

Abstract
We examined a large dataset of female metastatic breast cancers (MBCs) profiled with comprehensive genomic profiling (CGP) to identify the prevalence and distribution of immunotherapy responsiveness‐associated biomarkers. DNA was extracted from 3831 consecutive MBCs: 1237 (ERpos/HER2 neg), 1953 ERneg/HER2 amp, and 641 triple‐negative breast cancer (TNBC). CGP was performed using the FoundationOne® or FoundationOne®CDx NGS assay. Tumor mutational burden (TMB) and microsatellite instability (MSI) were determined in a subset of cases. PD‐L1 expression in immunocytes in a subset of cases was determined by immunohistochemistry using the companion diagnostic VENTANA PD‐L1 SP142 Assay. The median age of the cohort was 54 years (range 20–89). Genomic alterations (GAs)/tumor were similar (range: 5.9–7.3). Markers of potential immune checkpoint inhibitor (ICPI) benefit included: CD274 (PD‐L1) amplification (1%–3%), BRAF GA (1%–4%), TMB of ≥10 mutations/Mb (8%–12%), MSI‐high (0.1%–0.4%), PBRM1 GA (1%), and positive PD‐L1 staining of immunocytes ranging from 13% in ERpos/HER2 neg and 33% in ERneg/HER2 amp to 47% in the TNBC group. Potential markers of ICPI resistance included inactivating STK11 GA (1%–2%) and MDM2 amplification (3%–6%). MTOR pathway targets were common with lowest frequency in TNBC. ERBB2 short variant mutations were most frequent ERpos/HER2 neg and absent in TNBC. BRCA1/2 GA were least frequent in ERneg/HER2 amp. The demonstrations of clinical benefit of immunotherapy in MBC support the need for development and utilization of biomarkers to guide the use of ICPIs for these patients. In addition to guiding therapy selection, CGP shows potential to identify GA linked to response and resistance to ICPI in MBC.
Keywords: biomarkers, comprehensive genomic profiling, immunotherapy, metastatic breast cancer, microsatellite instability, PD‐L1, tumor mutational burden
Both our results and published literature suggest that in addition to guiding targeted therapy selection, comprehensive genomic profiling shows potential to identify genomic alterations linked to response and resistance to immune checkpoint inhibitor (ICPI) therapy in metastatic breast cancer (MBC). The demonstration of clinical benefit of immune checkpoint blockade in MBC supports the need for the development of biomarkers used to guide the use of ICPI drugs for these patients.

1. INTRODUCTION
The use of immune checkpoint inhibitors (ICPIs) targeting pathways that promote cancer immune evasion has growing momentum in the field of cancer treatment and research. 1 Each cancer has varying degrees of somatic mutations that are unique to the cancer type, and ICPIs have been found to be more effective against tumors with high mutational burden. 2 Exposure to carcinogens, such as smoking and UV light, has been linked to high mutation burden, and ICPIs were initially approved in cancers associated with these causes, including melanoma, non‐small cell lung cancer (NSCLC), and bladder cancer. 3 , 4
Over time, it was noted that ICPIs were effective only in a limited number of patients, and significant and severe immune‐related adverse effects (irAEs), such as pneumonitis, colitis, and thyroiditis, were being observed. 5 Despite the promising clinical effects of ICPIs, the overall response rates (ORRs) have been low. 6 For instance, pembrolizumab, which has been approved by the United States (US) Food and Drug Administration (FDA) for use in platinum refractory head and neck squamous cell carcinoma, has an ORR of only 13%–18%. It is also seen that across all cancers, resistance to ICPIs against PD‐1 and PD‐L1 approaches 60%. 7 Recent studies have been focusing on identifying biomarkers that can help delineate patients who may have a better response to ICPIs as well as to avoid unwanted irAEs. 8 At the same time, previous studies performed on a wide variety of malignancies suggested that PD‐L1 overexpression was a major biomarker for predicting benefit for anti‐PD‐1/PD‐L1 therapies. 9 , 10 The US FDA has approved the use of pembrolizumab in solid tumors with high microsatellite instability (MSI), based on a biomarker assessment of MSI status. High MSI is associated with increased number of mutations in tumoral DNA which correspond to higher levels of tumor mutational burden (TMB) and the increased presence of circulating antitumor lymphocytes and neoantigens. 11 Neoantigens are defined as tumor‐specific antigens that arise from non‐synonymous mutations and other genetic alterations, 12 and when processed into short peptides and presented by MHC molecules to T cells stimulate the immune system recognition of cancer cells as “non‐self” and enable subsequent immune‐mediated attack. 13 The more somatic mutations a tumor has, the more neoantigens are likely to form, thus, TMB can be considered as an indirect measure of tumor neoantigen load. Studies have shown that tumors featuring higher TMB levels have enhanced responsiveness to ICPIs especially when they are administered as monotherapies. 14 Based on data from the KEYNOTE‐158 trial, FDA granted regulatory approval for the use of FoundationOne CDx as the first companion diagnostic for the anti‐PD‐1 therapy pembrolizumab, to identify patients with unresectable or metastatic TMB high (≥10 mutations/megabase, mut/Mb) solid tumors that have progressed following prior treatment with no alternative treatment options. 15
More recently, single gene mutations have been reported as predictive biomarkers for ICPI response. BRAF mutations have been linked to improved ICPI response, and tumors such as renal cell carcinoma with loss of function genomic alterations (GAs) in the PBRM1 chromatin remodeling tumor suppressor gene may also be ICPI responsive. 16 , 17 Studies of KRAS‐mutant NSCLC have linked inactivating alterations in the STK11/LKB1 tumor suppressor gene with significant risk of PD‐1 and PD‐L1 inhibitor resistance. 18 PTEN alterations have also been associated with lower ORR and shorter progression‐free survival (PFS) independently of clinical factors and PD‐L1 status in triple‐negative breast cancer (TNBC) patients treated with ICPI. 19 In addition, studies have indicated that the MDM2 proto‐oncogene, a negative regulator of the TP53 gene, when amplified as seen in multiple tumors, may be associated with disease hyperprogression in patients treated with PD‐1/PD‐L1 inhibitors. 20
Both TNBC and HER2‐positive tumors have been recently recognized as being lymphocyte‐rich and are accompanied by abundant tumor‐infiltrating lymphocytes (TILs), also referred to as tumor‐infiltrating immune cells (ICs). PD‐L1 is expressed in the TILs of 20% of breast cancers, with TNBC and HER2‐positive tumors showing higher levels at 33% and 56%, respectively. 21 A number of trials evaluating pembrolizumab and atezolizumab, both as monotherapy and in combination with other conventional treatment options, are in various stages and have shown notable results. 22 In March 2019, the FDA approved atezolizumab for PD‐L1‐ positive (IC score based) unresectable locally advanced or metastatic TNBC employing the PD‐L1 assay device called VENTANA PD‐L1 (SP142) Assay as a companion diagnostic biomarker. 23 Based on this background information, we queried whether comprehensive genomic profiling (CGP) of the three major subtypes of metastatic breast cancer (MBC) could identify biomarkers that have been linked to responsiveness to ICPI treatment.
2. METHODS
Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817). DNA was extracted from formalin‐fixed, paraffin‐embedded tissue samples obtained from 3831 cases of clinically diagnosed MBC received between September 2012 and July 2018, including 1237 ER+/HER2 not amplified, 1953 ER‐/HER2 amplified (amp), and 641 TNBC cases. CGP was performed using either the FoundationOne® or FoundationOne®CDx assay in a Clinical Laboratory Improvement Amendments (CLIA)‐certified, CAP (College of American Pathologists)‐accredited laboratory (Foundation Medicine). The pathologic diagnosis of each case was confirmed on routine hematoxylin and eosin (H&E)‐stained slides and all samples forwarded for DNA extraction contained a minimum of 20% tumor nuclear area, compared with benign nuclear area.
Sequencing for the detection of base substitutions, insertions, deletions, copy number alterations (focal amplifications and homozygous deletions), and select gene fusions was performed using a hybrid capture‐based system as previously described. 24 TMB was determined on 0.8 to 1.1 Mb of sequenced DNA for each case based on the number of somatic base substitution or indel alterations per Mb after filtering to remove known somatic and deleterious mutations, as previously described. 25 MSI was calculated as previously described. 26 Patients were classified as MSI‐high (MSI‐H), MSI cannot be determined, or microsatellite stable. PD‐L1 expression was determined using the VENTANA PD‐L1 (SP142) Assay on 5‐micron tissue sections. Immunohistochemistry (IHC) slide evaluation was based on the tumor‐infiltrating IC score and did not include the tumor cell score (TC). In this study, an IC ≥1% was considered to be positive. The IC score is defined as the proportion of tumor‐infiltrating IC staining in the total tumor area. Tumor areas are defined as the TCs with intra‐ and peritumoral stroma. The Fisher's exact test was used to determine if the proportion of each metastatic sample site differed across the three MBC subgroups. A p < 0.05 is considered statistically significant.
3. RESULTS
The distribution of metastatic disease sites for the majority of samples provided for CGP among the ERpos/HER2 neg, ERneg/HER2 amp, and TNBC subgroups is summarized in Table S1. The most common metastatic sites across subgroups included liver, bone, and lung. The incidence of bone metastases was significantly higher in the ERpos/HER2 neg subgroup compared with the ERneg/HER2 amp (10.82% vs. 4.65%, p < 0.0001) and TNBC subgroup (10.82% vs. 3.27%, p = 0.0002). The ERneg/HER2 amp subgroup had a significantly higher percentage of brain metastases compared with the ERpos/HER2 neg subgroup (8.90% vs. 1.59%, p < 0.0001).
The clinical and CGP findings in the 3831 cases of MBC are shown in Table 1. The median ages and age ranges of the patients in each subtype of MBC were similar. The mean number of GA per tumor was also similar, ranging from 5.9 GA/tumor in the TNBC group to 7.3 GA/tumor in the HER2‐amplified/ER‐ group. The known and likely GA listed by alteration type (short variant (SV) substitutions including base substitutions, short indels and truncations, copy number changes including both amplifications and homozygous deletions and rearrangements/fusions), and the protein and transcript effects are provided for the three MBC tumor types in Table S2 (ERpos/HER2 neg subset of 1214 cases), Table S3 (ERneg/HER2 amp subset of 1927 cases), and Table S4 (TNBC subset of 633 cases).
TABLE 1.
Clinical features and biomarkers associated with immunotherapy responsiveness in metastatic breast cancer
| ERpos/HER2neg | ERneg/HER2amp | TNBC | |
|---|---|---|---|
| Number of cases | 1237 | 1953 | 641 |
| Age (range in years) | 55 (23–89) | 55 (20–89) | 53 (20–85) |
| GA/tumor | 6.3 | 7.3 | 5.9 |
| MTOR GA | PIK3CA 38% | PIK3CA 38% | PIK3CA 19% |
| PTEN 10% | PTEN 5% | PTEN 15% | |
| NF1 5% | NF1 7% | NF1 8% | |
| CDH1 GA | 7% | 4% | 3% |
| ESR1 GA | 15% | 6% | 0.5% |
| BRCA1/2 GA | 3%/6% | 2%/3% | 7%/3% |
| ERBB2 amp | 9% | 100% | 0% |
| ERBB2 SV | 9% | 7% | 0% |
| Other kinase targets | FGFR1 18% | FGFR1 11% | FGFR1 8% |
| FGFR2 2% | FGFR2 1% | FGFR2 4% | |
| EGFR 2% | EGFR 3% | EGFR 4% | |
| KIT 1% | KIT 2% | KIT 2% | |
| MET 0.4% | MET 1% | MET 1% | |
| BRAF 2% | BRAF 1% | BRAF 4% | |
| AR amp | 1% | 1% | 1% |
| MSI‐High | 0.2% | 0.1% | 0.4% |
| CD274 (PD‐L1) amp | 1% | 1% | 3% |
| TMB >10 mut/Mb | 8% | 12% | 9% |
| TMB >20 mut/Mb | 2% | 2% | 3% |
| Positive (≥1%) Immunocyte PD‐L1 IHC staining a | 13% | 33% | 47% |
| PBRM1 GA | 1% | 1% | 1% |
| STK11 GA | 1% | 1% | 2% |
| MDM2 amp | 6% | 5% | 3% |
Abbreviations: Amp, amplification; ERpos, estrogen receptor positive; GA, genomic alteration; IHC, immunohistochemistry; SV, short variant; TMB, tumor mutational burden; TNBC, triple‐negative breast cancer.
Subset of recent MBC cases that were stained for PD‐L1 immunocyte expression with the VENTANA PD‐L1 (SP142) Assay approved in March 2019 as a CDx for the atezolizumab‐nab‐paclitaxel approval for the treatment of TNBC.
As seen in Table 1 and the long tail plots (Figure 1A‐C), MTOR pathway genes (PIK3CA, PTEN, and NF1) were found altered in all three MBC groups, but representation was lowest in the TNBC patients. PTEN alterations, including SVs, deletions, and rearrangements, were detected across all three subtypes (5%–15%). CDH1 (7%) and ESR1 (15%) GA were most frequently identified in the ERpos/HER2 neg cases. ERBB2 SV mutations were most frequently found in the ERpos/HER2 neg (9%) and in the ERneg/HER2 amp cases (7%) and were not identified in TNBC cases. Other kinase targets were uncommon in all three groups except for the identification of FGFR1 GA in ERpos/HER2 neg tumors (18%). Potentially targetable kinase GA rarely encountered in any of the MBC groups included FGFR2 (1%–4%), EGFR (2%–4%), KIT (1%–2%), MET (0.4%–1%), and BRAF (1%–4%). At 7%, BRCA1 GA were most frequent in the TNBC cases and were rare in the ERpos/HER2 neg (3%) and ERneg/HER2 amp (2%) groups. At 6%, BRCA2 GA were most frequent in the ERpos/HER2 neg cases and less common in the ERneg/HER2 amp (3%) and TNBC (3%) cohorts. AR was amplified in 1% in all three groups of MBCs. AR protein expression was not assessed in this study.
FIGURE 1.
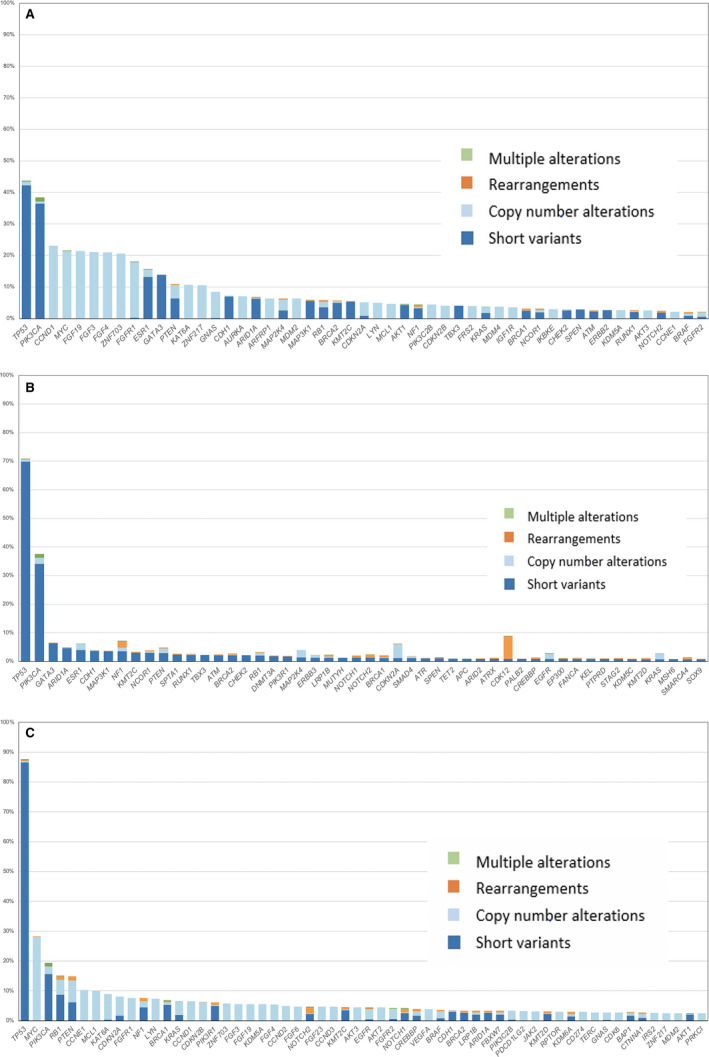
Most prevalent genomic alterations by gene and alteration type across breast cancer subtypes. (A) Genomic alterations in 1237 ERpos/HER2 neg metastatic breast cancers. (B). Genomic alterations in 1953 ERneg/HER2 amp metastatic breast cancers. (C) Genomic alterations in 641 triple‐negative breast cancers
When focused on potential biomarkers of ICPI responsiveness, this study includes individual GA, calculated MSI status, TMB derived from CGP, and PD‐L1 expression determined by IHC. Individual genomic markers that have been linked to responsiveness to ICPI in other tumor types were identified infrequently in MBC. This included markers of potential ICPI benefit such as CD274 (PD‐L1) amplification found in 1%–3% of the MBC (Figure 2). BRAF‐activating SV mutations are found in 1%–4% of MBC, and PBRM1‐inactivating SV mutations are found in 1% of MBC. MSI‐H status, predictive of efficacy for the ICPI pembrolizumab for all solid tumors including MBC, was uncommon in this study, ranging from 0.1% to 0.4%. The calculated TMB, also linked to responsiveness of advanced malignancies to ICPI in multiple studies, varied among the three MBC groups. When ≥10 mutations/Mb is used as a cut‐off, 8%–12% of MBC were positive. Using the VENTANA PD‐L1 SP142 Assay that was included in the approval of the atezolizumab‐nab‐paclitaxel combination treatment for TNBC in March of 2019, the frequency of positive staining ranged from 13% in the ERpos/HER2 neg subgroup to 33% in the ERneg/HER2 amp subgroup to 47% in the TNBC subgroup. This study also evaluated biomarkers associated with resistance and hyperprogression to ICPI and included inactivating GA in STK11 found in 1%–2% of MBC and amplification of the MDM2 gene encountered in 3%–6% of MBC (Figure 3).
FIGURE 2.
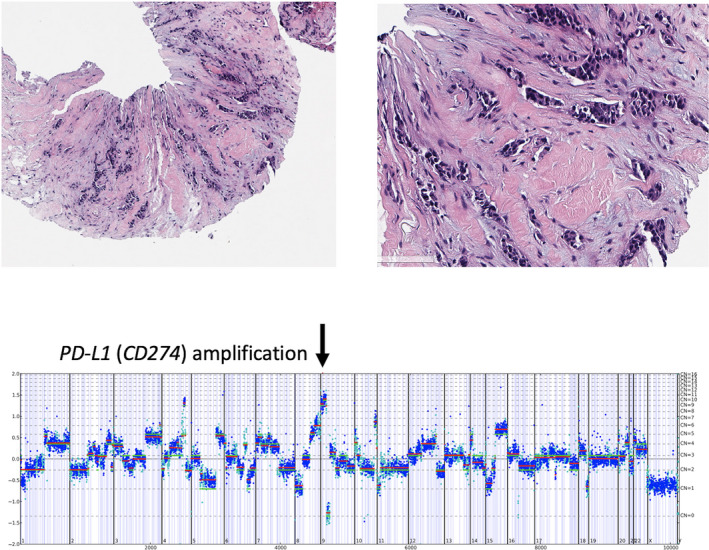
Case of stage IV triple‐negative breast cancer in a 72‐year‐old woman with a CD274 amplification. The genome‐wide copy number plot demonstrates an amplification of the CD274 gene at 12 copies per cell
FIGURE 3.
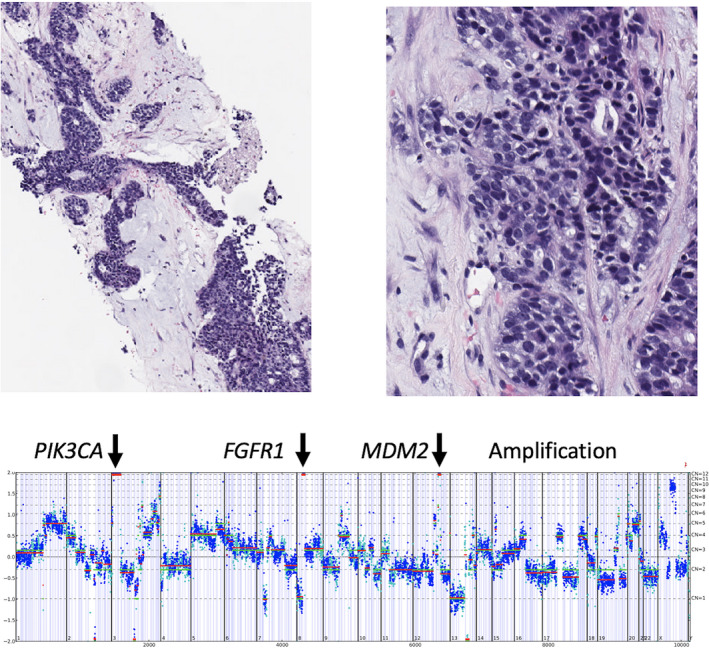
Case of stage IV triple‐negative breast cancer in a 57‐year‐old patient with MDM2 amplification. Comprehensive genomic profiling revealed multiple potentially targetable gene amplifications in FGFR1 and PIK3CA, but also featured amplification of MDM2
4. DISCUSSION
Currently, the US FDA‐approved companion diagnostic biomarkers linked to ICPI for breast cancer include PD‐L1 expression using the VENTANA PD‐L1 (SP142) IHC assay in TNBC for atezolizumab, TMB detected by CGP using the FoundationOne CDx assay for pembrolizumab, in addition to the identification of MSI/mismatch repair deficiency (dMMR) by multiple modalities for pembrolizumab. In this study, the metastatic tumor tissues of a significant percentage of patients with MBC were found to contain these GAs associated with response to ICPIs. Among patients in the ERpos/HER2 neg subgroup, 46.2% were found to harbor US FDA‐approved targetable alterations, including PIK3CA (38%), TMB ≥10 mut/Mb (8%) and MSI‐H (0.2%). Within the ER‐/HER2‐amplified cohort, 12.1% possessed approved targetable alterations, including TMB ≥10 mut/Mb (12%) and MSI‐H (0.1%). The highest percentage of actionable mutations was found in the historically difficult to treat TNBC group, with 56.4% of patients possessing GAs indicative of response to ICPIs including PD‐L1 SP142 staining ≥1% (47%), TMB ≥10 mut/Mb (9%), and MSI‐H (0.4%). PIK3CA frequency was 19% in the TNBC group.
The majority of studies involving breast cancer and ICPI have focused on PD‐1 and PD‐L1 blockades. 21 The KEYNOTE‐012 trial (phase 1b) studied pembrolizumab in advanced, heavily pretreated TNBC. In the 111 enrolled TNBC patients, 58.6% of their tumors had some level of PD‐L1 expression. Among 27 patients for whom response was analyzed, the ORR was 18.5% and the irAE was reportedly mild. 27 The phase Ib KEYNOTE‐028 trial showed that in PD‐L1+ ER+/HER2‐negative MBC, pembrolizumab had an ORR of 12%. 21 Another phase 1b/II trial showed that combination of pembrolizumab and eribulin had a favorable response, but the PD‐L1 status did not predict the response to treatment. 16 I‐SPY2 (Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging and molecular Analysis 2) and phase III KEYNOTE‐522 are some of the studies evaluating ICPIs, such as pembrolizumab, in combination with standard chemotherapy. 21 The results of the latter showed that the combination of pembrolizumab and chemotherapy led to a pathological complete response frequency of 64.8% when compared to 51.2% with placebo using chemotherapy. 28 It has been observed that HER2 expression can result in immune suppression, so trials like PANACEA and PembroMab are evaluating pembrolizumab, trastuzumab, and ado‐trastuzumab emtansine in HER2+ disease. 21 , 29 KEYNOTE‐119 is an ongoing phase 3 trial comparing monotherapy with pembrolizumab and chemotherapy in metastatic TNBC. 30 Pembrolizumab monotherapy was reported to not significantly improve OS as second‐ or third‐line treatment for mTNBC versus chemotherapy, although the pembrolizumab treatment effect increased as PD‐L1 enrichment increased. 31 Several trials have also been performed with the monoclonal antibody atezolizumab which prevents the interaction of PD‐L1 with PD‐1 and B7.1 (CD80), which in turn causes less suppression of tumor‐reactive lymphocytes. 32
PD‐L1 expression itself is seen as a promising biomarker for breast cancer in general. Its expression has been shown to be associated with lymph node spread, higher histological grades, ER receptor negativity, and TNBC. 33 In the IMpassion130 study by Schmidt et al, PD‐L1 expression was around 41%. 34 In most studies of TNBC IHC staining of ICs, PD‐L1 expression has ranged from 40% to 65%. 35 In 2014, Schalper et al reported that nearly 60% of breast cancers expressed PD‐L1 in ICs and that the IC PD‐L1 expression status was associated with better overall disease outcomes. 36 In the current study, PD‐L1 expression was 47% using the VENTANA PD‐L1 SP142 Assay. In both the current study and the study reported by Schmid et al, 34 a cut‐off of >1% expression in ICs was used to define positive PD‐L1 expression. Currently, there is no standard method of staining or cut‐off of positive PD‐L1 expression in ER+and HER2‐amplified MBC including whether to include the IHC expression in IC only, TC only, or the combination. 34 , 35 Since VENTANA PD‐L1 SP142 was approved as a CDx for treatment of TNBC with atezolizumab and nab‐paclitaxel, we decided to use the same assay for all MBC samples; hence, we are only scoring IC staining.
PD‐L1, the protein encoded by the CD274 gene, undergoes several genetic alterations including amplification, epigenetic regulation, transcriptional activation, glycosylation, phosphorylation, and ubiquitination. Copy number alterations (CNAs) resulting in amplification of CD274 in Hodgkin's lymphoma are associated with high response rates to PD‐L1 inhibitors. 36 Barrett et al evaluated 54 cases of TNBC and found that high levels of PD‐1 and PD‐L1 with high copy number variants (CNVs) were found in 20% and 37%, respectively. This rate contrasts with the current study where CD274 amplification was identified in only 1%–3% of MBC cases. A large analysis of 118 187 tumor samples showed that CD274 amplification was identified in 0.7% of the samples and did not correlate with PD‐L1 expression. 16
In this study, 5%–15% of MBC cases possessed PTEN alterations. Prior work has shown the loss of the tumor suppressor PTEN to be associated with poor responses to PD‐1 blockade in patients with melanoma, uterine sarcoma, and mTNBC, 19 and that partial PTEN deletions associate with worse OS in breast cancer. 37 In addition to the established role of PTEN in cancer progression, PTEN deficiency can lead to increased immunosuppressive cytokines and expression signatures that are unfavorable for effective antitumor responsiveness. 38 , 39 , 40 In breast cancer cell lines, Mittendorf et al showed that PTEN loss resulted in overexpression of PD‐1 and PD‐L1 which was downregulated after PI3K pathway inhibition and may increase antitumor adaptive processes. 41 Clinical trials are needed to confirm that PTEN‐altered breast cancer harbors ICPI resistance that may be reversed by PI3K/AKT/mTOR inhibitor mono‐ or combination therapy.
PTEN deletion and high TMB were not associated with PD‐L1 expression in the current analysis. 42 The JAK2 pathway also seems to play a role in regulating PD‐L1 expression and reports are emerging showing that JAK2 inhibitors may have a benefit in TNBCs with 9p.24.1 amplification. 43 Even though numerous studies have included the status of CD274 amplification, data on its impact on clinical outcomes and ICPI therapy are very limited, highlighting the need for more research on this topic in the future.
Findings from the various KEYNOTE trials in other tumor types led to the accelerated approval by the FDA of pembrolizumab in MSI‐H solid tumors. A large study of 39 cancer types has found an MSI‐H status in 1.53% of primary breast cancers, in comparison to our study where it was only 0.1%–0.4% in cases of clinically advanced and metastatic disease. 44 Given the low prevalence of MSI in breast cancer and lack of robust data on clinical outcomes, MSI, at least for now, seems to have limited utility in breast cancer.
Tumors with higher TMB have shown enhanced responses to ICPIs. In the recent CheckMate 026 trial, NSCLC patients with high TMB had a better ORR and PFS on treatment with nivolumab than with chemotherapy. 45 Thomas et al. found that immune subtypes with high TMB had better ORR to ICPI therapies than subtypes with low TMB levels in BRCA‐positive breast cancer. In this study, the TMB was measured by whole genome sequencing (WGS) and ranged from 0 to 115 mutations/Mb with a mean of 1.63 mutations/Mb, which was used as the cut‐off. 46 In WGS analysis by Park et al, 100 mutations/Mb was considered as high TMB. 47 TMB varies widely among other cancers also reflecting the technique used for measurement. For example, the CheckMate 026 trial in lung cancer used whole exome sequencing and a TMB score >243 mutations/Mb as the cut‐off. 45 In breast cancer, some reports suggest that TMB is higher in ER‐negative cancers when compared to ER‐positive cancers. 48 High TMB has also been associated with poor disease‐free survival. 49 More work is needed to understand the relationship of clinical response along the continuum of TMB scores and how discrete values can provide more clinical information beyond a simple dichotomous classification. Additionally, harmonization efforts are currently underway to ensure alignment and improve interchangeability between TMB estimates generated from different targeted gene panels, 47 an activity essential to ensure this biomarker provides consistent information to further evaluate its clinical implications in controlled prospective studies and to inform treatment decisions across diagnostic platforms.
BRAF mutations are very uncommon in breast cancer representing around 0%–1.2% in public databases of both localized curable primary and MBC and 1%–4% of clinically advanced MBC in the current study. Reports indicate that TMB in BRAF‐altered MBC is generally higher, and hence may be a potential biomarker of ICPI efficacy, but larger studies are lacking with regard to the predictive status of BRAF mutations in breast cancer. Inactivation of STK11 has been shown to cause a “cold” tumor immune microenvironment by blunting the action of cytotoxic T lymphocytes and has been associated with PD‐1/PD‐L1 inhibitor resistance in NSCLC. 18 Traditionally, STK11 alterations in breast cancer, observed at 1%–2% in this study, have been associated with Peutz‐Jeghers syndrome and have not, to date, been linked to resistance to ICPI treatments.
Finally, a single study analyzed the molecular profiles of 102 878 diverse cancer types and found that MDM2 amplification occurred in 3.5% of the patients. 46 Hyperprogression occurs in 10–30% of cancers treated with immunotherapy and has been associated with MDM2 amplification. Reports have shown significant correlation with failure of anti‐PD‐1/PD‐L1 agents and MDM2 amplification, but few of these have been specific to breast cancer. 46 , 50 More studies that analyze response to ICPIs in MBC of various types are needed to confirm if MDM2 amplification is a predictor of resistance and hyperprogression in MBC patients treated with ICPI agents.
Both our results and published literature suggest that in addition to guiding targeted therapy selection, CGP shows potential to identify GA linked to response and resistance to ICPI therapy in MBC. The demonstration of clinical benefit of immune checkpoint blockade in MBC supports the need for the development of biomarkers used to guide the use of ICPI drugs for these patients.
CONFLICT OF INTEREST
Abirami Sivapiragasam: No conflict to disclose. P. Ashok Kumar: No conflict to disclose. Ethan S. Sokol: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd. Lee A. Albacker: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd, J. Keith Killian: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd. Shakti Ramkissoon: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd. Richard Huang: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd. Patents with VENTANA (Roche). Eric A. Severson: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd. Natalie Danziger: Employment by Foundation Medicine Inc. Charlotte A. Brown: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd. Kimberly McGregor: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd. Jeffrey S. Ross: Employment by Foundation Medicine Inc. Stock in F. Hoffman La Roche Ltd.
Supporting information
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGMENTS
The authors thank Cui Guo for providing statistical analysis support.
Sivapiragasam A, Ashok Kumar P, Sokol ES, et al. Predictive Biomarkers for Immune Checkpoint Inhibitors in Metastatic Breast Cancer. Cancer Med. 2021;10:53–61. 10.1002/cam4.3550
Funding information
This study was supported by Foundation Medicine, a member of the Roche Group.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the supplementary material of this article.
REFERENCES
- 1. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pfeifer GP. Environmental exposures and mutational patterns of cancer genomes. Genome Med. 2010;2(8):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA‐approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29‐39. [DOI] [PubMed] [Google Scholar]
- 4. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winer A, Bodor JN, Borghaei H. Identifying and managing the adverse effects of immune checkpoint blockade. J Thorac Dis. 2018;10(suppl 3):S480‐S489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oliva M, Spreafico A, Taberna M, et al. Immune biomarkers of response to immune‐checkpoint inhibitors in head and neck squamous cell carcinoma. Ann Oncol. 2019;30(1):57‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366(26):2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kitano S, Nakayama T, Yamashita M. Biomarkers for immune checkpoint inhibitors in melanoma. Front Oncol. 2018;8:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gopalakrishnan D, Koshkin VS, Ornstein MC, Papatsoris A, Grivas P. Immune checkpoint inhibitors in urothelial cancer: recent updates and future outlook. Ther Clin Risk Manag. 2018;14:1019‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reck M, Rodríguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375(19):1823‐1833. [DOI] [PubMed] [Google Scholar]
- 11. Chang L, Chang M, Chang HM, Chang F. Microsatellite instability: a predictive biomarker for cancer immunotherapy. Appl Immunohistochem Mol Morphol. 2018;26(2):e15‐e21. [DOI] [PubMed] [Google Scholar]
- 12. Yarchoan M, Johnson BA III, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(9):569. [DOI] [PubMed] [Google Scholar]
- 13. Castle JC, Uduman M, Pabla S, Stein RB, Buell JS. Mutation‐derived neoantigens for cancer immunotherapy. Front Immunol. 2019;10:1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30(1):44‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marabelle A, Fakih MG, Lopez J, et al. Association of tumor mutational burden with outcomes in patients with select advanced solid tumors treated with pembrolizumab in KEYNOTE‐158. Ann Oncol. 2020;21:1353‐1365. [DOI] [PubMed] [Google Scholar]
- 16. Goodman AM, Piccioni D, Kato S, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018;4(9):1237‐1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miao D, Margolis CA, Gao W, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359(6377):801‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD‐1 inhibitor resistance in KRAS‐mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barroso‐Sousa R, Keenan TE, Pernas S, et al. Tumor mutational burden and PTEN alterations as molecular correlates of response to PD‐1/L1 blockade in metastatic triple‐negative breast cancer. Clin Cancer Res. 2020;26(11):2565‐2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adashek JJ, Subbiah IM, Matos I, et al. Hyperprogression and immunotherapy: fact, fiction, or alternative fact? Trends Cancer. 2020;6(3):181‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swoboda A, Nanda R. Immune checkpoint blockade for breast cancer. Cancer Treat Res. 2018;173:155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Solinas C, Gombos A, Latifyan S, Piccart‐Gebhart M, Kok M, Buisseret L. Targeting immune checkpoints in breast cancer: an update of early results. ESMO Open. 2017;2(5):e000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. U.S. Food & Drug Administration . FDA approves atezolizumab for PD‐L1 positive unresectable locally advanced or metastatic triple‐negative breast cancer. 2019. http://www.fda.gov/drugs/drug‐approvals‐and‐databases/fda‐approves‐atezolizumab‐pd‐l1‐positive‐unresectable‐locally‐advanced‐or‐metastatic‐triple‐negative [Accessed June 20, 2020]. [DOI] [PubMed]
- 24. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trabucco SE, Gowen K, Maund SL, et al. A novel next‐generation sequencing approach to detecting microsatellite instability and pan‐tumor characterization of 1000 microsatellite instability‐high cases in 67,000 patient samples. J Mol Diagn. 2019;21(6):1053‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nanda R, Chow LQM, Dees EC, et al. Pembrolizumab in patients with advanced triple‐negative breast cancer: phase Ib KEYNOTE‐012 study. J Clin Oncol. 2016;34(21):2460‐2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmid P, Dent R, O'Shaughnessy J. Pembrolizumab for Early triple‐negative breast cancer. Reply. N Engl J Med. 2020;382(26):e108. [DOI] [PubMed] [Google Scholar]
- 29. Gennari R, Menard S, Fagnoni F, et al. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin Cancer Res. 2004;10(17):5650‐5655. [DOI] [PubMed] [Google Scholar]
- 30. Winer EP, Dang T, Karantza V, Su S. KEYNOTE‐119: a randomized phase III study of single‐agent pembrolizumab (MK‐3475) vs single‐agent chemotherapy per physician's choice for metastatic triple‐negative breast cancer (mTNBC). J Clin Oncol. 2016;34(15_suppl):TPS1102‐TPS1102. [Google Scholar]
- 31. Cortés J, Lipatov O, Im S‐A, et al. LBA21 ‐ KEYNOTE‐119: phase III study of pembrolizumab (pembro) versus single‐agent chemotherapy (chemo) for metastatic triple negative breast cancer (mTNBC). Ann Oncol. 2019;30:v859‐v860. [Google Scholar]
- 32. Rosenberg JE, Hoffman‐Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang M, Sun H, Zhao S, et al. Expression of PD‐L1 and prognosis in breast cancer: a meta‐analysis. Oncotarget. 2017;8(19):31347‐31354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med. 2018;379(22):2108‐2121. [DOI] [PubMed] [Google Scholar]
- 35. Marra A, Viale G, Curigliano G. Recent advances in triple negative breast cancer: the immunotherapy era. BMC Med. 2019;17(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schalper KA, Velcheti V, Carvajal D, et al. In situ tumor PD‐L1 mRNA expression is associated with increased TILs and better outcome in breast carcinomas. Clin Cancer Res. 2014;20(10):2773‐2782. [DOI] [PubMed] [Google Scholar]
- 37. Lebok P, Kopperschmidt V, Kluth M, et al. Partial PTEN deletion is linked to poor prognosis in breast cancer. BMC Cancer. 2015;15:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Horton BL, Williams JB, Cabanov A, Spranger S, Gajewski TF. Intratumoral CD8(+) T‐cell apoptosis is a major component of T‐cell dysfunction and impedes antitumor immunity. Cancer Immunol Res. 2018;6(1):14‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell‐mediated immunotherapy. Cancer Discov. 2016;6(2):202‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao J, Chen AX, Gartrell RD, et al. Immune and genomic correlates of response to anti‐PD‐1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mittendorf EA, Philips AV, Meric‐Bernstam F, et al. PD‐L1 expression in triple‐negative breast cancer. Cancer Immunol Res. 2014;2(4):361‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barrett MT, Lenkiewicz E, Malasi S, et al. The association of genomic lesions and PD‐1/PD‐L1 expression in resected triple‐negative breast cancers. Breast Cancer Res. 2018;20(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen M, Pockaj B, Andreozzi M, et al. JAK2 and PD‐L1 amplification enhance the dynamic expression of PD‐L1 in triple‐negative breast cancer. Clin Breast Cancer. 2018;18(5):e1205‐e1215. [DOI] [PubMed] [Google Scholar]
- 44. Bonneville R, Krook MA, Kautto EA, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. 2017;2017:PO.17.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Melendez B, Van Campenhout C, Rorive S, Remmelink M, Salmon I, D'Haene N. Methods of measurement for tumor mutational burden in tumor tissue. Transl Lung Cancer Res. 2018;7(6):661‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kato S, Ross JS, Gay L, et al. Analysis of MDM2 amplification: next‐generation sequencing of patients with diverse malignancies. JCO Precis Oncol. 2018;2018:10.1200/PO.17.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Merino DM, McShane LM, Fabrizio D, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020;8(1):e000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thomas A, Routh ED, Pullikuth A, et al. Tumor mutational burden is a determinant of immune‐mediated survival in breast cancer. Oncoimmunology. 2018;7(10):e1490854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu J, Guo X, Jing M, Sun T. Prediction of tumor mutation burden in breast cancer based on the expression of ER, PR, HER‐2, and Ki‐67. Onco Targets Ther. 2018;11:2269‐2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ju W, Chen S, Wang G, Cai S, Xiong J, Xiang X. Association between MDM2/MDM4 amplification and PD‐1/PD‐L1 inhibitors‐related hyperprogressive disease: a pan‐cancer analysis. J Clin Oncol. 2019;37(15_suppl):2557. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.