

Abstract
COVID-19 has emerged as a rapidly escalating serious global health issue, affecting every section of population in a detrimental way. Present situation invigorated researchers to look for potent targets, development as well as repurposing of conventional therapeutic drugs. NSP12, a RNA polymerase, is key player in viral RNA replication and, hence, viral multiplication. In our study, we have screened a battery of FDA-approved drugs against SARS-CoV-2 RNA polymerase using in silico molecular docking approach. Identification of potent inhibitors against SARS-CoV-2 NSP12 (RNA polymerase) were screeened from FDA approved drugs by virtual screening for therapeutic applications in treatment of COVID-19. In this study, virtual screening of 1749 antiviral drugs was executed using AutoDock Vina in PyRx software. Binding affinities between NSP12 and drug molecules were determined using Ligplot+ and PyMOL was used for visualization of docking between interacting residues. Screening of 1749 compounds resulted in 14 compounds that rendered high binding affinity for NSP12 target molecule. Out of 14 compounds, 5 compounds which include 3a (Paritaprevir), 3d (Glecaprevir), 3h (Velpatasvir), 3j (Remdesivir) and 3l (Ribavirin) had a binding affinity of − 10.2 kcal/mol, −9.6 kcal/mol, − 8.5 kcal/mol, − 8.0 kcal/mol and − 6.8 kcal/mol, respectively. Moreover, a number of hydrophobic interactions and hydrogen bonding between these 5 compounds and NSP12 active site were observed. Further, 3l (Ribavirin) was docked with 6M71 and molecular dynamic simulation of the complex was also performed to check the stability of the conformation. In silico analysis postulated the potential of conventional antiviral drugs in treatment of COVID-19. However, these finding may be further supported by experimental data for its possible clinical application in present scenario.
Keywords: NSP12, Antiviral drugs, Drug targets, Molecular docking, RdRp, Drug repurposing
Introduction
COVID-19 (coronavirus disease) emerged as grave health threat which started from Wuhan (China) in early December 2019 and rapidly spread across the world in few weeks (Loeffelholz and Tang 2020; Bogoch et al. 2020). As of, 19 January 2021 over 93 million cases and 2 million deaths have been reported globally, with over 4.7 million new cases and nearly 93,000 new deaths reported over the past week. (WHO 2021). SARS-CoV-2 belongs to Coronaviridae family, which causes infection in birds, mammals and humans (Rodriguez-Morales et al. 2020). In the last two decades, Coronaviruses were responsible for three huge outbreaks, which includes the SARS epidemic (2002–03) (Drosten et al. 2003), the MERS epidemic (2012) (Zaki et al. 2012) and the present COVID-19 outbreak (Chan et al. 2020).
SARS-CoV-2 is highly similar to other coronaviruses in terms of their genomic organization, which consist of single-stranded RNA as their genetic material (Mousavizadeh and Ghasemi 2020). Proteins of Human coronaviruses are characterized into structural proteins and non-structural proteins. Structural proteins are further classified as S (spike), E (envelope), M (membrane), and N (nucleocapsid) proteins; whereas, non-structural proteins are categorized as proteases (NSP3 and NSP5) and RNA-dependent RNA polymerase (RdRp, also called as NSP12) (Elfiky 2017; Wu et al. 2020). NSP12 catalyzes the viral RNA synthesis and, thus, plays a central role in viral replication as well as multiplication process, along with NSP7 and NSP8 proteins as co-factors (Gao et al. 2020). NSP12 plays an important role in viral replication and has been targeted for treatment of several viral infections, including Hepatitis C, Zika and other Coronaviruses (Ganesan and Barakat 2017; Elfiky 2019, 2020; Elfiky and Elshemey 2018; Hui et al. 2020).
Till date, no specific treatment against SARS-CoV-2 has been reported despite of ardent efforts. Newer strategies for development or repurposing of conventional drugs are dire need for the present grave situation. Number of FDA approved antiviral drugs could have potential therapeutic options for SARS-CoV-2 infection. Also, a number of FDA-approved drugs are under investigation in randomized controlled clinical trials, including remdesivir (GS-5734) (Li et al. 2020; Gordon et al. 2020). Nowadays, computational biology and molecular simulation studies open new possibilities of drug screening and may aid in the development of COVID-19 treatment (Dong et al. 2020).
In this study, virtual screening of 1749 antiviral drugs was performed with NSP12 protein template by molecular docking and molecular dynamics simulation approach. The interactions of drug and protein moiety were also studied to understand the molecular interaction between drugs and proteins. The results have been found to be promising and indicate potential antiviral activity of selected drugs against SARS-CoV-2, which can be further validated through experimental settings.
Materials and methods
Datasets and sequence alignment
The sequences corresponding to SARS-CoV nsp12 and SARS-CoV-2 nsp12 were retrieved from the NCBI GeneBank database. The related structural information of the SARS-CoV (PDB ID: 6NUR) and SARS-CoV-2 (PDB ID: 6M71) were collected from RCSB database (https://www.rcsb.org/). Pairwise sequence comparisons of SARS-CoV-2 nsp12 and SARS-CoVnsp12 were performed using Clustal Omega webserver. The nsp12 protein from SARS-CoV exhibits 97% identity with SARS-CoV-2 nsp12.
Pocket detection and selection
For determining the specific functional part of protein, active site prediction is necessary. Potential active site of the target protein SARS-Cov-2 RNA-dependent RNA polymerase (6M71) was determined using PrankWeb (Jendele et al. 2019).
Ligand selection and preparation
FDA-approved drugs dataset from Zinc15 library (Sterling and Irwin 2015) and antiviral agents (Accession Number: DBCAT000066) from DrugBank (Wishart et al., 2018) were retrieved for virtual screening. These molecules were downloaded in Structure Data File (SDF) format. Open Babel (http://openbabel.org) tool was used to convert various file formats. PyRx (Dallakyan and Olson 2015) was initially used to minimize compounds energy and convert all molecules to AutoDock Ligand (PDBQT) format.
Virtual screening
Virtual screening was executed using the published crystal structure of SARS-CoV-2 nsp12 (PDB ID: 6M71). AutoDock Vina (Trott and Olson 2010) in PyRx 0.8 was used to perform virtual screening. The compounds without any specified binding sites were docked against whole surface of the protein. The value of the grid box was set to center_x = 121.8, center_y = 123.5, center_z = 127.0 while size_x = 74.8, size_y = 84.5, and size_z = 106.0 with the default exhaustiveness value of 8. The outcomes of docking results were reported in the form of binding energy. LigPlot+ program was used for the analysis of post-docking results (Laskowski and Swindells 2011). Using PyMOL, the docked complexes with lowest binding affinity values were further analyzed for hydrogen and hydrophobic bond interaction analysis. The resulting ligands have high potential to be used as drug candidates.
Molecular features analyses
The ADME and drug-likeness predictions of compounds were carried out using SwissADME (http://www.swissadme.ch/) (Daina et al. 2017). The SMILES of compounds have been used in SwissADME web tool as input. Further, ADMET and the pharmacokinetic properties were evaluated using admetSAR (http://lmmd.ecust.edu.cn/admetsar2) web server to ensure the druggability potential of compounds (Yang et al. 2019).
Molecular docking and simulation
The structure of SARS-Cov-2 RNA-dependent RNA polymerase (PDB_ID: 6M71) was prepared by automated protein preparation protocol in Discovery Studio 4.1 (DS). Pre-processing of the selected structure in terms of fixing the missing atoms in the incomplete residues, deletion of co-crystallized H2O molecules, modeling of loops and protonation of selected residues were performed by applying CHARM force-filed. Side-by-side, the ligand Ribavirin (DB00811), a synthetic guanosine analog broad-spectrum antiviral effective against a number of RNA viruses, was processed using ‘Prepare Ligands’ protocol in DS 4.1 to generate protonation state, and have molecular geometry for docking and simulation studies.
Molecular docking and molecular dynamics (MD) analysis of these pre-processed molecules were performed by ‘define and edit binding site’ tool to specify the active site. Ligand molecule was allowed to dock into the active site of 6M71 structure using CDOCKER tool of Discovery studio. Selection of the poses was done on the basis of interaction energy of these conformations. To check the stability of the docked complex, molecular dynamics simulation was performed for the period of 100 ns using academic version of “Desmond” program. An orthorhombic water box shape of 10 × 10 × 10 Å3 dimensions was used as the solvent system using TIP3P. Neutralization of the system was done by adding Na + ions and 0.15 M salt concentration. Molecular dynamics simulations using NPT ensemble was performed with the temperature at 310 K and pressure at 1 bar using Nose–Hoover Chain as thermostat and Martyna–Tobias–Klein as barostat, respectively. The simulations were run for a period of 100 ns with time step of 1.0 femtoseconds (fs).
Results and discussion
In Coronaviridae, the protein subunit containing the domain RdRp is recognized as non-structural protein NSP12. NSP12 is one of the most powerful enzymes essential to genome replication and transcription success of RNA viruses (Venkataraman et al. 2018). SARS-CoV-2 nsp12 is nearly similar to SARS-CoV (97% identity and 98% similarity) (Fig. 1). Most of the residues which were not identical have similar properties (strongly or weakly) and also overall architecture of nsp12–nsp7–nsp8 complex in both are almost similar.
Fig. 1.

Alignment of the amino acid sequences of SARS-Cov-2 and SARS-Cov NSP12 ‘*’ fully conserved residues; ‘:’ strongly similar properties; ‘.’ weakly similar properties
The SARS-CoV-2 nsp12 structure includes a right-hand RdRp domain, and an N-terminal extension domain that is unique to nidovirus (Lehmann et al. 2015). Architecture of polymerase domain is conserved in viral polymerase family (McDonald 2013) and it contains three subdomains; a finger (L366-A581 and K621-G679), a palm (residues T582–P620 and T680–Q815) and a thumb sub-domain (H816–E920) (Fig. 2). The active site is established in the palm domain by conserved polymerase motifs (A–G) (Gao et al. 2020).
Fig. 2.

Sequence of SARS-CoV-2 NSP12 showing different domains
Virtual screening (VS) has undeniably changed and enhanced the process of drug discovery, and has been considered as one of the most promising drug design techniques for identifying hit molecules as starting points in medicinal chemistry (Lavecchia et al. 2013; Lionta et al. 2014). Recently in various research, FDA-approved drugs were repurposed against SARS-CoV-2 (Chandel et al. 2020a, 2020b). Compounds were downloaded in Structure Data File (SDF) format either from ZINC database or DrugBank. Energy of each compound was minimized and all converted to (PDBQT) format for virtual screening. Virtual screening was executed using the published electron microscopic structure of SARS-CoV-2 NSP12 PDB ID: 6M71 (Gao et al. 2020) with a resolution 2.90 Å. Table 1 describes the docking results which have been shown to be strong binding affinities. We studied the interactions between SARS-CoV-2 NSP12 and these strongest inhibitors. The graphical representation of the 14 best screened compounds is depicted in the pictorial form in Fig. 3 (Fig. 3).
Table 1.
Docking results of different poses showing best binding affinities
| Compounds | Binding affinity (kcal/mol) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pose 1 | Pose 2 | Pose 3 | Pose 4 | Pose 5 | Pose 6 | Pose 7 | Pose 8 | Pose 9 | |
| CID 45110509 | − 10.2 | − 10 | − 9.9 | − 9.7 | − 9.7 | − 9.6 | − 9.6 | − 9.5 | − 9.5 |
| CID 71171 | − 10.1 | − 9.6 | − 9.5 | − 9.5 | − 9.4 | − 9.2 | − 9.2 | − 9.1 | − 9 |
| CID 8223 | − 10.1 | − 9.4 | − 9.4 | − 9.2 | − 9.2 | − 9.1 | − 8.9 | − 8.9 | − 8.6 |
| CID 66828839 | − 9.6 | − 9.3 | − 9 | − 9 | − 8.9 | − 8.7 | − 8.4 | − 8.4 | − 8.3 |
| CID 24873435 | − 9.4 | − 9.1 | − 8.8 | − 8.7 | − 8.6 | − 8.6 | − 8.6 | − 8.6 | − 8.5 |
| CID 60152109 | − 9.4 | − 9 | − 8.7 | − 8.4 | − 8.4 | − 8.3 | − 8.3 | − 8.1 | − 8.1 |
| CID 11167602 | − 9.2 | − 9.1 | − 8.4 | − 8.3 | − 8.2 | − 8.2 | − 8.1 | − 8 | − 7.8 |
| CID 91936863 | − 9.2 | − 8.6 | − 8.6 | − 8.2 | − 8.2 | − 8.2 | − 8.1 | − 8.1 | − 7.9 |
| CID 57379345 | − 9.2 | − 8.4 | − 8.4 | − 8.3 | − 8.1 | − 8.1 | − 8 | − 7.9 | − 7.9 |
| CID 67683363 | − 8.5 | − 8.4 | − 8.3 | − 8.1 | − 8.1 | − 8 | − 8 | − 8 | − 7.8 |
| CID 54671008 | − 8.2 | − 8.1 | − 8.1 | − 8.1 | − 8 | − 7.8 | − 7.8 | − 7.8 | − 7.8 |
| CID 121304016 | − 8 | − 8 | − 7.7 | − 7.3 | − 7.1 | − 7.1 | − 7 | − 6.7 | − 6.7 |
| CID 9853053 | − 8 | − 7.8 | − 7.6 | − 7.5 | − 7.3 | − 7.3 | − 7.2 | − 7.2 | − 7.1 |
| CID 45375808 | − 7.7 | − 7.2 | − 7.1 | − 7.1 | − 6.8 | − 6.8 | − 6.7 | − 6.7 | − 6.7 |
| CID 37542 | − 6.8 | − 6.2 | − 6.2 | − 6 | − 6 | − 6 | − 6 | − 6 | − 5.9 |
| CID 10445549 | − 6.4 | − 6.4 | − 6.3 | − 6.3 | − 6.2 | − 6.2 | − 6.1 | − 6 | − 6 |
| CID 464205 | − 6.4 | − 6.3 | − 6.1 | − 6 | − 6 | − 6 | − 5.9 | − 5.9 | − 5.9 |
Fig. 3.

Graphical representations of the best screened compounds
Compound 3a (CID 45110509), compound 3d (CID 66828839), compound 3 h (CID 67683363), compound 3j (CID 121304016) and compound 3 l (CID 37542) exhibit hydrogen bond interactions in the palm sub-domain which contain active site residues shown in Table 2. Compound 3b, compound 3c, compound 3e, compound 3f, compound 3 g, compound 3i and compound 3 k were showing good binding affinity, hydrogen bond and hydrophobic interaction but not in the active site containing palm sub-domain.
Table 2.
Hydrogen and hydrophobic bond interaction between some ligands and the receptor
| # | Compounds | Binding affinity (kcal/mol) | Hydrogen bonds and its length | Hydrophobic bond |
|---|---|---|---|---|
| 3a | CID 45110509 | − 10.2 | Arg553A (2.80, 2.92, 3.28), Asp623A (3.08), Thr687A (3.08), Ser759A (3.33) | Tyr455, Tyr619, Pro620, Lys621, Cys622, Ser681, Ser682, Ala688, Asn691, Asp760 |
| 3b | CID 71171 | − 10.1 | Phe321A (3.28) | Arg249, Ser255, Ile266, Trp268, Thr319, Val320, Pro322, Pro323, Arg349, Pro461 |
| 3c | CID 8223 | − 10.1 | Arg553A (2.98), Lys621A (2.84), Cys622A (2.91), Asp623A (2.71, 3.22) | Tyr455, Lys551, Asp618, Tyr619, Pro620, Asp760, Lys798 |
| 3d | CID 66828839 | − 9.6 | Arg553A (3.06), Arg555A (3.19), Tyr619A (2.86), Cys622A (2.80) | Thr556, Asp618, Pro620, Lys621, Asp623, Ser682, Thr687, Asp760, Asp761 |
| 3e | CID 60152109 | − 9.4 | Thr206A (2.93), Asn209A (2.88) | Phe35, Asp36, Ile37, Lys50, Arg116, Thr120, Lys121, Val71, Asp208, Tyr217, Asp218, Asp221 |
| 3f | CID 11167602 | − 9.2 | Asp36A (3.27), Asn209A (2.97) | Phe35, Ile37, Tyr38, Val204, Thr206, Asp208, Tyr217, Asp218, Asp221, Tyr728 |
| 3 g | CID 57379345 | − 9.2 | Asp208A (3.12) | Arg33, Ala34, Phe35, Ile37, Thr120, Lys121, Thr123, Asp126, Val71, Thr206, Asn209, Tyr217, Asp218, Asp221 |
| 3 h | CID 67683363 | − 8.5 | Arg33A (3.28), Tyr129A (2.89), Thr141A (3.10), Lys780A (3.23) | Val31, Tyr32, Lys47, Tyr122, Asn138, Asp140, Ser709, Thr710, Gly774, Asn781 |
| 3i | CID 54671008 | − 8.2 | Tyr32A (2.79), Arg33A (3.14), Asp126A (3.07), Asp140A (2.86, 3.01), Thr141A (2.81, 3.09) | Lys47, Tyr122, Tyr129, Ala130, His133, Asp135, Asn138, Cys139 |
| 3j | CID 121304016 | − 8 | Lys47A (2.85), Tyr129A (3.05, 3.31), Ser709A (2.85), Asp711A (2.93), Asn781A (3.15) | Tyr32, Ala46, His133, Asp135, Thr710, Gln773, Gly774, Lys780, Ser784 |
| 3 k | CID 45375808 | − 7.7 | Ser255A (3.00) | Arg249, Thr252, Tyr265, Val315, Thr319, Arg349, Cys395, Pro461, Val675, Pro677 |
| 3 l | CID 37542 | − 6.8 | Trp617A (2.86), Asp761A (2.74, 2.77), Lys798A (3.28), His810A (3.18), Glu811A (2.95) | Trp800, Phe812, Cys813 |
| 3 m | CID 10445549 | − 6.4 | Glu23A (3.18, 3.23), Ala162A (2.82), Ser164A (2.85), Asn404A (2.84, 2.90) | Ser26, Asp163, Val405, Ala406, Phe407, Thr409 |
| 3n | CID 464205 | − 6.4 | Ser384A (3.04, 3.22), Asn386A (2.91) | Thr93, Lys97, Met94, His381, Gly385, Leu401, Thr402 |
The drug-likeness predictions of 14 compounds were carried out using SwissADME. Lipinski 's rule of 5 states that a molecule should meet the following conditions for any ligand to be considered drug-like: molecular weight < 500 Dalton, number of donors of H-bonds < 5, number of acceptors of H-bonds < 10 and LogP < 5 (Lipinski 2004). Out of 14 FDA-approved compounds, only 3 (3f, 3 l and 3n) followed the Lipinski’s rule of five and are provided in Table 3.
Table 3.
Drug-likeness prediction of selected inhibitors of Nsp12
| # | Molecular formula | Molecular weight | H− bond acceptors | H− bond donors | TPSA (Topological surface area) | iLOGP | Lipinski violations |
|---|---|---|---|---|---|---|---|
| 3a | C40H43N7O7S | 765.88 | 10 | 3 | 198.03 | 2.07 | 2 |
| 3b | C34H41N5O8S | 679.78 | 9 | 4 | 180.96 | 0.82 | 2 |
| 3c | C33H35N5O5 | 581.66 | 6 | 3 | 118.21 | 3.28 | 1 |
| 3d | C38H46F4N6O9S | 838.87 | 15 | 3 | 203.6 | 4.07 | 2 |
| 3e | C42H62N2O4S | 691.02 | 6 | 2 | 95.09 | 5.23 | 2 |
| 3f | C21H15ClF4N4O3 | 482.82 | 8 | 3 | 92.35 | 3.51 | 0 |
| 3 g | C28H36ClN5O3S | 558.14 | 6 | 3 | 113.62 | 4.96 | 1 |
| 3 h | C49H54N8O8 | 883 | 10 | 4 | 193.1 | 5.14 | 2 |
| 3i | C20H21FN6O5 | 444.42 | 9 | 3 | 152.24 | 2.71 | 1 |
| 3j | C27H35N6O8P | 602.58 | 12 | 4 | 213.36 | 3.24 | 2 |
| 3 k | C22H29FN3O9P | 529.45 | 11 | 3 | 167.99 | 3.23 | 2 |
| 3 l | C8H12N4O5 | 244.2 | 7 | 4 | 143.72 | 0.13 | 0 |
| 3 m | C11H15N5O3 | 265.27 | 6 | 6 | 140.31 | 0.33 | 1 |
| 3n | C9H14N5O4P | 287.21 | 7 | 3 | 146.19 | 0.41 | 0 |
Further, all these 14 best docked ligands were further subjected to admetSAR for evaluation of ADMET properties (Table 4). Blood–Brain Barrier penetration was shown by all except 3c. Hepatotoxicity was only shown by 3e. The acute oral toxicity for the all compounds was estimated as class III. All compounds demonstrated no Caco-2 permeability. Similarly, intestinal absorption (human) was observed in all compounds. Honey bee toxicity was detected in 3b, 3c, 3e and 3 h compounds. Most of the ligands were CYP3A4 substrate except 3f, 3 k, 3 l, 3 m and 3n compounds, which were non-substrates. Only four ligands (3i, 3 l, 3 m and 3n) were non-inhibitors of CYP3A4, while rest were inhibitors. The water solubility of most of the docked compounds is greater than −3.
Table 4.
ADMET properties of potential drug candidates
| 3a | 3b | 3c | 3d | 3e | 3f | 3 g | 3 h | 3i | 3j | 3 k | 3 l | 3 m | 3n | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ames mutagenesis | − | − | − | − | − | − | − | − | − | − | − | − | − | + |
| Acute oral toxicity (c) | III | III | III | III | III | II | III | III | III | III | III | III | III | III |
| Blood Brain Barrier | + | + | − | + | + | + | + | + | + | + | + | + | + | + |
| Caco-2 | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CYP3A4 inhibition | + | − | + | + | + | − | + | + | − | − | + | − | − | − |
| CYP3A4 substrate | + | + | + | + | + | + | + | + | + | + | + | − | − | − |
| Fish aquatic toxicity | + | + | + | + | + | + | + | + | + | + | + | − | − | − |
| Honey bee toxicity | − | + | + | − | + | − | − | + | − | − | − | − | − | − |
| Hepatotoxicity | + | + | + | + | − | + | + | + | + | + | + | + | + | + |
| Human Intestinal Absorption | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| P-glycoprotein inhibitor | + | + | + | + | + | + | + | + | − | + | + | − | − | − |
| P-glycoprotein substrate | + | + | + | + | + | − | + | + | + | + | − | − | − | − |
| Subcellular localization | M | PM | N | L | L | M | L | M | M | L | M | M | N | L |
| Tetrahymena pyriformis | 1.74 | 1.28 | 1.69 | 2.10 | 0.99 | 1.06 | 1.53 | 1.25 | 1.42 | 1.64 | 0.25 | 0.03 | 0.69 | − 0.69 |
| Water solubility | − 3.69 | − 3.34 | − 2.68 | − 3.84 | − 3.50 | − 4.58 | − 3.90 | − 3.73 | − 2.88 | − 3.47 | − 3.75 | − 1.17 | − 1.79 | − 2.97 |
+ represents the presence, + represents the absence, PM Plasma membrane, M Mitochondria, L Lysosome
Compound 3a (Paritaprevir) is an antiviral drug used to treat chronic hepatitis C as part of the combination treatment (Wilkins et al. 2015). This drug was also studied against nsp 15 protein by Khan et al. (2020) and against nsp13 helicase and nsp14 of SARS-CoV-2 by Gurung (2020). The compound 3a showed the highest binding affinity (− 10.2 kcal/mol) and had hydrogen bonds with amino acids Arg553, Asp623, Thr687, Ser759; whereas hydrophobic interaction with Tyr455, Tyr619, Pro620, Lys621, Cys622, Ser681, Ser682, Ala688, Asn691 and Asp760 residues (Fig. 4a, b).
Fig. 4.

Molecular docking interactions between compound 3a and SARS-CoV-2 NSP12. a 2D model of the interactions between 3a and SARS-CoV-2 NSP12; b 3D model of the interactions between 3a and SARS-CoV-2 NSP12
Compound 3d (Glecaprevir) has been reported to have the binding affinity (− 10.1 kcal/mol) to the receptor via hydrogen bond interaction with Arg553, Arg555, Tyr619, Cys622; while, the hydrophobic interaction was with the residues Thr556, Asp618, Pro620, Lys621, Asp623, Ser682, Thr687, Asp760 and Asp761 (Fig. 5a, b). Glecaprevir is an antiviral agent and NS3/4A protease inhibitor of the Hepatitis C virus (HCV) that inhibits the replication of viral RNA (Zeuzem et al. 2018). Glecaprevir is a useful therapy in conjunction with Pibrentasvir for patients who have encountered clinical failure from other inhibitors of the NS3/4A protease (Zeuzem et al. 2018). Glecaprevir was also investigated for the treatment of COVID-19 infection (Mahdian et al. 2020; Chtita et al. 2020).
Fig. 5.

Molecular docking interactions between compound 3d and SARS-CoV-2 NSP12. a 2D model of the interactions between 3d and SARS-CoV-2 NSP12; b 3D model of the interactions between 3d and SARS-CoV-2 NSP12
Compound 3 h (Velpatasvir), a NS5A inhibitor is a part of combination therapy to treat chronic hepatitis C (Heo and Deeks 2018). With limited side effects, usually fatigue and headache, Velpatasvir may be attractive as a therapeutic to fight the new coronavirus (Chen et al. 2020). It was reported that velpatasvir has the binding affinity (− 8.5 kcal/mol) and bound to the receptor through hydrogen bond interaction with Arg33, Tyr129, Thr141, Lys780; whereas, the hydrophobic interaction was with Val31, Tyr32, Lys47, Tyr122, Asn138, Asp140, Ser709, Thr710, Gly774 and Asn781 residues (Fig. 6a, b).
Fig. 6.

Molecular docking interactions between compound 3 h and SARS-CoV-2 NSP12. a 2D model of the interactions between 3 h and SARS-CoV-2 NSP12; b 3D model of the interactions between 3 h and SARS-CoV-2 NSP12
Compound 3j (Remdesivir) is a promising candidate based on existing evidence, and clinical trials of remdesivir are now underway among adults with COVID-19 in hospitals (NIH 2020). Remdesivir is a direct-acting antiviral agent used for the treatment of serious 2019 coronavirus disease (COVID-19) patients (Tchesnokov et al. 2020; Baby et al. 2020). It showed a mixed outcome with a reasonable side effect in COVID-19 patients (Singh et al. 2020). It was found that Remdesivir has the binding affinity (− 8.0 kcal/mol), and residues Lys47, Tyr129, Ser709, Asp711 and Asn781 were connected with hydrogen bonds; while, residues Tyr32, Ala46, His133, Asp135, Thr710, Gln773, Gly774, Lys780 and Ser784 had hydrophobic interactions (Fig. 7a, b).
Fig. 7.

Molecular docking interactions between compound 3j and SARS-CoV-2 NSP12. a 2D model of the interactions between 3j and SARS-CoV-2 NSP12; b 3D model of the interactions between 3j and SARS-CoV-2 NSP12
Compound 3 l (Ribavirin) is a synthetic guanosine analog broad-spectrum antiviral effective against a number of RNA viruses (Galli et al. 2018). Our results of molecular docking and molecular dynamic simulation have confirmed that the Ribavirin have best interactions with 6M71 and also showed energetically stable conformations along with best druggability properties (Fig. 8). In silico screening, molecular mechanics, and molecular dynamics simulation (MDS) research indicate that at the receptor-binding domain that recognizes hACE-2 (RBD–hACE2) interface, ribavirin has strong interaction (Tiwari et al. 2020). It has the binding affinity (− 6.8 kcal/mol) and formed 5 hydrogen bonds (Trp617, Asp761, Lys798, His810 and Glu811) and 3 hydrophobic contacts (Trp800, Phe812, Cys813) (Fig. 9a, b).
Fig. 8.
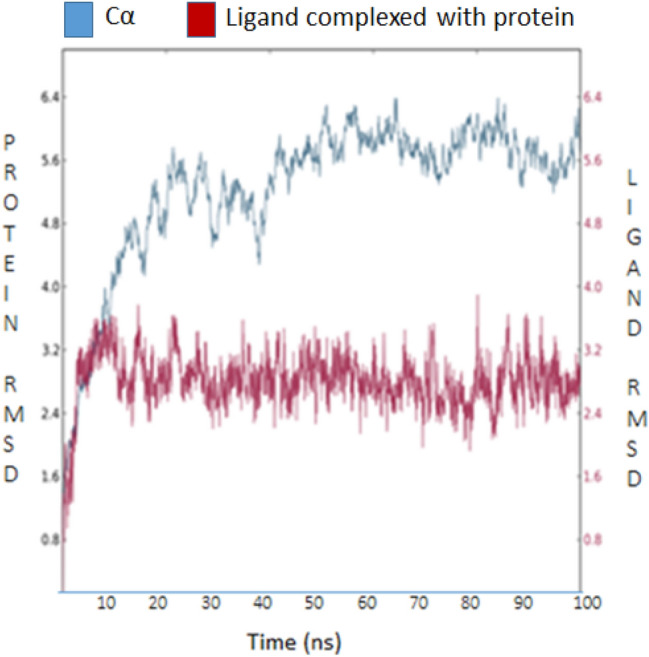
RMSD plots of Ribavirin (DB00811) for the time period of 100 ns (100,000 ps): in complex with SARS-CoV-2 RNA-dependent RNA Polymerase
Fig. 9.

Molecular docking interactions between compound 3 l and SARS-CoV-2 NSP12. a 2D model of the interactions between 3 l and SARS-CoV-2 NSP12; b 3D model of the interactions between 3 l and SARS-CoV-2 NSP12
Conclusions
Many studies are underway in establishing conventional antiviral drugs to counter SARS-CoV-2 infection. In a short time, a number of potential drug inhibitors have been screened and identified through docking-based virtual screening using various in silico approaches. In this study, docking studies were performed for NSP12 protein and 1615 FDA-approved antiviral compounds from ZINC15 database and 149 molecules of known antiviral activities. Out of 1764 screened compounds, 14 compounds exhibited strong binding affinities with NSP12 protein. Based on binding affinity, 5 compounds which include 3a (Paritaprevir), 3d (Glecaprevir), 3 h (Velpatasvir), 3j (Remdesivir) and 3 l (Ribavirin) were selected for further analysis. These five drugs showed high binding affinity, and also exhibited hydrophobic interactions and hydrogen bonding in the palm sub-domain of NSP12. 3 l (Ribavirin) was docked with 6M71 and molecular dynamic simulation of the complex was also performed to check the stability of the conformation. This study established the potential of conventional antiviral drugs in treatment of SARS-CoV-2 infection. The resulting compounds have high potential to be used as drug candidates. However, more extensive screening along with experimental results may also support the results of present finding and can open new future possibilities for drug discovery against SARS-CoV-2 infection.
Acknowledgements
Authors acknowledge Department of Biotechnology, Thapar Institute of Engineering & Technology, Patiala, India for providing resources to conduct this research.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
No ethical approval required.
Contributor Information
Sanjay Kumar Singh, Email: sanjaybiosoft@gmail.com.
Atul Kumar Upadhyay, Email: atul.upadhyay@thapar.edu.
M. Sudhakara Reddy, Email: msreddy@thapar.edu.
References
- Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y. Targeting SARS-CoV-2 main protease: a computational drug repurposing study. Arch Med Res. 2020 doi: 10.1016/j.arcmed.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoch II, Watts A, Thomas-Bachli A, Huber C, Kraemer MU, Khan K. Pneumonia of unknown etiology in Wuhan, China: potential for international spread via commercial air travel. J Travel Med. 2020 doi: 10.1093/jtm/taaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, Yuen KY. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel V, Sharma PP, Raj S, Choudhari R, Rathi B, Kumar D. Structure-based drug repurposing for targeting Nsp9 replicase and spike proteins of severe acute respiratory syndrome coronavirus 2. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1811773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel V, Raj S, Rathi B, Kumar D. In silico identification of potent FDA approved drugs against Coronavirus COVID-19 main protease: a drug repurposing approach. Chem Biol Lett. 2020;7(3):166–175. [Google Scholar]
- Chen YW, Yiu CB, Wong KY (2020) Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res 9:129. 10.12688/f1000research.22457.2. [DOI] [PMC free article] [PubMed]
- Chtita S, Belhassan A, Aouidate A, Belaidi S, Bouachrine M, Lakhlifi T. Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs via docking screening. Comb Chem High Throughput Screen. 2020 doi: 10.2174/1386207323999200730205447. [DOI] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Dong S, Sun J, Mao Z, Wang L, Lu YL, Li J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV) J Med Virol. 2020 doi: 10.1002/jmv.25768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C, Günther S, Preiser W, Van Der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Elfiky AA. Zika virus: novel guanosine derivatives revealed strong binding and possible inhibition of the polymerase. Fut Virol. 2017;12(12):721–728. doi: 10.2217/fvl-2017-0081. [DOI] [Google Scholar]
- Elfiky AA (2019) Novel guanosine derivatives as anti-HCV NS5b polymerase: a QSAR and molecular docking study. Med Chem 15(2):130–137. 10.2174/1573406414666181015152511. [DOI] [PubMed]
- Elfiky AA. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020;248:117477. doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky AA, Elshemey WM. Molecular dynamics simulation revealed binding of nucleotide inhibitors to ZIKV polymerase over 444 nanoseconds. J Med Virol. 2018;90(1):13–18. doi: 10.1002/jmv.24934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli A, Mens H, Gottwein JM, Gerstoft J, Bukh J. Antiviral effect of ribavirin against HCV associated with increased frequency of G-to-A and C-to-U transitions in infectious cell culture model. Sci Rep. 2018;8(1):1–13. doi: 10.1038/s41598-018-22620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A, Barakat K. Applications of computer-aided approaches in the development of hepatitis C antiviral agents. Expert Opin Drug Discov. 2017;12(4):407–425. doi: 10.1080/17460441.2017.1291628. [DOI] [PubMed] [Google Scholar]
- Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020 doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CJ, Tchesnokov EP, Woolner E, Perry JK, Feng JY, Porter DP, Götte M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J Biol Chem. 2020;295(20):6785–6797. doi: 10.1074/jbc.RA120.013679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung AB. In silico structure modelling of SARS-CoV-2 Nsp13 helicase and Nsp14 and repurposing of FDA approved antiviral drugs as dual inhibitors. Gene Rep. 2020;21:100860. doi: 10.1016/j.genrep.2020.100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo YA, Deeks ED. Sofosbuvir/Velpatasvir/Voxilaprevir: a review in chronic hepatitis C. Drugs. 2018;78(5):577–587. doi: 10.1007/s40265-018-0895-5. [DOI] [PubMed] [Google Scholar]
- Hui DS, Azhar EI, Madani TA, Ntoumi F, Koch R, Dar O. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health: the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;91:264–266. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendele L, Krivak R, Skoda P, Novotny M, Hoksza D. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019;47(W1):W345–W349. doi: 10.1093/nar/gkz424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan RJ, Jha RK, Singh E, Jain M, Amera GM, Singh RP, Muthukumaran J, Singh AK. Identification of promising antiviral drug candidates against non-structural protein 15 (NSP15) from SARS-CoV-2: an in silico assisted drug-repurposing study. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1814870. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. 2013;20(23):2839–2860. doi: 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
- Lehmann KC, Gulyaeva A, Zevenhoven-Dobbe JC, Janssen GM, Ruben M, Overkleeft HS, van Veelen PA, Samborskiy DV, Kravchenko AA, Leontovich AM, Sidorov IA. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015;43(17):8416–8434. doi: 10.1093/nar/gkv838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhou Y, Zhang M, Wang H, Zhao Q, Liu J (2020) Updated approaches against SARS-CoV-2. Antimicrob Agents Chemother. 10.1128/AAC.00483-20. [DOI] [PMC free article] [PubMed]
- Lionta E, Spyrou GK, Vassilatis D, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem. 2014;14(16):1923–1938. doi: 10.2174/1568026614666140929124445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Loeffelholz MJ, Tang YW. Laboratory diagnosis of emerging human coronavirus infections—the state of the art. Emerg Microbes Infect. 2020;9(1):747–756. doi: 10.1080/22221751.2020.1745095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdian S, Ebrahim-Habibi A, Zarrabi M. Drug repurposing using computational methods to identify therapeutic options for COVID-19. J Diabetes Metab Disord. 2020 doi: 10.1007/s40200-020-00546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SM. RNA synthetic mechanisms employed by diverse families of RNA viruses. Wiley Interdiscip Rev RNA. 2013;4(4):351–367. doi: 10.1002/wrna.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J Microbiol Immunol Infect. 2020 doi: 10.1016/j.jmii.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH (2020) National Institutes of Health, NIH Clinical Trial of Remdesivir to Treat COVID-19 Begins, Available from: https://www.nih.gov/news-events/news-releases/nih-clinical-trial-remdesivir-treat-covid-19-begins (Accessed on June 4, 2020).
- Rodriguez-Morales AJ, Bonilla-Aldana DK, Balbin-Ramon GJ, Rabaan AA, Sah R, Paniz-Mondolfi A, Pagliano P, Esposito S. History is repeating itself: probable zoonotic spillover as the cause of the 2019 novel coronavirus epidemic. Infez Med. 2020;28(1):3–5. [PubMed] [Google Scholar]
- Singh AK, Singh A, Singh R, Misra A. Remdesivir in COVID-19: a critical review of pharmacology, pre-clinical and clinical studies. Diabetes Metab Syndr. 2020;14(4):641–648. doi: 10.1016/j.dsx.2020.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T, Irwin JJ. ZINC 15—ligand discovery for everyone. J Chem Inf Model. 2015;55(11):2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchesnokov EP, Gordon CJ, Woolner E, Kocincova D, Perry JK, Feng JY, Porter DP, Gotte M. Template-dependent inhibition of coronavirus RNA-dependent RNA polymerase by remdesivir reveals a second mechanism of action. J Biol Chem. 2020 doi: 10.1074/jbc.AC120.015720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V. Denovo designing, retrosynthetic analysis, and combinatorial synthesis of a hybrid antiviral (VTAR-01) to inhibit the interaction of SARS-CoV2 spike glycoprotein with human angiotensin-converting enzyme 2. Biol Open. 2020 doi: 10.1242/bio.054056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman S, Prasad BV, Selvarajan R. RNA dependent RNA polymerases: insights from structure, function and evolution. Viruses. 2018;10(2):76. doi: 10.3390/v10020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2021) Coronavirus disease 2019 (COVID-19) Weekly Epidemiological Update. January 19, 2021. https://www.who.int/publications/m/item/weekly-epidemiological-update---19-january-2021. Accessed 21 Jan 2021
- Wilkins T, Akhtar M, Gititu E, Jalluri C, Ramirez J. Diagnosis and management of hepatitis C. Am Fam Physician. 2015;91(12):835–842. [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, Assempour N (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46(D1):D1074-D1082. 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed]
- Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020 doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z, et al. admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2019;35:1067–1069. doi: 10.1093/bioinformatics/bty707. [DOI] [PubMed] [Google Scholar]
- Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zeuzem S, Foster GR, Wang S, Asatryan A, Gane E, Feld JJ, Asselah T, Bourlière M, Ruane PJ, Wedemeyer H, Pol S. Glecaprevir–pibrentasvir for 8 or 12 weeks in HCV genotype 1 or 3 infection. N Engl J Med. 2018;378(4):354–369. doi: 10.1056/NEJMoa1702417. [DOI] [PubMed] [Google Scholar]