


Abstract
Codon usage bias is a universal feature of all genomes. Although codon usage has been shown to regulate mRNA and protein levels by influencing mRNA decay and transcription in eukaryotes, little or no genome-wide correlations between codon usage and mRNA levels are detected in mammalian cells, raising doubt on the significance of codon usage effect on gene expression. Here we show that gene-specific regulation reduces the genome-wide codon usage and mRNA correlations: Constitutively expressed genes exhibit much higher genome-wide correlations than differentially expressed genes from fungi to human cells. Using Drosophila S2 cells as a model system, we showed that the effect of codon usage on mRNA expression level is promoter-dependent. Regions downstream of the core promoters of differentially expressed genes can repress the codon usage effects on mRNA expression. An element in the Hsp70 promoter was identified to be necessary and sufficient for this inhibitory effect. The promoter-dependent codon usage effects on mRNA levels are regulated at the transcriptional level through modulation of histone modifications, nucleosome densities and premature termination. Together, our results demonstrate that promoters play a major role in determining whether codon usage influences gene expression and further establish the transcription-dependent codon usage effects on gene expression.
INTRODUCTION
Although 18 of the 20 amino acids can be encoded by two to six synonymous genetic codons, these codons are not used with the same frequencies, and every organism has its own preferred synonymous codons, a phenomenon called codon usage bias (1–3). Although synonymous codon changes were previously thought to be silent, accumulating genetic, biochemical, molecular and bioinformatic evidence now demonstrates that gene codon usage regulates gene expression. Codon usage controls translation elongation rate and co-translational protein folding process and has also been shown to be a major determinant of protein expression levels (3–8). Genes that encode highly expressed proteins are strongly enriched for preferred codons, and codon optimization has been shown to increases endogenous and heterologous gene expression in diverse eukaryotes and prokaryotes (5,6,8–14). Moreover, genome-wide correlations between codon usage bias and protein levels and synthesis rates have been observed (15–17). Codon usage regulates translation efficiency because rare codons cause ribosome stalling during translation, which, in eukaryotes, can result in premature translation termination (7,18,19).
Although codon usage was previously thought to mediate its effect on protein expression mainly due to regulation of translation efficiency, recent evidence demonstrates that the influence of codon usage on mRNA levels is an important, and in many cases the main, mechanism that determines the effect of codon usage on protein expression (15,20–23). Genome-wide expression profiling studies have uncovered positive correlations between codon usage biases and mRNA levels in both prokaryotic and lower eukaryotic organisms (15,16,24,25). Codon usage influences mRNA levels post-transcriptionally by affecting co-translational mRNA decay in a variety of systems ranging fungi to human cells (18,20,23,26–28).
The effect of codon usage on mRNA levels cannot be explained by its impact on mRNA half-lives alone. We have previously shown that in the filamentous fungus, Neurospora crassa, and in human cells, codon optimization has dramatic effects on some mRNA levels but little effect on mRNA stability (15,21). Effects of codon usage on mRNA levels are mainly due to an influence on transcription caused by changes in chromatin structures and transcription factor binding independent of translation. Consistent with these results, high GC content within open reading frames, which correlates with codon optimality, also increases mRNA levels and transcription without altering mRNA decay rates in mammalian cells (22,29,30). In addition, codon usage bias also positively correlates with RNA polymerase II (Pol II) activation and predicted mRNA synthesis rates genome-wide in fungi (15,17). Although these results indicate that the effect of codon usage on transcription is conserved in eukaryotes, the underlying mechanism is not known.
In mammalian cells, numerous studies have shown that codon usage optimization and increase of GC content in coding regions of selected genes robustly increase protein and mRNA production; these methods are frequently used to enhance gene expression (9,21,22,29,31,32). However, little or no genome-wide correlation between codon usage or GC content and gene expression level is observed in mammalian cells, which implies that codon usage does not have a significant role regulating gene expression (33–35). These conflicting results suggest the existence of unknown mechanisms that regulate the codon usage effect on gene expression. In this study, we show that the effect of codon usage on gene expression is promoter-dependent. We discovered that for constitutively expressed genes the correlation between codon usage biases and gene expression levels is much higher than for differentially expressed genes from fungal to human cells. Using Drosophila Schneider 2 (S2) cells as a model system, we demonstrated that promoter-dependent effects on gene expression are mediated by elements immediately downstream of core promoters. Codon optimality regulates gene expression at the level of transcription by affecting histone modifications, nucleosome densities, and premature termination in a promoter-dependent manner. Together, these results demonstrate that promoter and coding sequencing jointly determine the codon usage effect on gene expression.
MATERIALS AND METHODS
Codon manipulation and plasmid construction
The codons of firefly luciferase were optimized (Opt-luc) or deoptimized (De-luc) based on the Drosophila melanogaster codon usage frequency table (https://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=7227). The Opt-Luc and De-Luc genes were described previously (7,18). Opt-YFP is the commonly used eYFP sequence. De-YFP codons were deoptimized based on the Drosophila codon usage frequency table.
All plasmids were generated based on the Ac5-STABLE1-neo plasmid. A multiple cloning site was first inserted between Ac5 promoter and SV40 poly(A) signal between KpnI and BamHI sites. DNA encoding eYFP, SV40 terminator, gypsy insulator, 5xUAS, and 5xMyc-luciferase were inserted into the multiple cloning sites. Different promoters were amplified by PCR from the genome and inserted between 5xUAS and 5xMyc-luciferase coding regions at NotI/XhoI sites. A Gal4 expression vector was generated by inserting the Gal4 gene after the Ac5 promoter using EcoRI/HindIII sites, followed by another cassette expressing a gene to confer puromycin resistance. For YFP expression plasmids, the region from the Ac5 promoter to the gypsy insulator was replaced by linker DNA. 5xMyc-luciferase was replaced by Opt-YFP or De-YFP. For luciferase reporter plasmids used in generating stable cell lines, upstream eYFP was replaced by hph, the gene that results in hygromycin (hph) resistance. Nucleotide sequences of the reporter genes and promoters used in this study are shown in Supplementary Table S1.
Bioinformatic analyses of correlations between codon usage and mRNA levels in different organisms
mRNA-seq results in cells or tissues of Saccharomyces cerevisiae, Neurospora crassa, Drosophila melanogaster, Mus musculus and Homo sapiens were used in the analyses (15,40–42). For each genome, the tRNA adaptation index (tAI) and codon bias index (CBI) of each gene were calculated (43,44). Pearson correlation coefficients between mRNA levels and codon usage were determined. For human, 53 human tissue mRNA-seq data of the Genotype-Tissue Expression (GTEx) Project was used (Supplementary Table S2). RNA levels of each gene in each sample were compared with that in all the other samples. Constitutive 1: Genes are differentially expressed in no more than five samples (the threshold of differential expression being up- or down-regulated 2 folds). Constitutive 2: Genes are differentially expressed in no more than seven samples (threshold of differential expression being up- or down-regulated 3 folds).
The D. melanogaster data was obtained from NCBI Gene Expression Omnibus database (accession number: GSE64108) and the 72 samples were used to assess mRNA levels at different time points in a diurnal cycle in head, periphery (entire body except the head) and the heart at 3, 5 or 7 weeks of age under ad libitum feeding or time-restricted feeding (37) (Supplementary Table S2). Constitutive 1: Genes were differentially expressed in no more than five samples (threshold of differential expression being up- or down-regulated 2-fold). Constitutive 2: Genes were differentially expressed in no more than seven samples (threshold of differential expression being up- or down-regulated 3-fold). Regulated-2: The rest of total genes subtracted by constitutive two genes.
For the 71 RNA-seq samples of Neurospora crassa cultured under different culture conditions was obtained from Joint Genome Institute Genome Portal (38) (Supplementary Table S2). mRNA levels of each gene transcriptional levels in each sample were compared to that in all the other samples. Constitutive: Genes were differentially expressed in at most nine samples, with the threshold of differential expression being up- or down-regulated 3-fold.
S2 cell culture, transfection, and generation of stable cell lines
Drosophila S2 cells were cultured at 25°C in Schneider's Drosophila medium (Thermo Fisher Scientific), supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher Scientific) and 1% (v/v) penicillin-streptomycin (Sigma). For transfection, cells were plated at 1 × 106 cells per well of a 24-well plate. Gal4 expression plasmid and reporter plasmid were mixed at 1:1 molar ratio, and 500 ng total plasmid was transfected into each well using Lipofectamine 3000 (Thermo Fisher Scientific) following the manufacturer's instructions. Cells were harvested after 48 h for further analysis.
To generate stable cell lines, cells were plated at 2 × 106 cells per well in 12-well plates. Cells were transfected with Gal4 and luciferase expression plasmids at 1:1 molar ratio using Lipofectamine 3000 (Thermo Fisher Scientific) following the manufacturer's instructions. After 48 h, cells were passaged and selected with 10 ng/ml puromycin (Sigma) and 300 ng/ml hygromycin B (Thermo Fisher Scientific) for a week.
To determine mRNA stability, S2 cells transfected with plasmids were first cultured for 48 h. The cells were then passaged and split equally into four wells. Transcription was arrested by adding actinomycin D into the cell culture at a final concentration of 10 μg/ml. Cells were harvested at 0, 40, 80, 120 min after actinomycin D addition for RNA extraction.
RNAi
Templates of double-stranded RNAs from the DRSC (Drosophila RNAi Screening Center) were first PCR-amplified from genomic DNA using primers with T7 promoter sequence on both ends. dsRNAs were synthesized using HiScribe T7 High Yield RNA Synthesis Kit (NEB). Gene silencing was performed in 24-well plates by incubating 4 × 105 cells with 5 μg dsRNA in 200 μl serum free medium for 30 min. Then 600 μl medium supplied with 10% fetal bovine serum was added. Cells were harvested after 3 days for further analysis. The primers used for template amplification were Ars2 (5′-TAATACGACTCACTATAGGGCTAGCTCAGATGAAGAGAAAC-3′, 5′-TAATACGACTCACTATAGGGATACCAACGACGCTCCAC-3′), Mtr4 (5′-TAATACGACTCACTATAGGGGTGCTCACCGAGGAGGAT-3′, 5′-TAATACGACTCACTATAGGGCAGTGCAGCTTGATTTTGG-3′), ZCCHC8 (5′-TAATACGACTCACTATAGGGTCAGCCTTCGAAAGAAGTAG-3′, 5′-TAATACGACTCACTATAGGGTGCATTAAACAGAGCTATGC-3′), Rrp40 (5′-TAATACGACTCACTATAGGGCAGCCTCCATATCGTATCTC-3′, 5′-TAATACGACTCACTATAGGGCGAGTTGACGCAGACCA-3′).
Nuclear run-on
Cells were lysed in lysis buffer (10 mM Tris–HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.5% NP-40). Nuclei were isolated by centrifugation at 2000 × g for 5 min and were suspended in 40 μl freezing buffer (50 mM Tris–HCl, pH 7.4, 5 mM MgCl2, 30% glycerol and 1 mM DTT) and 60 μl transcriptional buffer (20 mM Tris–HCl, pH 8.0, 5 mM MgCl2, 150 mM KCl, 2 mM DTT, 500 μM ATP, 500 μM CTP, 500 μM GTP, 500 μM BrUTP and 200 U/ml Superase-in (Thermo Fisher)).After incubation at 25°C for 30 min, 500 μl TRIzol (Invitrogen) was added to each reaction to stop transcription. RNA was isolated and resuspended in 100 μl IP buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.05% NP-40 and 1 mM EDTA). Anti-BrU antibody (Santa Cruz Biotechnology, IIB5) and Protein G beads (Thermo Fisher Scientific) were incubated with RNA for 2 h at 4°C, followed by washing beads with IP buffer and isolation of RNA using TRIzol. Levels of newly transcribed RNA were measured by quantitative real-time PCR (RT-qPCR). RT-qPCR primers amplified the 5′ end of the Myc-tag sequence to measure Luc levels (5′-TGATATCATCGATTTAAAGCA-3′, 5′-CATGTCGCCCAAGCTCTCCAT-3′), and an actin intron (5′-GAGAAAAGCCGCGGAAAATGTGTG-3′, 5′-TCAATACAATAACTCTTTAGCTCG-3′) for normalization.
Chromatin immunoprecipitation assay
Cells were suspended and fixed with 1% formaldehyde in PBS buffer for 5 min at room temperature with gentle agitation. Fixation was quenched by adding glycine to a final concentration of 125 mM and incubation for 5 min at room temperature. Cells were pelleted by centrifugation at 1000 × g and resuspended in sonication buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 0.5% SDS). Chromatin was fragmented to 200–500 bp using a Bioruptor (Diagenode) with 7 cycles of 30 s on/off at high power. Samples were precleared using Protein G beads at 4°C for 1 h. An aliquot of 1/20 volume of sample was used as input. Antibodies were incubated with sample overnight at 4°C. The following antibodies were used: Pol II CTD (Abcam, ab26721), H3 (Active Motif, 39763), H3K27ac (Active Motif, 39133), H3K9me3 (Active Motif, 61013) and H3K27me3 (Active Motif, 61017). Protein G (Thermo Fisher Scientific, 10003D) or Protein A beads (Thermo Fisher Scientific, 10001D) were incubated with sample for 2 h at 4°C. Samples were washed with RIPA buffer (10 mM Tris–HCl, pH 8.0, 140 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate), high salt buffer (10 mM Tris–HCl, pH 8.0, 500 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate), LiCl buffer (10 mM Tris–HCl, pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate) and TE buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA). Chromatin was eluted with elution buffer (0.1 M NaHCO3, 1% SDS), de-crosslinked, and extracted with phenol. Immunoprecipitated DNA was quantified by RT-qPCR using primers to the 5′ end of the Myc-tag sequence (5′-TGATATCATCGATTTAAAGCA-3′, 5′-CATGTCGCCCAAGCTCTCCAT-3′) to measure Luc levels and to an actin intron (5′-GAGAAAAGCCGCGGAAAATGTGTG-3′, 5′-TCAATACAATAACTCTTTAGCTCG-3′) for normalization of Pol II CTD and H3K27ac. For normalization of H3K27me3, primers to 3L:10228701 were used (5′-GCACACGGTAATTGCTTATT-3′, 5′-TCGCATCTCGGTTCTTTC-3′). Primers to F22 (5′-CAGTTGATGGGATGAATTTGG -3′, 5′-TGCCTGTGGTTCTATCCAAAC-3′) were used for normalization of H3K9me3.
Northern blotting
Northern blot analyses were performed as previously described using [32P] UTP-labeled riboprobes (36). Riboprobes were transcribed in vitro from PCR products using the MEGAscript T7 Transcription Kit (Thermo Fisher Scientific) following the manufacturer's protocol. The primer sequences used for the template amplification are listed in Supplementary Table S3.
RESULTS
mRNA levels of constitutively expressed genes are more strongly correlated with codon usage than are levels from differentially expressed genes from fungi to human
We analyzed mRNA-seq results from previous studies to determine the genome-wide Pearson correlation coefficients between mRNA levels and codon usage in cells or tissues of S. cerevisiae, N. crassa, D. melanogaster, M. musculus and H. sapiens (15,37–39). Genome-wide mRNA levels exhibit a strong correlation with gene codon usage in both fungal species (S. cerevisiae and N. crassa) and a modest correlation in different Drosophila tissues (Figure 1A and Supplementary Figure S1). In mouse and human tissues, however, there is little or no correlation (0.02–0.07) between mRNA levels and codon usage genome-wide (Figure 1A and Supplementary Figure S1). Such lack of correlation is in sharp contrast with robust effects of codon usage and GC content on mRNA and protein levels observed in heterologous gene expression in mammalian cells (21,22,29).
Figure 1.
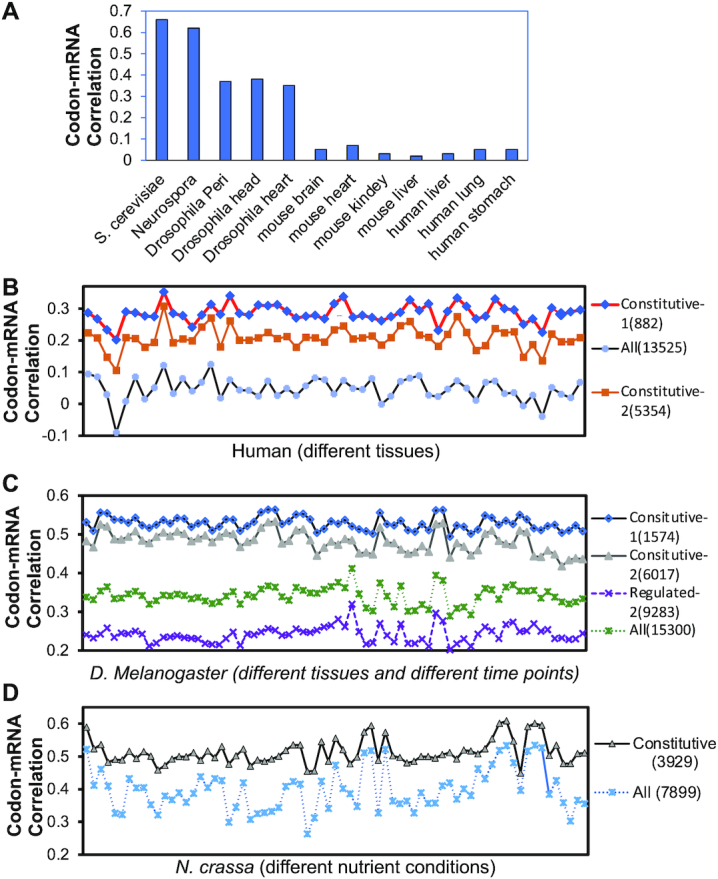
Differential genome-wide correlations between codon usage and mRNA levels between constitutive and regulated genes from fungi to human cells and tissues. (A) Pearson correlation coefficients between genome-wide gene codon usage and mRNA levels in the indicated organisms and tissues. (B–D) Pearson correlation coefficients between genome-wide gene codon usage and mRNA levels in B) various human tissue samples, C) Drosophila tissue samples collected at various time points, and D) Neurospora samples collected in different nutrient conditions. tAI was used to determine gene codon usage of human and Drosophila genes (B, C) and CBI was used to determine codon usage of N. crassa genes (D). The number of genes per category are indicated in parentheses. Each symbol indicates one tissue, one time points or one nutrient condition. The RNA-seq samples in B–D are listed in Supplementary Table S2.
Because the heterologous gene expression in mammalian cells is usually driven by a constitutive promoter, we wondered whether promoter-dependent regulation of gene expression contributes to the codon usage effect. We determined the genome-wide correlations between mRNA levels (FPKM (fragments per kilobase of transcript per million mapped reads) > 1) and codon usage in 53 human tissues (37). There is little or no genome-wide correlation between mRNA levels and codon usage in any tissues tested (Figure 1B and Supplementary Table S2). We next identified two categories of constitutively expressed genes: Constitutive 1 genes are those that are up- or downregulated by >2-fold in no more than five tissues. Constitutive 2 genes that those that are up- or downregulated by >3-fold in no more than seven tissues. The correlation between mRNA levels and codon usage for Constitutive 2 genes was ∼0.2, whereas the correlation for the Constitutive 1 genes was approximately 0.3, which is much higher than that of the all genes in all tissues (Figure 1B). This result suggests that codon usage does have a broad effect on gene expression in mammalian cells but that this correlation is likely masked by gene-specific regulation.
To determine whether this phenomenon is conserved across different eukaryotic organisms, we identified constitutively expressed genes in D. melanogaster and N. crassa by analyzing previous mRNA-seq results. For Drosophila, we analyzed mRNA-seq (FPKM > 1) results (NCBI Gene Expression Omnibus accession number: GSE64108) from 72 different tissue and different time points (Supplementary Table S2) using the same criteria as above. As expected, the constitutively expressed genes showed much higher correlations between codon usage and mRNA levels than the total genes for all samples (Figure 1C). In contrast, non-constitutively expressed genes showed much lower correlations (Figure 1C). For Neurospora, we analyzed mRNA-seq (FPKM > 1) results from 71 samples that were grown under different nutrient conditions (42) (Supplementary Table S2). Constitutive genes were defined as those up- or down-regulated by >2-fold in fewer than seven samples. In N. crassa, as in human and fly, the constitutively expressed genes had a higher codon usage-mRNA correlation than did all genes in all samples (Figure 1D). Together, these results indicate that the codon usage effect on mRNA levels is not universal, and it is likely differentially regulated by gene-specific promoters.
Promoter-dependent codon usage effects on mRNA levels are independent of mRNA stability in Drosophila S2 cells
To evaluate how promoter regions regulate the codon usage effect in animals, we used Drosophila S2 cells as a model system due to extensive characterization of Drosophila gene promoters and the availability of Drosophila promoter database (43,44)(https://labs.biology.ucsd.edu/Kadonaga/DCPD.htm). Epitope-tagged (5x c-Myc) codon-optimized (Opt) or codon-deoptimized (De) firefly luciferase (Luc) genes (18) controlled by different Drosophila promoters (including core promoter sequence from –40 to +40 nucleotides (nt) relative to the transcription start site plus 80–150 nt downstream region) were cloned into an expression vector regulated by upstream activation sites (UAS) that are recognized by Gal4 protein (Figure 2A). We selected promoters of both constitutive (Act5C and Tubulin) and facultative genes (the heat shock inducible Hsp70, the development and cell growth regulated Myc, and the circadian clock gene Per). In addition, a Drosophila Synthetic Core Promoter (DSCP) that is commonly used to drive gene expression was also used. A separate cassette for expression of eYFP was used for normalization of transfection efficiency, and a Gypsy insulator was inserted between the two expression cassettes to avoid transcriptional interference (Figure 2A). The resulting construct was transfected into S2 cells along with a Gal4-expression vector.
Figure 2.
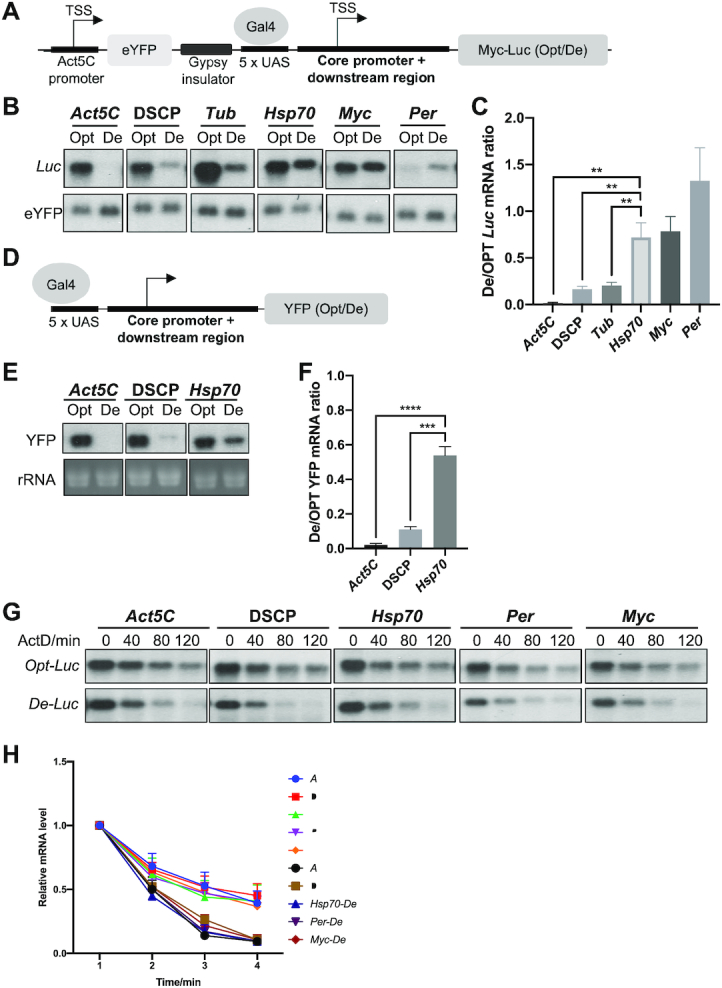
The codon usage effect on gene expression is dependent on promoter. (A) Diagram of the Luc reporter construct. (B) Northern blot analysis of Luc mRNA levels produced from reporter genes driven by indicated promoters. The probe was designed to hybridize to the common 5′ Myc-tag sequence of Luc. (C) Quantification of ratio of De-Luc to Opt-Luc luciferase mRNA ratios normalized by eYFP levels for experiment shown in panel B (n = 3). (D) Diagram of the YFP reporter construct. (E) Northern blot analysis of the YFP mRNA levels produced from reporter genes driven by indicated promoters (upper panel). The probe hybridizes to the common 3′ UTR sequence of YFP genes. rRNA level was used as a loading control. (F) Quantification of the De-YFP/Opt-YFP ratio normalized by rRNA for experiment shown in panel E (n = 3). (G) Northern blot analyses of mRNAs produced from Opt-Luc and De-Luc reporter genes driven by indicated promoters. Actinomycin D was added to the culture at 10 ng/mL at time 0. RNA was isolated at indicated time points, and RNA levels were normalized to eYFP levels. (H) Quantification of Opt-Luc and De-Luc mRNA decay rates for experiment shown in panel G (n = 3). Data are means ± SD. **P < 0.01. ***P < 0.001. ****P < 0.001. Two-sided Student's t-tests were used.
Steady-state levels of Opt-Luc and De-Luc were detected by northern blot using a probe to the common 5′ Myc-tag sequence. Opt-Luc mRNA levels were ∼5–20 times higher than those of the De-Luc for Act5C, DSCP, and Tubulin promoter constructs, indicating a robust effect of codon usage on mRNA levels (Figure 2B-C). This suggests that when gene expression is driven by a constitutive promoter, codon optimization increases mRNA levels, whereas codon-deoptimization represses gene expression. For the genes driven by Hsp70 and Myc promoters, however, the difference between Opt-Luc and De-Luc mRNA levels were modest (Figure 2B, C). Under the control of the Per promoter, the De-Luc mRNA level was higher than that of the Opt-Luc mRNA (Figure 2B, C). Ruling out the possibility that this promoter-dependent codon usage effect is due to the presence of a Luc-specific regulatory sequence, similar results were obtained using YFP as a reporter (Figure 2D–F). Thus, consistent with our genome-wide bioinformatic analysis, these results demonstrate that the codon usage effect on mRNA levels is dependent on gene promoters.
Due to the known role of codon usage on mRNA decay (18,20,26,45), we examined whether the observed promoter-dependent codon usage effect is due to a promoter-dependent influence on mRNA decay rates. We quantified decay rates of Luc mRNA expressed under the control of different promoters after the addition of the transcription inhibitor actinomycin D. As expected, the Opt-Luc mRNA was significantly more stable than De-Luc mRNA (Figure 2G, H). However, the decay rate of the Opt-Luc and De- Luc mRNA under different promoter was similar, indicating that the effect of codon usage on mRNA decay is promoter-independent. Because the steady state mRNA levels are determined by synthesis rates and mRNA decay rates, these results suggest that the promoter-dependent effect of codon usage on mRNA levels is regulated by transcription.
Identification of the regulatory element responsible for the promoter-dependent codon usage effect
The core promoters in Drosophila usually have a TATA box, an initiator, and a downstream core promoter element in an approximately 80-nt region. To determine whether the core promoter or an element in the 80–150 nt immediately downstream in the 5′ UTR is the main determinant of the promoter-dependent codon usage effect on mRNA levels, we replaced the immediate downstream region (IDR) of the DSCP construct with that from the Hsp70, the Myc or the Per promoter (Figure 3A). The IDR regions of Hsp70, Myc and Per all resulted in reduced codon usage effect on Luc mRNA mainly due to a dramatic increase of the De-Luc mRNA level (Figure 3B, C). The replacement of the immediate downstream region of the Hsp70 or Myc constructs with that of the DSCP construct rescued the codon-usage effect on Luc mRNA (Figure 3D, E and S2A). These results indicate that the immediate downstream region of the core promoter rather than the core promoter itself regulates the promoter-dependent codon usage effect.
Figure 3.
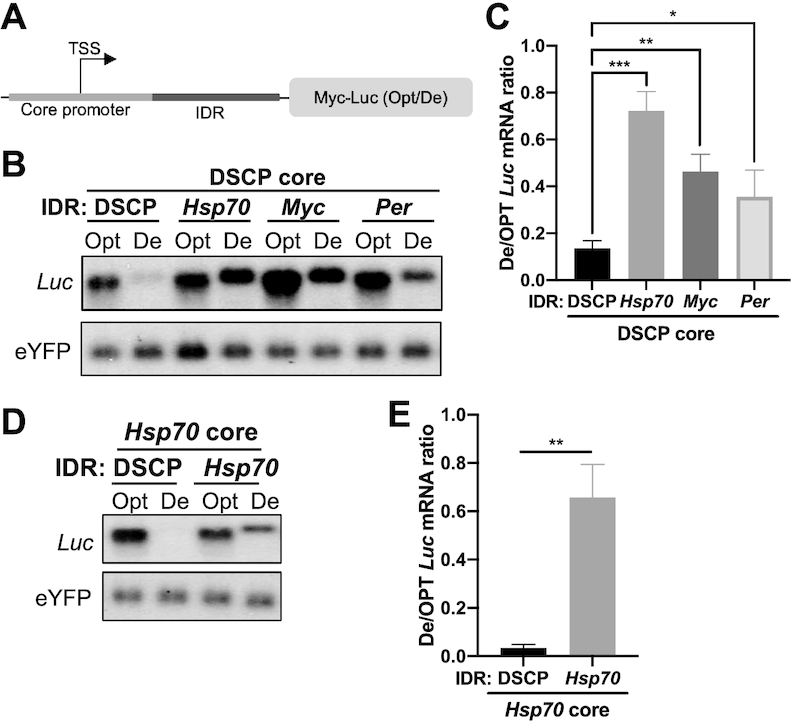
Promoter-dependent codon usage effect is regulated by the region downstream of the core promoter. (A) Diagram of promoter region. The core promoter is centered over the transcription start site (TSS). (B) Northern blot analysis of Luc mRNAs expressed from constructs with indicated chimeric promoters. The probes hybridize to the 5′ Myc-tag sequence for Luc and 5′ region of eYFP. (C) Quantification of De-Luc/Opt-Luc ratio normalized to eYFP for experiment shown in panel B (n = 3). (D) Northern blot analysis of Luc mRNAs expressed under the control of the Hsp70 core promoter and the IDR from either DSCP or Hsp70. (E) Quantification of De-Luc/Opt-Luc mRNA ratio normalized to eYFP for experiment shown in panel D (n = 3). Data are means ± SD. *P < 0.05. **P < 0.01. ***P < 0.001. Two-sided Student's t-tests were used.
To identify the element or elements in the Hsp70 IDR that regulates the codon usage effect, we swapped 20-nt regions (Mut1–4) within the immediate downstream region (84nt) of the Hsp70 core promoter into the corresponding regions of the DSCP in vectors for expression of Opt-Luc and De-Luc mRNAs (Figure 4A). Only one of these swaps, Mut-3, completely abolished the upregulation of De-Luc mRNA and resulted in a robust codon usage effect (Figure 4B and C). To confirm this finding, we inserted the corresponding 20-nt region of DSCP into the Hsp70 promoter (Figure 4A). This swap abolished the upregulation of De-Luc mRNA (Figure 4B and C), making the Hsp70 promoter resemble the DSCP in its codon usage-dependent effect on mRNA expression. Furthermore, mutating the left, middle, and right 10 nt of this 20-nt domain in the Hsp70 promoter only resulted in a partial increase in De-Luc mRNA, indicating that the entire 20-nt domain is involved in the regulation (Supplementary Figure S2B). Together these results demonstrate that this region of the Hsp70 IDR is both necessary and sufficient for the upregulation of the De-Luc mRNA and suppression of the codon usage effect. Because of its role in regulating codon usage-dependent gene expression, we term the 20-nt region of Hsp70 (5′-GTAAAGTGCAAGTTAAAGTG-3′) the codon usage-dependent element (CDE). Sequences with homology to this element were not found in Myc and Per promoters, suggesting that different promoters have different CDEs.
Figure 4.
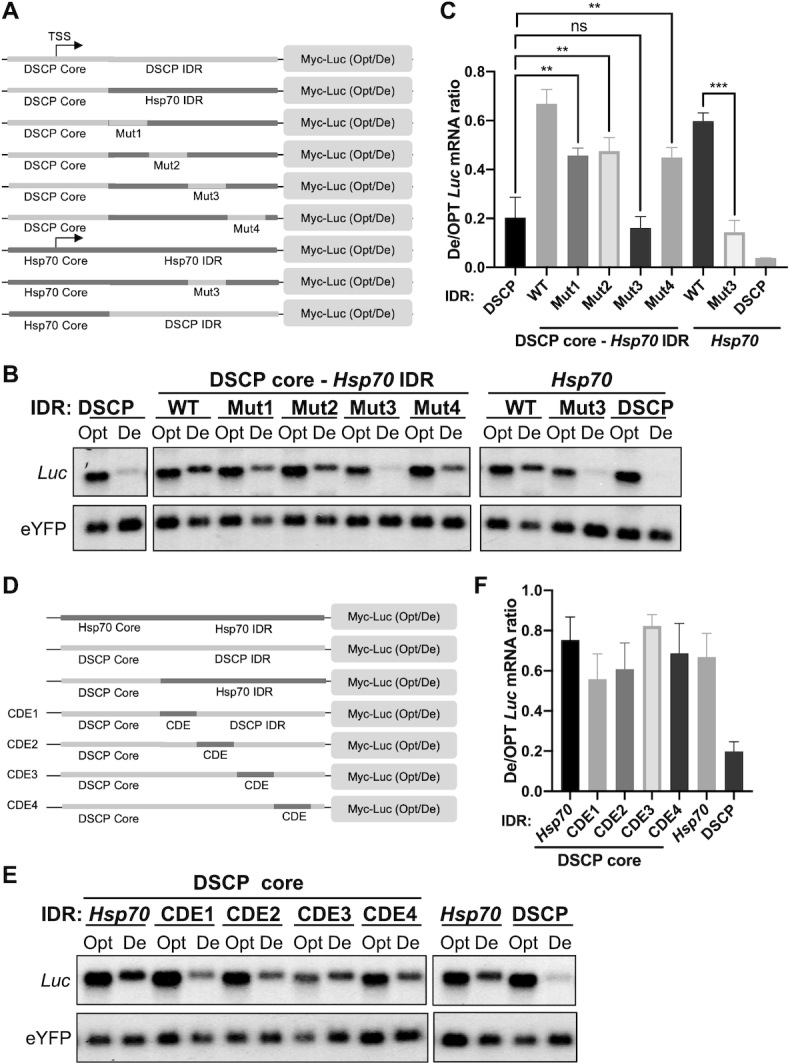
Identification of the regulatory element in the Hsp70 promoter that is necessary and sufficient to inhibits the codon usage effect on suppression of De-Luc expression. (A) Diagrams of the promoter regions of the reporter constructs used to identify the regulatory element in the Hsp70 5′ UTR. The mutated regions (Mut) were 20 nt long. (B) Northern blot analysis of De-Luc and Opt-Luc mRNA obtained from indicated constructs. Probes hybridize to the 5′ Myc-tag sequence for Luc and 5′ region of eYFP. (C) Quantification of De- Luc/Opt- Luc mRNA ratio normalized by eYFP for experiment shown in panel B (n = 3). (D) Diagram of the promoter region of the reporter constructs. The 20-nt CDE sequence was placed at different distances from the core DSCP core promoter. (E) Northern blot analysis of De-Luc and Opt-Luc mRNA levels expressed under control of the indicated promoters. (F) Quantification of De- Luc/Opt- Luc mRNA ratio normalized by eYFP for experiment shown in panel E (n = 3). Data are means ± SD. **P < 0.01. ***P < 0.001. ns: not significant. Two-sided student t-tests were used.
Within Drosophila core promoters, spacing between the TATA box, initiator, and downstream core promoter elements are functionally important for transcriptional regulation (46). To determine whether the spacing between core promoter and the CDE is important, we prepared reporter constructs in which the distance between the DSCP core promoter IDR and the Hsp70 CDE was varied from 0 to 80 nt (Figure 4D). We found that the presence of the CDE at any position in the De-Luc construct impaired the codon usage effect (Figure 4E, F). Similarly, the CDE placed downstream from the Act5C promoter enhanced De-Luc expression (Supplementary Figure S2C). These results indicate that the exact spacing between the CDE and core promoter is not critical for its function.
Promoter-dependent codon usage effects influence transcription
Although codon optimality affects mRNA decay, this effect is not promoter dependent (Figure 2G, H). This suggests that the promoter-dependent codon usage effects regulate transcription. To test this, the Act5C promoter, the DSCP, or the Hsp70 promoter was used to drive Luc expression in S2 cells. We first examined whether codon usage influenced transcription by purifying nuclei from S2 cells and quantifying the nuclear Opt-Luc and De-Luc mRNA levels. Northern blot analyses using probes to Gapdh (enriched in the total RNA) and U6 RNA (enriched in the nucleus) confirmed the purity of the nuclear preparations (Supplementary Figure S3A). Northern blot analysis of the Opt-Luc and De-Luc mRNAs showed that at the level of total RNA, the Act5C promoter and DSCP resulted in a robust codon usage effect on nuclear Luc mRNA (Figure 5A, B), suggesting that codon usage has a major impact on gene transcription in Drosophila cells. In contrast, the codon usage effect on the nuclear Luc mRNA was dramatically reduced for the Hsp70 promoter-driven mRNAs due to upregulation of the nuclear De-Luc mRNA level (Figure 5A, B). This suggests that the promoter-dependent codon usage effect is due to regulation at the level of transcription.
Figure 5.
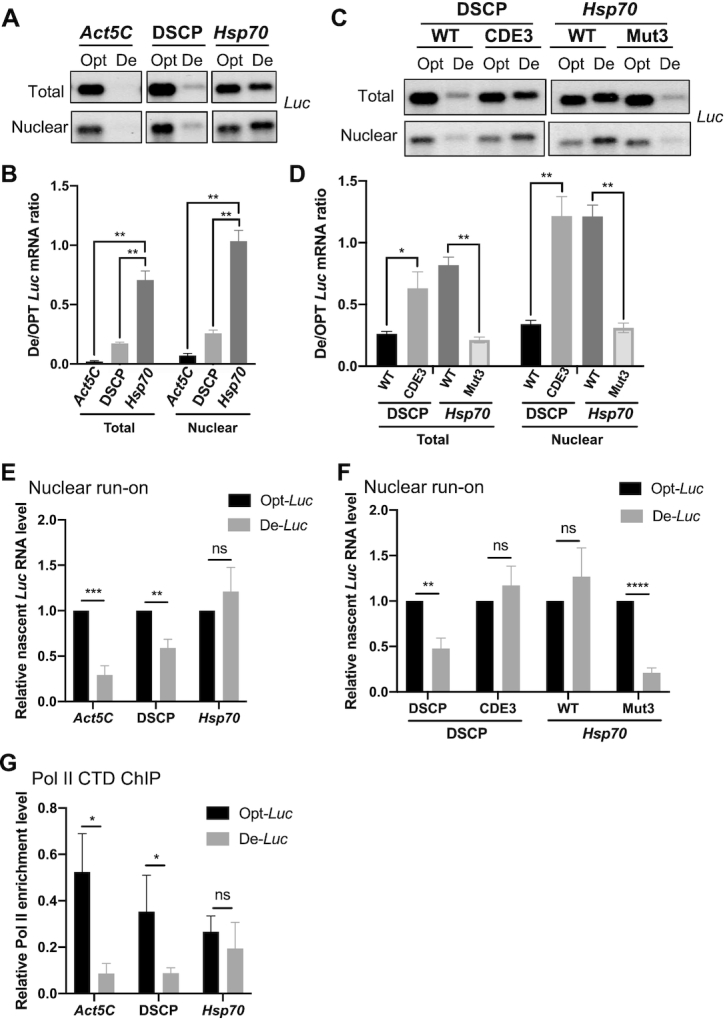
Promoter-dependent codon usage effect is controlled at the level of transcription. (A) Northern blot analysis of De-Luc and Opt-Luc mRNAs expressed under control of indicated promoters in total and nuclear RNA fractions. The probe hybridizes to the 5′ Myc-tag. (B) Quantification of De-Luc and Opt-Luc ratio for experiment shown in panel A (n = 3). (C) Northern blot analysis of De-Luc and Opt-Luc mRNAs expressed under control of DSCP or Hsp70 promoters with wild-type (WT) or mutant CDEs in nuclear and total RNA fraction. (D) Quantification of De-Luc/Opt-Luc ratio for experiment shown in panel C (n = 3). (E) Relative nascent De-Luc and Opt-Luc levels expressed under control of indicated promoters detected by qPCR in nuclear run-on assay. qPCR primers amplify the 5′ Myc-tag sequence, and expression was normalized to Act5C (n = 4). (F) Relative nascent De-Luc and Opt-Luc levels expressed under control of indicated promoters detected by qPCR in nuclear run-on assay (n = 4)). (G) Pol II CTD ChIP assay detecting Pol II levels in the 5′ Myc-tag region of different constructs (n = 3). Data are means ± SD. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.001. ns: not significant. Two-sided Student's t-tests were used.
To determine whether the Hsp70 CDE is responsible for the observed nuclear mRNA effect, we examined Luc mRNA expression under the control of the Hsp70 promoter with an impaired CDE (Mut3) or the DSCP IDR, which contains the CDE (Figure 4A). Mutation of CDE abolished the upregulation of nuclear De-Luc mRNA, whereas the presence of the CDE increased the nuclear De-Luc mRNA level (Figure 5C, D). This result suggests that the CDE rather than the core promoter is responsible for suppressing the codon usage effect at the transcriptional level.
To confirm these results, we also performed nuclear run-on assays to detect the nascent transcript levels. S2 cell nuclei were purified, and in vitro transcription was performed to label all nascent RNA transcripts with BrU. Labeled transcripts were then purified by immunoprecipitation using an anti-BrU antibody and quantified by qRT-PCR using primers targeting the 5′ end of the transcripts. As expected, transcription driven by the Act5C promoter and the DSCP was affected by codon usage, but levels of De-Luc and Opt-Luc levels expressed under control of the Hsp70 promoter were similar (Figure 5E). In addition, the mutation of CDE in the Hsp70 promoter construct (Mut3) caused a dramatic decrease of the nascent De-Luc mRNA, whereas addition of the CDE to the DSCP abolished the difference in levels between Opt-Luc and De-Luc mRNAs (Figure 5F).
Next, we examined whether codon usage directly affects transcription by influencing RNA polymerase II (Pol II) recruitment. We generated stable cell lines expressing Opt-Luc and De-Luc under control of different promoters and performed a chromatin immunoprecipitation assay (ChIP) using an antibody recognizing the C-terminal domain (CTD) of RNA Pol II. The previously used eYFP gene was replaced with the gene that impart hygromycin resistance (hph) to allow selection for stable cell lines. We confirmed that the expression levels of Gal4 and hph are similar among all cell lines (Supplementary Figure S3B–D). After ChIP, qPCR detecting the shared 5′ end of the Opt-Luc and De-Luc genes was performed. Pol II enrichment level was normalized to the first intron of the Act5C gene (Supplementary Figure S3E). When expressed under control of the Act5C promoter or the DSCP, Pol II enrichment at Opt-Luc locus was much higher than that at the De-Luc locus (Figure 5G), indicating that codon optimization greatly enhanced Pol II recruitment. In contrast, the Pol II recruitment was comparable between Opt-Luc and De-Luc loci when expression was driven by the Hsp70 promoter (Figure 5G). Together, these results demonstrate that codon usage plays an important role regulating transcription in Drosophila cells and that this regulation is dependent on a CDE downstream of the core promoter.
Codon usage and promoter jointly affect histone acetylation and nucleosome density
To understand how codon usage regulates transcription, we performed ChIP assays to examine histone modification and nucleosome levels in cell lines stably expressing Opt-Luc or De-Luc under the control of DSCP or Hsp70 promoters. ChIP for histone 3 (H3) lysine 27 acetylation (H3K27ac), an active chromatin mark, showed that for DSCP-driven reporter genes, the relative H3K27ac level was dramatically lower at the De-Luc locus than at the Opt-Luc locus (Figure 6A and Supplementary Figure S4A), consistent with levels of transcription and Pol II enrichment (Figure 5). For the reporter genes under the control of the Hsp70 promoter, the relative H3K27ac levels over Opt-Luc and De-Luc loci were comparable (Figure 6A), consistent with their transcriptional levels. ChIP assays showed that repressive histone modification marks H3 lysine 9 trimethylation and lysine 27 trimethylation were not significantly affected by codon optimization or promoter (Supplementary Figure S4B–E). Thus, codon usage regulates transcription by affecting chromatin structure through an activating histone modification.
Figure 6.
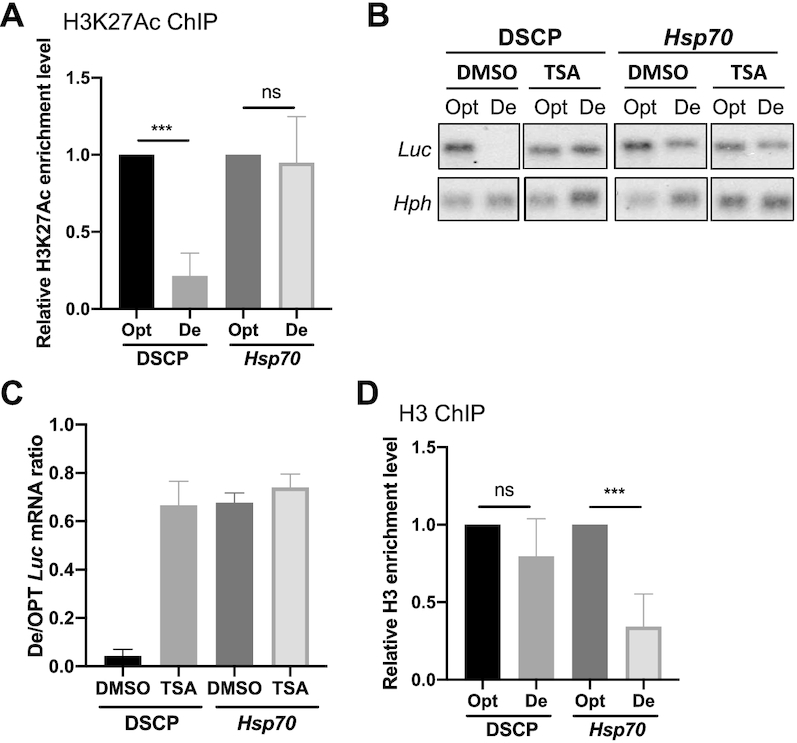
Codon usage and promoter influence H3K27ac levels and histone density, respectively. (A) H3K27ac ChIP assay over the 5′ Myc-tag region of indicated constructs in stably transfected cells. qPCR primers amplify the 5′ Myc-tag sequence, and data were normalized to Act5C (n = 4). (B) Northern blot analysis of De-Luc and Opt-Luc mRNAs expressed under the control of indicated promoters in stably transfected cells treated with DMSO or 150 μM TSA. Probes hybridized to the 5′ Myc-tag for Luc constructs and 5′ region for hph. The hph level was used as a control for transfected plasmid level. (C) Quantification of De-Luc/Opt-Luc ratio normalized to hph for experiment shown in panel C (n = 3). (D) H3 ChIP assay for nucleosome levels at the 5′ Myc-tag region of indicated constructs in stably transfected cells. qPCR primers amplified the 5′ Myc-tag sequence, and data were normalized to a transcription-inactivate region 3L1 (n = 4). Data are means ± SD. **P < 0.01. ns: not significant. Two-sided Student's t-tests were used.
To confirm the role of H3K27ac in regulating the codon usage-dependent effect, we treated S2 cells with trichostatin A (TSA), a specific inhibitor of class I and II histone deacetylases. When reporters were under control of DSCP, TSA treatment did not affect Opt-Luc mRNA levels but did increase the level of De-Luc mRNA to that of the Opt-Luc (Figure 6B, C), completely abolishing the codon usage effect. This result suggests that the codon usage-dependent effect on transcription is mainly regulated by histone acetylation: Optimal codons promote histone acetylation, whereas non-optimal codons inhibit it. TSA treatment had little effect on Opt-Luc or De-Luc mRNA levels when expressed under the control of the Hsp70 promoter, suggesting that the histone acetylation levels for Opt-Luc and De-Luc loci were already near their peaks and could not be further increased by TSA treatment.
Levels of H3 reflect nucleosome density on chromatin. An H3 ChIP assay showed that H3 binding was comparable at Opt-Luc and De-Luc loci when expression was driven by DSCP (Figure 6D and Supplementary Figure S4F), suggesting that despite dramatic differences in transcription level, codon usage does not regulate transcription by influencing nucleosome density. When expression of Opt-Luc and De-Luc mRNAs were driven by the Hsp70 promoter, however, H3 enrichment was significantly lower over the De-Luc gene locus than over the Opt-Luc locus (Figure 6D). This result suggests that the Hsp70 promoter specifically reduce nucleosome density at genes enriched for rare codons. It should be noted that because of low level of H3 over the De-Luc locus expressed under control of the Hsp70 promoter, the H3K27ac level normalized to the H3 level is higher for the De-Luc locus than for the Opt-Luc locus. Thus, despite of the transcriptional inhibitory effect of codon usage at the De-Luc locus, its low nucleosome density results in open chromatin that can counter the inhibitory effect of poor codon usage, and as a result, codon usage has a minimal effect on transcription driven by the Hsp70 promoter.
Codon usage and promoter influence premature transcription termination mediated by the Ars2-NEXT pathway
To further understand the how codon usage regulates gene expression, we performed a screen using double-stranded RNA (dsRNA) targeting factors known to be involved in transcriptional and post-transcriptional regulation. When Ars2-specific dsRNA was introduced into cell lines that stably express Opt-Luc and De-Luc constructs under the control of the DSCP, De-Luc mRNA levels, but not Opt-Luc levels, were upregulated (Figure 7A and Supplementary Figure S5A). Ars2 promotes premature termination of transcription of many RNA species (47–49). Thus, our data suggest that Ars2 specifically promotes the premature transcription termination of De-Luc. Ars2 recruits the NEXT complex, which interacts with nuclear exosome and causes degradation of early terminated transcripts (48,50). The silencing of genes that encode Mtr4 and ZCCHC8, two components of the NEXT complex (51), resulted in accumulation of a smear of short De-Luc but not Opt-Luc mRNA products (Figure 7A and Supplementary Figure S5B, C). Similarly, the silencing of Rrp40, which encodes a component of the nuclear exosome, also caused the accumulation of short De-Luc mRNA products (Figure 7A and Supplementary Figure S5D). These results indicate that the premature termination products of De-Luc are degraded by the nuclear exosome mediated by the NEXT complex.
Figure 7.
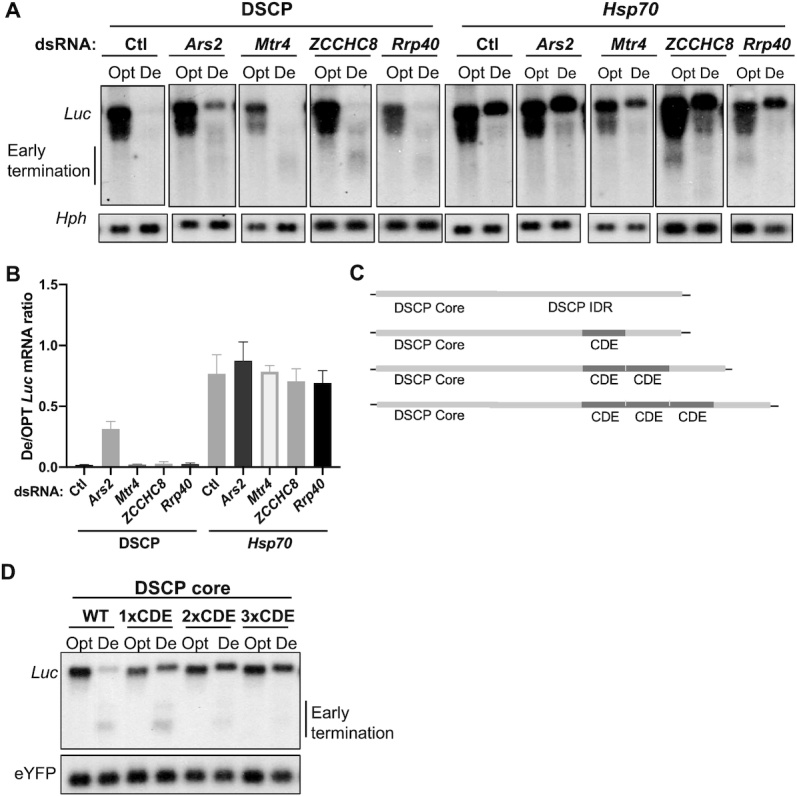
Codon usage and CDE affect premature transcription mediated by the Ars2-NEXT-exosome pathway. (A) Northern blot analysis of De-Luc and Opt-Luc stably expressed under the control of DSCP or Hsp70 promoter in cells treated with dsRNAs targeting Ars2, Mtr4, ZCCHC8, Rrp40, or no dsRNA as a control (Ctl). Probes hybridize to the 5′ Myc-tag sequence for Luc and 5′ region for hph. The hph level was used as a control for transfected plasmid level. (B) Quantification of De-Luc/Opt-Luc ratio normalized by hph for experiment shown in panel A (n = 3). (C) Diagrams of the promoter regions of the reporter constructs. Different numbers of copies of the CDE sequence were placed in the 5′ UTR of reporters driven by DSCP. (D) Northern blot analysis of De-Luc and Opt-Luc expressed under control of indicated promoters. Probes hybridize to 5′ Myc-tag sequence for Luc and 5′ region for eYFP. Data are means ± SD.
When Opt-Luc and De-Luc mRNAs were expressed under the control of Hsp70 promoter, silencing of Ars2 did not result in significant changes in De-Luc mRNA levels (Figure 7B). The silencing of Mtr4, ZCCHC8, and Rrp40 also did not cause the accumulation of De-Luc mRNA degradation products (Figure 7A). This suggests that Hsp70 promoter also inhibits the Ars2-mediated premature transcriptional termination of De-Luc. To examine whether the CDE of Hsp70 is responsible for suppressing premature transcriptional termination, we created Opt-Luc and De-Luc constructs with one to three copies of the CDE downstream of the DSCP core promoter (Figure 7C). With the addition of CDEs, De-Luc full-length mRNA levels were upregulated, but amount of premature termination products decreased (Figure 7D). This result suggests that the CDE can inhibit premature transcriptional termination of genes enriched for rare codons.
DISCUSSION
Although codon usage has been proposed to an important mechanism that determines levels of gene expression, how it broadly influences endogenous gene expression, especially in animal cells, is still unclear. Despite the robust codon usage effects on gene expression previously shown for many reporter genes, the lack of genome-wide correlation between codon usage and gene expression in mammalian cells called into question the physiological importance of codon usage on gene expression (33–35). We showed in this study that the lack of genome-wide correlation between codon usage and gene expression is partly due to gene-specific regulation: Constitutively expressed genes exhibited a much higher correlation between codon usage and gene expression than differentially expressed genes. This phenomenon was observed in mammalian cells, fungi, and Drosophila cells, indicating its conservation in eukaryotes. Using Drosophila S2 cells as the model system, we showed that the gene-specific codon usage effect is promoter-dependent. When reporter genes were expressed under the control of constitutive promoters such as Act5C and Tubulin, we observed robust codon usage effects on mRNA levels. In contrast, when reporters were driven by promoters of genes such as Hsp70, Myc, and Per, which are facultatively expressed, we observed weak or no codon usage effects. Although codon usage influenced mRNA decay in Drosophila cells as previously reported (18,20,26), the promoter-dependent effect was independent of the codon usage effect on mRNA decay.
Transcription levels are mainly determined by enhancer–promoter interactions and core promoter strengths (46,52). Little is known about how sequences downstream of core promoters regulate transcription. Our identification and characterization of a CDE critical for the promoter-dependent codon usage effect in the Hsp70 promoter revealed that the sequences downstream of a core promoter are necessary and sufficient to mediate the codon usage effect on mRNA. The lack of sequence homologous to the Hsp70 CDE in the Myc and Per promoters suggests that different elements downstream of these promoters likely regulate the codon usage effect on expression of these genes. Our findings indicate a previously unappreciated role for the region downstream of the core promoter in transcriptional regulation.
We and others previously showed that codon usage and GC content can regulate gene expression at the level of transcription independent of translation in fungi and human cells (15,17,21,22,29,30,53). Here we demonstrated that the codon usage effect regulates gene expression in Drosophila S2 cells, indicating that the transcriptional effect of codon usage is conserved in eukaryotes. In S2 cells, codon optimality influenced H3K27ac levels, and the codon usage-dependent effect on mRNA levels was mostly abolished when cells were treated with a histone deacetylase inhibitor, suggesting that codon usage influences transcription by modulating H3K27ac levels. When the reporter genes were expressed under the control of the Hsp70 promoter, however, the CDE specifically countered the effect of poor codon usage by decreasing nucleosome density, resulting in upregulation of the codon de-optimized mRNA. In addition to the influence on chromatin, codon usage also impacted premature transcription termination mediated by the Ars2-NEXT-exosome pathway. The fact that both transcriptional level and premature termination depend on promoter context suggests that these two aspects of the codon usage function might be coupled. It is possible that premature transcription termination events are influenced by chromatin structure and/or transcription rate. Modulation of Pol II activity can alter the extent of premature transcription termination (54). Interestingly, promoters can influence transcription termination mechanisms by changing configurations of Pol II complexes in Caenorhabditis elegans (55).
The influence of codon usage on chromatin structure in S2 cells is consistent with our previous results in Neurospora and human cells (15,21), suggesting that a common mechanism underlies the effects of codon usage on eukaryotic transcription. How codon usage results in chromatin modification changes and how elements downstream of core promoters modulate nucleosome density are not known, however. Our results suggest that codon usage is evolved to adapt to both translation and transcription processes. The codon usage information is likely recognized by the transcription or chromatin regulatory machinery in forms of DNA elements, which are used to regulate chromatin structure to suppress or activate transcription. Although most known transcriptional regulatory elements reside in the promoter regions, our results in fungal and animal systems demonstrate that the coding sequences can also play a major role in transcriptional regulation. In agreement with this conclusion, a significant portion of transcription factor recognition sites were previously found to be in exonic regions, which was proposed to be a force that drives codon usage biases (56,57). It is likely that the DNA elements within ORFs leading to gene activation show enrichment of nucleotide sequences found in preferred codons whereas those causing gene repression are enriched for nucleotide sequences preferentially found in rare codons. Our demonstration of the importance of the promoter element in determining the codon usage effect suggests that the nucleotide elements around core promoters can influence the chromatin regulatory effect of nucleotide elements in the open reading frames, resulting in promoter-dependent codon usage effects on transcription. The understanding of the mechanisms involved should be a major focus of the future codon usage research.
The promoter-dependent codon usage effect on gene expression demonstrated here provides an explanation for the lack of correlation between codon usage and gene expression in mammalian cells. However, it should be noted that mammalian genes are different from fungal genes in their intron numbers and sizes, which could also influence or mask the codon usage effect. A very recent study showed that intron-mediated splicing events can specifically promote nuclear export of AU-rich mRNAs and thus can regulate the codon usage effects on gene expression (33). Therefore, other mechanisms also contribute to the observed low correlation between codon optimality and gene expression in mammalian cells.
DATA AVAILABILITY
All data are available from the corresponding author upon request. The nucleotide sequences of the reporter genes and promoters used in this study can be found in Supplementary Table S1. The primer sequences used for the template amplification used for making probesfor Northern blot analysis are described in Supplementary Table S3.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of our laboratory for assistance and discussion.
Contributor Information
Qian Yang, Department of Physiology, University of Texas Southwestern Medical Center, Dallas, TX 75390-9040, USA.
Xueliang Lyu, Department of Physiology, University of Texas Southwestern Medical Center, Dallas, TX 75390-9040, USA; State Key Laboratory of Agricultural Microbiology, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, Hubei 430070, China.
Fangzhou Zhao, Department of Physiology, University of Texas Southwestern Medical Center, Dallas, TX 75390-9040, USA.
Yi Liu, Department of Physiology, University of Texas Southwestern Medical Center, Dallas, TX 75390-9040, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35 GM118118]; Welch Foundation [I-1560 to Y.L.]; X.L. is partially supported by National Natural Science Foundation of China [31701735]; International Postdoctoral Exchange Fellowship Program 2017 by the Office of China Postdoctoral Council ([2017]32). Funding for open access charge: National Institutes of Health [R35 GM118118].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985; 2:13–34. [DOI] [PubMed] [Google Scholar]
- 2. Sharp P.M., Tuohy T.M., Mosurski K.R.. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986; 14:5125–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Plotkin J.B., Kudla G.. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 2011; 12:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chaney J.L., Clark P.L.. Roles for synonymous codon usage in protein biogenesis. Annu. Rev. Biophys. 2015; 44:143–166. [DOI] [PubMed] [Google Scholar]
- 5. Hanson G., Coller J.. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018; 19:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quax T.E., Claassens N.J., Soll D., van der Oost J.. Codon bias as a means to fine-tune gene expression. Mol. Cell. 2015; 59:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu C.H., Dang Y., Zhou Z., Wu C., Zhao F., Sachs M.S., Liu Y.. Codon usage influences the local rate of translation elongation to regulate Co-translational protein folding. Mol. Cell. 2015; 59:744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou M., Guo J., Cha J., Chae M., Chen S., Barral J.M., Sachs M.S., Liu Y.. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 2013; 495:111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lampson B.L., Pershing N.L., Prinz J.A., Lacsina J.R., Marzluff W.F., Nicchitta C.V., MacAlpine D.M., Counter C.M.. Rare codons regulate KRas oncogenesis. Curr. Biol. 2013; 23:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu Y., Ma P., Shah P., Rokas A., Liu Y., Johnson C.H.. Non-optimal codon usage is a mechanism to achieve circadian clock conditionality. Nature. 2013; 495:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carlini D.B., Stephan W.. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics. 2003; 163:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kimchi-Sarfaty C., Oh J.M., Kim I.W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M.. A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science. 2007; 315:525–528. [DOI] [PubMed] [Google Scholar]
- 13. Buhr F., Jha S., Thommen M., Mittelstaet J., Kutz F., Schwalbe H., Rodnina M.V., Komar A.A.. Synonymous codons direct cotranslational folding toward different protein conformations. Mol. Cell. 2016; 61:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weinberg D.E., Shah P., Eichhorn S.W., Hussmann J.A., Plotkin J.B., Bartel D.P.. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 2016; 14:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou Z., Dang Y., Zhou M., Li L., Yu C.H., Fu J., Chen S., Liu Y.. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E6117–E6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jeacock L., Faria J., Horn D. Codon usage bias controls mRNA and protein abundance in trypanosomatids. Elife. 2018; 7:e32496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harigaya Y., Parker R.. Analysis of the association between codon optimality and mRNA stability in Schizosaccharomyces pombe. BMC Genomics. 2016; 17:895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao F., Yu C.H., Liu Y.. Codon usage regulates protein structure and function by affecting translation elongation speed in Drosophila cells. Nucleic. Acids. Res. 2017; 45:8484–8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Q., Yu C.H., Zhao F., Dang Y., Wu C., Xie P., Sachs M.S., Liu Y.. eRF1 mediates codon usage effects on mRNA translation efficiency through premature termination at rare codons. Nucleic Acids Res. 2019; 47:9243–9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Presnyak V., Alhusaini N., Chen Y.H., Martin S., Morris N., Kline N., Olson S., Weinberg D., Baker K.E., Graveley B.R. et al.. Codon optimality is a major determinant of mRNA stability. Cell. 2015; 160:1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu J., Dang Y., Counter C., Liu Y.. Codon usage regulates human KRAS expression at both transcriptional and translational levels. J. Biol. Chem. 2018; 293:17929–17940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kudla G., Lipinski L., Caffin F., Helwak A., Zylicz M.. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006; 4:e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Q., Medina S.G., Kushawah G., DeVore M.L., Castellano L.A., Hand J.M., Wright M., Bazzini A.A.. Translation affects mRNA stability in a codon-dependent manner in human cells. Elife. 2019; 8:e45396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coghlan A., Wolfe K.H.. Relationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae. Yeast. 2000; 16:1131–1145. [DOI] [PubMed] [Google Scholar]
- 25. dos Reis M., Wernisch L., Savva R.. Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res. 2003; 31:6976–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burow D.A., Martin S., Quail J.F., Alhusaini N., Coller J., Cleary M.D.. Attenuated codon optimality contributes to neural-specific mRNA decay in drosophila. Cell Rep. 2018; 24:1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buschauer R., Matsuo Y., Sugiyama T., Chen Y.H., Alhusaini N., Sweet T., Ikeuchi K., Cheng J., Matsuki Y., Nobuta R. et al.. The Ccr4-Not complex monitors the translating ribosome for codon optimality. Science. 2020; 368:eaay6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bazzini A.A., Del Viso F., Moreno-Mateos M.A., Johnstone T.G., Vejnar C.E., Qin Y., Yao J., Khokha M.K., Giraldez A.J. Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J. 2016; 35:2087–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Newman Z.R., Young J.M., Ingolia N.T., Barton G.M.. Differences in codon bias and GC content contribute to the balanced expression of TLR7 and TLR9. Proc. Natl. Acad. Sci. U. S. A. 2016; 113:E1362–E1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pouyet F., Mouchiroud D., Duret L., Semon M.. Recombination, meiotic expression and human codon usage. eLife. 2017; 6:e27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pershing N.L., Lampson B.L., Belsky J.A., Kaltenbrun E., MacAlpine D.M., Counter C.M.. Rare codons capacitate Kras-driven de novo tumorigenesis. J. Clin. Invest. 2015; 125:222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fath S., Bauer A.P., Liss M., Spriestersbach A., Maertens B., Hahn P., Ludwig C., Schafer F., Graf M., Wagner R.. Multiparameter RNA and codon optimization: a standardized tool to assess and enhance autologous mammalian gene expression. PLoS One. 2011; 6:e17596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mordstein C., Savisaar R., Young R.S., Bazile J., Talmane L., Luft J., Liss M., Taylor M.S., Hurst L.D., Kudla G.. Codon usage and splicing jointly influence mRNA localization. Cell Syst. 2020; 10:351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rudolph K.L., Schmitt B.M., Villar D., White R.J., Marioni J.C., Kutter C., Odom D.T.. Codon-driven translational efficiency is stable across diverse mammalian cell states. PLos Genet. 2016; 12:e1006024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Semon M., Mouchiroud D., Duret L.. Relationship between gene expression and GC-content in mammals: statistical significance and biological relevance. Hum. Mol. Genet. 2005; 14:421–427. [DOI] [PubMed] [Google Scholar]
- 36. Xue Z., Ye Q., Anson S.R., Yang J., Xiao G., Kowbel D., Glass N.L., Crosthwaite S.K., Liu Y.. Transcriptional interference by antisense RNA is required for circadian clock function. Nature. 2014; 514:650–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Consortium G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huntley M.A., Lou M., Goldstein L.D., Lawrence M., Dijkgraaf G.J., Kaminker J.S., Gentleman R.. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genomics. 2016; 17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Katta S.S., Chen J., Gardner J.M., Friederichs J.M., Smith S.E., Gogol M., Unruh J.R., Slaughter B.D., Jaspersen S.L.. Sec66-Dependent regulation of yeast spindle-pole body duplication through Pom152. Genetics. 2015; 201:1479–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. dos Reis M., Savva R., Wernisch L.. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic. Acids. Res. 2004; 32:5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bennetzen J.L., Hall B.D.. Codon selection in yeast. J. Biol. Chem. 1982; 257:3026–3031. [PubMed] [Google Scholar]
- 42. Wu V.W., Thieme N., Huberman L.B., Dietschmann A., Kowbel D.J., Lee J., Calhoun S., Singan V.R., Lipzen A., Xiong Y. et al.. The regulatory and transcriptional landscape associated with carbon utilization in a filamentous fungus. PNAS. 2020; 117:6003–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoskins R.A., Landolin J.M., Brown J.B., Sandler J.E., Takahashi H., Lassmann T., Yu C., Booth B.W., Zhang D., Wan K.H. et al.. Genome-wide analysis of promoter architecture in Drosophila melanogaster. Genome Res. 2011; 21:182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vo Ngoc L., Kassavetis G.A., Kadonaga J.T.. The RNA polymerase II core promoter in drosophila. Genetics. 2019; 212:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Espinar L., Schikora Tamarit M.A., Domingo J., Carey L.B.. Promoter architecture determines cotranslational regulation of mRNA. Genome Res. 2018; 28:509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smale S.T., Kadonaga J.T.. The RNA polymerase II core promoter. Annu. Rev. Biochem. 2003; 72:449–479. [DOI] [PubMed] [Google Scholar]
- 47. Giacometti S., Benbahouche N.E.H., Domanski M., Robert M.C., Meola N., Lubas M., Bukenborg J., Andersen J.S., Schulze W.M., Verheggen C. et al.. Mutually exclusive CBC-containing complexes contribute to RNA fate. Cell Rep. 2017; 18:2635–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hallais M., Pontvianne F., Andersen P.R., Clerici M., Lener D., Benbahouche Nel H., Gostan T., Vandermoere F., Robert M.C., Cusack S. et al.. CBC-ARS2 stimulates 3′-end maturation of multiple RNA families and favors cap-proximal processing. Nat. Struct. Mol. Biol. 2013; 20:1358–1366. [DOI] [PubMed] [Google Scholar]
- 49. Iasillo C., Schmid M., Yahia Y., Maqbool M.A., Descostes N., Karadoulama E., Bertrand E., Andrau J.C., Jensen T.H.. ARS2 is a general suppressor of pervasive transcription. Nucleic Acids Res. 2017; 45:10229–10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lubas M., Andersen P.R., Schein A., Dziembowski A., Kudla G., Jensen T.H.. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep. 2015; 10:178–192. [DOI] [PubMed] [Google Scholar]
- 51. Lubas M., Christensen M.S., Kristiansen M.S., Domanski M., Falkenby L.G., Lykke-Andersen S., Andersen J.S., Dziembowski A., Jensen T.H.. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell. 2011; 43:624–637. [DOI] [PubMed] [Google Scholar]
- 52. Field A., Adelman K.. Evaluating enhancer function and transcription. Annu. Rev. Biochem. 2020; 89:213–234. [DOI] [PubMed] [Google Scholar]
- 53. Zhou Z., Dang Y., Zhou M., Yuan H., Liu Y.. Codon usage biases co-evolve with transcription termination machinery to suppress premature cleavage and polyadenylation. Elife. 2018; 7:e33569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kamieniarz-Gdula K., Proudfoot N.J.. Transcriptional control by premature termination: a forgotten mechanism. Trends Genet. 2019; 35:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Miki T.S., Carl S.H., Grosshans H.. Two distinct transcription termination modes dictated by promoters. Genes Dev. 2017; 31:1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stergachis A.B., Haugen E., Shafer A., Fu W., Vernot B., Reynolds A., Raubitschek A., Ziegler S., LeProust E.M., Akey J.M. et al.. Exonic transcription factor binding directs codon choice and affects protein evolution. Science. 2013; 342:1367–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sullivan A.M., Arsovski A.A., Lempe J., Bubb K.L., Weirauch M.T., Sabo P.J., Sandstrom R., Thurman R.E., Neph S., Reynolds A.P. et al.. Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep. 2014; 8:2015–2030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the corresponding author upon request. The nucleotide sequences of the reporter genes and promoters used in this study can be found in Supplementary Table S1. The primer sequences used for the template amplification used for making probesfor Northern blot analysis are described in Supplementary Table S3.