2C, the mutant tRNAIle transcripts (G37) were modified with m1G37 modification by the Mj-Trm5 but less efficiently as cytoplasmic tRNALeu(CAG) transcripts (G37). In contrast, the modification was not detected in the wild type tRNAIle transcripts (A37) in the presence of Mj-Trm5. As controls, human cytoplasmic tRNALeu(CAG) transcripts (G37) were modified by the Mj-Trm5, while human cytoplasmic tRNAThr transcripts (A37) were not modified in the presence of Mj-Trm5. These results indicated that the substitution of A to G at position 37 introduced the m1G37 modification of tRNAIle.
Impaired the 5′ end processing of tRNAIle precursor
We explored an in vitro processing system to assess the effect of m.4295A>G mutation on 5′ end processing of tRNAIle. As shown in Figure 3A, the wild type and mutant tRNAIle precursors corresponding to mtDNA at positions 4235 to 4350 were incubated with mitochondrial RNase P, which was reconstituted from purified recombinant proteins MRPP1, MRPP2 and MRPP3, at various time courses (53,55,65). No qualitative processing alterations of the mutant tRNAIle precursors were observed, but the processing efficiencies of the mutant tRNAIle transcripts were mildly decreased, as compared with those of wild type counterparts (Figure 3B). The processing efficiencies of mutant tRNAIle transcripts catalyzed by RNase P were 46.8% of those in their wild type counterparts (Figure 3C). These results demonstrated that the m.4295A>G mutation perturbed the 5′ end processing of tRNAIle precursors.
Figure 3.

In vitro assay for the processing of mitochondrial tRNAIle precursors. (A) Mitochondrial tRNAIle precursors. Twenty-nine nucleotides (nt) of 5′ end leaders of tRNAIle were shown, including the m.4295A>G substitution. (B) In vitro processing assays. Processing assays with mitochondrial RNase P were carried out in parallel for wild type and mutant substrates. Samples were withdrawn and stopped after 5, 10, 15, 20 or 25 min, respectively. Reaction products were resolved by denaturing polyacrylamide gel electrophoresis and reacted with a chemiluminescent substrate CDP-Star™ to detect the chemiluminescent. The graph shows the results of a representative experiment of 25 min reaction. (C) Relative processing efficiencies of tRNAIle precursors catalyzed by RNase P. The relative processing efficiencies were calculated from the initial phase of the reaction. The calculations were based on three independent determinations.
Perturbed flexibility and conformation of tRNAIle
To experimentally test the impact of m.4295A>G mutation on the stability of tRNAIle, we measured the melting temperatures (Tm) of wild type and mutant tRNAIle transcripts by calculating the derivatives of absorbance against a temperature curve. As shown in Figure 4A, the Tm values for wild-type (A37) and mutant (unmodified G37, m1G37) transcripts were 41.67 ± 0.56°C, 45.05 ± 0.06°C and 45.08 ± 0.03°C, respectively. These data were in a good agreement with MD data, indicating that the m.4295A>G mutation caused the less flexibility of tRNAIle.
Figure 4.

Analysis of conformation and stability of tRNAIle. (A) Thermal stability of wild type (A37), mutant (unmodified G37 and m1G37) tRNAIle. ΔTm indicates the difference of Tm values between wild type (A37) and mutant (G37) tRNAIle. The calculations were based on three independent experiments. (B) Northern blot analysis of tRNA under native condition. Two microgram of total mitochondrial RNA from various cell lines were electrophoresed through native polyacrylamide gel, electroblotted and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAIle, tRNAMet and tRNASer(AGY), respectively. (C) Northern blot analysis of tRNA under denaturing condition. Two microgram of total mitochondrial RNA from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAIle, tRNAMet, tRNAAsp, tRNAGlu, tRNALys and 12S rRNA, respectively. (D) Quantification of tRNA levels. Average relative tRNAs content per cell, was normalized to the average content per cell of reference 12S rRNA in three cybrid cell lines derived from one hearing-impaired subject (III-8) and three cybrid cell lines derived from one control subject (C59). The values for the latter are expressed as percentages of the average values for the control cell lines. The calculations were based on three independent determinations of each tRNA content in each cell line and three determinations of the content of reference tRNA marker in each cell line. The error bars indicate two standard deviations. P indicates the significance, according to the t-test, of the differences between mutant and control cell lines.
To examine if the m.4295A>G mutation affected the conformation of tRNAIle, total mitochondrial RNAs from mutant and control cell lines were electrophoresed through 10% native polyacrylamide gel in Tris–glycine buffer and then electroblotted onto a positively charged nylon membrane for hybridization analysis with DIG-labelled probes for tRNAIle, tRNAMet and tRNASer(AGY), respectively. As shown in Figure 4B, electrophoretic patterns showed that the tRNAIle in three mutant cybrid cell lines carrying the m.4295A>G mutation migrated faster than those of control cybrid cell lines lacking this mutation. These data suggested that the structural alterations by the m.4295A>G mutation caused the conformation change of tRNAIle.
Reductions in the steady state levels of tRNAIle
To further evaluate the effect of m.4295A>G mutation on the stability of tRNAIle, we subjected mitochondrial RNAs from mutant and control cybrids to Northern blots under a denaturing condition and hybridized them with DIG-labeled oligodeoxynucleotide probes for tRNAIle, tRNAMet, tRNAAsp and tRNALys and 12S rRNA as loading control, respectively. As shown in Figure 4C, the steady-state levels of tRNAIle in the mutant cybrids were markedly decreased as compared to those in control cybrids. For comparison, the average levels of each tRNA in the various control or mutant cybrid cell lines were normalized to the average levels of 12S rRNA in the same cell lines for reference. As shown in Figure 4D, the average levels of tRNAIle in the mutant cybrid cell lines were 36.8% (P<0.001) of mean values of three control cybrids. However, the average levels of tRNAMet, tRNAAsp, tRNAGlu and tRNALys in the mutant cybrid cell lines were comparable with those in control cell lines, respectively.
Aberrant aminoacylation of tRNAIle
To investigate whether the m.4295A>G mutation affected the aminoacylation of tRNAIle, we examined the aminoacylation levels of tRNAIle as well as tRNAThr, tRNALys, tRNAMet and tRNASer(AGY) by the use of electrophoresis in an acidic urea PAGE system to separate uncharged tRNA species from the corresponding charged tRNA, electroblotting and hybridizing with the tRNA probes described above. As shown in Figure 5A, the upper and lower bands represented the charged and uncharged tRNA, respectively. The electrophoretic mobility of either charged or uncharged tRNAIle in mutant cell lines migrated faster than those of control cell lines. To further distinguish nonaminoacylated tRNA from aminoacylated tRNA, samples of tRNAs were deacylated by being heated for 10 min at 60°C at pH 8.3 and then run in parallel. As shown in Figure 5B, only one band (uncharged tRNA) was present in both mutant and control cell lines after deacylating. Notably, the efficiencies of aminoacylated tRNAIle in the mutant cell lines were 85.6% of the average values of control cell lines (P < 0.001) (Figure 5C). However, there were no obvious differences in the electrophoretic mobility and levels of tRNAThr, tRNALys, tRNAMet between mutant and control cell lines. These results suggested that the deficient nucleotide modification at position 37 affected the aminoacylation properties of tRNA.
Figure 5.

In vivo aminoacylation assays. (A) Various amount of total mitochondrial RNA purified from six cell lines under acid conditions were electrophoresed at 4°C through an acid (pH 5.0) 10% polyacrylamide–8 M urea gel, electroblotted and hybridized with a DIG-labeled oligonucleotide probe specific for the mt-tRNAIle. The blots were then stripped and rehybridized with mt-tRNAThr, tRNALys, tRNAMet and tRNASer(AGY), respectively. (B) The samples from one control (C59.32) and mutant (III-8.46) cell lines were deacylated (DA) by heating for 10 min at 60°C at pH.8.3 and electrophoresed as above. (C) Qualification of aminoacylated proportions of tRNAs in the mutant and control cell lines. The calculations were based on three independent determinations. Graph details and symbols are explained in the legend to the Figure 4.
Impairment of mitochondrial translation
To investigate whether the aberrant tRNA metabolism caused by m.4295A>G mutation impaired mitochondrial translation, mutant and control cybrids were labeled for 30 min with [35S]methionine–[35S]cysteine in methionine-free regular DMEM medium in the presence of 100 μg/ml of emetine to inhibit cytosolic protein synthesis (57). Figure 6A showed typical electrophoretic patterns of mitochondrial translation products of mutant and control cell lines. Patterns of the mtDNA-encoded polypeptides of the cybrids harboring the m.4295A>G mutation were qualitatively identical to those of the control cells, in terms of electrophoretic mobility of the various polypeptides. However, cell lines carrying the m.4295A>G mutation displayed significant decrease in the total rate of labeling of mitochondrial translation products relative to those of the control cell line. Figure 6B showed a quantification of the results of a large number of labeling experiments and electrophoretic runs. In particular, the overall levels of 13 mitochondrial translation products in the mutant cell lines were 72.5% (P < 0.001), relative to the mean values measured in the control cell lines. The average levels of ND1, ND2, ND3, ND4, ND4L, ND5, ND6, CYTB, CO1, CO2, CO3, ATP6 and ATP8 in the mutant cells were 66.3%, 63.5%, 84.3, 53.0%, 105.6%, 70.3%, 76.9%, 61.4%, 72.5%, 76.9%, 70.0%, 64.7% and 103.2% of those in the control cell lines, respectively.
Figure 6.

Analysis of mitochondrial translation. (A) Representative gel for electrophoretic patterns of the mitochondrial translation products of six cybrid cell lines labeled for 30 min with [35S]methionine in the presence of 100 μg/ml of emetine and corresponding Coomassie Brilliant Blue stained gel used as loading control. Samples containing equal amounts of protein (30 μg) were run in SDS/polyacrylamide gradient gels. CO1, CO2 and CO3 indicate subunits I, II, and III of cytochrome c oxidase; ND1, ND2, ND3, ND4, ND4L, ND5 and ND6, subunits 1, 2, 3, 4, 4L, 5 and 6 of the respiratory chain reduced nicotinamide-adenine dinucleotide dehydrogenase; A6 and A8, subunits 6 and 8 of the H+-ATPase; and CYTB, apocytochrome b. (B) Quantification of the rates of the mitochondrial translation labeling. The rates of mitochondrial protein labeling were expressed as percentages of the value for average values of three control cybrids in each gel, with error bars representing two SDs. A total of three independent labeling experiments and three electrophoretic analyses of each labeled preparation were performed on cell lines. (C) Twenty micrograms of total cellular proteins from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted and hybridized with 7 mtDNA encoded subunits in mutant and control cells with TOM20 as a loading control. (D) Quantification of mitochondrial protein levels. Average relative ND1, ND4, ND5, ND6, CO2, ATP8 and CYTB content per cell, were normalized to the average content per cell of TOM20 in three mutant cell lines carrying the m.4295A>G mutation and three control cell lines lacking the mutation.
Furthermore, a Western blot analysis was carried out to examine the levels of seven mtDNA encoded OXPHOS subunits in mutant and control cells lines with TOM20 as a loading control. As shown in Figure 6C and D, the overall levels of seven mtDNA encoded proteins in the mutant cybrids were 71.7% (P < 0.001), relative to the mean values measured in the control cybrids. The average levels of ND1, ND4, ND5, ND6, CO2, ATP8 and CYTB in the mutant cells were 65.3%, 31.8%, 68.5%, 69.7%, 79.1%, 102.1% and 85.3% of those in the control cell lines after normalization to TOM20, respectively. However, the levels of polypeptide synthesis in mutant cells, relative to those in control cells, showed no significant correlation with either the number or the proportion of the codons for isoleucine (Supplemental Table S3).
We then examined the levels of seven subunits (mtDNA-encoded CO2 and six nucleus-encoding proteins) of phosphorylation system (OXPHOS) in control and mutant cell lines by Western blotting analysis. As shown in Supplemental Figure 3, the average level of CO2 in the mutant cybrids was 76.0% (P = 0.001) of control cybrids. However, the levels of other six polypeptides, NDUFB8, NDUFS3, SDHB, UQCRC2, CO10 and ATP5A, encoded by nuclear genes, in mutant cell lines were comparable with those in control cell lines.
Blue native gel analysis of OXPHOS complexes
To investigate whether the m.4295A>G mutation induced-alterations affected the stability of OXPHOS complexes, we measured the steady-state levels of five OXPHOS complexes by Blue-native polyacrylamide gel electrophoresis (58,66,67). As shown in Figure 7A and B, the levels of complex I (CI), complex III (CIII), complex IV (CIV) and complex V (CV) in the mutant cybrids were 45.94% (P < 0.001), 82.08% (P < 0.001), 68.79% (P < 0.001) and 60.54% (P < 0.001) of those in the control cybrids after normalization to TOM20, respectively. However, the levels of complex II (CII) in mutant cell lines were comparable with those in control cell lines. The lower levels of the respiratory complexes I, III and IV may be due to the misfolded and/or misassembled these complexes.
Figure 7.

Blue-native gel analysis of OXPHOS complexes. (A) The steady-state levels of five OXPHOS complexes by Blue-Native gel electrophoresis. Thirty microgram of mitochondrial proteins from mutant and control cell lines were electrophoresed through a Blue-Native gel, electroblotted and hybridized with antibody cocktail specific for subunits of each OXPHOS complex as well as Tom20 and Coomassie staining as a loading control. (B) Quantification of levels of complexes I, II, III, IV and V in mutant and control cell lines. The calculations were based on three independent experiments. Graph details and symbols are explained in the legend to Figure 4.
Deficient activities of respiratory chain complexes
To evaluate the effect of the m.4295A>G mutation on the oxidative phosphorylation, we measured the activities of respiratory complexes by the use of isolating mitochondria from mutant and control cell lines. The activity of complex I (NADH ubiquinone oxidoreductase) was determined through the oxidation of NADH with ubiquinone as the electron acceptor. The activity of complex II (succinate ubiquinone oxidoreductase) was examined through the artificial electron acceptor DCPIP. The activity of complex III (ubiquinone cytochrome c oxidoreductase) was measured through the reduction of cytochrome c (III) by using D-ubiquinol-2 as the electron donor. The activity of complex IV (cytochrome c oxidase) was monitored through the oxidation of cytochrome c (II). As shown in Figure 8A, the average activities of complexes I, III, and IV in three mutant cell lines carrying the m.4295A>G mutation were 49.9% (P < 0.001), 88.1% (P < 0.001) and 70.2% (P < 0.001) of the mean values measured in three control cell lines, respectively. The activity of complex II in three mutant cell lines carrying the m.4295A>G mutation was 99.7% (P = 0.905) of the mean values measured in three control cell lines.
Figure 8.
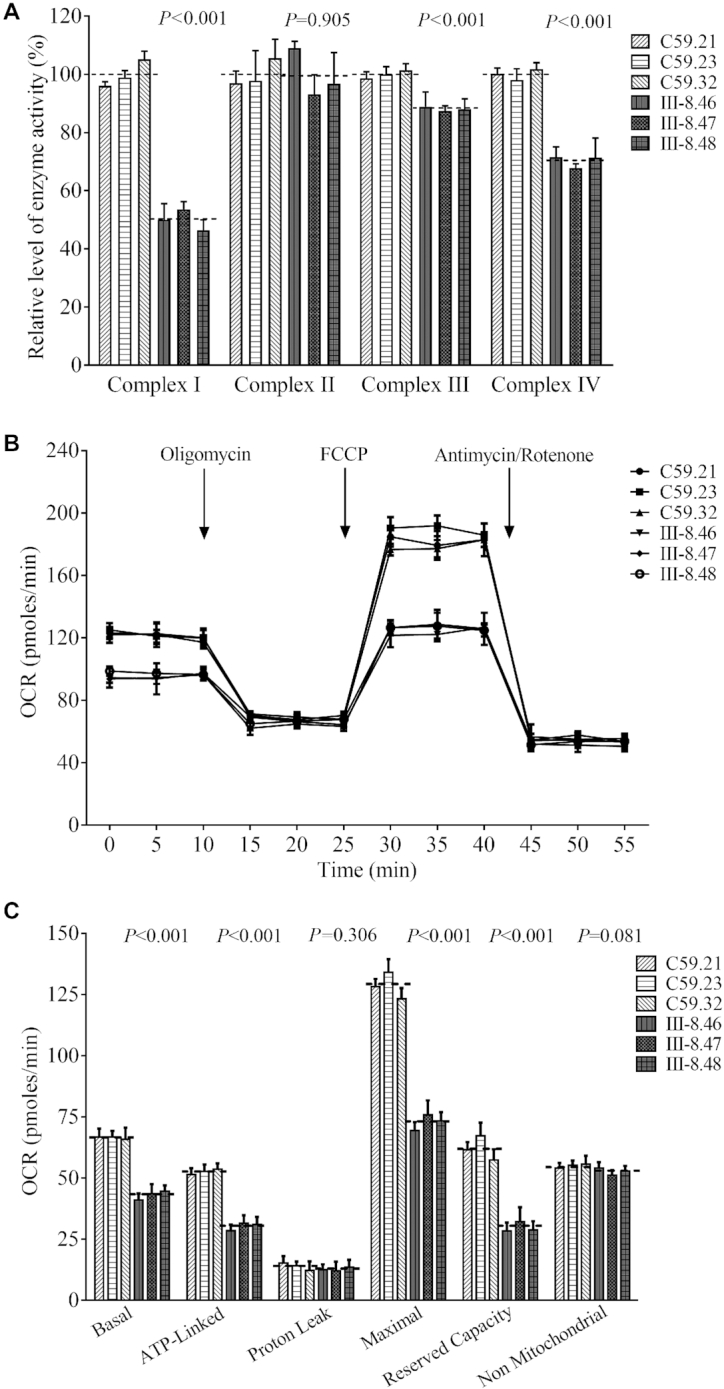
Respiration assays. (A) Enzymatic activities of respiratory chain complexes. The activities of respiratory complexes were investigated by enzymatic assay on complexes I, II, III, and IV in mitochondria isolated from various cell lines. (B) An analysis of O2 consumption in the various cell lines using different inhibitors. The rates of O2 (OCR) were first measured on 2 × 104 cells of each cell line under basal condition and then sequentially added to oligomycin (1.5 μM), carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) (0.5 μM), rotenone (1 μM) and antimycin A (1 μM) at indicated times to determine different parameters of mitochondrial functions. (C) Graphs presented the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity and non-mitochondrial OCR in mutant and control cell lines. Non-mitochondrial OCR was determined as the OCR after rotenone/antimycinA treatment. Basal OCR was determined as OCR before oligomycin minus OCR after rotenone/antimycin A. ATP-linked OCR was determined as OCR before oligomycin minus OCR after oligomycin. Proton leak was determined as Basal OCR minus ATP-linked OCR. Maximal was determined as the OCR after FCCP minus non-mitochondrial OCR. Reserve Capacity was defined as the difference between Maximal OCR after FCCP minus Basal OCR. OCR values were expressed in picomoles of oxygen/minute. The average of 4 determinations for each cell line is shown, the horizontal dashed lines represent the average value for each group. Graph details and symbols are explained in the legend to Figure 4.
The effect of m.4295A>G mutation on cellular bioenergetics was further evaluated by measurement of the oxygen consumption rates (OCR) of mutant and control cell lines using a Seahorse Bioscience XF-96 Extracellular Flux Analyzer (61). As shown in Figure 8B, the basal OCR in three mutant cell lines was 64.8% (P < 0.001) relative to the mean values measured in three control cell lines. To further investigate which complex of the respiratory chain was affected in the mutant cell lines, oligomycin (to inhibit the ATP synthase), FCCP (to uncouple the mitochondrial inner membrane and allow for maximum electron flux through the ETC), rotenone (to inhibit complex I) and antimycin A (to inhibit complex III) were added sequentially while measuring OCR. The difference between the basal OCR and the drug-insensitive OCR yields the amount of ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and non-mitochondrial OCR. The ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity and non-mitochondrial OCR in mutant cell lines were 57.7% (P < 0.001), 91.7% (P = 0.306), 56.7% (P < 0.001), 48.0% (P < 0.001) and 95.9% (P = 0.081), relative to the mean values measured in the control cell lines, respectively (Figure 8C).
Reduced levels in mitochondrial ATP production
Luciferin/luciferase assay was carried out to examine the capacity of oxidative phosphorylation in mutant and wild-type cell lines. Populations of cells were incubated in the media in the presence of glucose, and 2-deoxy-d-glucose with pyruvate. As shown in Figure 9A, the levels of ATP production in mutant cell lines in the presence of glucose (total cellular levels of ATP) were comparable to those measured in control cell lines. In contrast, the levels of ATP production in mutant cell lines, in the presence of 2-deoxy-d-glucose and pyruvate to inhibit the glycolysis (mitochondrial levels of ATP), varied from 64.0% to 64.5%, with an average of 64.2% relative to the mean values measured in the control cell lines (P < 0.001).
Figure 9.
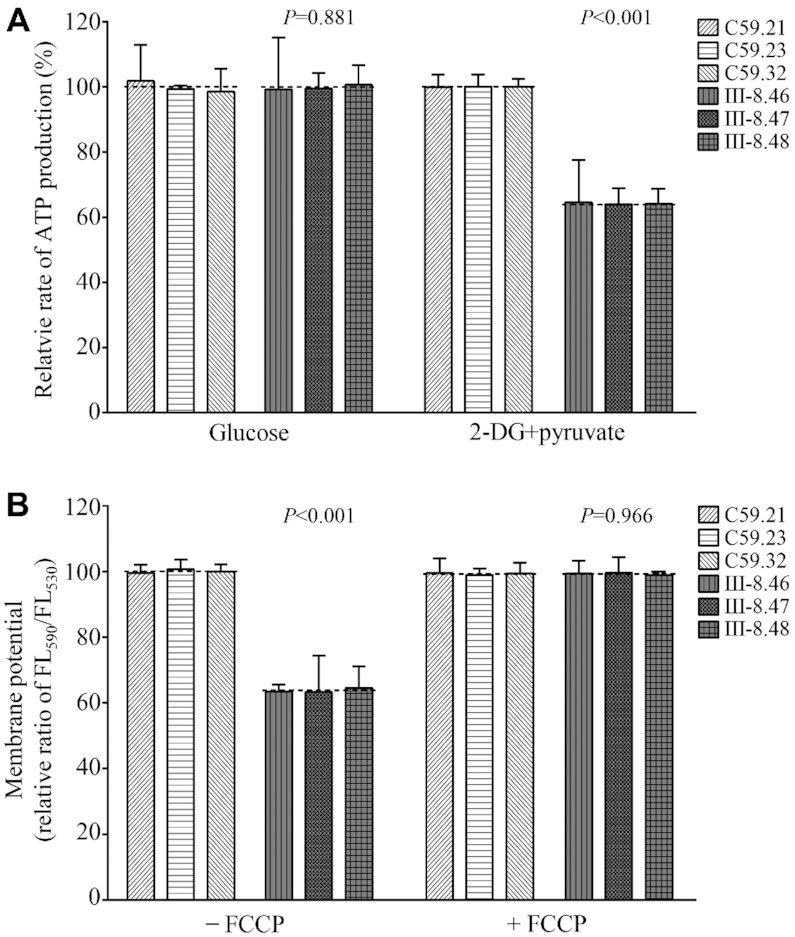
Measurement of mitochondrial ATP levels and membrane potential analysis. (A) Measurement of mitochondrial and cell ATP levels. Cells were incubated with 10 mM glucose or 5 mM 2-deoxy-d-glucose plus 5 mM pyruvate to determine ATP generation under mitochondrial ATP synthesis for 2 h. Average rates of mitochondrial and cellular ATP level per cell line are shown. (B) Mitochondrial membrane potential analysis. The mitochondrial membrane potential (ΔΨm) was measured in mutant and control cell lines using a fluorescence probe JC-10 assay system. The ratio of fluorescence intensities Ex/Em = 490/590 nm and 490/530 nm (FL590/FL530) were recorded to delineate the ΔΨm level of each sample. Relative ratios of JC-10 fluorescence intensities at Ex/Em = 490/525 and 490/590 nm in the absence and presence of 10 μM of FCCP in three control cell lines and three mutant cell lines were shown. The average of 3–4 determinations for each cell line is shown. Graph details and symbols are explained in the legend to Figure 4.
Decreases in mitochondrial membrane potential
The mitochondrial membrane potential (ΔΨm) generated by proton pumps (complexes I, III and IV) is an essential component in the process of energy storage during oxidative phosphorylation (63). Together with the proton gradient (ΔpH), ΔΨm forms the transmembrane potential of hydrogen ions which is harnessed to make ATP (66). As shown in Figure 9B and Supplemental Figure S4, the ΔΨm of three mutant cell lines carrying the m.4295A>G mutation ranged from 63.2% to 64.6%, with an average 63.7% (P < 0.001) of the mean values measured in three control cell lines. In contrast, the levels of ΔΨm in mutant cell lines in the presence of FCCP were comparable to those in the control cell lines.
The increase of mitochondrial ROS production
Mitochondrial ROS are gradually recognized as important signaling mediators in wide range of cellular processes, such as metabolic adaptation, adaptive responses to hypoxia, cellular differentiation, autophagy and regulation of innate or adaptive immunity (68). The levels of mitochondrial ROS among mutant and control cybrid cell lines were determined using MitoSOX assay via flow cytometry under normal conditions and then following H2O2 stimulation. Geometric mean intensity was recorded to measure the production rate of ROS of each sample. As shown in Figure 10A and B, the levels of ROS generation in the mutant cell lines harboring the m.4295A>G mutation ranged from 131.6% to 161.7%, with an average 142.9% (P < 0.001) of the mean values measured in three control cell lines under unstimulated conditions. As illustrated in Figure 10C and D, the levels of ROS generation in the mutant cell lines varied from 210.1% to 228.0%, with an average 220.5% (P < 0.001) of the mean values measured in the control cell lines under stimulation conditions.
Figure 10.

Measurement of mitochondrial ROS production. The levels of ROS generation by mitochondria in living cells from mutant and control cell lines were determined using the mitochondrial superoxide indicator MitoSOX-Red. Fluorescence was measured using a FACS Calibur instrument (BD Biosciences), with excitation at 488 nm and emission at 580 nm. The data were analyzed with Flow Jo software. (A, C) Flow cytometry histogram showing MitoSOX-Red fluorescence of control cybrids (C59.32) (Green) and mutant cybrids (III-8.48) (Red) with or without H2O2 stimulation. (B, D) Relative ratios of MitoSOX-Red fluorescence intensity of control cybrids (C59.32) and mutant cybrids (III-8.48) with or without H2O2 stimulation. (E) Western blotting analysis of antioxidative enzymes SOD1, SOD2 and catalase in six cell lines with β-actin as a loading control. (F) Quantification of SOD1, SOD2 and catalase. Average relative values of SOD1, SOD2 and catalase were normalized to the average values of β-actin in various cell lines. The values for the latter are expressed as percentages of the average values for the control cell lines. The average of three independent determinations for each cell lines is shown. Graph details and symbols are explained in the legend to Figure 4.
To test whether the m.4295A>G mutation-induced mitochondrial ROS production affected the expression of antioxidant systems, we examined the levels of three antioxidant enzymes: SOD2 in mitochondrion, SOD1 and catalase in cytosol in the various cell lines (48,68). As shown in Figure 10E and F, the mutant cell lines revealed marked increases in the levels of SOD2 (182.9%), SOD1 (248.6%) and catalase (201.7%), as compared with those in the control cell lines.
Alteration in autophagy
To investigate if the m.4295A>G mutation affected the autophagy, we evaluated the mitophagic states of mutant and control cell lines by flow cytometry using Cyto-ID, a cationic amphilic tracer dye that labeled mitophagic compartments (69,70). Geometric mean intensity was measured for assessment of autophagy in each sample. As shown in Figure 11A and B, the average levels of autophagy in three mutant cell lines was 129.0% (P < 0.001) of the mean values measured in three control cell lines. The status of autophagy in mutant and control cell lines were further examined using Western blot analysis with two markers: microtubule-associated protein 1A/1B light chain 3B (LC3) and sequestosome 1 (SQSTM1/p62). During autophagy, the cytoplasmic form (LC3-I) is processed into a cleaved and lipidated membrane-bound form (LC3-II), which is essential for membrane biogenesis and closure of the membrane. LC3-II is recleaved by cysteine protease (Atg4B) following completion of the autophagosome, and recycled. SQSTM1/p62, one of the best-known autophagic substrates, interacts with LC3 to ensure the selective delivery of these proteins into the autophagosome (71,72). As shown in Figure 11C, the increased levels of LC3 and reduced level of p62 were observed in mutant cybrids carrying the m.4295A>G mutation, compared to those in control cybrids. In particular, the average levels of LC3-II/(LC3-I+II) and p62 in three mutant cell lines carrying the m.4295A>G mutation were 153.8% (P = 0.001) and 27.3% (P < 0.001) of the mean values measured in three control cell lines, respectively (Figure 11D). These data suggested that the m.4295A>G mutation promoted autophagy in these cybrids.
Figure 11.

Assessment of autophagy. (A) Histogram of flow cytometric analysis of mutant cybrids (III-8.48) and control cybrids (C59.32) using CYTOID® Autophagy Detection Kit. Three control and three mutant cybrids were incubated with DMEM in the absence and presence of rapamycin (inducers of autophagy) and chloroquine (lysosomal inhibitor) at 37°C for 18 h, added to CYTO-ID®-Green dye and analyzed using a Novocyte flow cytometer (ACEA Biosciences). (B) Relative ratios of Cyto-ID fluorescence intensity from three mutant and three control cell lines. Three independent determinations were done in each cell line. (C) Western blot analysis for autophagic response proteins: LC3-I/II and p62. Twenty micrograms of total cellular proteins from various cell lines were electrophoresed, electroblotted and hybridized with LC3, p62 and with β-actin as a loading control. (D) Quantification of autophagy markers LC3 I/II and p62 in mutant and control cell lines were determined as described elsewhere (70). Three independent determinations were done in each cell line. G. Graph details and symbols are explained in the legend to Figure 4.
DISCUSSION
In the present study, we investigated the pathophysiology underlying deafness-associated tRNAIle 4295A>G mutation. This mutation was only present in the matrilineal relatives of the Chinese family with maternal inheritance of deafness. The occurrence of m.4295A>G mutation in several hearing-impaired families from different ethnic backgrounds strongly indicated that this mutation is involved in the pathogenesis of deafness (35,36). The m.4295A>G mutation affected a highly conserved adenosine (A37), adjacent to the 3′ end of the anticodon of tRNAIle, where the position is important for the stability and identity of tRNA, fidelity, and efficiency of translation (18–23). Here, we demonstrated that the m.4295A>G mutation-induced m1G37 had wide-ranging and significant ramifications for the structure and function of tRNAIle in mitochondrial translation, and remarkable effects on mitochondrial function. The creation of m1G37 modification caused by the m.4295A>G mutation was evidenced by the primer extension and enzymatic assays that M. jannaschii Trm5 catalyzed the m1G37 modification in the unmodified mutant (G37) but not wild type (A37) tRNAIle transcripts. In fact, t6A37 modification is responsible for codons starting with A (ANN codons), while the m1G37 modification decodes CNN codons (2,73). In particular, the t6A37 enhances the anticodon-codon base-pairing by cross-strand base-stacking of the t6A37 base with the first position of the codon, and stabilizes the structure of anticodon loop by preventing across the loop base-pairing between U33-A37, as well as maintains the efficiency and accuracy of translation (74–76). In contrast, the m1G37 modification stabilizes the anticodon of tRNA to prevent +1 ribosomal frameshift errors (19,22). MD stimulation revealed that mutant ASL structure of tRNAIle with m1G37 were less flexible than that in wild type counterpart, thereby causing the instability of tRNAIle. The altered structure and stability of tRNAIle caused by the m.4295A>G mutation were further supported by the increasing melting temperature and faster electrophoretic mobility of mutated tRNA with respect to the wild-type molecule, as in the case of tRNAMet 4435A>G mutation (32). The m.4295A>G mutation resulted in the aberrant 5' end processing of tRNAIle precursors, catalyzed by RNase P, and also reduced the 3' end processing efficiency of tRNAIle precursors, mediated by RNase Z (76). Moreover, the deficient nucleotide modifications of A37 or G37 often perturbed the aminoacylation properties of tRNA (32,77,78). In this study, the cell lines bearing the m.4295A>G mutation displayed reduced aminoacylation efficiency and faster electrophoretic mobility of tRNAIle, with respect to the wild-type cell lines, comparable with those with deficient t6A37 modification of tRNAThr (64). As a result, the pleiotropic effects of m.4295A>G mutation on tRNAIle metabolism including the processing of tRNA precursors, stability and aminoacylation of tRNAIle contributed to marked reductions in the steady-state level of tRNAIle, in contrast with mild reductions in tRNAAsp and tRNAMet observed in the cell lines bearing the m.7551A>G and m.4435A>G mutations (31,32). Notably, the reduced levels of tRNAIle in mutant cells harboring the m.4295A>G mutation approached the proposed threshold level, which is 30% of the control levels of tRNA, to support the normal rate of mitochondrial translation (62,79–81).
The m.4295A>G mutation-induced ramifications of tRNAIle metabolisms including the creation of m1G37 modification, shortage and inefficient aminoacylation impaired the mitochondrial translation and subsequently caused oxidative phosphorylation deficiency. Both pulse-labeling mitochondrial translation and Western blot assays showed ∼30% reductions in the levels of mitochondrial proteins in the cells harboring the m.4295A>G mutation. Notably, the mutant cell lines carrying the m.4295A>G mutation exhibited the variable reductions in the levels of 13 mtDNA-encoded polypeptides, ranging from marked reductions in the levels of ND4 with high content of isoleucine codons, to no reduction in the levels of ATP8 with extremely low content of isoleucine codons. However, the reduced levels of these polypeptides in mutant cybrids were not significantly correlated with the numbers or proportions of isoleucines (Supplemental Table S3), in contrast with what was previously shown in cells carrying the tRNASer(UCN) 7445A>G and tRNALys 8344A>G mutations (81,82). Alternatively, the reduced fidelity of translation may make these mitochondrial proteins to be metabolically less stable. Hence, these alterations in mitochondrial protein synthesis perturbed the stabilities and activities of complex I, III, IV and V in the cells bearing the m.4295A>G mutation. In particular, the defects in the synthesis of ND1, ND2, ND4, ND5 and ND6 resulted in the pronounced effects on the assembly and activity of complex I, as in the cases of mutations in the genes encoding complex I subunits (39,70,83). Furthermore, the impairment of mitochondrial translation yielded the respiratory and oxidative phosphorylation deficiencies, including decreased rates in the basal OCR, ATP-linked OCR, reserve capacity and maximal OCR as well as significant decreases in the level of mitochondrial ATP in the cell lines carrying the m.4295A>G mutation, as revealed by those in cell lines carrying the tRNAAsp 7551A>G and tRNAMet 4435A>G mutations (31,32). As a result, the defective oxidative phosphorylation diminished mitochondrial membrane potentials and elevated the production of reactive oxygen species and the subsequent failure of cellular energetic processes (84). In particular, the effect of ROS overproduction on cellular functions was evidenced by the increasing expression of anti-oxidant enzymes SOD1, SOD2 and catalase observed in the cells bearing m.4295A>G mutation (85,86). Furthermore, the m.4295A>G mutation-induced alterations may affect the mitophagic removal of damaged mitochondria (87,88). Using flow cytometry with Cyto-ID, a cationic amphilic tracer dye, the mutant cybrids bearing the m.4295A>G mutation displayed the increasing levels of autophagy. The effect of the m.4295A>G mutation on autophagy were further evidenced by the decreased levels of p62 which indicated the preferential reduction of autophagic substrates, and increased levels of LC3-II that implied the increasing generation of autophagosome in the mutant cybrids. These data indicated that the m.4295A>G mutation promoted the autophagic degradation of ubiquitinated proteins. These mitochondrial dysfunctions may lead to the dysfunction or death of hair cells and neurons in cochlea, thereby contributing to the development of hearing loss (89,90).
However, the incomplete penetrance of deafness and relatively mild biochemical defects indicated that the m.4295A>G mutation was necessary evident but not sufficient to produce a clinical phenotype. The other genetic or epigenetic factors may contribute to the development of clinical phenotype in the subjects carrying the m.4295A>G mutation (15,24,80). In particular, the hearing specific phenotypes of this tRNA mutation may be attributed to the tissue-specificity of OXPHOS via tRNA modification or involvement of nuclear modifier genes (91–93). However, the m.4295A>G mutation is also associated with progressive tissue-specific pathologies, including hypertrophic cardiomyopathy (37) and occipital stroke (94). Changes in tRNA methylation profiles may specify cellular metabolic states and efficiently adapt protein synthesis rates to cell stress in the different tissues (95). Alternatively, the tissue heterogeneity may arise from differential expression of tRNA genes, variable activation of the integrated stress response pathway, metabolic changes and the ability of certain tissues to respond to impaired mitochondrial translation (96–98).
In summary, our findings convincingly demonstrate the pathogenic mechanism underlying the tRNA modification deficiency caused by the deafness-associated tRNAIle 4295A>G mutation. The m.4295A>G mutation caused the substitution of t6A37 with the m1G37 modification, and impacted the tRNAIle structure and function, including the processing of tRNA precursor, stability and aminoacylation capacity of tRNAIle. The aberrant tRNA metabolism resulted in the defects in mitochondrial translation, respiratory deficiency, decreasing membrane potentials and ATP production, and finally increasing ROS production and promoting autophagy. As a result, mitochondrial dysfunctions caused by the m.4295A>G mutation manifested hearing loss. However, the tissue-specificity of this pathogenic mtDNA mutation is likely due to the involvement of nuclear modifier genes or tissue-specific differences in tRNA metabolism. Thus, our findings highlighted the essential role of deficient tRNA modifications in mitochondrial biogenesis and their pathogenic consequence of hearing loss. Furthermore, the approach utilized in this study provides a paradigm for understanding the pathogenic effects of other mitochondrial tRNA mutations.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to patients and their family members for their participation. We are grateful to Dr Shigeyuki Yokoyama (RIKEN Structural Biology Laboratory) for the pET26b-aTrm5 plasmid.
Contributor Information
Feilong Meng, Division of Medical Genetics and Genomics, The Children's Hospital, Zhejiang University School of Medicine and National Clinical Research Center for Child Health, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Mi Zhou, Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Yun Xiao, Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Xiaoting Mao, Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Jing Zheng, Division of Medical Genetics and Genomics, The Children's Hospital, Zhejiang University School of Medicine and National Clinical Research Center for Child Health, Hangzhou, Zhejiang 310058, China.
Jiaxi Lin, Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Tianxiang Lin, Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Zhenzhen Ye, Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Xiaohui Cang, Division of Medical Genetics and Genomics, The Children's Hospital, Zhejiang University School of Medicine and National Clinical Research Center for Child Health, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Yong Fu, Division of Otolaryngology-Head and Neck Surgery, The Children's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Meng Wang, Division of Medical Genetics and Genomics, The Children's Hospital, Zhejiang University School of Medicine and National Clinical Research Center for Child Health, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Min-Xin Guan, Division of Medical Genetics and Genomics, The Children's Hospital, Zhejiang University School of Medicine and National Clinical Research Center for Child Health, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; Zhejiang Provincial Key Lab of Genetic and Developmental Disorder, Hangzhou, Zhejiang 310058, China; Joint Institute of Genetics and Genome Medicine between Zhejiang University and University of Toronto, Hangzhou, Zhejiang 310058, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Science and Technology of Zhejiang Province [2018C03026]; National Key Technologies R&D Program [2018YFC1004802] from the Ministry of Science and Technology of China (to M.X.G.); Chinese National Science Foundation [82030028 to M.X.G., 82071063 to J.Z., 81700922 to F.M.]; Zhejiang Provincial Natural Science Foundation of China [LY19H130005 to M.W.]; Funding for open access charge: Ministry of Science and Technology of Zhejiang Province [2018C03026]; National Key Technologies R&D Program [2018YFC1004802] from the Ministry of Science and Technology of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Phizicky E.M., Hopper A.K.. tRNA biology charges to the front. Genes Dev. 2010; 24:1832–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. El Yacoubi B., Bailly M., de Crécy-Lagard V.. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012; 46:69–95. [DOI] [PubMed] [Google Scholar]
- 3. Bohnsack M.T., Sloan K.E.. The mitochondrial epitranscriptome: the roles of RNA modifications in mitochondrial translation and human disease. Cell Mol. Life Sci. 2018; 75:241–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asano K., Suzuki T., Saito A., Wei F.Y., Ikeuchi Y., Numata T., Tanaka R., Yamane Y., Yamamoto T., Goto T. et al.. Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res. 2018; 46:1565–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suzuki T., Nagao A., Suzuki T.. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011; 45:299–329. [DOI] [PubMed] [Google Scholar]
- 6. Abbott J.A., Francklyn C.S., Robey-Bond S.M.. Transfer RNA and human disease. Front. Genet. 2014; 5:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Haute L., Dietmann S., Kremer L., Hussain S., Pearce S.F., Powell C.A., Rorbach J., Lantaff R., Blanco S., Sauer S. et al.. Deficient methylation and formylation of mt- tRNAMet wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun. 2016; 7:12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang M., Liu H., Zheng J., Chen B., Zhou M., Fan W., Wang H., Liang X., Zhou X., Eriani G. et al.. A deafness- and diabetes-associated tRNA mutation causes deficient pseudouridinylation at position 55 in tRNAGlu and mitochondrial dysfunction. J. Biol. Chem. 2016; 291:21029–21041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kirino Y., Goto Y., Campos Y., Arenas J., Suzuki T.. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:7127–7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki T., Yashiro Y., Kikuchi I., Ishigami Y., Saito H., Matsuzawa I., Okada S., Mito M., Iwasaki S., Ma D. et al.. Complete chemical structures of human mitochondrial tRNAs. Nat.Commun. 2020; 11:4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boccaletto P., Machnicka M.A., Purta E., Piatkowski P., Baginski B., Wirecki T.K., de Crecy-Lagard V., Ross R., Limbach P.A., Kotter A. et al.. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018; 46:D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andrews R.M., Kubacka I., Chinnery P.F., Lightowlers R.N., Turnbull D.M., Howell N.. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999; 23:147. [DOI] [PubMed] [Google Scholar]
- 13. Hori H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 2014; 5:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Crécy-Lagard V., Boccaletto P., Mangleburg C.G., Sharma P., Lowe T.M., Leidel S.A., Bujnicki J.M.. Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res. 2019; 47:2143–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guan M.X., Yan Q., Li X., Bykhovskaya Y., Gallo-Teran J., Hajek P., Umeda N., Zhao H., Garrido G., Mengesha E. et al.. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 2006; 79:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Li R., Lin X., Guan M.X.. Isolation and characterization of the putative nuclear modifier gene MTO1 involved in the pathogenesis of deafness-associated mitochondrial 12S rRNA A1555G mutation. J. Biol. Chem. 2002; 277:27256–27264. [DOI] [PubMed] [Google Scholar]
- 17. Li X., Guan M.X.. A human mitochondrial GTP binding protein related to tRNA modification may modulate the phenotypic expression of the deafness-associated mitochondrial 12S rRNA mutation. Mol. Cell. Biol. 2002; 22:7701–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Helm M., Brulé H., Degoul F., Cepanec C., Lerouxm J.P., Giegé R., Florentz C.. The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res. 1998; 26:1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Allner O., Nilsson L.. Nucleotide modifications and tRNA anticodon-mRNA codon interactions on the ribosome. RNA. 2011; 17:2177–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drummond D.A., Wilke C.O.. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008; 134:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johansson M.J., Esberg A., Huang B., Bjork G.R., Bystrom A.S.. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 2008; 28:3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bjork G.R. Genetic dissection of synthesis and function of modified nucleosides in bacterial transfer RNA. Prog. Nucleic Acid Res. Mol. Biol. 1995; 50:263–338. [DOI] [PubMed] [Google Scholar]
- 23. Wang X., Yan Q., Guan M.X.. Combination of the loss of cmnm5U34 with the lack of s2U34 modifications of tRNALys, tRNAGlu, and tRNAGln altered mitochondrial biogenesis and respiration. J. Mol. Biol. 2010; 395:1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meng F., Cang X., Peng Y., Li R., Zhang Z., Li F., Fan Q., Guan A.S., Fischel-Ghosian N., Zhao X. et al.. Biochemical evidence for a nuclear modifier allele (A10S) in TRMU (methylaminomethyl-2-thiouridylate-methyltransferase) related to mitochondrial tRNA modification in the phenotypic manifestation of deafness-associated 12S rRNA mutation. J. Biol. Chem. 2017; 292:2881–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kopajtich R., Nicholls T.J., Rorbach J., Metodiev M.D., Freisinger P., Mandel H., Vanlander A., Ghezzi D., Carrozzo R., Taylor R.W., Marquard K. et al.. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am. J. Hum. Genet. 2014; 95:708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghezzi D., Baruffini E., Haack T.B., Invernizzi F., Melchionda L., Dallabona C., Strom T.M., Parini R., Burlina A.B., Meitinger T. et al.. Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am. J. Hum. Genet. 2012; 90:1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zeharia A., Shaag A., Pappo O., Mager-Heckel A.M., Saada A., Beinat M., Karicheva O., Mandel H., Ofek N., Segel R. et al.. Acute infantile liver failure due to mutations in the TRMU gene. Am. J. Hum. Genet. 2009; 85:401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schweizer U., Bohleber S., Fradejas-Villar N.. The modified base isopentenyladenosine and its derivatives in tRNA. RNA Biol. 2017; 14:1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin H., Miyauchi K., Harada T., Okita R., Takeshita E., Komaki H., Fujioka K., Yagasaki H., Goto Y.I., Yanaka K. et al.. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun. 2018; 9:1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Powell C.A., Kopajtich R., D'Souza A.R., Rorbach J., Kremer L.S., Husain R.A., Dallabona C., Donnini C., Alston C.L., Griffin H. et al.. TRMT5 mutations cause a defect in post-transcriptional modification of mitochondrial tRNA associated with multiple respiratory-chain deficiencies. Am. J. Hum. Genet. 2015; 97:319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang M., Peng Y., Zheng J., Zheng B., Jin X., Liu H., Wang Y., Tang X., Huang T., Jiang P. et al.. A deafness-associated tRNAAsp mutation alters the m1G37 modification, aminoacylation and stability of tRNAAsp and mitochondrial function. Nucleic Acids Res. 2016; 44:10974–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou M., Xue L., Chen Y., Li H., He Q., Wang B., Meng F., Wang M., Guan M.X.. A hypertension-associated mitochondrial DNA mutation introduces an m1G37 modification into tRNAMet, altering its structure and function. J. Biol. Chem. 2018; 293:1425–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qu J., Li R., Tong Y., Lu F., Qian Y., Hu Y., Mo J.Q., West C.E., Guan M.X.. The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation. Invest. Ophthalmol. Vis. Sci. 2006; 7:475–483. [DOI] [PubMed] [Google Scholar]
- 34. Liu Y., Li R., Li Z., Wang X.J., Yang L., Wang S., Guan M.X.. Mitochondrial transfer RNAMet 4435A>G mutation is associated with maternally inherited hypertension in a Chinese pedigree. Hypertension. 2009; 53:1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng J., Bai X., Xiao Y., Ji Y., Meng F., Aishanjiang M., Gao Y., Wang H., Fu Y., Guan M.X.. Mitochondrial tRNA mutations in 887 Chinese subjects with hearing loss. Mitochondrion. 2020; 52:163–172. [DOI] [PubMed] [Google Scholar]
- 36. Gutierrez Cortes N., Pertuiset C., Dumon E., Borlin M., Hebert-Chatelain E., Pierron D., Feldmann D., Jonard L., Marlin S., Letellier T. et al.. Novel mitochondrial DNA mutations responsible for maternally inherited nonsyndromic hearing loss. Hum. Mutat. 2012; 33:681–689. [DOI] [PubMed] [Google Scholar]
- 37. Merante F., Myint T., Tein I., Benson L., Robinson B.H.. An additional mitochondrial tRNAIle point mutation (A-to-G at nucleotide 4295) causing hypertrophic cardiomyopathy. Hum. Mutat. 1986; 8:216–222. [DOI] [PubMed] [Google Scholar]
- 38. King M.P., Attadi G.. Mitochondria-mediated transformation of human ρ° cells. Methods Enzymol. 1996; 264:313–334. [DOI] [PubMed] [Google Scholar]
- 39. Zhang J., Ji Y., Lu Y., Fu R., Xu M., Liu X., Guan M.X.. Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T>C mutation altered the assembly and function of complex I, apoptosis and autophagy. Hum. Mol. Genet. 2018; 27:1999–2011. [DOI] [PubMed] [Google Scholar]
- 40. Tang X., Zheng J., Ying Z., Cai Z., Gao Y., He Z., Yu H., Yao J., Yang Y., Wang H., Chen Y., Guan M.X.. Mitochondrial tRNASer(UCN) variants in 2651 Han Chinese subjects with hearing loss. Mitochondrion. 2015; 23:17–24. [DOI] [PubMed] [Google Scholar]
- 41. Zhao H., Li R., Wang Q., Yan Q., Deng J.H., Han D., Bai Y., Young W.Y., Guan M.X.. Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am. J. Hum. Genet. 2004; 74:139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan X., Wang X., Wang Z., Sun S., Chen G., He Y., Mo J.Q., Li R., Jiang P., Lin Q. et al.. Maternally transmitted late-onset non-syndromic deafness is associated with the novel heteroplasmic T12201C mutation in the mitochondrial tRNAHis gene. J. Med. Genet. 2011; 48:682–690. [DOI] [PubMed] [Google Scholar]
- 43. Rieder M.J., Taylor S.L., Tobe V.O., Nickerson D.A.. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 1998; 26:967–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Z., Liu Y., Yang L., Wang S., Guan M.X.. Maternally inherited hypertension is associated with the mitochondrial tRNAIle A4295G mutation in a Chinese family. Biochem. Biophys. Res. Commun. 2008; 367:906–911. [DOI] [PubMed] [Google Scholar]
- 45. Miller G., Lipman M.. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc. Nat. Acad. Sci. U.S.A. 1973; 70:190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera? A visualization system for exploratory research and analysis. J. Computat. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 47. Case D.A., Thomas E.C., Tom D., Holger G., Ray L., Kenneth M.M., Alexey O., Carlos S., Bing W., Robert J.W.. The Amber biomolecular simulation programs. J. Computat. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gong S., Wang X., Meng F., Cui L., Yi Q., Zhao Q., Cang X., Cai Z., Mo J.Q., Liang Y. et al.. Overexpression of mitochondrial histidyl-tRNA synthetase restores mitochondrial dysfunction caused by a deafness-associated tRNAHis mutation. J. Biol. Chem. 2020; 295:940–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. King M.P., Attardi G.. Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J. Biol. Chem. 1993; 268:10228–10237. [PubMed] [Google Scholar]
- 50. Hao R., Yao Y.N., Zheng Y.G., Xu M.G., Wang E.D.. Reduction of mitochondrial tRNALeu(UUR) aminoacylation by some MELAS-associated mutations. FEBS Lett. 2004; 578:135–139. [DOI] [PubMed] [Google Scholar]
- 51. Goto-Ito S., Ito T., Ishii R., Muto Y., Bessho Y., Yokoyama S.. Crystal structure of archaeal tRNA m1G37 methyltransferase aTrm5. Proteins. 2008; 72:1274–1289. [DOI] [PubMed] [Google Scholar]
- 52. Goto-Ito S., Ito T., Yokoyama S.. Trm5 and TrmD: two enzymes from distinct origins catalyze the identical tRNA modification, m1G37. Biomolecules. 2017; 7:E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiao Y., Wang M., He Q., Xu L., Zhang Q., Meng F., Jia Z., Zhang F., Wang H., Guan M.X.. Asymmetrical effects of deafness-associated mitochondrial DNA 7516delA mutation on the processing of RNAs in the H-strand and L-strand polycistronic transcripts. Nucleic Acids Res. 2020; 48:11113–11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang S., Li R., Fettermann A., Li Z., Qian Y., Liu Y., Wang X., Zhou A., Mo J.Q., Yang L. et al.. Maternally inherited essential hypertension is associated with the novel 4263A>G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ. Res. 2011; 108:862–870. [DOI] [PubMed] [Google Scholar]
- 55. Zhao X., Cui L., Xiao Y., Mao Q., Aishanjiang M., Kong W., Liu Y., Chen H., Hong H., Jia Z. et al.. Hypertension-associated mitochondrial DNA 4401A>G mutation caused the aberrant processing of tRNAMet, all 8 tRNAs and ND6 mRNA in the light-strand transcript. Nucleic Acids Res. 2019; 47:10340–10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Enriquez J.A., Attardi G.. Analysis of aminoacylation of human mitochondrial tRNAs. Methods Enzymol. 1996; 264:183–196. [DOI] [PubMed] [Google Scholar]
- 57. Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 1996; 264:197–211. [DOI] [PubMed] [Google Scholar]
- 58. Jha P., Wang X., Auwerx J.. Analysis of mitochondrial respiratory chain super-complexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 2016; 6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen D., Zhang Z., Chen C., Yao S., Yang Q., Li F., He X., Ai C., Wang M., Guan M.X.. Deletion of Gtpbp3 in zebrafish revealed the hypertrophic cardiomyopathy manifested by aberrant mitochondrial tRNA metabolism. Nucleic Acids Res. 2019; 47:5341–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Birch-Machin M.A., Turnbull D.M.. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods. Cell. Biol. 2001; 65:97–117. [DOI] [PubMed] [Google Scholar]
- 61. Dranka B.P., Benavides G.A., Diers A.R., Giordano S., Zelickson B.R., Reily C., Zou L., Chatham J.C., Hill B.G., Zhang J. et al.. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 2011; 51:1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gong S., Peng Y., Jiang P., Wang M., Fan M., Wang X., Zhou H., Li H., Yan Q., Huang T. et al.. A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 2014; 42:8039–8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Reers M., Smiley S.T., Mottola-Hartshorn C., Chen A., Lin M., Chen L.B.. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995; 260:406–417. [DOI] [PubMed] [Google Scholar]
- 64. Jia Z., Zhang Y., Li Q., Ye Z., Liu Y., Fu C., Cang X., Wang M., Guan M.X.. A coronary artery disease-associated tRNAThr mutation altered mitochondrial function, apoptosis and angiogenesis. Nucleic Acids Res. 2019; 47:2056–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Holzmann J., Frank P., Loffler E., Bennett K.L., Gerner C., Rossmanith W.. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008; 135:462–474. [DOI] [PubMed] [Google Scholar]
- 66. Li Y., D’Aurelio M., Deng J.H., Park J.S., Manfredi G., Hu P., Lu J., Bai Y.. An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J. Biol. Chem. 2007; 282:17557–17562. [DOI] [PubMed] [Google Scholar]
- 67. Yu J., Liang X., Ji Y., Ai C., Liu J., Zhu L., Nie Z., Jin X., Wang C., Zhang J. et al.. PRICKLE3 linked to ATPase biogenesis manifested Leber's Hereditary Optic Neuropathy. J. Clin, Invest. 2020; 130:4935–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sena L.A., Chandel N.S.. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012; 48:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Anding A.L., Baehrecke E.H.. Cleaning house: selective autophagy of organelles. Dev. Cell. 2017; 41:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ji Y., Zhang J., Lu Y., Yi Q., Chen M., Xie S., Mao X., Xiao Y., Meng F., Zhang M. et al.. Complex I mutations synergize to worsen the phenotypic expression of Leber's hereditary optic neuropathy. J. Biol.Chem. 2020; 295:13224–13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Korolchuk V.I., Menzies F.M., Rubinsztein D.C.. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy. 2009; 5:862–863. [DOI] [PubMed] [Google Scholar]
- 72. Zhu Y., Chen G., Chen L., Zhang W., Feng D., Liu L., Chen Q.. Monitoring autophagy in mammalian cells. Meth. Enzymol. 2014; 547:39–55. [DOI] [PubMed] [Google Scholar]
- 73. Lin H., Miyauchi K., Harada T., Okita R., Takeshita E., Komaki H., Fujioka K., Yagasaki H., Goto Y.I. et al.. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun. 2018; 9:1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lescrinier E., Nauwelaerts K., Zanier K., Poesen K., Sattler M., Herdewijn P.. The naturally occurring N6-threonyl adenine in anticodon loop of Schizosaccharomyces pombe tRNA causes formation of a unique U-turn motif. Nucleic Acids Res. 2006; 34:2878–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Murphy F.V.T., Ramakrishnan V., Malkiewicz A., Agris P.F.. The role of modifications in codon discrimination by tRNA(Lys)UUU. Nat. Struct. Mol. Biol. 2004; 11:1186–1191. [DOI] [PubMed] [Google Scholar]
- 76. Levinger L., Giegé R., Florentz C.. Pathology-related substitutions in human mitochondrial tRNAIle reduce precursor 3' end processing efficiency in vitro. Nucleic Acids Res. 2003; 31:1904–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Miyauchi K., Kimura S., Suzuki T.. A cyclic form of N6-threonylcarbamoyladenosine as a widely distributed tRNA hypermodification. Nat. Chem. Biol. 2013; 9:105–111. [DOI] [PubMed] [Google Scholar]
- 78. Yarus M., Cline S.W., Wier P., Breeden L., Thompson R.C.. Actions of the anticodon arm in translation on the phenotypes of RNA mutants. J. Mol. Biol. 1986; 192:23–255. [DOI] [PubMed] [Google Scholar]
- 79. Fan W., Zheng J., Kong W., Cui L., Aishanjiang M., Yi Q., Wang M., Cang X., Tang X., Chen Y. et al.. Contribution of a mitochondrial tyrosyl-tRNA synthetase mutation to the phenotypic expression of the deafness-associated tRNASer(UCN) 7511A>G mutation. J. Biol. Chem. 2019; 294:19292–19305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chomyn A., Enriquez J.A., Micol V., Fernandez-Silva P., Attardi G.. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 2000; 275:19198–19209. [DOI] [PubMed] [Google Scholar]
- 81. Guan M.X., Enriquez J.A., Fischel-Ghodsian N., Puranam R.S., Lin C.P., Maw M.A., Attardi G.. The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol. Cell. Biol. 1998; 18:5868–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Enriquez J.A., Chomyn A., Attardi G.. MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nat. Genet. 1995; 10:47–55. [DOI] [PubMed] [Google Scholar]
- 83. Ji Y., Zhang J., Yu J., Wang Y., Lu Y., Liang M., Li Q., Jin X., Wei Y., Meng F. et al.. Contribution of mitochondrial ND1 3394T>C mutation to the phenotypic manifestation of Leber's hereditary optic neuropathy. Hum Mol. Genet. 2019; 28:1515–1529. [DOI] [PubMed] [Google Scholar]
- 84. Wallace D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005; 39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hayashi G., Cortopassi G.. Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 2015; 88A:10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Raimundo N., Song L., Shutt T.E., McKay S.E., Cotney J., Guan M.X., Gilliland T.C., Hohuan D., Santos-Sacchi J., Shadel G.S.. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell. 2012; 148:716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Melser S., Lavie J., Bénard G.. Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta. 2015; 1853:2812–2821. [DOI] [PubMed] [Google Scholar]
- 88. Youle R.J., Narendra D.P.. Mechanisms of autophagy. Nat. Rev. Mol. Cell Biol. 2011; 12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Keithley E.M. Pathology and mechanisms of cochlear aging. J. Neurosci. Res. 2020; 98:1674–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Guan M.X. Molecular pathogenetic mechanism of maternally inherited deafness. Ann. N.Y. Acad. Sci. 2004; 1011:259–271. [DOI] [PubMed] [Google Scholar]
- 91. Dittmar K.A., Goodenbour J.M., Pan T.. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006; 2:e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang Q., Zhang L., Chen D., He X., Yao S., Zhang Z., Chen Y., Guan M.X.. Deletion of Mtu1 (Trmu) in Zebrafish revealed the essential role of tRNA modification in mitochondrial biogenesis and hearing function. Nucleic Acids Res. 2018; 46:10930–10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tischner C., Hofer A., Wulff V., Stepek J., Dumitru I., Becker L., Haack T., Kremer L., Datta A.N., Sperl W. et al.. MTO1 mediates tissue specificity of OXPHOS defects via tRNA modification and translation optimization, which can be bypassed by dietary intervention. Hum. Mol. Genet. 2015; 24:2247–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Finnilä S., Hassinen I.E., Majamaa K.. Phylogenetic analysis of mitochondrial DNA in patients with an occipital stroke: Evaluation of mutations by using sequence data on the entire coding region. Mutat. Res. 2001; 458:31–39. [DOI] [PubMed] [Google Scholar]
- 95. Gkatza N.A., Castro C., Harvey R.F., Heiß M., Popis M.C., Blanco S., Bornelöv S., Sajini A.A., Gleeson J.G., Griffin J.L. et al.. Cytosine-5 RNA methylation links protein synthesis to cell metabolism. PLoS Biol. 2019; 17:e3000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Torres A.G., Reina O., Stephan-Otto Attolini C., Ribas de Pouplana L.. Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:8451–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Agnew T., Goldsworthy M., Aguilar C., Morgan A., Simon M., Hilton H., Esapa C., Wu Y., Cater H., Bentley L. et al.. A Wars2 mutant mouse model displays OXPHOS deficiencies and activation of tissue-specific stress response pathways. Cell Rep. 2018; 25:3315–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dogan S.A., Pujol C., Maiti P., Kukat A., Wang S., Hermans S., Senft K., Wibom R., Rugarli E.I., Trifunovic A.. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014; 19:458–469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.